

Abstract
We here review the extraordinary mineralogical properties of green rusts and their naturally occurring form, fougerite, and discuss the pertinence of these properties within the alkaline hydrothermal vent (AHV) hypothesis for life's emergence. We put forward an extended version of the AHV scenario which enhances the conformity between extant life and its earliest progenitor by extensively making use of fougerite's mechanistic and catalytic particularities.
Keywords: emergence of life, alkaline vent hypothesis, fougerite, bioenergetics
1. In lieu of an introduction: eat your soup and keep quiet
Studies into the origin of life have over the last two decades grown into a vast global research machinery predominantly institutionalized under the label ‘astrobiology’. Sifting through the various research projects promoted and funded by these entities, one will invariably come across the theme of ‘prebiotic synthesis of organic molecules' and the firm conviction that once all the simple organic building blocks are present, the progression to living cells will be spontaneous; life from a soup or snow of organics or ‘cyanosulfidic chemistry’ (sic) [1–4] (but see [5]). Just as you only need all the letters of a good research article piled up on your desk. You can then go home and wait until they spontaneously self-assemble into the final manuscript, can't you? Of course, you can't as we all know and you can't because of the second law of thermodynamics. The same is true for the origin of life as tirelessly pointed out by thermodynamically minded scientists for more than half a century [6,7]. Being painfully aware of this basic truth, Jacques Monod concluded that ‘Man is alone in the universe’ [8].
To push the self-assembling article metaphor even further, we can also ask ourselves whether we actually need specific letters. Is not the essence of the article in ideas, experiments and conclusions which may just as well be rendered in Cyrillic, Hanzi, Hiragana, Devanagari or whatever other kinds of writing? Ideas, experiments and conclusions (if appropriate) can certainly be likened to a decrease in entropy in the subsystem of humanity (a generation of a more ordered, less likely state) which obviously needs to be driven by an increase in entropy of a larger system and this increase in entropy is obviously mediated by the eating and breathing scientist, his/her discarded manuscript drafts that litter the screen, pushed out of site in the ‘trash’ or those that lie crumpled in (or in the vicinity of) the waste paper basket. These basic thermodynamic ramifications as applied to life's emergence on our planet have been spelled out in detail in several articles over the last few decades [9–15]. We find the strictures from thermodynamics to be insurmountable and therefore feel obliged to search for reasons for life's emergence in thermodynamic disequilibria and converters thereof [12] rather than in the ways an organic soup may have been generated [1,16–20].
2. The top-down approach: gleaning insights from extant biology
2.1. The driving disequilibrium is electrochemical
As pointed out in the past [10], the environmental thermodynamic disequilibrium permitting life to exist is a redox tension. Electron transfer from reducing environmental compounds (substrates) to oxidizing ones collapses this electrochemical disequilibrium but while doing so generates chemical mass-action disequilibria in the form of high ATP/ADP ratios, life's immediate source of free energy. As evidenced by the universality of ion-motive membrane potentials in all cellular life, this process was almost certainly ‘chemiosmotic’ in the beginning [9] with substrate-level phosphorylation likely emerging later as an adaptation to very low environmental free energy. The precise make-up and functioning of the mechanisms transforming redox free energy into ion-motive force (IMF), i.e. of the disequilibrium converters [12], is what the field of bioenergetics deals with. In the following, we will not consider the final step of this process, that is, IMF-driven ATP synthesis, but will come back to this problem towards the end of this article. Just as is true for the nature of the driving environmental disequilibrium itself, we believe that the processes life performs today cannot be fundamentally different from those operating in the beginning and that we can therefore obtain information on the founding disequilibrium converters from looking at specific bioenergetic processes in extant life.
2.2. The catalysts and disequilibrium converters are inorganic entities
An intriguing message emerging from the ensemble of bioenergetic (free energy converting) reactions is that they are almost exclusively performed by transition metals or clusters thereof harboured as active centres by larger polypeptide scaffolds. Obvious exceptions are quinones and flavins which play pivotal roles in the conservation of redox energy due to their two-electron redox properties. We have speculated in the past that in earliest life, these roles may have been played by the transition metals molybdenum and tungsten [21,22]. Bioenergetics therefore appears indeed to be a job done predominantly by inorganic entities. Extrapolating this empirical fact back to the times of life's emergence and keeping in mind that it is bioenergetics that maintains life within the boundaries of the second law, of course, takes the onus of enabling life off the back of organic molecules. Transition metals have obviously been present on our planet and have been deposited as minerals on its surface since the very beginning. The structural affinities between specific minerals and the reactive metal clusters of certain bioenergetic enzymes are striking and have been discussed before [23,24]. The leads from extant bioenergetics have in the past been interpreted to, in particular, point towards the minerals mackinawite and greigite as potentially important mediators in the transition of inorganic free energy conversion to fully fledged bioenergetics and led to the catchphrase notion of the ‘rocky roots of biochemistry’ [25]. While this image certainly appealed to the geochemists, the physicists and other mineralogists, it now seems to us that it may actually have increased the barrier for biologists to embrace the notion of a thermodynamically driven emergence of life. Is not life all about movements, structural flexibility and fluxes of metabolites? And is not all that antithetical to what one expects to happen on a mineral, solid as a rock?
In this contribution, we will try to convey that rock-solid minerals are only part of the story and that the vastly underestimated soft-matter aspects of certain minerals may provide the missing link between inorganic free energy converting processes and life's foundational bioenergetic mechanisms.
2.3. The top-down lead to green rust
The mineral we will highlight is known to the mineralogists as green rust (GR) and its naturally occurring form as fougerite ([(Fe2+, Mg2+)1−x Fe3+x (OH)2]x+[x/nAn− ·mH2O]x−) [26–29]. There are many avenues leading to the recognition that GR is potentially extremely pertinent to life's emergence within an inorganic context [30]. We will here follow and describe the approach already extensively exploited above which is to retrodict from extant biology likely inorganic precursor catalysts and reactions. The quest for promising types of environmental disequilibria tapped into by nascent life has led some of the authors of this article to envisage hydrothermally exhaled methane as reductant (fuel) in a cascade of redox reactions resembling those found in present-day methanotrophic bacteria [31,32]. The first and chemically most challenging step in this cascade is the oxidation of methane to methanol. In extant life, this reaction is frequently performed by a di-iron enzyme called soluble methane monooxygenase (sMMO) [33]. sMMOs are actually only one small subfamily within the vast superfamily of di-iron enzymes [34]. Di-iron hydrolases are certainly among the most versatile and widely distributed enzymes in all domains of the tree of life, likely even surpassing the already impressive versatility of Mo-bis-pterin enzymes [35,36]. The di-iron centre within its immediate protein environment is depicted in figure 1a. Restricting this view to the first coordination sphere of the iron atoms, we find the structure shown in figure 1b. The octahedral coordination by predominantly oxygen atoms seen for both irons and in particular their bis-μ-hydroxo-bridging is strongly reminiscent of what a di-iron unit (figure 1d) of the crystal lattice in GR (figure 1c) looks like.
Figure 1.
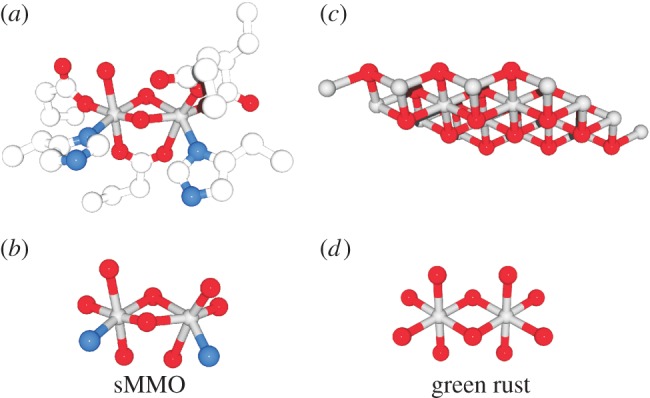
Structural proximity of the active centre in biological di-iron hydrolases. (a) Di-iron centre together with its ligating amino acids in the enzyme sMMO (pdb-entry: 1FZ1). (b) The first coordination sphere of iron atoms in sMMO. (c) Lattice structure of the Fe2+/Fe3+-oxyhydroxide sheet of GR. (d) Individual di-iron site from within (c). Colour coding: grey, iron; red, oxygen; blue, nitrogen; white, carbon. (Online version in colour.)
Synthetic GRs have been studied by the mineralogists at least since Bernal et al. [26], long before the discovery of fougerite which demonstrated that GRs exist naturally in the environment (under appropriate conditions [27]). A plethora of excellent reviews on various forms of GR have been published by these colleagues [37–39]. All these articles discuss the outstanding catalytic versatility of GRs but mainly from a technological or environmental (mostly depollution) perspective. In this contribution, we will therefore focus on the potential relevance of GRs as redox catalysts and disequilibrium converters in life's emergence and for more information of the mineral side of GR, we refer readers to the above cited literature.
3. Green rust, a far shot from your middle-of-the-road mineral
GR crystals do not grow to macroscopic sizes. The mineral is rather composed of hexagonal nanocrystals with typical dimensions as indicated in figure 2b. a-dimensions as low as a few nanometres and as high as 2 µm have been observed while the height (c-dimension) of the nanocrystals is much less variable in the range of 6–40 nm [41]. The aggregate of nanocrystals features a chaotic aspect containing numerous voids, grooves and canyons as seen in the scanning electron microscopic (SEM) image of figure 2a (and see [42]; figures 3–5) resulting in structures with fractal geometry. These aggregates do not form a solid bulk but rather behave like clays. X-ray diffraction (XRD) and Mössbauer experiments have elucidated the atomic structure of single nanocrystals as layered Fe2+/Fe3+ oxyhydroxides (see figures 1c and 2c) with the plane of the layers containing the crystallographic a and b axes (figure 1b). The interstitial space contains counter-anions and water molecules and its height varies between 7.9 Å and 40 Å [39], meaning that a typical nanocrystal on average contains about 20 layers. Two forms have been particularly well characterized by the mineralogists and are dubbed GR1 and GR2 [26,41,45,46]. GR1 harbours mono- and divalent anions with spherical (e.g. Cl−) or ‘flat’ (e.g. carbonate) shapes, while GR2 contains molecules featuring tetrahedral geometry (for example, ). Figure 3a,b compares the differences in interstitial size and arrangement of anions in the interlayer space between the two forms. The wider interstitial space of GR2 is correlated to the fact that two anion planes are superposed in between Fe oxyhydroxide layers (giving rise to three interstitial Miller planes with indices (002) and (003) observable by XRD) rather than only a single anion plane in the middle between layers as observed in GR1. With even bulkier interstitial anions [44], layer spacings of up to 40 Å have been observed (figure 3c).
Figure 2.

The structure of GR. (a) SEM image of GR as synthesized according to [40]. (b) Crystallographic shape of a single GR nanocrystal. (c) Layer structure of GR1 (grey, iron; red, oxygen; green, chloride). The oxyhydroxide layers run parallel to the ab-plane of (b). (Online version in colour.)
Figure 3.

Structural flexibility of GR in response to the chemical nature of interstitial anions. (a) GR1 with a single intercalating plane of anions is formed in the presence of spherical and ‘flat’ anions such as chloride, carbonate and nitrite. The shown structure contains chloride ions marked in green. (b) GR2 features two intercalating planes containing anions of tetragonal geometry such as sulfate (yellow, sulfur). The exact orientation of the sulfate molecules is still controversial with [43] proposing the apical oxygen to point towards an Fe3+ site and [41] considering an inversed configuration where three basal oxygens of sulfate approach hydroxide oxygens surrounding this Fe3+ site. We have chosen to depict the configuration favoured in [43] but hold no opinion as to which geometry is more appropriate. To the considerations outlined in the text, the exact orientation is unimportant and what matters is the double-layered arrangement of the intercalating anions. (c) GR in the presence of alkyl-carboxylates as intercalating anions. Only the layer spacing is specified while the exact arrangement of the alkyl-carboxylate chains has not been reported [44]. (Online version in colour.)
Figure 5.
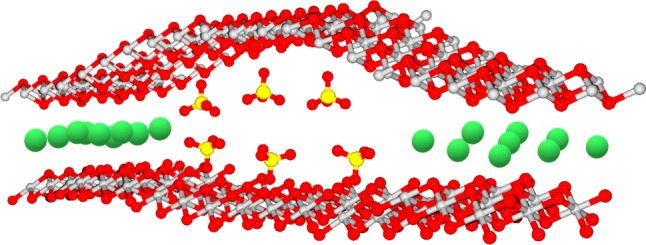
Schematic of the layer and interlayer local geometry in a putative GR1–GR2 mixed mineral. Colour coding as in figures 1 and 2. (Online version in colour.)
Not only can GR swell and shrink in its c-direction in response to different interstitial anions, the iron oxyhydroxide layers do also readily slide with respect to each other. This is nicely demonstrated by the XRD-derived structures of GR1 and GR2 as viewed along a normal to the layers (figure 4). In GR1, individual layers are shifted with respect to each other and only the fourth layer superimposes on the first one (figure 4a). In GR2, by contrast, all layers perfectly superimpose (figure 4b). These arrangements rationalize the fact that the nominal crystallographic unit cell of GR1 is much larger than that of GR2 since it comprises four rather than two layers as is the case for GR2 [41,43,47]. The observation that mineral synthesized as GR1 can be transformed to GR2 [43,48] shows that layer stacking is flexible and that layers can move laterally.
Figure 4.
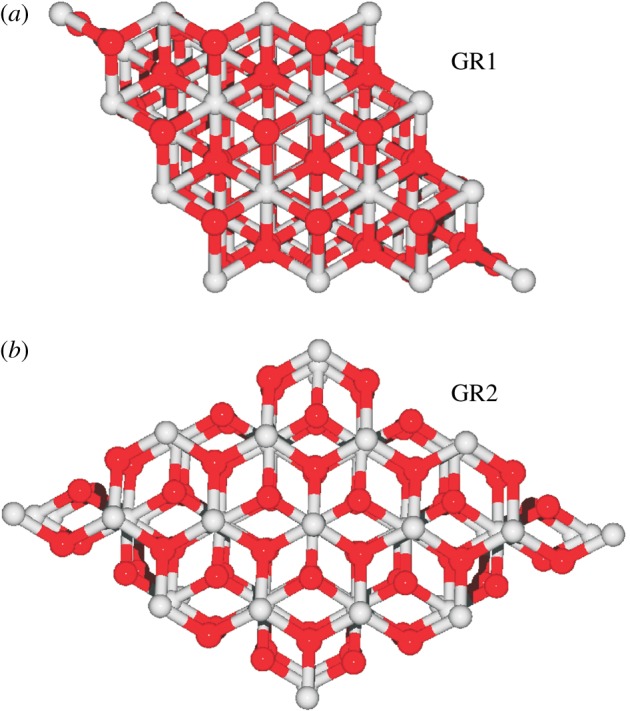
Top view of the iron oxyhydroxide layer arrangement in GR1 (a) and GR2 (b). In GR1, subsequent layers are shifted with respect to each other and only the fourth layer superimposes on the first one, while in all GR2 layers, respective atoms are arranged similarly [41]. (Online version in colour.)
While the endpoints, that is, GR1 and GR2, are thoroughly characterized, the atomic details of interconversions have so far not been addressed. In this context, it seems particularly interesting to us to understand what ‘mixed-state’ GR, e.g. GR1 exposed to varying concentrations of GR2-inducing intercalating anions, such as sulfate, would look like. Would the GR nanocrystals phase-separate into chloride- and sulfate-containing subpopulations or would single nanocrystals contain both anions in the same layer resulting in structures resembling that shown in figure 5? Preliminary results from XRD data on mixed-anion grown crystals indicate that structures as indicated in figure 5 do indeed occur (data to be published in a future paper). As we will discuss below, GR nanocrystals containing high amounts of carbonate (yielding GR1) and substoichiometric concentrations of phosphate (resulting in GR2) would be particularly pertinent within the alkaline hydrothermal vent (AHV) hypothesis for life's emergence. To date, detailed information on phosphate-containing GR is limited. While Hansen & Poulsen [49] have found roughly half of the intercalated sulfate ions replaced by the added phosphate, Ruby et al. [48] reported phosphate ions predominantly accumulating at the lateral faces of GR1 nanocrystals and no significant exchange in the interlayer space. In the so-called ferric GR (see below), by contrast, strong uptake of phosphate ions was observed [50]. In this context, it is noteworthy that phosphate is stereochemically identical to sulfate. Its higher negative charge, however, may require higher oxidation states of GR (i.e. tending towards the ‘ferric’ form) to allow perfect compensation between the charges present in the positive Fe oxyhydroxide layer and the interlayer anions. These considerations obviously apply to the bulk state of the mineral while localized charge unbalance is very likely tolerated in the nanocrystals. The pertinence of these considerations to emergence-of-life scenarios will be discussed below.
The astonishing structural flexibility of GR is on a par with its unparalleled redox versatility. Naturally occurring GR, that is, fougerite, has been shown to be stable at Fe2+ abundancies ranging from 1/3 to 2/3 of total iron (figure 6b,c). At higher values, GR dissolves into soluble iron species while at lower values, it transforms into more rock-like minerals such as magnetite, haematite or lepidocrocite [39]. These transformations, however, occur on timescales of several hours to days. For shorter periods of time, GR has been taken both to the fully reduced (figure 6a) and fully oxidized (‘ferric GR’, figure 6d) states. The change in charge due to variable Fe2+/Fe3+ ratios is compensated by partial deprotonation of the hydroxide bridges in the layers [51]. Conserved features in the XRD diffractograms show that even at these redox extremes, the layered structure is preserved [51–53]. It is noteworthy that hours to days sound like geological timescales to biological redox reactions which occur in the range of micro- to milliseconds.
Figure 6.

Crystallographic distribution of Fe2+ and Fe3+ ions in GRs of varying oxidation states. The long-term stability range for fougerite extends from 1/3 (b) to 2/3 (c) of total iron. The fully oxidized form represented in (d) is frequently referred to as ferric fougerite.
4. Green rust; poor man's palladium
All reviews from the mineralogy community on GR tirelessly emphasize the extraordinary catalytic versatility of this mineral which obviously results from the combination of its structural softness, low water activity in the interlayer space and widely amenable redox properties. Among the most iconic reactions are the eight-electron reduction of nitrate to ammonia [54] and the reduction of nitrite to the gases NO, N2O and N2 [55]. Not only are these reactions of interest to technological applications of GR [39], they also are extremely intriguing to emergence of life research since they represent precisely those catalytic reactions which have been proposed as major players in specific types of primordial metabolism [31,32]. The most pertinent bioenergetic reaction pathway of the oxidation of methane to methanol and on to formaldehyde initiated by activated O2 (in the Archaean possibly derived from disproportionation of two NO molecules) or directly by NO [31] has not been studied in GR so far but is now actively investigated in our laboratories. At any rate, we strongly suspect that the full space of possible coupled processes mediated by GR presently remains widely unexplored.
5. Fougerite, green rust on steroids
Synthetic GRs are surely already astonishingly versatile catalysts. However, for the sake of experimental rigour and accessibility, these synthetic minerals represent relatively ‘pure’ cases in which the mineralogists mainly play with the nature of the intercalating anions. Fougerite, GR's naturally occurring cousin, is not restrained by researchers' quest for simplicity and reproducibility. Fougerite can and does partially replace iron by other cationic metals such as Mg [28,56] and Ni [57]. Since Mg, for example, replaces specifically Fe2+ sites, the effective redox potential of the so-obtained fougerite variants will differ substantially from their Mg-free form [28]. Nickel-containing fougerite is particularly interesting since Ni/Fe sites are involved in biological oxidation of molecular hydrogen, a fact already noted when considering a potential role of the mineral mackinawite in life's emergence [23,58]. The analysis of extant life furthermore suggests a prime importance of the two-electron redox metals molybdenum and tungsten already in early life [36] due to their versatile electrochemical properties [21]. A possible intercalation of molybdate/tungstate and thiomolybdate/thiotungstate ions between iron oxyhydroxide layers is therefore an intriguing possibility which has to our knowledge not yet been assessed experimentally. A plethora of so far unexplored catalytic abilities of ‘chaotically’ constituted fougerite, that is, containing fluctuating mixtures of both transition metals in the layers and anions in the interlayer spaces, therefore seems inevitable to us and the lack of empirical data desperately begs for intense research efforts into fougerite's true versatility.
6. The alkaline vent hypothesis, evolving towards increasing congruency with extant life
The inclusion of fougerite into the AHV hypothesis can substantially diminish the gap between our ideas of the inorganic precursor entity and a living cell. The effluents in AHVs are loaded with the reductants H2 and CH4 [59] while the ocean waters adjacent to the precipitated chimneys may in the Archaean have carried the oxidants and [60]. These same redox substrates are frequently involved in providing free energy to certain prokaryotes. Nitrite may in fact only have been present in low amounts due to reduction by abundant Fe2+. Nitrate, by contrast, is a two-electron redox compound which will be reduced by Fe2+ only extremely slowly due to kinetic inhibition. Fougerite, however, will reduce nitrate through several intermediate redox states to ultimately yield ammonia (figure 7, top interlayer), that is, via processes indeed observed in synthetic GR [61]. A crucial intermediate in this reaction is NO [55] which is required for the activation of methane and eventually will allow its oxidation to methanol, a reaction observed in extant anaerobic methanotrophs but so far untested in GR. The specific reaction we propose [32] will yield NH2, the amino radical (formerly called amidogen [69]), together with methanol.
Figure 7.

Sketch of a fougerite nano- to micro-crystallite situated to hold disequilibria between the chemistries of an alkaline and highly reduced hydrothermal solution against the acidulous carbonic ocean bearing the high potential electron acceptor, nitrate [60]. Fougerite, a redox and physically flexible hydrous mineral, is shown here as a putative combined proto-enzyme (nano-engine) with the capacities of (i) nitrate reductase [46,61], (ii) methane monooxygenase? [31,32], (iii) aminase [62], and (iv) peptidase? [24]. Amyloid peptide comprising a mixture of apolar α-sheets and polar β-sheets is extruded from the hydrous interlayers to form (i) a protoferredoxin (p-fd) [63], (ii) a P-loop (P-l) [64,65], (iii) Ni-tetra-glycine residue nest (Ni-[G]4) [66,67], (iv) ion channels (ion-channel) [64], and ultimately (v) aggregating to form a dielectric and osmotic barrier both encapsulating and embedding fougerite nanocrystals as speculated upon in figure 8 [64,68]. (Online version in colour.)
6.1. Organics from fougerite
Amino radicals are highly reactive and therefore short-lived. They react readily with organic molecules [70] and have been shown to produce amino acids [69] (second interlayer from top in figure 7). Amino acids may furthermore form from ammonia and carboxyls as shown in the third interlayer of figure 7 [62]. Two NH2 radicals at sufficiently high concentrations produce hydrazine (N2H4), a compound which is instrumental in the synthesis of heterocyclic compounds from simple linear hydrocarbons. The bifunctionality conferred by its two amines likely is the key to hydrazine's propensity to form heterocycles such as pyrazoles [71] or imides via the Einhorn–Brunner reaction [72]. Heterocycles obviously play a crucial role in extant life both as fundamental redox molecules (NAD(P), flavins, quinones, etc.) and as nucleobases. The most fundamental organic molecular entities of a living cell involved in both redox metabolism and inheritance may thus have been furnished and self-ordered by the multi-proto-enzyme nanoengines of fougerite at the very emergence of nascent life according to the reactions depicted in figure 7 (cf. [73–79]).
6.2. Towards compartmentation
The simple amino acids produced will have condensed to more or less extended polypeptide chains. Some of these may have started to coordinate clusters of transition metals [80], while the bulk of them likely aggregated as amyloids eventually coating the insides of voids in the bulk mineral of the chimneys and occasionally forming closed vesicles [64] as indicated in figure 8. Amyloid vesicles have been observed in extant life and stand out by their high degree of impermeability to electrical charges. Marine bacteria even manage to render their amyloid vesicles gas-tight by using ‘one of the most hydrophobic proteins known’ [82].
Figure 8.

Cartoon of a ‘community’ of discrete amyloidal globules (protocells) comprising variable valence fougerite (green, brown and red to illustrate the trend towards oxidation), and mackinawite (yellow) as the proto-enzymes, mainly situated at the globule margins. The discrete structures result from hydropathic forces (cf. figure 7 and [81]). (Online version in colour.)
The thickness of the amyloid wall in buoyancy vesicles has been determined at 2 nm [82] which corresponds to about half the thickness of cytoplasmic membranes in living cells. An immediate criticism to our likening of amyloid walls to cellular membranes will be that the latter obviously are lipid-based. We consider the notion of lipidic cytoplasmic membranes resembling the so-called liposomes as fundamentally flawed. The initial ‘fluid mosaic model’ proposed in 1972 [83] imagined a sea of lipid bilayer with an occasional floating membrane–integral protein complex. Over the years, empirical data forced the recognition that membranes are ‘more mosaic than fluid’ [84] due to ‘the high density of proteins in the bilayer, that make the bilayer a molecularly crowded space’ and that ‘hardly leave a fraction of the bilayer unperturbed’ [85]. Recent atomic force microscopy and electron microscopy studies unambiguously show that protein in fact fully dominates the internal volume of both prokaryotic and eukaryotic membrane layers with almost 80% being proteinaceous matter [86,87], while the surface is almost entirely covered by protein [87]. Cell membranes (as well as organellar ones) thus appear to be essentially protein with lipids only serving as lubricants to avoid cloaking of proteins into too large superstructures. We would therefore emphasize that real-life cytoplasmic membranes are indeed much more similar to our amyloid-based vesicles than to the older notion of lipidic membranes.
6.3. …and on to disequilibrium-converting entities
For all we know about amyloid barriers in extant life, such vesicles would likely have been extraordinarily tight both with respect to ions and to electrical charges. This would rapidly insulate the interior from supply of thermodynamic disequilibria, i.e. reductants, oxidants and pH gradients, resulting in all fougerite-, mackinawite- and greigite-mediated reactions to reach equilibrium and hence in still-born nascent life. However, it seems conceivable to us that while the amyloid barrier forms, it would not always grow around mineral nanocrystals but rather embed a few of these within the growing wall. Given the observed size distribution of fougerite nanocrystals and assuming the appropriate orientation for some, these may stick out on both sides of the amyloid wall (some not by much since a-dimensions as low as 5–10 nm have been observed). Those embedded nanocrystals would allow transport of both electrons and ions, yet crucially not in a passive/diffusive way but strongly coupling mass and charge transport due to the spatial correlation of Fe3+ sites and interlayer anions. We emphasize that the idea of an integration of fougerite nanocrystals in the form of electrically gated ion pores into protein or lipid barriers can be tested by experimental approaches. The manifold possible reactions which may occur under the considered conditions within the interlayer channels seem almost unimaginable to us and beg for empirical research on such systems. However, one specific reaction immediately comes to the minds of bioenergeticists: if indeed phosphate can substitute for sulfate in GR2-type interlayers then it seems almost inevitable to us (given the arrangement of the sulfate anions in two superimposed strata of the bipartite GR2 interlayer, figure 6) that the equilibrium between phosphates (Pi) diffusing into the interlayers and their condensed form pyrophosphate (PPi) will be shifted towards PPi as a result of the much lower water activity (PPi + H2O ↔ 2Pi + 2H+) within fougerite when compared with bulk water. In sulfate-containing GR2, the sulfate anions are located beneath Fe3+ sites and the same must be true for the strongly negatively charged Pi and PPi molecules. If we now imagine a current flowing through the Fe oxyhydroxide layers, it will pull the interlayer anions in the same direction as the electron holes within the iron oxyhydroxide layer will move. Which direction may that be? The answer comes straightforwardly from the fact that the oxidants are dissolved salts (i.e. nitrate and nitrite), that is, they will not be able to cross the amyloid barrier apart from going through fougerite pores and, as they do, they will be reduced by Fe2+ in the oxyhydroxide layers. The reductants, by contrast, are gases which will likely diffuse through the amyloid barrier. In this context, it is noteworthy that most gases are about twice as soluble in cytoplasmic membranes of extant organisms than in the peri- and the cytoplasm and these membranes therefore are permeable to gaseous molecules just as we propose for the amyloid walls. There will therefore necessarily be a net excess of reducing equivalents in the interior of the amyloid vesicles which will tend to flow outwards, implying that the positively charged electrons–holes will go inwards—and pull the out-of-bulk-water-equilibrium PPi/Pi mixture with it. Since PPi is metastable of the order of tens to hundreds of hours in aqueous solutions [88], the vesicle interior will thus be supplied with a chemical (group-transfer) disequilibrium. Both the mechanism and the outcome of these reactions are eerily reminiscent of what the enzyme H+-translocating pyrophosphatase does and is for in many extant prokaryotic and eukaryotic cells [89,90].
7. Conclusion
Vitalism, the attribution of complexity- and eventually life-generating properties to organic molecules, although almost unanimously abandoned almost a hundred years ago [91], is in fact alive and well and thriving in the plethora of organic soup hypotheses dominating the origin-of-life paradigm to the present day [13,14]. However, we feel that a variant of vitalism even haunts those metabolism-first-type approaches which imagine life's beginning to be very different from what it is now and that, once a primitive but somewhat alien type of metabolism was established, this nascent life would ‘want’ to explore all options and finally evolve into present-day kinds of cellular organisms. We do not share this view but hold that a ‘desire to become something’ is an emergent phenomenon contingent on the appearance of complex, neuron-containing (sentient) life over the last few hundred million years. At life's emergence and for a long period of time, life was force-fed by environmental sources of free energy and only could adapt to toxic and/or stressful conditions or to tolerate environmental perturbations via happenstance mutational events. To our minds, this implies that nascent life could not have been ‘as we don't know it’ [92], but that there must be a continuous and contiguous stem of features characterizing life from its earliest beginnings to the present day.
The scenario outlined above intends to progress one step further in the direction of deriving a potential pathway producing life which was, in the beginning, as we know it now. The inclusion of the mineral fougerite in our considerations is crucial for being able to make this step. That fougerite, already a self-assembled highly complex but also highly flexible and malleable redox-active mineral containing bound water in its interlayers, may have been instrumental in the emergence of life, would go a long way to easing the pain of those that find even the complexity of a single cell unnerving. Many aspects of our scenario are open to empirical research and possibly falsification.
Acknowledgements
S.D. and W.N. thank Vasile Heresanu (CINaM/Luminy France) for performing XRD experiments on diverse samples of GR and M.J.R. thanks James Milner-White (Glasgow/UK) for discussions on abiotic peptides.
Data accessibility
This article has no additional data.
Competing interests
We declare we have no competing interests.
Funding
This work was supported by the CNRS (Défi Origines, grant SIAM) and by the NASA Astrobiological Institute under agreement no. NNH13ZDA017C (Icy Worlds).
References
- 1.Haldane JBS. 1929. The origin of life. Rationalist Annual 3, 3–10. [Google Scholar]
- 2.Miller SL. 1987. Which organic compounds could have occurred on the prebiotic earth? In Cold Spring Harbor symposia on quantitative biology, vol. 52, pp. 17–27. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [DOI] [PubMed] [Google Scholar]
- 3.Szostak JW. 2009. Origins of life: systems chemistry on early Earth. Nature 459, 171–172. ( 10.1038/459171a) [DOI] [PubMed] [Google Scholar]
- 4.Sutherland JD. 2017. Opinion: studies on the origin of life—the end of the beginning. Nat. Rev. Chem. 1, 0012 ( 10.1038/s41570-016-0012) [DOI] [Google Scholar]
- 5.Christie A. 1945. Sparkling cyanide. London, UK: Collins Crime Club. [Google Scholar]
- 6.Schrödinger E. 1944. What is life? The physical aspect of the living cell. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 7.Nicolis G, Prigogine Y. 1989. Exploring complexity. New York, NY: St Martin's Press. [Google Scholar]
- 8.Monod J. 1974. On chance and necessity. In Studies in the philosophy of biology, pp. 357–375. London, UK: Palgrave. [Google Scholar]
- 9.Lane N, Allen JF, Martin W. 2010. How did LUCA make a living? Chemiosmosis in the origin of life. Bioessays 32, 271–280. ( 10.1002/bies.200900131) [DOI] [PubMed] [Google Scholar]
- 10.Schoepp-Cothenet B, et al. 2013. On the universal core of bioenergetics. Biochim. Biophys. Acta Bioenergetics 1827, 79–93. ( 10.1016/j.bbabio.2012.09.005) [DOI] [PubMed] [Google Scholar]
- 11.Ducluzeau A-L, Schoepp-Cothenet B, Baymann F, Russell MJ, Nitschke W. 2014. Free energy conversion in the LUCA: quo vadis? Biochim. Biophys. Acta Bioenergetics 1837, 982–988. ( 10.1016/j.bbabio.2013.12.005) [DOI] [PubMed] [Google Scholar]
- 12.Branscomb E, Biancalani T, Goldenfeld N, Russell MJ. 2017. Escapement mechanisms and the conversion of disequilibria: the engines of creation. Phys. Rep. 677, 1–60. ( 10.1016/j.physrep.2017.02.001) [DOI] [Google Scholar]
- 13.Branscomb E, Russell MJ. 2018. Frankenstein or a submarine alkaline vent: who is responsible for abiogenesis? Part 1: what is life—that it might create itself? Bioessays 40, 1700179 ( 10.1002/bies.201700179) [DOI] [PubMed] [Google Scholar]
- 14.Branscomb E, Russell MJ. 2018. Frankenstein or a submarine alkaline vent: who is responsible for abiogenesis? Part 2: as life is now, so it must have been in the beginning. Bioessays 40, 1700182 ( 10.1002/bies.201700182) [DOI] [PubMed] [Google Scholar]
- 15.Branscomb E, Russell MJ. 2018. Why the submarine alkaline vent is the most reasonable explanation for the emergence of life. Bioessays 41, 1800208 ( 10.1002/bies.201800208) [DOI] [PubMed] [Google Scholar]
- 16.Oparin AI. 1938. Origin of life. New York, NY: McMillan. [Google Scholar]
- 17.Levy M, Miller SL. 1998. The stability of the RNA bases: implications for the origin of life. Proc. Natl Acad. Sci. USA 95, 7933–7938. ( 10.1073/pnas.95.14.7933) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bada JL. 2004. How life began on Earth: a status report. Earth Planet. Sci. Lett. 226, 1–15. ( 10.1016/j.epsl.2004.07.036) [DOI] [Google Scholar]
- 19.Damer B, Deamer D. 2015. Coupled phases and combinatorial selection in fluctuating hydrothermal pools: a scenario to guide experimental approaches to the origin of cellular life. Life 5, 872–887. ( 10.3390/life5010872) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Kranendonk MJ, Deamer DW, Djokic T. 2017. Life springs. Sci. Am. 317, 28–35. ( 10.1038/scientificamerican0817-28) [DOI] [PubMed] [Google Scholar]
- 21.Duval S, Santini JM, Lemaire D, Chaspoul F, Russell MJ, Grimaldi S, Nitschke W, Schoepp-Cothenet B. 2016. The H-bond network surrounding the pyranopterins modulates redox cooperativity in the molybdenum-bisPGD cofactor in arsenite oxidase. Biochim. Biophys. Acta Bioenergetics 1857, 1353–1362. ( 10.1016/j.bbabio.2016.05.003) [DOI] [PubMed] [Google Scholar]
- 22.Baymann F, Schoepp-Cothenet B, Duval S, Guiral M, Brugna M, Baffert C, Russell MJ, Nitschke W. 2018. On the natural history of flavin-based electron bifurcation. Front. Microbiol. 9, 1357 ( 10.3389/fmicb.2018.01357) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nitschke W, McGlynn SE, Milner-White EJ, Russell MJ. 2013. On the antiquity of metalloenzymes and their substrates in bioenergetics. Biochim. Biophys. Acta Bioenergetics 1827, 871–881. ( 10.1016/j.bbabio.2013.02.008) [DOI] [PubMed] [Google Scholar]
- 24.Russell MJ. 2018. Green rust: the simple organizing ‘seed’ of all life? Life 8, 35 ( 10.3390/life8030035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell MJ, Martin W. 2004. The rocky roots of the acetyl coenzyme-A pathway. Trends Biochem. Sci. 24, 358–363. ( 10.1016/j.tibs.2004.05.007) [DOI] [PubMed] [Google Scholar]
- 26.Bernal JD, Dasgupta DR, Mackay AL. 1959. The oxides and hydroxides of iron and their structural inter-relationships. Clay Min. Bull. 4, 15–30. ( 10.1180/claymin.1959.004.21.02) [DOI] [Google Scholar]
- 27.Trolard F, Abdelmoula M, Bourrié G, Humbert B, Génin J-MR. 1996. Evidence of the occurrence of a ‘Green Rusts’ component in hydromorphic soils. Proposition of the existence of a new mineral: ‘Fougerite’. C. R. Acad. Sci. Ser. II 323, 1015–1022. [Google Scholar]
- 28.Bourrié G, Trolard F, Refait P, Feder F. 2004. A solid-solution model for Fe (II)-Fe (III)-Mg (II) green rusts and fougerite and estimation of their Gibbs free energies of formation. Clays Clay Miner. 52, 382–394. ( 10.1346/CCMN.2004.0520313) [DOI] [Google Scholar]
- 29.Trolard F, Bourrié G, Abdelmoula M, Refait P, Feder F. 2007. Fougerite, a new mineral of the pyroaurite–iowaite group: description and crystal structure. Clays Clay Miner. 55, 323–334. ( 10.1346/CCMN.2007.0550308) [DOI] [Google Scholar]
- 30.Arrhenius GO. 2003. Crystals and life. Helv. Chim. Acta 86, 1569–1586. ( 10.1002/hlca.200390135) [DOI] [Google Scholar]
- 31.Nitschke W, Russell MJ. 2013. Beating the acetyl coenzyme-A pathway to the origin of life. Phil. Trans. R. Soc. B 368, 20120258 ( 10.1098/rstb.2012.0258) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell MJ, Nitschke W. 2017. Methane: fuel or exhaust at the emergence of life. Astrobiology 17, 1053–1066. ( 10.1089/ast.2016.1599) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tinberg CE, Lippard SJ. 2011. Dioxygen activation in soluble methane monooxygenase. Acc. Chem. Res. 44, 280–288. ( 10.1021/ar1001473) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janiewski AJ, Que L Jr. 2018. Dioxygen activation by nonheme diiron enzymes: diverse dioxygen adducts, high-valent intermediates, and related model complexes. Chem. Rev. 118, 2554–2592. ( 10.1021/acs.chemrev.7b00457) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarz G, Mendel RR, Ribbe MW. 2009. Molybdenum cofactors, enzymes and pathways. Nature 460, 839–847. ( 10.1038/nature08302) [DOI] [PubMed] [Google Scholar]
- 36.Schoepp-Cothenet B, van Lis R, Philippot P, Magalon A, Russell MJ, Nitschke W. 2012. The ineluctable requirement for the trans-iron elements molybdenum and/or tungsten in the origin of life. Sci. Rep. 2, 263 ( 10.1038/srep00263) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mills SJ, Christy AG, Génin J-MR, Kameda T, Colombo F. 2012. Nomenclature of the hydrotalcite supergroup: natural layered double hydroxides. Mineral. Mag. 76, 1289–1336. ( 10.1180/minmag.2012.076.5.10) [DOI] [Google Scholar]
- 38.Ruby C, Usman M, Naille S, Hanna K, Carteret C, Mullet M, François M, Abdelmoula M. 2010. Synthesis and transformation of iron-based layered double hydroxides. Appl. Clay Sci. 48, 195–202. ( 10.1016/j.clay.2009.11.017) [DOI] [Google Scholar]
- 39.Usman M, Byrne JM, Chaudhary A, Orsetti S, Hanna K, Ruby C, Kappler A, Haderlein SB. 2018. Magnetite and green rust: synthesis, properties, and environmental applications of mixed-valent iron minerals. Chem. Rev. 118, 3251–3304. ( 10.1021/acs.chemrev.7b00224) [DOI] [PubMed] [Google Scholar]
- 40.Géhin A, Ruby C, Abdelmoula M, Benali O, Ghanbaja J, Ph R, Génin J-MR. 2002. Synthesis of Fe(II-III) hydroxysulphate green rust by coprecipitation. Solid State Sci. 4, 61–66. ( 10.1016/S1293-2558(01)01219-5) [DOI] [Google Scholar]
- 41.Christiansen BC, Balic-Zunic T, Petit P-O, Frandsen C, Mørup S, Geckeis H, Katerinopoulou A, Svane Stipp SL. 2009. Composition and structure of an iron-bearing, layered double hydroxide (LDH)—green rust sodium sulphate. Geochim. Cosmochim. Acta 73, 3579–3592. ( 10.1016/j.gca.2009.03.032) [DOI] [Google Scholar]
- 42.Mielke RE, Robinson KJ, White LM, McGlynn SE, McEachern KR, Kanik I, Russell MJ. 2011. Iron-sulfide-bearing chimneys as potential catalytic energy traps at life's emergence. Astrobiology 11, 933–950. ( 10.1089/ast.2011.0667) [DOI] [PubMed] [Google Scholar]
- 43.Simon L, François M, Refait P, Renaudin G, Lelaurain M, Génin J-MR. 2003. Structure of the Fe(II-III) layered double hydroxysulphate green rust two from Rietveld analysis. Solid State Sci. 5, 327–334. ( 10.1016/S1293-2558(02)00019-5) [DOI] [Google Scholar]
- 44.Ayala-Luis KB, Koch CB, Hansen HCB. 2010. One-pot synthesis and characterization of FeII–FeIII hydroxide (green rust) intercalated with C9–C14 linear alkyl carboxylates. Appl. Clay Sci. 50, 512–519. ( 10.1016/j.clay.2010.10.002) [DOI] [Google Scholar]
- 45.Génin J-MR, Refait P, Bourrié G, Abdelmoula M, Trolard F. 2001. Structure and stability of the Fe(II)–Fe(III) green rust ‘fougerite’ mineral and its potential for reducing pollutants in soil solutions. Appl. Geochem. 16, 559–570. ( 10.1016/S0883-2927(00)00043-3) [DOI] [Google Scholar]
- 46.Génin J-MR, Renard A, Ruby C. 2008. Fougérite FeII − III oxyhydroxycarbonate in environmental chemistry and nitrate reduction. Hyperfine Interact. 186, 31–37. ( 10.1007/s10751-008-9837-z) [DOI] [Google Scholar]
- 47.Génin J-MR, Aïssa R, Géhin A, Abdelmoula M, Benali O, Ernstsen V, Ona-Nguema G, Upadhyay C, Ruby C. 2005. Fougerite and FeII–III hydroxycarbonate green rust; ordering, deprotonation and/or cation substitution; structure of hydrotalcite-like compounds and mythic ferrosic hydroxide Fe(OH)(2+x). Solid State Sci. 7, 545–572. ( 10.1016/j.solidstatesciences.2005.02.001) [DOI] [Google Scholar]
- 48.Ruby C, Géhin A, Aissa R, Ghanbaja J, Abdelmoula M, Génin J-MR. 2006. Chemical stability of hydroxysulphate green rust synthetised in the presence of foreign anions: carbonate, phosphate and silicate. Hyperfine Interact. 167, 803–807. ( 10.1007/s10751-006-9361-y) [DOI] [Google Scholar]
- 49.Hansen H-CB, Poulsen IF. 1999. Interaction of synthetic sulphate ‘Green Rust’ with phosphate and the crystallization of vivianite. Clays Clay Miner. 47, 312–318. [Google Scholar]
- 50.Barthélémy K, Naille S, Despas C, Ruby C, Mallet M. 2012. Carbonated ferric green rust as a new material for efficient phosphate removal. J. Colloid Interface Sci. 384, 121–127. ( 10.1016/j.jcis.2012.06.038) [DOI] [PubMed] [Google Scholar]
- 51.Ph R, Benali O, Abdelmoula M, Génin J-MR. 2003. Formation of ferric green rust and/or ferrihydrite by fast oxidation of iron(II–III) hydroxychloride green rust. Corros. Sci. 45, 2435–2449. ( 10.1016/S0010-938X(03)00073-8) [DOI] [Google Scholar]
- 52.Génin J-MR, Ruby C, Upadhyay C. 2006. Structure and thermodynamics of ferrous, stoichiometric and ferric oxyhydroxycarbonate green rusts; redox flexibility and fougerite mineral. Solid State Sci. 8, 1330–1343. ( 10.1016/j.solidstatesciences.2006.05.010) [DOI] [Google Scholar]
- 53.Antony H, Legrand L, Chaussé A. 2008. Carbonate and sulphate green rusts—mechanisms of oxidation and reduction. Electrochim. Acta 53, 7146–7156. ( 10.1016/j.electacta.2008.05.008) [DOI] [Google Scholar]
- 54.Hansen HCB, Koch CB, Nancke-Krogh H, Borggaard OK, Sørensen J. 1996. Abiotic nitrate reduction to ammonium: key role of green rust. Environ. Sci. Technol. 30, 2053–2056. ( 10.1021/es950844w) [DOI] [Google Scholar]
- 55.Guerbois D, Ona-Nguema G, Morin G, Abdelmoula M, Laverman AM, Mouchel JM, Barthelemy K, Maillot F, Brest J. 2014. Nitrite reduction by biogenic hydroxycarbonate green rusts: evidence for hydroxy-nitrite green rust formation as an intermediate reaction product. Environ. Sci. Technol. 48, 4505–4514. ( 10.1021/es404009k) [DOI] [PubMed] [Google Scholar]
- 56.Refait P, Abdelmoula M, Trolard F, Génin J-MR, Ehrhardt JJ, Bourrié G. 2001. Mössbauer and XAS study of a green rust mineral; the partial substitution of Fe2+ by Mg2+. Am. Mineral. 86, 731–739. ( 10.2138/am-2001-5-613) [DOI] [Google Scholar]
- 57.Chaves LHG, Curry JE, Stone DA, Carducci MD, Chorover J. 2009. Nickel incorporation in Fe(II,III) hydroxysulfate green rust: effect on crystal lattice spacing and oxidation products. Rev. Bras. Cienc. Solo 33, 1115–1123. ( 10.1590/S0100-06832009000500005) [DOI] [Google Scholar]
- 58.Russell MJ, Nitschke W, Branscomb E. 2013. The inevitable journey to being. Phil. Trans. R. Soc. B 368, 20120254 ( 10.1098/rstb.2012.0254) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Proskurowski G, Lilley MD, Seewald JS, Früh-Green GL, Olson EJ, Lupton JE, Sylva SP, Kelley DS. 2008. Abiogenic hydrocarbon production at lost city hydrothermal field. Science 319, 604–607. ( 10.1126/science.1151194) [DOI] [PubMed] [Google Scholar]
- 60.Wong ML, Charnay BD, Gao P, Yung YL, Russell MJ. 2017. Nitrogen oxides in early Earth's atmosphere as electron acceptors for life's emergence. Astrobiology 17, 975–983. ( 10.1089/ast.2016.1473) [DOI] [PubMed] [Google Scholar]
- 61.Hansen HCB, Guldberg S, Erbs M, Koch CB. 2001. Kinetics of nitrate reduction by green rusts—effects of interlayer anion and Fe (II): Fe (III) ratio. Appl. Clay Sci. 18, 81–91. ( 10.1016/S0169-1317(00)00029-6) [DOI] [Google Scholar]
- 62.Barge LM, Flores E, Baum MM, VanderVelde DG, Russell MJ. 2019. Redox and pH gradients drive amino acid synthesis in iron oxyhydroxide mineral systems. Proc. Natl Acad. Sci. USA 116, 4828–4833. ( 10.1073/pnas.1812098116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Milner-White EJ, Russell MJ. 2005. Nests as sites for phosphates and iron-sulfur thiolates in the first membranes: 3 to 6 residue anion-binding motifs. Orig. Life Evol. Biosph. 35, 19–27. ( 10.1007/s11084-005-4582-7) [DOI] [PubMed] [Google Scholar]
- 64.Milner-White EJ, Russell MJ. 2008. Predicting the conformations of peptides and proteins in early evolution. Biol. Direct 3, 3 ( 10.1186/1745-6150-3-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bianchi A, Giorgi C, Ruzza P, Toniolo C, Milner-White EJ. 2012. A synthetic hexapeptide designed to resemble a proteinaceous P-loop nest is shown to bind inorganic phosphate. Proteins 80, 1418–1424. ( 10.1002/prot.24038) [DOI] [PubMed] [Google Scholar]
- 66.Martin RB, Chamberlin M, Edsal JT. 1960. The association of nickel(II) ion with peptides. J. Am. Chem. Soc. 82, 495–498. ( 10.1021/ja01487a064) [DOI] [Google Scholar]
- 67.Ma NWH, White DA, Martin RB. 1967. Metal ion exchange of square-planar nickel(II) tetraglycine with poly-dentate amines. Inorg. Chem. 6, 1632–1636. ( 10.1021/ic50055a004) [DOI] [Google Scholar]
- 68.Zhang S, Holmes T, Lockshin C, Rich A. 1993. Spontaneous assembly of a self-complimentary oligopeptide to form a stable macroscopic membrane. Proc. Natl Acad. Sci. USA 90, 3334–3338. ( 10.1073/pnas.90.8.3334) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cook RL, Sammells AF. 1989. Electrochemical amination reaction for amino acid synthesis. J. Electrochem. Soc. 136, 1845–1846. ( 10.1149/1.2097048) [DOI] [Google Scholar]
- 70.Danen WC, Neugebauer FA. 1975. Aminyl free radicals. Angew. Chem. Int. Ed. 14, 783–789. ( 10.1002/anie.197507831) [DOI] [Google Scholar]
- 71.Wiley RH, Hexner PE. 1951. 3,5-Dimethylpyrazole. Org. Synth. 31, 43 ( 10.15227/orgsyn.031.0043) [DOI] [Google Scholar]
- 72.Einhorn A, Bischkopff E, Szelinski B, Schupp G, Spröngerts E, Ladisch C, Mauermayer T. 1905. Über die N-Methylolverbindungen der Säureamide. Justus Liebig's Annalen der Chemie 343, 207–305. ( 10.1002/jlac.19053430207) [DOI] [Google Scholar]
- 73.Sowerby SJ, Heckl WM. 1998. The role of self-assembled monolayers of the purine and pyrimidine in the emergence of life. Orig. Life Evol. Biosph. 28, 283–310. ( 10.1023/A:1006570726326) [DOI] [PubMed] [Google Scholar]
- 74.Russell MJ, Hall AJ, Mellersh AR. 2003. On the dissipation of thermal and chemical energies on the early Earth: the onsets of hydrothermal convection, chemiosmosis, genetically regulated metabolism and oxygenic photosynthesis. In Natural and laboratory-simulated thermal geochemical processes (ed. Ikan R.), pp. 325–388. Dordrecht, The Netherlands: Kluwer Academic Publishers. [Google Scholar]
- 75.Carny O, Gazit E. 2005. A model for the role of short self-assembled peptides in the very early stages of the origin of life. FASEB J. 19, 1051–1055. ( 10.1096/fj.04-3256hyp) [DOI] [PubMed] [Google Scholar]
- 76.Yarus M. 2011. Getting past the RNA-world: the initial Darwinian ancestor. Cold Spring Harb. Perspect. Biol. 3, a003590 ( 10.110/cshperspect.a003590) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yarus M. 2017. The genetic code and RNA-amino acid affinities. Life 7, 13 ( 10.3390/life7020013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aldersley MF, Joshi PC, Price JD, Ferris JP. 2011. The role of montmorillonite in its catalysis of RNA synthesis. Appl. Clay Sci. 54, 1–14. ( 10.1016/j.clay.2011.06.011) [DOI] [Google Scholar]
- 79.Biondi E, Furukawa Y, Kawai J, Benner SA. 2017. Adsorption of RNA on mineral surfaces and mineral precipitates. Beilstein J. Org. Chem. 13, 393–404. ( 10.3762/bjoc.13.42) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Milner-White EJ, Russell MJ. 2011. A peptide era heralding the emergence of life. Genes 2, 671–688. ( 10.3390/genes2040671) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Phillips JC. 2014. Fractals and self-organized criticality in proteins. Physica A 415, 440–448. ( 10.1016/j.physa.2014.08.034) [DOI] [Google Scholar]
- 82.Bayro MJ, Daviso E, Belenky M, Griffin RG, Herzfeld J. 2008. An amyloid organelle, solid-state NMR evidence for cross-assembly of gas vesicles. J. Biol. Chem. 287, 3479–3484. ( 10.1074/jbc.M111.313049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Singer SJ, Nicolson GL. 1972. The fluid mosaic model of the structure of cell membranes. Science 175, 720–731. ( 10.1126/science.175.4023.720) [DOI] [PubMed] [Google Scholar]
- 84.Engelman DM. 2005. Membranes are more mosaic than fluid. Nature 438, 578–580. ( 10.1038/nature04394) [DOI] [PubMed] [Google Scholar]
- 85.Goni FM. 2014. The basic structure and dynamics of cell membranes: an update of the Singer-Nicolson model. Biochim. Biophys. Acta 1838, 1467–1476. ( 10.1016/j.bbamem.2014.01.006) [DOI] [PubMed] [Google Scholar]
- 86.Kumar S, Cartron ML, Mullin N, Qian P, Leggett GJ, Hunter CN, Hobbs JK. 2017. Direct imaging of protein organization in an intact bacterial organelle using high resolution atomic force microscopy. ACS Nano 11, 126–133. ( 10.1021/acsnano.6b05647) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Casella S, Huang F, Mason D, Zhao G-Y, Johnson GN, Mullineaux CW, Liu L-N. 2017. Dissecting the native architecture of cyanobacteria photosynthetic machinery. Mol. Plant 10, 1434–1448. ( 10.1016/j.molp.2017.09.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.vanWazer JR, Griffith EJ, McCullough JF. 1955. Structure and properties of the condensed phosphates. Hydrolytic degradation of pyro- and tripolyphosphates. J. Am. Chem. Soc. 77, 287–291. ( 10.1021/ja01607a011) [DOI] [Google Scholar]
- 89.Baltscheffsky M, Schultz A, Baltscheffsky H. 1999. H+-PPases: a tightly membrane-bound family. FEBS Lett. 457, 527–533. ( 10.1016/S0014-5793(99)90617-8) [DOI] [PubMed] [Google Scholar]
- 90.Tsai J-Y, Tang K-Z, Li K-M, Hsu B-L, Chiang Y-W, Goldman A, Sun Y-J. 2019. Roles of the hydrophobic gate and exit channel in Vigna radiata pyrophosphatase ion translocation. J. Mol. Biol. 431, 1619–1632. ( 10.1016/j.jmb.2019.03.009) [DOI] [PubMed] [Google Scholar]
- 91.Mayr E. 2010. The decline of vitalism. In The nature of life: classical and contemporary perspectives from philosophy and science (eds Bedau MA, Cleland CE), pp. 93–95. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 92.Wächtershäuser G. 2000. Life as we don't know it. Science 289, 1307–1308. ( 10.1126/science.289.5483.1307) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.