

Abstract
This protocol describes how to prepare mouse brain tissue for quantification of multiple inflammatory mediators using a multiplex bead-based immunoassay. It is important to have methods that allow quantification of multiple analytes from small amounts of tissue. Bio-Plex is a Luminex xMAP-based multiplex bead-based immunoassay technology that permits simultaneous analysis of up to 100 analytes from a single tissue sample. This assay has been used extensively to investigate analytes in plasma and serum samples as well as cultured and primary cells. Here, we describe a method for simultaneous analysis of 33 different inflammatory cytokines and chemokines from mouse brain tissue using the Bio-Plex Pro Mouse Chemokine Panel 33-Plex.
Keywords: Bio-Plex, Mouse brain, Cerebral Malaria, Plasmodium berghei, Chemokine, Cytokine
1. Introduction
1.1. Protocol Overview
Since the discovery of the first cytokine, interferon-alpha, more than 60 years ago [1], >300 additional cytokines, chemokines, and growth factors have been discovered and studied extensively [2]. However, the complexities of intracellular and extracellular signaling networks generated by these immune mediators are not entirely understood. It is therefore important to conduct more in-depth research into how these mediators function, especially in combination, under both steady state and inflammatory conditions. Simultaneous analysis of multiple immune mediators offers a more comprehensive understanding of disease processes, immune responses, and therapeutic interventions [3,4,5]. This type of analysis is now feasible with commercially available multiplex cytokine and chemokine assays. These assays allow researchers to analyze an entire network of cytokines / chemokines in a single sample, which conserves tissue, streamlines workflows, and accelerates research.
Another advantage of multiplex assays is evident when tissue samples are limited, such as when working with small animal models. Use of a conventional ELISA-based approach to analyze multiple immune mediators one-by-one requires large sample volumes. These large samples are often not available when processing small tissues or specific anatomical regions within a tissue. It is therefore important to have methods that allow simultaneous analysis of immune networks with high reproducibility using small sample volumes. Multiplex immunoassays offer a solution to this problem by surveying dozens of analytes simultaneously. These assays can be used to quantify analytes in plasma / serum samples, cultured / primary cells, tissue samples, cerebrospinal fluid, saliva, and sputum, among others. In addition to saving time and money, multiplex immunoassays help advance vaccine development, drug discovery, basic research, and clinical trials by providing a quantitative snapshot of immune mediators expressed in samples of interest [6].
1.2. Advantages and challenges of the protocol.
Multiplex ELISA platforms can measure various protein quantities from a single, small volume of sample. Here, we describe the analysis of 33 different cytokines and chemokines from inflamed brain tissue harvested from mice infected with the parasite Plasmodium berghei – a pathogen that induces a disease referred to as cerebral malaria. While we focus here on the preparation of mouse brain tissue samples, we anticipate that this protocol can be utilized for the preparation of other samples and tissue types.
Proper planning and timing are crucial for the proper execution of this protocol. For optimal results, it is important to plan and allow sufficient time to perform instrument validation / calibration, design plate layouts, and perform mixing / dispensing steps with precision. We cannot overstate the importance of using calibrated pipettors (preferably multichannel) when dispensing the small volumes required for this assay.
2. Before you begin running the assay
-
2.1.High-Level Workflow and Reagents Needs Overview:
-
2.1.1.Add 50 μl 1x beads to wells
-
2.1.2.Wash buffer: 2 x 100 μl
-
2.1.3.Add 50 μl standards, samples and controls; incubate on shaker at 850 rpm for 30 min
-
2.1.4.Wash buffer: 3 x 100 μl
-
2.1.5.Add 25 μl 1x detection antibody; incubate on shaker at 850 rpm for 30 min
-
2.1.6.Wash buffer: 3 x 100 μl
-
2.1.7.Add 50 μl 1x streptavidin-PE; incubate on shaker at 850 rpm for 10 min
-
2.1.8.Wash buffer: 3 x 100 μl
-
2.1.9.Resuspend in 125 μl assay buffer; shake for 30 seconds
-
2.1.10.Acquire data on Bio-Plex system.
-
2.1.1.
-
2.2.Plan the Plate Layout.
-
2.2.1.A standard plate layout can be set-up as follows, which allows 39 samples in duplicate:
-
2.2.1.
-
2.3.Instrument Validation and Calibration
-
2.3.1.Check Sheath Fluid.
-
2.3.1.1.Ensure sufficient volume of approximately 1 liter per assay.
-
2.3.1.1.
-
2.3.2.Bio-Plex 200 Instrument Validation.
-
2.3.2.1.Run the Bio-Rad Validation Kit 4.0 monthly.
-
2.3.2.1.
-
2.3.3.Turn on the Bio-Plex 200 and allow the laser to warm up at least 30 minutes before performing any readings.
-
2.3.4.Bio-Plex 200 Instrument Calibration.
-
2.3.4.1.Run the Bio-Rad Calibration Kit daily. Allow for approximately 30 minutes to run the Calibration Kit.
-
2.3.4.1.
-
2.3.1.
-
2.4.Bio-Plex Pro Wash Station Setup and Preparation.
-
2.4.1.Prepare Wash Solution.
-
2.4.1.1.The Bio-Plex Wash Buffer is supplied at 10x.
-
2.4.1.2.Dilute 60 ml of the 10x wash buffer with 540 ml of deionized water.
-
2.4.1.1.
-
2.4.2.Prepare Wash Station.
-
2.4.2.1.Fill Liquid Bottle 1 with 600 ml of 1x Bio-Plex wash buffer.
-
2.4.2.2.Fill Liquid Bottle 2 with 600 ml of deionized water.
-
2.4.2.3.Empty Waste Bottle if necessary.
-
2.4.2.4.Prime Channel 1.
-
2.4.2.1.
-
2.4.1.
3. Materials and Methods
-
3.1.Mouse Treatment
-
3.1.1.Adult 8-week-old C57BL/6J (B6) mice used in this study were purchased from the Jackson Laboratories (Bar Harbor, ME). Plasmodium berghei ANKA (PbA) was maintained as previously reported [7]. Animals were infected intraperitoneally with 106 parasitized red blood cells. Parasitemia in each animal was measured by staining 1 μl of blood with Hoechst (1:1000) as previously described [7].
-
3.1.2.To deplete CD8+ T cells, mice were injected intraperitoneally with 500 μg of anti-CD8 depleting antibody (clone: YTS 169.4; BioXcell) prior to infection with PbA.
-
3.1.3.On day 6 post-infection, mice received an intracardiac perfusion with saline. A mouse brain hemisphere (~0.2g) was flash frozen in 2 ml microtubes until processing.
-
3.1.1.
-
3.2.Tissue Homogenization and Lysis for Bio-Plex.
-
3.2.1.Prepare the Total Lysis Buffer (TLB). There are three components:
-
3.2.1.1.The first component is the lysis buffer, supplied at 1x (or near 1x).
-
3.2.1.2.The second component is PMSF (phenylmethylsulfonyl fluoride, a serine protease inhibitor).
-
3.2.1.2.1.Prepare a solution of 500 mM PMSF by dissolving 0.436 g PMSF in 5 ml DMSO.
-
3.2.1.2.2.Only 200 μl is required per 50 ml of lysis buffer, so store the remaining aliquots at −20°C or scale down appropriately.
-
3.2.1.2.1.
-
3.2.1.3.The third component is Cell Lysis Factor QG. This is supplied as a lyophilized powder. Two vials are required to prepare 50 ml of lysis buffer.
-
3.2.1.3.1.Resuspend each vial with 250 μl of deionized water and vortex for 15 seconds to mix. This yields a 100x solution.
-
3.2.1.3.1.
-
3.2.1.4.Finally, mix all reagents:
-
3.2.1.4.1.Add 500 μl of the reconstituted Cell Lysis Factor QG
-
3.2.1.4.2.Add 200 μl of 500 mM PMSF solution (2 mM final concentration of PMSF)
-
3.2.1.4.3.Add to 49.3 ml of lysis buffer. Vortex gently and set aside on ice.
-
3.2.1.4.4.Label the buffer as total lysis buffer (TLB).
-
3.2.1.4.1.
-
3.2.1.1.
-
3.2.2.Homogenization
-
3.2.2.1.Place brain hemisphere (~0.2g) into 2 ml microtube with a cap.
-
3.2.2.2.Add 3 to 4 2.3 mm Zirconia/Silica beads and 1 ml of TLB for homogenization.
-
3.2.2.3.Place tubes into MP Biomedicals FastPrep-24 5G Homogenizer.
-
3.2.2.4.Use recommended homogenization settings for mouse brain (6.0m/sec for 40 seconds).
-
3.2.2.5.Remove samples from homogenizer and place into 4°C centrifuge.
-
3.2.2.6.Centrifuge samples at 12,000 rpm for 15 minutes. For this protocol, the samples were then assayed immediately.
-
3.2.2.1.
-
3.2.1.
-
3.3.Running the Bio-Plex Multiplex Assay
-
3.3.1.Prepare the Bio-Plex Quality Control
-
3.3.1.1.Reconstitute the vial of Quality Control with 250 μl of TLB with 0.5% Fetal Bovine Serum (FBS).[See section 7.3].
-
3.3.1.2.Incubate the reconstituted control on ice for precisely 30 minutes.
-
3.3.1.3.The Quality Control does not require further dilution.
-
3.3.1.1.
-
3.3.2.Prepare the Bio-Plex Sample Standards by generating a four-fold standard dilution series.
-
3.3.2.1.Reconstitute the vial of Sample Standards with 250 μl of TLB with 0.5% FBS.
-
3.3.2.2.Vortex for 5 seconds.
-
3.3.2.3.Incubate the reconstituted sample standard on ice for precisely 30 minutes.
-
3.3.2.4.Make a four-fold dilution series by mixing 50 μl of control with 150 μl of standard diluent buffer, such as in the following scheme:
-
3.3.2.5.Vortex at medium speed for 5 seconds between liquid transfers.
-
3.3.2.6.Make 8 standard dilutions (for samples with low analyte abundance; 7 dilutions is typical).
-
3.3.2.1.
-
3.3.3.Add Bio-Plex Capture Antibody/magnetic bead conjugates.
-
3.3.3.1.The beads are supplied at 10x.
-
3.3.3.2.First vortex the beads for 30 seconds.
-
3.3.3.3.For 96 wells, dilute 570 μl of the beads at 10x with 5130 μl of Assay Buffer solution to make 5700 μl of 1x solution.
-
3.3.3.4.Vortex the 1x beads for 10 seconds.
-
3.3.3.5.Pour the 1x Bio-Plex capture antibody/magnetic bead conjugate solution into a reagent reservoir and add 50 μl to each well using a multichannel pipettor.
-
3.3.3.6.Wash the plate twice with 100 μl of wash buffer by selecting [MAG x2] in the wash station settings.
-
3.3.3.1.
-
3.3.4.Add samples, standards, blank and controls.
-
3.3.4.1.Vortex samples for 10 seconds prior to dispensing.
-
3.3.4.2.Add 50 μl of each sample to the appropriate wells.
-
3.3.4.3.Seal the plate with the supplied sealing tape to protect from spillage.
-
3.3.4.4.Incubate/mix the plate on a plate shaker at 850 rpm for 30 minutes at room temperature (RT).
-
3.3.4.5.Remove the sealing tape and wash the plate three times with 100 μl wash buffer by selecting [MAG x3] in the wash station settings.
-
3.3.4.1.
-
3.3.5.Add Bio-Plex Detection Antibodies.
-
3.3.5.1.The detection antibody solution is supplied at 10x.
-
3.3.5.2.For 96 wells, dilute 300 μl of the 10x detection antibody with 2700 μl of Detection Ab Diluent HP to make 3000 μl of 1x solution.
-
3.3.5.3.Vortex the 1x detection antibody solution for 10 seconds.
-
3.3.5.4.Pour the 1x Bio-Plex detection antibody solution into a reagent reservoir and add 25 μl to each well using a multichannel pipettor.
-
3.3.5.5.Seal the plate with fresh sealing tape to protect from spillage.
-
3.3.5.6.Incubate/mix the plate on a plate shaker at 850 rpm for 30 minutes at RT.
-
3.3.5.7.Remove the sealing tape and wash the plate three times with 100 μl wash buffer by selecting [MAG x3] in the wash station settings.
-
3.3.5.1.
-
3.3.6.Add Bio-Plex Fluorophore (Phycoerythrin)-Streptavidin Conjugate
-
3.3.6.1.Bio-Plex SA-PE (Streptavidin-Phycoerythrin) is supplied at 100x.
-
3.3.6.2.Vortex the 100x SA-PE solution for 5 seconds.
-
3.3.6.3.For 96 wells, dilute 60 μl of the 100x SA-PE with 5940 μl of the Bio-Plex Assay Buffer solution to make 6000 μl of 1x solution.
-
3.3.6.4.Vortex the 1x Bio-Plex SA-PE solution for 5 seconds.
-
3.3.6.5.Pour the 1x Bio-Plex SA-PE solution into a reagent reservoir and add 50 μl to each well using a multichannel pipettor.
-
3.3.6.6.Seal the plate with fresh sealing tape to protect from spillage and photobleaching.
-
3.3.6.7.Incubate/mix the plate on a plate shaker at 850 rpm for 10 minutes at RT.
-
3.3.6.8.Remove the sealing tape and wash the plate three times with 100 μl wash buffer by selecting [MAG x3] in the wash station settings.
-
3.3.6.1.
-
3.3.7.Prepare plate for reading.
-
3.3.7.1.Pour the 1x Bio-Plex Assay Buffer solution into a reagent reservoir and add 125 μl to each well using a multichannel pipettor.
-
3.3.7.2.Seal the plate with fresh sealing tape to protect from spillage and photobleaching.
-
3.3.7.3.Incubate/mix the plate on a plate shaker at 850 rpm for 10 seconds at RT.
-
3.3.7.4.Remove the sealing tape from the plate.
-
3.3.7.5.The assay plate is now ready to be read by the instrument.
-
3.3.7.1.
-
3.3.8.Instrument Settings
-
3.3.8.1.For readings with the Bio-Plex 200 instrument, use the following settings:
-
3.3.8.1.1.RP1 (PMT) Low
-
3.3.8.1.2.DD Gates (low) 5,000
-
3.3.8.1.3.DD Gates (high) 25,000
-
3.3.8.1.4.Bead Events 50
-
3.3.8.1.1.
-
3.3.8.1.
-
3.3.1.
4. Results
For this study, we infected two cohorts of mice (non-depleted and CD8+ T-cell depleted mice) with Plasmodium berghei and evaluated chemokine and cytokine expression using the Bio-Plex Luminex xMAP-based multiplex bead-based immunoassay. This assay was performed to identify the changes in chemokine and cytokine levels within the brain during experimental cerebral malaria (ECM). We also wanted to determine the role of CD8+ T cells in driving the inflammatory expression profile. Presented here are the results from 33 cytokines / cytokines from non-depleted and CD8+ T-cell depleted mice infected with P. berghei. All samples are normalized to naive (uninfected) control brain tissue for each analyte (Figure 3).
Figure 3.
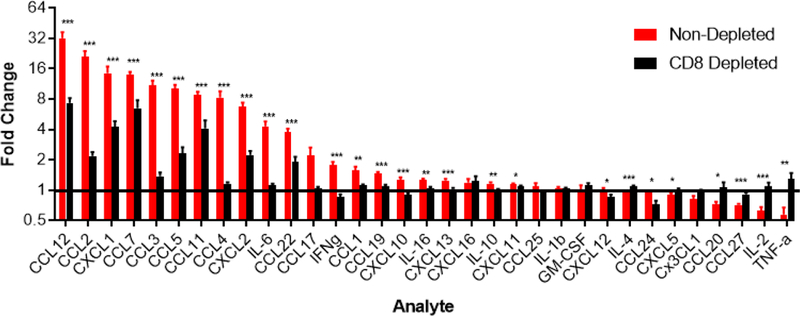
Expression levels of 33 cytokines and chemokines in non-depleted (red; n=12; 396 total data points) vs. CD8+ T cell-depleted (black; n=11; 363 total data points) mice at day 6 following P. berghei infection. All samples were normalized to uninfected control brain samples (n=11; black line denotes naive group). Data represents two combined independent experiments. Asterisks denote statistical significance (*p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001) of non-depleted vs. CD8 depleted mice as determined by a Student’s t-test or Mann-Whitney U test. A Shapiro Wilk Test was used to determine normality of the data. Outliers were removed via a Grubbs’ test for outliers (15 of 396 and 7 of 363 data points were removed from the non-depleted and depleted groups, respectively).
During ECM, mice without T cell depletion showed statistically significant (p < 0.05) upregulation of 20 pro-inflammatory chemokines and cytokines when compared to the naive control group. Interestingly, all these chemokines and cytokines were significantly reduced in CD8+ T cell depleted mice (Figure 3). In addition, there was a strong positive correlation between the fold-changes obtained in the first and second experiment (Figure 4). Collectively, these data indicate that CD8+ T cells promote upregulation of the denoted inflammatory mediators in the brain during ECM in a reproducible manner.
Figure 4.
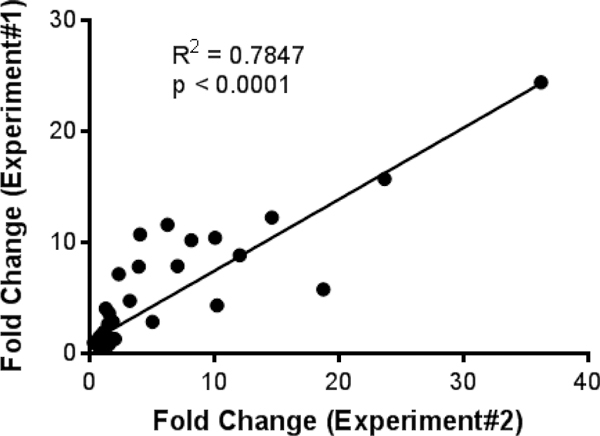
Reproducibility of the 33 cytokine / chemokine expression patterns between two independent experiments. Graph shows the average fold change of upregulated cytokines and chemokines plotted for depleted and non-depleted data from two independent experiments. Linear regression analysis was performed to the best fit line, R2, and p value.
Of note, there is a known frameshift mutation [8] of the CXCL11 gene in inbred C57BL/6 mice, resulting in either a lack of expression or the expression of a truncated form of the CXCL11 protein. In this assay, we did observe CXCL11 protein signal, which can be attributed to possible antibody cross-reactivity in the assay or the detection of a truncated form of the CXCL11 protein.
5. Conclusions
Here, we describe a method for preparing mouse brain tissue samples to determine the concentrations of multiple cytokine / chemokine analytes using the Bio-Plex mouse 33-plex panel. For this study, we evaluated three cohorts of mice (naive, PbA-infected and PbA-infected with CD8 T-cell depletion) and evaluated the inflammatory profile within these mice. We observed elevated chemokine and cytokine levels in the brains of infected mice during development of cerebral malaria. Twenty different analytes were upregulated, with the majority being chemokines involved in the recruitment immune cells [9]. Importantly, depletion of CD8+ T cells resulted in a significant reduction in all upregulated cytokines and chemokines, demonstrating the importance of CD8+ T cells in promoting CNS inflammation during cerebral malaria. Future studies will focus on development of methods to perform multiplex analyses with sorted cells, which should help identify cellular source(s) of the different cytokines and chemokines.
There is tremendous value in performing research using Bio-Plex assays. Currently, there are thousands of research publications [10] describing the use of multiplex bead-based immunoassays, primarily from serum, plasma, or cell culture samples. Additionally, protocols are available for other biological sample sources (e.g. sputum, lavage, etc.) through online resources at www.bio-rad.com. In this study, we describe a simple method to prepare samples from mouse brain tissue, but we expect that this same methodology can be used for many different tissues. The low sample volume requirements in this protocol allow multiple assays to be performed on a single tissue preparation. Only 50 μl of tissue sample per mouse from a 1 ml preparation was required to analyze the full panel of cytokines/chemokines. The remainder of the sample can be used for other assays or to repeat experiments from the same biological samples. One can also reduce the buffer volume during the homogenization step if preparing smaller tissues or a higher protein concentration is required. One can expect single digit pg/mL levels of sensitivity for each cytokine and chemokine target, depending on the quality of the samples. Exact sensitivity values are provided in the product data sheet supplied with the kits. With respect to specificity, one should expect less than 10% cross-talk between targets, unless otherwise specified. The product data sheet will report if targets share significant homology in amino acid sequence and have cross-reactivity greater than 10%. Importantly, we observed that the Bio-Rad 33-plex assay is highly reproducible despite biological variability between experimental animals and treatments in this study. Development of technology to analyze protein analytes in smaller, defined cell populations should aid in characterizing inflammatory reactions that develop throughout the body.
6. Appendices (Materials List)
-
6.1.Instrumentation
-
6.1.1.MP Biomedical FastPrep-24 5G Homogenizer
-
6.1.2.Bio-Rad Bio-Plex 200 System
-
6.1.3.Bio-Plex Pro Wash Station (Bio-Rad Cat. #30034376)
-
6.1.4.Eppendorf Thermomixer C with plate insertion (Cat. #2231000574).
-
6.1.5.Eppendorf Centrifuge 5415 R (Cat. #EP-5415R)
-
6.1.6.Pipet-Lite Multi Pipette L8–200XLS+ 20 μl - 200 μl multichannel pipette (Rainin Cat. #17013805)
-
6.1.1.
-
6.2.Reagents and Consumables
-
6.2.1.Bio-Plex Sheath Fluid (Bio-Rad Cat #171000055)
-
6.2.2.Bio-Plex Validation Kit 4.0 (Bio-Rad Cat #171203001)
-
6.2.3.Bio-Plex Calibration Kit (Bio-Rad Cat #171203060)
-
6.2.4.Bio-Plex Pro Mouse Chemokine Panel 33-plex (Bio-Rad Cat #12002231)
-
6.2.5.Bio-Plex Pro Cell Signaling Reagent Kit (Bio-Rad Cat #171304006M)
-
6.2.6.Phenylmethylsulfonyl fluoride (PMSF, Sigma-Aldrich Cat #78830)
-
6.2.7.Dimethyl sulfoxide (DMSO, Sigma-Aldrich Cat #D2650)
-
6.2.8.Fetal Bovine Serum, ultra-low IgG (ThermoFisher Cat #16250086)
-
6.2.9.Zirconia/Silica Beads, 2.3 mm (Biospec Cat. #11079125z)
-
6.2.10.Titertube Micro Test Tubes (Bio-Rad Cat #2239391)
-
6.2.11.Reagent Reservoirs, 25 ml (VistaLab Cat #3054–1002 or #3054–1004)
-
6.2.12.Reagent Reservoirs, 50 ml (VistaLab Cat #3054–1006)
-
6.2.13.Eppendorf Tubes, 1.5 ml
-
6.2.14.Eppendorf Tubes, 2.0 ml screw-cap
-
6.2.1.
-
6.3.Software
-
6.3.1.Bio-Plex Manager Software, Standard Edition, Version 6.2 (Bio-Rad Cat #171STND01)
-
6.3.2.Bio-Plex Data Pro Software, Version 1.2 (Bio-Rad Cat #171001513)
-
6.3.1.
7. Additional Suggestions
-
7.1.
Remember to incubate the reconstituted Standards on ice for 30 minutes. Time is critical in this step for consistency.
-
7.2.Recall two diluent solutions provided in the assay kit:
-
7.2.1.Standard Diluent. Used for making dilutions with the standards.
-
7.2.2.Sample Diluent. Used for making sample dilutions with serum and plasma samples. The goal here is to match the sample matrix to a first approximation.
-
7.2.1.
-
7.3.
For the preparation of the Bio-Plex Quality Control (section 1), FBS is used here as a carrier protein/stabilizer to match, to a first approximation, the total protein concentration in the brain homogenate samples.
-
7.4.Additional buffer used in this assay:
-
7.4.1.Tissue Lysis Buffer. Used for making samples from tissues. It is currently supplied in a separate kit: Bio-Plex Pro Cell Signaling Reagent Kit (Bio-Rad Cat #171304006M).
-
7.4.1.
-
7.5.
For ease of handling multiple samples and simultaneous preparation, we suggest preparing samples in 1 ml Titertubes (Bio-Rad Cat #2239391).
-
7.6.
We strongly suggest using a calibrated 20 – 200 μl multichannel pipettor as well as reagent reservoirs during the assay.
Figure 1.
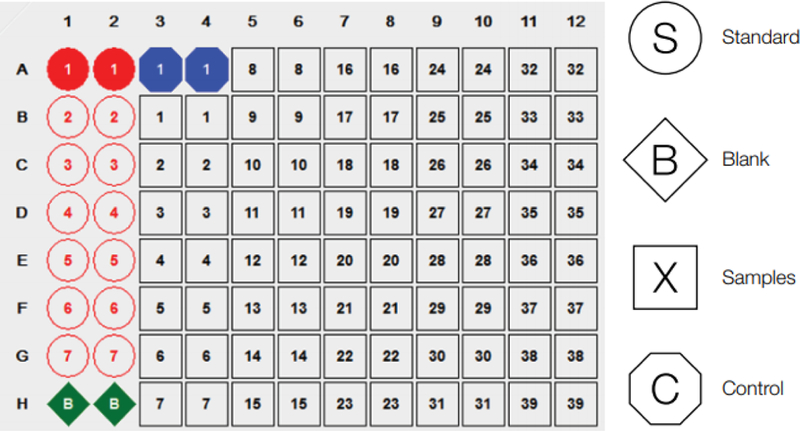
Recommended plate layout for sample standards, samples, blanks and controls.
Figure 2.
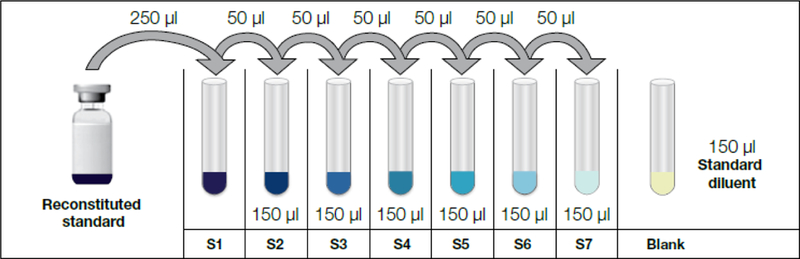
Dilution scheme for sample standards. Here 7 dilutions are shown. In this procedure, we performed 8 four-fold dilutions to maximize the sensitivity of the assay.
8. Funding
This work was supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH), and through collaboration with Bio-Rad Laboratories.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
10. References
- [1].Isaacs A and Lindenmann J (1957). Virus Interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci 147, 927, 258–267. [DOI] [PubMed] [Google Scholar]
- [2].Turner MD et al. (2014). Cytokines and chemokines: At the crossroads of cell signaling and inflammatory disease. Biochim Biophys Acta 1843, 2,563–2,582. [DOI] [PubMed] [Google Scholar]
- [3].Etzioni R et al. (2003). Combining biomarkers to detect disease with application to prostate cancer. Biostatistics 4, 523–538. [DOI] [PubMed] [Google Scholar]
- [4].Hsu MJ et al. (2014). Biomarker selection for medical diagnosis using the partial area under the ROC curve. BMC Res Notes 7:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Williams FMK (2009). Biomarkers: in combination they may do better. Arthritis Res Ther 11, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tan W and Huang I (2018). Multiplex Cytokine Immunoassays - Minimizing Effort, Maximizing Results. Bioradiations. Online article originally published July 10, 2018. [Google Scholar]
- [7].Swanson et al. (2016). CD8+ T Cells Induce Fatal Brainstem Pathology during Cerebral Malaria via Luminal Antigen-Specific Engagement of Brain Vasculature. PLOS Pathogens. 12(12):e1006022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Muller M et al. (2010). Review: The chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity - a tale of conflict and conundrum. Neuropathy and Applied Neurobiology, 36, 368–387. [DOI] [PubMed] [Google Scholar]
- [9].Zlotnik A and Yoshie O (2012). The Chemokine Superfamily Revisited. Immunity, 36(5), 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Houser B (2012). Bio-Rad’s Bio-Plex suspension array system, xMAP technology overview. Arch Physiol Biochem. 118 (4); 192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]