

Abstract
Molecular cloning is a cornerstone of biomedical, biotechnological, and synthetic biology research. As such, improved cloning methodologies can significantly advance the speed and cost of research projects. Whereas current popular cloning approaches use in vitro assembly of DNA fragments, in vivo cloning offers potential for greater simplification. It is generally assumed that bacterial in vivo cloning requires Escherichia coli strains with enhanced recombination ability; however, this is incorrect. A widely present, bacterial RecA-independent recombination pathway is re-emerging as a powerful tool for molecular cloning and DNA assembly. This poorly understood pathway offers optimal cloning properties (i.e. seamless, directional, and sequence-independent) without requiring in vitro DNA assembly or specialized bacteria, therefore vastly simplifying cloning procedures. Although the use of this pathway to perform DNA assembly was first reported over 25 years ago, it failed to gain popularity, possibly due to both technical and circumstantial reasons. Technical limitations have now been overcome, and recent reports have demonstrated its versatility for DNA manipulation. Here, we summarize the historical trajectory of this approach and collate recent reports to provide a roadmap for its optimal use. Given the simplified protocols and minimal requirements, cloning using in vivo DNA assembly in E. coli has the potential to become widely employed across the molecular biology community.
Keywords: cloning, recombination, DNA recombination, homologous recombination, synthetic biology, biotechnology
Introduction
DNA manipulation has revolutionized biomedical research and synthetic biology. The ability to design, modify, and assemble DNA pieces with almost limitless creativity has allowed unprecedented investigation of molecular and cellular physiology as well as the creation of synthetic life (1, 2). Restriction enzymes were historically groundbreaking for this aim (3), but carry some limitations. These enzymes rely on the presence (or absence) of specific recognition sequences for their action, and so inevitably leave sequence “scars” in the final DNA products. Coupled with the laborious protocols involved, the constraints of this approach have driven the development of novel cloning techniques offering greater flexibility and simplicity. Currently, the most popular approaches are “homology-based” cloning methods. These techniques use small homologous sequences at the termini of DNA fragments, normally introduced using PCR, to drive the assembly of desired circular products. The majority of homology-based approaches perform DNA fragment assembly in a sequence-independent, directional, and seamless (scar-free) manner, offering complete flexibility on DNA construct design. A plethora of different homology-based methodologies have been developed, employing a diverse range of mechanisms for DNA assembly (e.g. LIC (ligation-independent cloning) (4), Gibson assembly (5), In-fusion cloning (6), SLIC (7), SLiCE (8, 9), yeast gap-repair cloning (10–12), RecET, λ phage (7, 13), and USER (14) among others). Current state-of-the-art methods predominantly involve in vitro assembly of DNA fragments, such as by enzymatic action or ssDNA3 annealing, prior to transformation into Escherichia coli for plasmid propagation. DNA assembly can also be performed in vivo, circumventing the requirement for in vitro treatments. Bacterial in vivo recombination (or recombineering) offers greater simplicity than in vitro assembly approaches. These techniques involve transformation of exogenous DNA fragments into E. coli, which are ligated in vivo into desired sequences by the action of recombinase enzymes. Recombinase enzymes, such as endogenous RecA or phage-derived RecE/RecT, form new DNA sequences by fusing fragments using homologous sequences between different DNA molecules (7, 13, 15). However, the ability of these enzymes to recombine DNA also causes plasmid instability, inducing deletions, multimerization, or genomic integration of the plasmid sequence (16–18). It is for this reason that “recombination-deficient” bacterial strains, lacking RecA or RecE/RecT expression (19, 20), are ubiquitously used for the growth and maintenance of plasmid DNA across the molecular biology community.
However, widely used laboratory E. coli strains are not in fact recombination-deficient. An E. coli RecA-independent recombination (RAIR) exists and was first exploited as a cloning tool almost 30 years ago (21). Although never gaining popularity in the intervening years, this pathway has recently re-emerged as a powerful method for simplified plasmid cloning (22–26). By using in vivo assembly of DNA fragments, driven by short homologous sequences, this approach allows versatile cloning to be performed using standard laboratory E. coli strains, without the need for in vitro DNA assembly and avoiding the plasmid instability of recombineering strains. This promising technique offers a simple and powerful cloning approach, which is readily accessible to any molecular biologist. Currently there is poor awareness of the RecA-independent recombination pathway's existence, and confusion remains over the need for specialized recombination cloning strains. Here, we aim to clarify this confusion, offering a historical perspective of the technique's trajectory, a summary of current mechanistic understanding, and a roadmap for optimal employment as a molecular cloning and DNA assembly tool.
Using E. coli RecA-independent recombination for DNA assembly
In this review, we focus on the in vivo assembly of DNA using a recombination pathway that is endogenous to standard laboratory E. coli, including those that are deficient in RecA. We refer to this pathway as RAIR. Strains expressing alternative recombinases such as RecE/T or λRed (27) have also been used for in vivo recombination cloning. Whereas these pathways could also be considered “RecA-independent recombination,” the cloning methodology that we discuss here uses common laboratory bacterial strains and does not require enhanced recombinase activity.
Upon transformation of linear dsDNA, the RAIR pathway joins DNA fragments containing homologous sequences at their termini to form a circular plasmid product that will undergo propagation. The pathway efficiently joins short homologous sequences of around 15–30 bp, which can be conveniently introduced by PCR or through DNA synthesis. Using specific primer design, as detailed in Fig. 1, all plasmid DNA modifications can be performed. These include both simple protocols (insertion or deletion of sequences, point mutagenesis (Fig. 1A), and subcloning (Fig. 1B) (22, 28–35) and more complex procedures, such as performing multiple insertions, deletions, or mutations, simultaneous assembly of multiple DNA fragments (22, 25, 36), library creation (22, 32), or combinations of different modifications (22) (Fig. 2). Generally, all procedures follow three steps: homology generation (predominantly PCR), DNA cleanup, and transformation for DNA assembly. The simplest option for DNA cleanup is post-PCR incubation with the methylation-sensitive restriction enzyme DpnI, which cuts methylated template DNA at the abundant dam site “Gm6ATC,” but does not cut the newly amplified PCR products (37). Multiple protocol variations are possible for cloning using the RAIR pathway, with advantages dependent on the specific aims of the required cloning project. These details will be discussed below in greater depth. Overall, use of this recombination pathway offers the benefits of the most versatile homology-based cloning methods while requiring no specialized bacteria or commercial kits, thus vastly simplifying molecular cloning procedures.
Figure 1.

Primer design for generation of homologous regions. A, insertions, deletions, and mutagenesis require vector amplification to produce a linear product, re-circularized by a single recombination event on transformation. Primers bind astride the modification site, with homologous regions encoded at 5′ primer ends. B, subcloning of insert DNA requires generation of distinct homology arms (colored) to drive directional assembly of two fragments. C, a length of at least 15 bp (top) and increased binding strength (bottom) of homologous DNA regions enhances recombination efficiency (based on data from Ref. 22). D, PCR Protocols textbook cover, published 1993, depicting “Recombination PCR” cloning by Jones and Howard. Reproduced with permission from Springer Nature.
Figure 2.
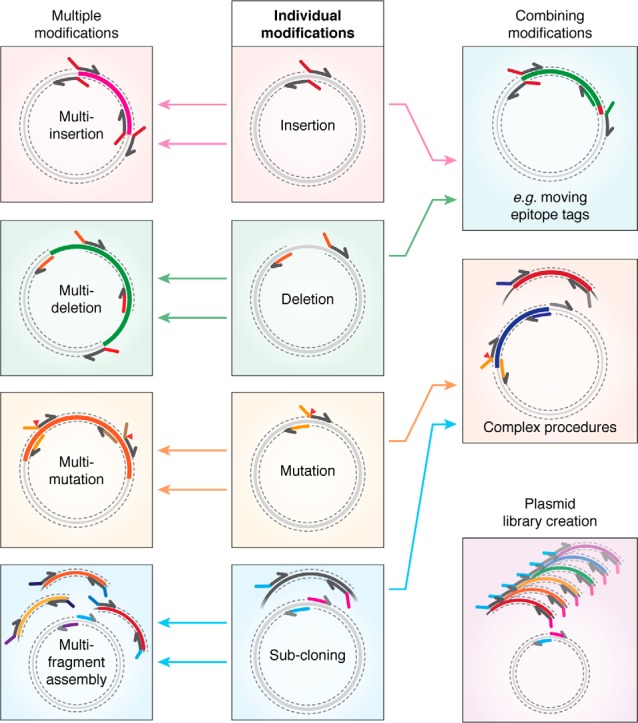
The capabilities and versatility of the RAIR cloning approach. Primers to perform individual modifications (insertions, deletions, mutagenesis, and subcloning; center) can be combined to perform multiple modifications of the same type (left) or complex procedures from multiple modifications of different types (right).
A historical perspective: Why was this technique neglected?
The employment of RecA-independent recombination for molecular cloning was first reported almost 3 decades ago. In the first study, Douglas Jones and Bruce Howard (21) described their “recombination PCR” method for plasmid mutagenesis or subcloning. The method required PCR amplification of a template DNA using primers to introduce homologous regions, which directed DNA assembly by recombination upon transformation into the DH5α E. coli strain. Despite its inclusion in molecular biology textbooks on multiple occasions (38–40), including representation on the cover of PCR Protocols in 1993 (Fig. 1D), the technique never gained widespread popularity. The original report (21) has received 32 citations to date since 1991, whereas a more popular recent cloning method, Gibson assembly (5), has accumulated 1725 citations since its publication in 2009 (PubMed records until July 2019). Early improvements to the initial protocol mainly focused on post-PCR removal of template DNA, employing enzymatic digestion (33, 41), isolation of desired products by gel purification (42), or reduction of the quantity of template DNA used (34). Further approaches to enhance method efficiency included the use of a nonmutagenic primer pair within the antibiotic resistance gene, so that amplification of this essential sequence in two halves will forcibly enhance selection of correctly recombined clones (33, 35). Post-PCR DNA clean-up has been achieved by generation of DpnI-expressing E. coli strains for in vivo destruction of template DNA (30), yet this approach significantly complicates plasmid maintenance, offering little experimental advantage. Since the original study, cloning using the RAIR pathway has been only sporadically reported, with its main use in high-throughput applications, such as cloning of thousands of open reading frames from human liver samples (31) or Campilobacter jejuni (29).
It was only in recent years that this method really gained traction, with a number of reports demonstrating the potential of the approach to simplify all plasmid cloning protocols (22–26, 32, 36, 43). Why has this technique failed to gain popularity for so long? There are several reasons that are likely to have delayed its widespread use. First, the technique relies on PCR amplification and was first reported at a time when the fidelity and processivity of polymerases were limited (44). Accurate and successful amplification of a large plasmid sequence would have been difficult, and the laborious protocols for gene sequencing required at the time, would have made this unappealing. Second, introduction of overlapping sequences by PCR requires longer primers, which would have added substantial synthesis costs compared with restriction enzyme–mediated cloning. Indeed, multiple reports cited the cost of oligonucleotides as the major caveat of PCR-based approaches (45–47). Third, two recombination cloning reports, Bubeck et al. (28) and Oliner et al. (48), were coincidentally published. Despite the former study using RAIR and the latter using the RecET pathway, both reports have often been referred to indistinguishably. Since then, confusion has clouded the use of RAIR, and cloning using in vivo E. coli recombination was predominantly attributed to the use of specialized strains with enhanced recombination activity (15, 48–50). Finally, having been referred to as “black box” cloning, the lack of mechanistic understanding will have hindered the uptake and appreciation of this molecular cloning approach.
In the last few decades, the fidelity and processivity of DNA polymerases have dramatically improved, and the cost of oligonucleotide synthesis and DNA sequencing have become easily affordable. Taq polymerase has been replaced by engineered enzymes, such as Phusion and Q5, with ∼50-fold improved error rate and higher processivity (51). Oligonucleotides can be purchased cheaply for at least 110-bp sequences, allowing considerable-sized sequences to be introduced using PCR primers. With these developments in hand, use of the bacterial RecA-independent pathway for molecular cloning can finally realize its potential.
The recombination mechanism: Opening the black box
Despite its use as a cloning tool, the mechanism of RecA-independent recombination is poorly characterized, with many enzymes involved in the pathway and with the mechanisms by which they act yet to be identified. Active research was conducted 3 decades ago, but it was only recently, with development of cloning applications, that further mechanistic studies have been conducted. First insights into the recombination mechanism came from observing the re-circularization of linearized plasmid after transformation into E. coli (52, 53). Re-circularization in recA− strains was observed frequently, with fusion occurring at sites with sequence similarities, suggesting a dependence on homology. Preferential re-circularization occurred at sites closer to linear DNA ends, an observation later confirmed by others (54). Reported homology requirements for the RAIR pathway are very low, suggested to be as little as 10 bp (28), 6 bp (55), or even just 4 bp (21), yet there is a significant increase in efficiency with longer stretches of homology (22, 23, 32) (Fig. 1C). Further mechanistic characterization using engineered homology regions demonstrated that successful recombination requires linear DNA ends (28), and for insertion of a DNA fragment, recombination could not take place without linearization between the vector's homologous sequences. Homology regions do not need to be at the termini of the fragments, as recombination can occur using sequences at least 180 bp from fragment ends (23). In such cases, the intervening nonhomologous sequence between homologous regions and linear DNA termini is lost from the final product (21, 28, 52). From their work on plasmid re-circularization, Conley et al. (53) proposed that RAIR occurred through a single-strand annealing mechanism. In this model, in vivo 3′ to 5′ exonuclease activity would produce ssDNA at linear ends, which can anneal through short regions of homology to be subsequently repaired by polymerases and ligases (Fig. 3) (53). Such a mechanism would explain the preference for recombination at homology sites close to linear termini, as those more distant are less likely to be converted to ssDNA. It also explains the loss of nonhomologous DNA termini, which would be excluded after DNA repair. From analysis of recombination in mutant E. coli strains, Conley et al. (52) demonstrate a dramatic reduction in plasmid re-circularization in xth1 mutant bacteria, a gene encoding exonuclease III (ExoIII). This protein was proposed to be the factor producing single-stranded ends by action on double-stranded linear DNA, allowing homologous sequence annealing (52). The single-strand annealing hypothesis was supported by a study showing that the efficiency of recombination was increased by almost 2 orders of magnitude in bacterial strains deficient for ssDNA exonucleases (56). Such proteins would degrade the single-stranded homologous regions that are required for annealing to direct re-circularization.
Figure 3.

Current understanding of the RecA-independent recombination mechanism. Homologous regions at the termini of linear DNA fragments (1) are converted to ssDNA by 3′ to 5′ action of exonuclease III (2) before homology-directed annealing (3) and repair of dsDNA through contribution of DNA polymerase I (4). Further contributions of protein interactions, such as homology recognition, ssDNA-binding proteins, or DNA ligases, are current unclear (question marks, red type).
A recent thorough analysis of RAIR cloning ability in bacterial mutant strains showed that cloning is specifically impaired in ExoIII mutants (xthA) (57), elegantly confirming the observations of Conley et al (53). The impaired recombination in xthA strains can be circumvented by transformation of DNA with homology arms already exposed as ssDNA (57), further supporting the single-strand annealing hypothesis. Interestingly, ExoIII has previously been employed ex vivo as a tool for DNA assembly (58, 59) in a familiar procedure where incubation of homology containing DNA with ExoIII yields sequences that could be annealed in vitro. The possibility of in vivo enzymatic action or of cloning without enzymatic treatment was not considered or assessed by the authors.
As the single-strand annealing mechanism of RAIR uses DNA with free termini (28), with no requirement for “repair template” DNA, it has been proposed to act endogenously for emergency repair of dsDNA breaks (60). This mechanism would result in mutagenic repair, deleting the intervening genomic sequence between homologies. The report by Nozaki and Niki (57) shows little requirement for other exonucleases, such as RecBCD, which had previously been implicated in this recombination pathway (61). The lack of RecBCD dependence of RAIR cloning is interesting, given that RecA-independent but RecBCD-dependent repair of double-strand breaks has been described in multiple bacterial species, such as Mycobacterium smegmatis (62, 63), Deinococcus radiodurans (64), and E. coli (61, 65). In a study of microhomology-mediated plasmid re-circularization, with a strong resemblance to RAIR, Chayot et al. (65) implicate exonuclease activity of RecBCD in ssDNA production and the DNA ligase LigA in final repair. Recombination cloning ability is not lost in mutants of RecB, RecC, or RecD (57), so this likely represents a DNA repair pathway distinct from that which is required for plasmid circularization. Exonuclease exposure of ssDNA would require gap-filling and ligation (Fig. 3). Nozaki et al. (57) further demonstrate a substantial contribution of DNA polymerase I (polA) activity for RAIR cloning, which had also previously been suggested as a mechanistic player (53). It remains to be seen whether plasmid assembly is completed by LigA when cloning using the RAIR pathway.
It now seems clear that upon transformation, double-stranded linear DNA molecules are the substrate for 3′-5′-exonuclease action by ExoIII, which yields single-stranded homologous DNA sequences. These sequences can anneal, and the resulting gaps are filled, likely by DNA polymerase I, to complete plasmid circularization (Fig. 3). Beyond this, further details of the recombination mechanism remain unclear. Are single-stranded binding proteins required for DNA stability? Does single-strand annealing require homology recognition factors, as other recombination mechanisms do (66), or does it simply rely on annealing by conventional base-pairing? Which enzymes are required for full repair of circular DNA? Although the mechanism of recombination is now becoming clearer, the picture is not yet fully complete.
An unappreciated role for RAIR in other cloning approaches
The homologous regions engineered to assemble DNA segments using in vivo assembly are virtually identical to those employed by in vitro homology-based cloning methods such as In-fusion (6), SLiCE (8, 9), or Gibson assembly (5). As all cloning methods end with transformation into E. coli, the efficiency of these in vitro homology-based methods is likely to be enhanced by in vivo RAIR. A recent study showed that the majority of DNA fragments are left unassembled using in vitro enzymatic assembly methods (67) and therefore are substrates for in vivo assembly. Therefore, the RAIR pathway potentially offers unappreciated support to many homology-based methods. Indeed, qPCR comparison of in vitro assembly efficiency between Gibson and restriction/ligation shows that similar levels of successfully assembled products prior to transformation yield a greater number of colonies for Gibson than for restriction enzymes (31). As Gibson assembly produces homology-containing linear DNA, a substrate for RAIR, whereas restriction enzymes do not, it is very possible that in vivo assembly is supporting this technique. This consideration should not be overlooked when assessing the efficiency of homology-based cloning methods (68), and true calculation of method efficiency would require a RAIR-deficient strain or quantification of in vitro assembly (67).
A number of homology-based methodologies have unclear assembly mechanisms, which most likely rely on the RAIR pathway. Without direct evidence for successful in vitro DNA assembly, such as gel electrophoresis visualization of intact circular DNA prior to transformation, it is difficult to be sure that such techniques do not predominantly rely on the RAIR pathway. For example, two homology-based methods have been proposed to occur by in vitro DNA annealing but appear most likely to rely on recombination in vivo. The first, FastCloning (43), is a homology-based cloning method requiring no in vitro assembly step. After PCR amplification to introduce homologous overlaps between DNA fragments and DpnI destruction of template DNA, amplified products are transformed into E. coli for propagation. However, the authors suggested that the 3′-5′-exonuclease activity of proofreading DNA polymerases in the final cycles of the PCR, enhanced by depletion of nucleotides in the reaction, would leave ssDNA available for annealing in vitro. Similarly, the polymerase incomplete primer extension (PIPE) method (69) follows the same procedure, and the authors suggested that the incomplete action of DNA polymerases during PCR would produce single-stranded termini for annealing prior to transformation. It has now been demonstrated that neither proofreading enzymes nor incomplete PCR are required. The “polymerase incomplete” notion, which is the basis of PIPE, has its roots in a historic observation that Taq polymerase can produce incompletely amplified DNA fragments (70). PIPE, among other studies (22, 23, 25, 43), uses newly engineered polymerases with greater processivity, which are unlikely to leave incomplete termini. Second, both Taq (nonproofreading) and Phusion (proofreading) enzymes are sufficient for successful cloning using the RAIR pathway (22, 69); therefore, 3′-5′-exonuclease activity of polymerases in vitro is not needed for a successful assembly. Last, DNA assembly is efficient even if vector DNA is linearized directly with restriction enzymes, which do not leave single-stranded homologous termini (22, 23, 28). Therefore, these techniques likely use in vivo assembly through the RAIR pathway.
Finally, even routine protocols that are widely employed in molecular biology laboratories have been demonstrated to rely on in vivo recombination. The QuikChangeTM mutagenesis approach was originally suggested to produce a nicked circular plasmid containing the desired mutation, with nicks being repaired in vivo. Developments of this approach have shown protocols to perform not just mutations, but also insertions and deletions, assuming a similar mechanism (71, 72). A “nicked plasmid” PCR would theoretically amplify with linear rather than exponential DNA accumulation, as it would be reliant on template DNA for amplification in all cycles. Amplification, however, has been shown to be nonlinear, and PCR products seem to be linear dsDNA with homologous sequences at the termini, where the desired point mutation is introduced (73). Through observations that enhanced efficiency and improved PCR amplification can be achieved using offset rather than fully overlapping primers (74), and observing that the linear DNA fragment has, in fact, homology-containing dsDNA, it is evident that the mechanism of QuikChangeTM mutagenesis relies on RAIR rather than nicked plasmid formation (73).
A roadmap for the use of the RAIR pathway in molecular cloning
Recent reports reviving the use of the RAIR pathway for molecular cloning have offered both proof-of-concept studies highlighting the power of the approach (23, 25, 26) and development of complete systems that allow complex cloning strategies to be achieved (22, 32). We have synthesized reported protocols to offer a method overview, with optimal procedures for different cloning scenarios. We focus predominantly on recent reports, because technical developments make the procedures employed in early studies of less relevance.
While all employ RAIR as a common mechanism for DNA assembly, different routes for generation of homology-containing DNA can be pursued (Fig. 4), with all protocols following a similar scheme: (a) production of homologous sequences, (b) removal of residual plasmid DNA, and (c) transformation into E. coli.
Figure 4.

A roadmap of protocols for application of cloning by in vivo assembly. Cloning can proceed through four routes for generation of homologous regions. DNA cleanup is required prior to transformation in each case to remove original template circular DNA. Fragments can be amplified either using a single-tube (1) or separate tube (2) PCR, with template DNA destroyed by DpnI incubation. Unamplifiable vectors can be linearized by restriction digestion (3) and co-transformed with PCR-amplified insert containing homologous regions. Finally, direct co-transformation of synthesized dsDNA with target vector DNA linearized by PCR or restriction digestion can be employed (4).
Route 1: Single-tube PCR
The simplest method for cloning using RAIR consists of a single-tube, single-step PCR where all primers and vectors are added to a single reaction. After amplification and DpnI incubation, all fragments will be directly co-transformed into competent E. coli for assembly (22). This approach can be used for the majority of cloning procedures. Although unconventional, multiple vectors can be reliably amplified simultaneously, greatly simplifying complex procedures (Fig. 2) (22). Designing homologous regions with a melting temperature (Tm) lower than that of template-binding regions will prevent potential primer-dimer–induced issues. However, if homologous regions are extended (e.g. for enhanced efficiency during complex assemblies), there is a possibility that homologous regions could bind together, hindering PCR amplification. Troubleshooting such issues can be achieved by confirmation of separate fragment amplification using independent PCRs (i.e. Route 2).
Route 2: Multi-tube PCR
Individual fragments are amplified in separate reactions and mixed prior to transformation (23, 25, 32). This approach is particularly useful either when the use of multiple primers limits DNA amplification or when nonspecific amplification occurs and DNA fragments would require purification. Amplification products are similarly treated with DpnI and co-transformed into bacteria.
Route 3: Vector digestion
Some vector backbones cannot be amplified by PCR due to high GC content or repeat sequences (e.g. particular expression vectors (75) or viral backbone plasmids (22)). In such cases, vectors can be linearized by restriction digestion before co-transformation with PCR-amplified inserts containing regions homologous to the termini of linearized vector DNA (22, 23, 28, 32). Although more laborious than vector amplification (Routes 1 and 2), this approach allows cloning when PCR of vector DNA cannot be conducted.
Route 4: Gene synthesis insertion
Synthesis of custom linear dsDNA fragments is now commercially available (e.g. gBlocks®). This offers a simple and versatile alternative for the generation of novel custom sequences that can be directly transformed into bacteria if they contain appropriate homologous regions to recombine with a linearized vector, as has been demonstrated for PCR-based gene synthesis (76). This is a particularly versatile approach to assemble several custom DNA fragments, which is of high value to synthetic biology. Although currently more expensive that PCR-based approaches, this route may see greater adoption as DNA synthesis services become increasingly affordable or where PCR amplification of insert sequences is not possible.
Optimization of cloning efficiency using the RAIR pathway
The RAIR pathway is very efficient for DNA assembly. Up to 6 fragments have been assembled in vivo using the RAIR pathway (25, 32), but as with any other method, the efficiency of cloning decreases with increasing complexity, resulting in fewer colonies (22). The decrease in efficiency likely results from the need to co-transform and successfully recombine several DNA fragments. Because a number of variables have been reported to impact on in vivo assembly, tailoring experimental conditions can enhance experimental success.
Primer design
Unique primer design allows a variety of plasmid modifications to be performed (Figs. 1 and 2). Insertion of small sequences, deletions, or point mutagenesis all involve full plasmid amplification using primers binding astride the desired modification site, with homologous sequences encoded at their 5′ ends (Fig. 1A). Small insertions can be introduced by inclusion in both forward and reverse primers to form the homologous regions, whereas deletions simply exclude the undesired plasmid regions from amplification. Subcloning is performed by amplifying both vector and insert DNA with homologous sequences encoded in primers for either vector or insert (Fig. 1B). A major advantage of this primer design approach is its modularity, where primers for individual modifications can be used in a combinatorial manner, allowing simultaneous and complex plasmid modification (Fig. 2).
Homology design
The properties of homologous sequences have been optimized to yield maximum efficiency. The length of homologous regions is proportional to the efficiency of assembly, with at least 15 bp offering reliable cloning (22, 23, 32). Bearing this in mind, it is not the length per se that improves cloning efficiency, but the “strength of homology annealing” (Tm) (22) (Fig. 1C). This is important to consider because regions of relatively low GC content may require longer homology arms for successful cloning. The primary limitation on longer homology is the cost of additional base pairs on primer purchasing, and homologies longer than 30 bp are likely to cause unnecessary cost increases. When performing one or two recombination events, homologous regions with lower annealing temperatures are sufficient (Tm around 50 °C), but when performing three or more recombination events, Tm of these overlaps can be increased (Tm = 60–65 °C) to aid assembly. However, up to five simultaneous mutations have been performed with primers containing homologous regions with Tm of 48–52 °C (22).
Optimizing PCR
Although fragment assembly is independent of the PCR amplification method, successful PCR amplification is the primary prerequisite for successful cloning. In difficult cases, the use of PCR additives, such as DMSO (3%) or betaine (1 m), and other additives, such as 1,2-propanediol and ethylene glycol (77, 78), can facilitate amplification. As discussed previously, alternative cloning routes are available to circumvent PCR amplification if required. General PCR-cycling parameters are dependent on the required amplification. Using high-fidelity DNA polymerases such as Phusion or Q5 will minimize PCR-introduced errors, in particular during vector amplification. Similarly, minimizing cycle numbers to around 18–20 yields sufficient DNA product while avoiding accumulation of mutations (22, 32, 43).
Post-PCR DNA handling
Prior to transformation, original vector DNA must be removed to limit false positive colony formation. Although greatly reducing the level of template DNA is a possibility (34), the required increase in PCR cycles for sufficient product formation dramatically increases the likelihood of introducing mutations during amplification. Using around 1 ng of template plasmid and minimal PCR cycles (22, 24) is the best compromise between yield and background noise. Post-PCR, incubation with DpnI is widely used to successfully remove the majority of unwanted template DNA (22, 25, 43) and is the simplest method for this purpose (Fig. 4). Post-PCR purification of DNA fragments by gel extraction has been suggested (23, 32, 34), but this laborious approach does not offer any advantages over treatment with DpnI unless PCR amplification produces nonspecific products. Incubation of separately amplified PCR products together prior to transformation has been reported (32) but is widely accepted to be unnecessary.
Transformation and bacterial strains
When transforming multiple fragments, the optimal insert/vector ratio has been examined, but there is little consensus about the ideal parameters. Whereas Jacobus and Gross (23) show little influence of this factor, Kostylev et al. (25) show that increasing insert/vector ratio can enhance colony formation, up to a ceiling at 5:1. This ratio cannot be controlled when performing single-tube cloning procedures (Fig. 4, Route 1), but a higher stoichiometry of insert to vector is normally achieved coincidentally, by the higher amplification efficiency of shorter insert fragments than the lengthier vector backbone.
The vast majority of reports to date use chemically competent cells, as is widely standard for laboratory plasmid maintenance. Because the RAIR pathway is ubiquitous in common laboratory bacteria, the strain employed does not appear to be critical, but note that some variations in efficiency have been demonstrated (57). Successful cloning has been reported in the commonly used DH5a (22, 23, 25), XL10-Gold (22), TOP10, NEB5α, NEB10β, and BL21 (DE3) (32) strains, among others. A direct comparison of bacteria strains with equivalent transformation efficiency has not been reported and could allow further method optimization. Whereas strain choice is flexible, cell competency is important. For procedures requiring single or double recombination events, homemade cells of lower competency (106–107 cfu/μg of DNA) are sufficient, but more complex procedures with three or more recombination events require highly competent cells (109 cfu/μg) (22).
One caveat of RAIR cloning is the incorporation of small fragments. A number of reports have highlighted poor cloning efficiency when assembling small DNA fragments of less than 200 bp (25, 32). This may arise due to poor transformation efficiency of small fragments or, possibly, their destruction by complete single-stranded exonuclease activity. As an alternative, with synthesis of 110 bp oligonucleotides routinely possible at relatively low cost, modifications up to around 150 bp apart can be introduced using a single pair of long primers.
Conclusion and future outlook
The simplification of molecular cloning procedures and reduced cost and versatility of plasmid modification offered by cloning using RecA-independent recombination (Fig. 5) give this approach great potential to become the method of choice for any laboratory in biomedical, biotechnological, and synthetic biology research. The recently revived interest in the use of RAIR for molecular cloning has also stimulated interest in the mechanism of recombination. With a full mechanistic understanding and identification of the proteins involved will come the ability to develop RAIR-enhanced strains, allowing greater cloning complexity to be achieved. RAIR-enhanced strains could avoid the plasmid instability issues of current “recombination enhanced” bacteria, given the apparent lack of RAIR action on circularized plasmids. Initial attempts have been made toward enhanced-RAIR bacteria (57, 79, 80) but do not appear yet to offer advantages great enough to warrant widespread adoption. Of course, a major advantage of this technique is that “cloning-compatible” strains already exist in the freezers of molecular biology laboratories across the world.
Figure 5.

Method overview. Through PCR amplification of DNA fragments, plasmid modifications of many varieties and homologous sequences to direct DNA assembly are introduced. PCR template DNA can be destroyed by DpnI digestion. Circular propagative plasmid products are assembled by homologous recombination in vivo.
The approach holds promise not only for standard cloning procedures, but also for high-throughput screening applications, where the versatility of homology-based approaches can be employed in plasmid library construction, as proof-of-concept studies have begun to suggest (22, 29, 32). With tumbling costs for DNA synthesis and its suitability for use with the RAIR pathway, future cloning approaches are likely to become simpler and more versatile yet. It has been many years since cloning using this elusive recombination pathway first stepped on stage, but it seems that the time is now right for it to enjoy the spotlight.
Acknowledgments
We thank Beatriz Herguedas, Alexandra Pinggera, Hinze Ho, Julia Morud Lekholm, Rafael Fernández-Leiro, and Javier Coloma for critical reading of the manuscript and insightful comments.
This work is supported by Medical Research Council Grant MC_U105174197 (to J. F. W.); Ministerio de Ciencia, Innovación y Universidades Grant RTI2018-095629-J-I00 (to J. G.-N.); and the Fondo Europeo de Desarrollo Regional (FEDER) (to J. G.-N.). The authors declare that they have no conflicts of interest with the contents of this article.
- ssDNA
- single-stranded DNA
- RAIR
- RecA-independent recombination
- ExoIII
- exonuclease III
- PIPE
- polymerase incomplete primer extension
- dsDNA
- double-stranded DNA.
References
- 1. Green M., and Sambrook D. W. J. (2012) Molecular Cloning: A Laboratory Manual, 4th Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 2. Jewett M. C., and Forster A. C. (2010) Update on designing and building minimal cells. Curr. Opin. Biotechnol. 21, 697–703 10.1016/j.copbio.2010.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith H. O., and Wilcox K. W. (1970) A restriction enzyme from Hemophilus influenzae: I. Purification and general properties. J. Mol. Biol. 51, 379–391 10.1016/0022-2836(70)90149-X [DOI] [PubMed] [Google Scholar]
- 4. Aslanidis C., and de Jong P. J. (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074 10.1093/nar/18.20.6069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 6. Benoit R. M., Wilhelm R. N., Scherer-Becker D., and Ostermeier C. (2006) An improved method for fast, robust, and seamless integration of DNA fragments into multiple plasmids. Protein Expr. Purif. 45, 66–71 10.1016/j.pep.2005.09.022 [DOI] [PubMed] [Google Scholar]
- 7. Li M. Z., and Elledge S. J. (2007) Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4, 251–256 10.1038/nmeth1010 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Y., Werling U., and Edelmann W. (2012) SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 40, e55 10.1093/nar/gkr1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Motohashi K. (2015) A simple and efficient seamless DNA cloning method using SLiCE from Escherichia coli laboratory strains and its application to SLiP site-directed mutagenesis. BMC Biotechnol. 15, 47 10.1186/s12896-015-0162-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Joska T. M., Mashruwala A., Boyd J. M., and Belden W. J. (2014) A universal cloning method based on yeast homologous recombination that is simple, efficient, and versatile. J. Microbiol. Methods 100, 46–51 10.1016/j.mimet.2013.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mashruwala A. A., and Boyd J. M. (2016) De novo assembly of plasmids using yeast recombinational cloning. Methods Mol. Biol. 1373, 33–41 10.1007/7651_2015_275 [DOI] [PubMed] [Google Scholar]
- 12. van Leeuwen J., Andrews B., Boone C., and Tan G. (2015) Rapid and efficient plasmid construction by homologous recombination in yeast. Cold Spring Harb. Protoc. 2015, pdb.prot085100 10.1101/pdb.prot085100 [DOI] [PubMed] [Google Scholar]
- 13. Zhang Y., Muyrers J. P., Testa G., and Stewart A. F. (2000) DNA cloning by homologous recombination in Escherichia coli. Nat. Biotechnol. 18, 1314–1317 10.1038/82449 [DOI] [PubMed] [Google Scholar]
- 14. Geu-Flores F., Nour-Eldin H. H., Nielsen M. T., and Halkier B. A. (2007) USER fusion: a rapid and efficient method for simultaneous fusion and cloning of multiple PCR products. Nucleic Acids Res. 35, e55 10.1093/nar/gkm106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trehan A., Kiełbus M., Czapinski J., Stepulak A., Huhtaniemi I., and Rivero-Müller A. (2016) REPLACR-mutagenesis, a one-step method for site-directed mutagenesis by recombineering. Sci. Rep. 6, 19121 10.1038/srep19121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peijnenburg A. A. C. M., Bron S., and Venema G. (1987) Structural plasmid instability in recombination- and repair-deficient strains of Bacillus subtilis. Plasmid 17, 167–170 10.1016/0147-619X(87)90023-0 [DOI] [PubMed] [Google Scholar]
- 17. Casali N. (2003) Escherichia coli host strains. Methods Mol. Biol. 235, 27–48 10.1385/1-59259-409-3:27 [DOI] [PubMed] [Google Scholar]
- 18. Biek D. P., and Cohen S. N. (1986) Identification and characterization of recD, a gene affecting plasmid maintenance and recombination in Escherichia coli. J. Bacteriol. 167, 594–603 10.1128/jb.167.2.594-603.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hall S. D., Kane M. F., and Kolodner R. D. (1993) Identification and characterization of the Escherichia coli RecT protein, a protein encoded by the recE region that promotes renaturation of homologous single-stranded DNA. J. Bacteriol. 175, 277–287 10.1128/jb.175.1.277-287.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y., Buchholz F., Muyrers J. P. P., and Stewart A. F. (1998) A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 20, 123–128 10.1038/2417 [DOI] [PubMed] [Google Scholar]
- 21. Jones D. H., and Howard B. H. (1991) A rapid method for recombination and site-specific mutagenesis by placing homologous ends on DNA using polymerase chain reaction. BioTechniques 10, 62–66 [PubMed] [Google Scholar]
- 22. García-Nafría J., Watson J. F., and Greger I. H. (2016) IVA cloning: a single-tube universal cloning system exploiting bacterial in vivo assembly. Sci. Rep. 6, 27459 10.1038/srep27459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jacobus A. P., and Gross J. (2015) Optimal cloning of PCR fragments by homologous recombination in Escherichia coli. PLoS One 10, e0119221 10.1371/journal.pone.0119221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y., Liu Y., Chen J., Tang M. J., Zhang S. L., Wei L. N., Li C. H., and Wei D. B. (2015) Restriction-ligation-free (RLF) cloning: a high-throughput cloning method by in vivo homologous recombination of PCR products. Genet. Mol. Res. 14, 12306–12315 10.4238/2015.October.9.19 [DOI] [PubMed] [Google Scholar]
- 25. Kostylev M., Otwell A. E., Richardson R. E., and Suzuki Y. (2015) Cloning should be simple: Escherichia coli DH5α-mediated assembly of multiple DNA fragments with short end homologies. PLoS One. 10, e0137466 10.1371/journal.pone.0137466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang F., Spangler J. R., and Huang A. Y. (2017) In vivo cloning of up to 16 kb plasmids in E. coli is as simple as PCR. PLoS One 12, e0183974 10.1371/journal.pone.0183974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sharan S. K., Thomason L. C., Kuznetsov S. G., and Court D. L. (2009) Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 4, 206–223 10.1038/nprot.2008.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bubeck P., Winkler M., and Bautsch W. (1993) Rapid cloning by homologous recombination in vivo. Nucleic Acids Res. 21, 3601–3602 10.1093/nar/21.15.3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parrish J. R., Limjindaporn T., Hines J. A., Liu J., Liu G., and Finley R. L. (2004) High-throughput cloning of Campylobacter jejuni ORFs by in vivo recombination in Escherichia coli. J. Proteome Res. 3, 582–586 10.1021/pr0341134 [DOI] [PubMed] [Google Scholar]
- 30. Li J., Li C., Xiao W., Yuan D., Wan G., and Ma L. (2008) Site-directed mutagenesis by combination of homologous recombination and DpnI digestion of the plasmid template in Escherichia coli. Anal. Biochem. 373, 389–391 10.1016/j.ab.2007.10.034 [DOI] [PubMed] [Google Scholar]
- 31. Zhu D., Zhong X., Tan R., Chen L., Huang G., Li J., Sun X., Xu L., Chen J., Ou Y., Zhang T., Yuan D., Zhang Z., Shu W., and Ma L. (2010) High-throughput cloning of human liver complete open reading frames using homologous recombination in Escherichia coli. Anal. Biochem. 397, 162–167 10.1016/j.ab.2009.10.018 [DOI] [PubMed] [Google Scholar]
- 32. Beyer H. M., Gonschorek P., Samodelov S. L., Meier M., Weber W., and Zurbriggen M. D. (2015) AQUA cloning: a versatile and simple enzyme-free cloning approach. PLoS One 10, e0137652 10.1371/journal.pone.0137652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yao Z., Jones D. H., and Grose C. (1992) Site-directed mutagenesis of herpesvirus glycoprotein phosphorylation sites by recombination polymerase chain reaction. PCR Methods Appl. 1, 205–207 10.1101/gr.1.3.205 [DOI] [PubMed] [Google Scholar]
- 34. Martin A., Toselli E., Rosier M. F., Auffray C., and Devignes M. D. (1995) Rapid and high efficiency site-directed mutagenesis by improvement of the homologous recombination technique. Nucleic Acids Res. 23, 1642–1643 10.1093/nar/23.9.1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Howorka S., and Bayley H. (1998) Improved protocol for high-throughput cysteine scanning mutagenesis. BioTechniques 25, 764–772 10.2144/98255bm03 [DOI] [PubMed] [Google Scholar]
- 36. Wu Y., You L., Li S., Ma M., Wu M., Ma L., Bock R., Chang L., and Zhang J. (2017) In vivo assembly in Escherichia coli of transformation vectors for plastid genome engineering. Front. Plant Sci. 8, 1454 10.3389/fpls.2017.01454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lacks S., and Greenberg B. (1975) A deoxyribonuclease of Diplococcus pneumoniae specific for methylated DNA. J. Biol. Chem. 250, 4060–4066 [PubMed] [Google Scholar]
- 38. Jones D. H., and Winistorfer S. C. (1993) Use of polymerase chain reaction for making recombinant constructs. Methods Mol. Biol. 15, 241–250 10.1385/0-89603-244-2:241 [DOI] [PubMed] [Google Scholar]
- 39. Jones D. H., and Winistorfer S. C. (1997) Recombination and site-directed mutagenesis using recombination PCR. Methods Mol. Biol. 67, 131–140 10.1385/0-89603-483-6:131 [DOI] [PubMed] [Google Scholar]
- 40. Jones D. H., and Winistorfer S. C. (2003) Recombination and site-directed mutagenesis using recombination PCR. Methods Mol. Biol. 226, 517–524 10.1385/1-59259-384-4:517 [DOI] [PubMed] [Google Scholar]
- 41. Jones D. H., and Winistorfer S. C. (1992) Recombinant circle PCR and recombination PCR for site-specific mutagenesis without PCR product purification. BioTechniques 12, 528–530, 532,, 534–535 [PubMed] [Google Scholar]
- 42. Jones D. H. (1994) PCR mutagenesis and recombination in vivo. PCR Methods Appl. 3, S141–S148 10.1101/gr.3.6.S141 [DOI] [PubMed] [Google Scholar]
- 43. Li C., Wen A., Shen B., Lu J., Huang Y., and Chang Y. (2011) FastCloning: a highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 11, 92 10.1186/1472-6750-11-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hultman T., Murby M., Ståhl S., Hornes E., and Uhlén M. (1990) Solid phase in vitro mutagenesis using plasmid DNA template. Nucleic Acids Res. 18, 5107–5112 10.1093/nar/18.17.5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lai D., Zhu X., and Pestka S. (1993) A simple and efficient method for site-directed mutagenesis with double-stranded plasmid DNA. Nucleic Acids Res. 21, 3977–3980 10.1093/nar/21.17.3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zaret K. S., Liu J. K., and DiPersio C. M. (1990) Site-directed mutagenesis reveals a liver transcription factor essential for the albumin transcriptional enhancer. Proc. Natl. Acad. Sci. 87, 5469–5473 10.1073/pnas.87.14.5469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schulga A. A., Levichkin I. V., Kurkbanov F. T., Okorokov A. L., Pozmogova G. E., and Kirpichnikov M. P. (1994) An approach to construction of hybrid polypeptide molecules: homologue recombination method. Nucleic Acids Res. 22, 3808–3810 10.1093/nar/22.18.3808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oliner J. D., Kinzler K. W., and Vogelstein B. (1993) In vivo cloning of PCR products in E. coli. Nucleic Acids Res. 21, 5192–5197 10.1093/nar/21.22.5192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Petit M. (2005) Mechanisms of homologous recombination in bacteria. In The Dynamic Bacterial Genome (Mullany P., ed) pp. 3–32, Cambridge University Press, Cambridge, UK: 10.1017/CBO9780511541544.001 [DOI] [Google Scholar]
- 50. Lee J., Rha E., Yeom S.-J., Lee D.-H., Choi E.-S., and Lee S.-G. (2013) Generating in vivo cloning vectors for parallel cloning of large gene clusters by homologous recombination. PLoS One 8, e79979 10.1371/journal.pone.0079979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McInerney P., Adams P., and Hadi M. Z. (2014) Error rate comparison during polymerase chain reaction by DNA polymerase. Mol. Biol. Int. 2014, 287430 10.1155/2014/287430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Conley E. C., Saunders V. A., and Saunders J. R. (1986) Deletion and rearrangement of plasmid DNA during transformation of Escherichia coli with linear plasmid molecules. Nucleic Acids Res. 14, 8905–8917 10.1093/nar/14.22.8905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Conley E. C., Saunders V. A., Jackson V., and Saunders J. R. (1986) Mechanism of intramolecular recyclization and deletion formation following transformation of Escherichia coli with linearized plasmid DNA. Nucleic Acids Res. 14, 8919–8932 10.1093/nar/14.22.8919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bi X., and Liu L. F. (1994) recA-independent and recA-dependent intramolecular plasmid recombination: differential homology requirement and distance effect. J. Mol. Biol. 235, 414–423 10.1006/jmbi.1994.1002 [DOI] [PubMed] [Google Scholar]
- 55. Nakano Y. J., and Kuramitsu H. K. (1992) Mechanism of Streptococcus mutans glucosyltransferases: hybrid-enzyme analysis. J. Bacteriol. 174, 5639–5646 10.1128/jb.174.17.5639-5646.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dutra B. E., Sutera V. A. Jr., and Lovett S. T. (2007) RecA-independent recombination is efficient but limited by exonucleases. Proc. Natl. Acad. Sci. U.S.A. 104, 216–221 10.1073/pnas.0608293104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nozaki S., and Niki H. (2019) Exonuclease III (XthA) enforces in vivo DNA cloning of Escherichia coli to create cohesive ends. J. Bacteriol. 201, e00660–18 10.1128/JB.00660-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hsiao K. (1993) Exonuclease III induced ligase-free directional subcloning of PCR products. Nucleic Acids Res. 21, 5528–5529 10.1093/nar/21.23.5528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kaluz S., Kölble K., and Reid K. B. M. (1992) Directional cloning of PCR products using exonuclease III. Nucleic Acids Res. 20, 4369–4370 10.1093/nar/20.16.4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Morrical S. W. (2015) DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb. Perspect. Biol. 7, a016444 10.1101/cshperspect.a016444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lovett S. T., Hurley R. L., Sutera V. A. Jr., Aubuchon R. H., and Lebedeva M. A. (2002) Crossing over between regions of limited homology in Escherichia coli: RecA-dependent and RecA-independent pathways. Genetics 160, 851–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gupta R., Ryzhikov M., Koroleva O., Unciuleac M., Shuman S., Korolev S., and Glickman M. S. (2013) A dual role for mycobacterial RecO in RecA-dependent homologous recombination and RecA-independent single-strand annealing. Nucleic Acids Res. 41, 2284–2295 10.1093/nar/gks1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gupta R., Barkan D., Redelman-Sidi G., Shuman S., and Glickman M. S. (2011) Mycobacteria exploit three genetically distinct DNA double-strand break repair pathways. Mol. Microbiol. 79, 316–330 10.1111/j.1365-2958.2010.07463.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xu G., Lu H., Wang L., Chen H., Xu Z., Hu Y., Tian B., and Hua Y. (2010) DdrB stimulates single-stranded DNA annealing and facilitates RecA-independent DNA repair in Deinococcus radiodurans. DNA Repair 9, 805–812 10.1016/j.dnarep.2010.04.006 [DOI] [PubMed] [Google Scholar]
- 65. Chayot R., Montagne B., Mazel D., and Ricchetti M. (2010) An end-joining repair mechanism in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 107, 2141–2146 10.1073/pnas.0906355107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Verma P., and Greenberg R. A. (2016) Noncanonical views of homology-directed DNA repair. Genes Dev. 30, 1138–1154 10.1101/gad.280545.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ma X., Liang X., and Huo Y.-X. (2019) Developing a transformation-independent and unbiased qPCR assay to rapidly evaluate the determinants of DNA assembly efficiency. Engineering 5, 803–810 10.1016/j.eng.2019.06.002 [DOI] [Google Scholar]
- 68. Fisher A. B., Canfield Z. B., Hayward L. C., Fong S. S., and McArthur G. H. 4th (2013) Ex vivo DNA assembly. Front. Bioeng. Biotechnol. 1, 12 10.3389/fbioe.2013.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Klock H. E., Koesema E. J., Knuth M. W., and Lesley S. A. (2008) Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins 71, 982–994 10.1002/prot.21786 [DOI] [PubMed] [Google Scholar]
- 70. Olsen D. B., and Eckstein F. (1989) Incomplete primer extension during in vitro DNA amplification catalyzed by Taq polymerase; exploitation for DNA sequencing. Nucleic Acids Res. 17, 9613–9620 10.1093/nar/17.23.9613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liu H., and Naismith J. H. (2008) An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91 10.1186/1472-6750-8-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Qi D., and Scholthof K.-B. G. (2008) A one-step PCR-based method for rapid and efficient site-directed fragment deletion, insertion, and substitution mutagenesis. J. Virol. Methods 149, 85–90 10.1016/j.jviromet.2008.01.002 [DOI] [PubMed] [Google Scholar]
- 73. Xia Y., Chu W., Qi Q., and Xun L. (2015) New insights into the QuikChangeTM process guide the use of Phusion DNA polymerase for site-directed mutagenesis. Nucleic Acids Res. 43, e12 10.1093/nar/gku1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zheng L., Baumann U., and Reymond J. L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 10.1093/nar/gnh110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Aricescu A. R., Lu W., and Jones E. Y. (2006) A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D Biol. Crystallogr. 62, 1243–1250 10.1107/S0907444906029799 [DOI] [PubMed] [Google Scholar]
- 76. Marsic D., Hughes R. C., Byrne-Steele M. L., and Ng J. D. (2008) PCR-based gene synthesis to produce recombinant proteins for crystallization. BMC Biotechnol. 8, 44 10.1186/1472-6750-8-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Henke W., Herdel K., Jung K., Schnorr D., and Loening S. A. (1997) Betaine improves the PCR amplification of GC-rich DNA sequences. Nucleic Acids Res. 25, 3957–3958 10.1093/nar/25.19.3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mousavian Z., Sadeghi H. M., Sabzghabaee A. M., and Moazen F. (2014) Polymerase chain reaction amplification of a GC rich region by adding 1,2 propanediol. Adv. Biomed. Res. 3, 65 10.4103/2277-9175.125846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Murphy K. C., and Marinus M. G. (2010) RecA-independent single-stranded DNA oligonucleotide-mediated mutagenesis. F1000 Biol. Rep. 2, 56 10.3410/B2-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kingston A. W., Roussel-Rossin C., Dupont C., and Raleigh E. A. (2015) Novel recA-independent horizontal gene transfer in Escherichia coli K-12. PLoS One 10, e0130813 10.1371/journal.pone.0130813 [DOI] [PMC free article] [PubMed] [Google Scholar]