

Abstract
T-cell activation requires stimulation of specific intracellular signaling pathways in which protein-tyrosine kinases, phosphatases, and adapter proteins interact to transmit signals from the T-cell receptor to the nucleus. Interactions of LCK proto-oncogene, SRC family tyrosine kinase (LCK), and the IL-2–inducible T cell kinase (ITK) with the T cell-specific adapter protein (TSAD) promotes LCK-mediated phosphorylation and thereby ITK activation. Both ITK and LCK interact with TSAD's proline-rich region (PRR) through their Src homology 3 (SH3) domains. Whereas LCK may also interact with TSAD through its SH2 domain, ITK interacts with TSAD only through its SH3 domain. To begin to understand on a molecular level how the LCK SH3 and ITK SH3 domains interact with TSAD in human HEK293T cells, here we combined biochemical analyses with NMR spectroscopy. We found that the ITK and LCK SH3 domains potentially have adjacent and overlapping binding sites within the TSAD PRR amino acids (aa) 239–274. Pulldown experiments and NMR spectroscopy revealed that both domains may bind to TSAD aa 239–256 and aa 257–274. Co-immunoprecipitation experiments further revealed that both domains may also bind simultaneously to TSAD aa 242–268. Accordingly, NMR spectroscopy indicated that the SH3 domains may compete for these two adjacent binding sites. We propose that once the associations of ITK and LCK with TSAD promote the ITK and LCK interaction, the interactions among TSAD, ITK, and LCK are dynamically altered by ITK phosphorylation status.
Keywords: Src homology 3 domain (SH3 domain), adaptor protein, T-cell, protein phosphorylation, protein structure, protein-protein interaction, protein kinase, cell signaling, NMR, nuclear magnetic resonance, SH2D2A, tyrosine-protein kinase, LCK proto-oncogene SRC family tyrosine kinase, IL-2-inducible T cell kinase (ITK), T cell-specific adapter protein (TSAD), immunity
Introduction
T-cell activation relies upon stimulation of specific intracellular signal-transduction pathways, where protein-tyrosine kinases (PTKs),3 phosphatases, and adapter molecules interact to convey the signal from the T-cell receptor (TCR) to the nucleus. Adapter molecules present specific protein interaction sites but lack intrinsic enzymatic activity (1). These molecules bridge interactions between other proteins in the signaling cascade, such as the PTKs and their substrates. PTKs in T cells include at least four families: the Src family, the SYK family, the CSK family, and the TEC family (2). The TEC family IL-2–inducible tyrosine kinase (ITK) is recruited to the membrane after TCR triggering and is activated by the SRC family PTK lymphocyte–specific kinase (LCK). This results in activation of phospholipase C γ1, eventually leading to gene transcription via intracellular Ca2+ release and transcription factor activation (reviewed in Ref. 3).
The T cell–specific adapter protein (TSAD), encoded by the SH2D2A gene, is an adapter molecule containing a SRC homology 2 (SH2) domain and a proline-rich region (PRR) in addition to several tyrosine phosphorylation sites (4, 5). The gene was first isolated from an activated CD8+ T-cell cDNA library by differential screening (4) and was later identified as an interaction partner to other signaling molecules, including LCK (6) and ITK (7).
TSAD influences T-cell signaling via its interaction with the tyrosine kinases LCK and ITK (8). Both ITK and TSAD are expressed at low levels in naive T cells but are induced upon activation of T cells (4, 9, 10). Absence of TSAD results in deficient polarization of actin to the immunological synapse (8) as well as deficient activation-induced cell death (11).
Both LCK and ITK consist of multiple domains, including one SH2 and one SRC homology 3 (SH3) domain (12). SH3 domains are regions of ∼60–70 amino acids (aa) present in many signaling molecules. The general structure of this domain consists of two short anti-parallel β-sheets packed against each other, resulting in a fold with a relatively flat hydrophobic surface containing at least one conserved aromatic (often a tryptophan) residue (13). SH3 domains bind to PRRs that can contain the prototypic SH3-binding motif PXXP belonging to one of two classes. Class I motifs have the consensus sequence +pΨPpΨP, and class II motifs have the consensus sequence ΨPpΨPp+. Ψ represents an aliphatic residue, + indicates a basic residue, uppercase positions are conserved residues that contact the SH3 domain and provide the specificity, and lowercase residues represent scaffolding positions, usually prolines (14). Ligand orientation is generally determined by the position of the basic residue, often an arginine, relative to the core proline residues (15).
We have previously mapped the interaction site of ITK on TSAD and showed that ITK binds with its SH3 domain to the PRR of TSAD (aa 239–274) (10), in a region coincident with that of LCK SH3 interacting with TSAD (16). Interaction of TSAD with ITK promoted LCK-mediated tyrosine phosphorylation of ITK as well as migration of T cells upon CXCL12 stimulation. T cells lacking TSAD failed to induce tyrosine phosphorylation of ITK as well as migration of T cells upon CXCL12 stimulation (10). Whereas ITK interacts with TSAD exclusively through its SH3 domain (10), LCK interacts with TSAD through both its SH3 and its SH2 domain (17). Thus, promotion of LCK-mediated phosphorylation of ITK by TSAD could be either due to simultaneous docking and interaction of LCK and ITK on TSAD or simply due to binding of LCK to TSAD activating LCK catalytic activity, thus facilitating phosphorylation of ITK.
In an effort to begin to understand the molecular details defining the association of TSAD, LCK, and ITK, we performed peptide array binding experiments, biochemical analyses, and 2D (1H, 15N) heteronuclear single quantum coherence (HSQC) NMR experiments to examine the interaction between TSAD, ITK SH3, and LCK SH3. Our data support a model whereby LCK and ITK are both recruited to TSAD via their SH3 domains, allowing LCK-mediated phosphorylation of ITK. Our results point to a complex protein assembly, where the ITK SH3 and LCK SH3 domains can both compete for and simultaneously bind to adjacent binding sites on TSAD encompassing aa 242–268. Our findings also suggest that, once active, ITK autophosphorylation of its own SH3 domain disrupts the ITK/TSAD interaction, possibly creating an open binding site on TSAD for recruiting and activation of another ITK molecule by LCK.
Results
Interaction of TSAD with ITK depends on the intact ITK SH3 domain
Expression of the adapter molecule TSAD (Fig. 1A) is induced in human T cells upon TCR triggering (9) concomitant with ITK (10). Murine TSAD was first cloned as an ITK/RLK interaction partner; thus, an alternative designation for TSAD is RLK/ITK-binding protein (RIBP) (7). Following up on this observation, we have previously found that the interaction of TSAD with ITK depends on the PRR of TSAD (10), indicating a role for the ITK SH3 domain in mediating this interaction. To reveal whether an intact ITK SH3 domain is required for interaction of ITK with TSAD, we expressed full-length Myc-tagged ITK constructs in 293T cells and performed co-immunoprecipitation experiments. Immunoprecipitation of ITK led to co-immunoprecipitation of TSAD only in the presence of WT ITK, whereas ITK containing the W208K mutation, which is known to abolish binding of the ITK SH3 domain to its ligands (18), did not co-immunoprecipitate with TSAD (Fig. 1B).
Figure 1.

Nonphosphorylated ITK SH3 domain is required for ITK binding to TSAD. A, schematic drawing of TSAD including the SH2 domain, the PRR and the three C-terminal tyrosines. The core sequences encoded by the TSAD cDNA constructs and the TSAD peptides used for transfection of cells and for NMR experiments, respectively, in this paper are also indicated. B, co-immunoprecipitation experiment showing the dependence of ITK-TSAD interaction on the SH3 domain of ITK. 293T cells were transfected with the indicated cDNA plasmids. Myc-tagged ITK proteins were immunoprecipitated from the cell lysates, followed by immunoblotting with the indicated antibodies. The result is one representative of two experiments. C and D, ITK SH3 domains mutated for Tyr180 display reduced interaction with TSAD. C, pulldown experiment using ITK SH3 domains with the indicated mutations was performed using lysates of 293T cells transiently transfected with HA-tagged WT and mutated TSAD cDNA. Pulled down proteins were immunoblotted with the indicated antibodies. D, the graph represents relative amount of TSAD interacting with ITK SH3 in the experiment shown in C. Signals were quantified by ImageJ analysis (n = 3, mean ± S.D. (error bars)).
We previously also showed that TSAD promotes LCK-mediated phosphorylation of ITK on Tyr511 (10). This phosphorylation event activates the ITK kinase and leads to autophosphorylation of ITK Tyr180 in the ITK SH3 domain (19). It has been shown that the specificity of ITK SH3 for its ligands may be altered by Tyr180 phosphorylation (20). To examine whether binding of ITK SH3 to TSAD was affected by phosphorylation of ITK Tyr180, we generated Tyr → Phe and Tyr → Glu mutated versions of the ITK SH3 domain.
Both the ITK SH3 Y180F and the Y180E mutant proteins displayed significantly reduced interaction with TSAD compared with the WT ITK SH3 domain (Fig. 1, C and D). Also, compared with the ITK SH3 Y180F mutant, the Y180E mutant, where the negative charge and the size of the glutamate in the ITK-Y180E mutant may mimic ITK phosphorylated (p)Tyr180, displayed significantly reduced binding to TSAD (Fig. 1D). Taken together, these data show that interaction of TSAD with ITK is mediated by ITK SH3; furthermore, they suggest that the physiological association between the two molecules is regulated by the phosphorylation status of ITK.
LCK SH3 and ITK SH3 bind to overlapping peptides in TSAD PRR
Having established that ITK SH3 binds to TSAD, we performed in silico motif analysis (https://scansite4.mit.edu/4.0/#home)4 (21) using the human TSAD amino acid sequence as input (GenBank number Q9NP31) and the default matrix for ITK SH3 ligands. This analysis revealed possible ITK SH3–binding sites at TSAD Pro263 and Pro247 (Fig. 2A). TSAD Pro247 was also indicated as a potential SRC-SH3 interaction site (data not shown). We then went on to dissect the interaction between the ITK SH3 domain and the PRR of TSAD compared with that of the LCK SH3 domain, which is known to bind to TSAD (17). A series of 14 overlapping peptides of 20 aa with a sliding window of three aa was synthesized and fixed onto a cellulose membrane. This peptide array covered most of the PRR of TSAD (aa 233–291) (Fig. 2B). The membrane was probed with recombinant GST-ITK SH3 and GST-LCK SH3 domains and immunoblotted with anti-GST and anti-mouse-HRP antibodies. The results show that ITK SH3 and LCK SH3 domains bind to overlapping peptides in the PRR of TSAD. However, the peptide array blotting analysis indicates that ITK SH3 prefers a binding site that is C-terminal relative to that preferred by LCK SH3 (Fig. 2A). The mutant ITK SH3 domain W208K (ITK-K) did not bind to any of the TSAD peptides.
Figure 2.

LCK SH3 and ITK SH3 bind to overlapping peptides in TSAD PRR. A, Scansite prediction of ITK SH3-binding sites on TSAD. B, peptide array mapping of LCK SH3- and ITK SH3-binding sites on TSAD. The amino acid sequence of the peptide in each of the spots is given to the left. The right panels show the results of probing the arrays with the indicated recombinant proteins GST-ITK SH3, GST-LCK SH3, GST-ITK SH3 mutated (W208K or ITK-K), or GST alone, respectively. Results from 12 of the 14 tested peptides are shown. Pro247 and Pro263 are indicated in boldface type. The brackets define the borders of TSAD aa 239–274. C and D, mutations of the predicted prolines to alanines fail to abolish binding of ITK SH3 to TSAD. The pulldown experiment was performed using GST-ITK SH3 and lysates of 293T cells transiently transfected with HA-tagged WT and mutated TSAD cDNA. Pulled down protein and lysate were immunoblotted with the indicated antibodies. D, the graph represents the relative amount of TSAD interacting with ITK SH3 in the experiment shown in C. Signals were quantified by ImageJ analysis (n = 3, mean ± S.D. (error bars)). E and F, deletion of either the Pro247 (aa 239–256) or the Pro263 (aa 257–274) region reduces binding of ITK SH3 to TSAD. The pulldown experiment was performed as in C. F, the graph represents ImageJ analysis as in D of the data shown in E (n = 3 or 6, mean ± S.D.). G and H, additional mutation of Pro263 further reduces binding of ITK SH3 to TSAD 257–274. The pulldown experiment was performed as in C. H, the graph represents ImageJ analysis as in D of data shown in G. The relative amount of TSAD interacting with ITK SH3 WT in each experiment is set to 1 (not shown) (n = 3, mean ± S.D.).
The ITK SH3-binding motif in TSAD PRR is focused on Pro263
The motif formed by the TSAD sequence flanking Pro263 (PΨPXXRP) is also found in the ITK SH3-binding motifs of SLP-76 and in CBL-B (18, 22, 23) as well as putative ITK SH3 ligands, as predicted by Scansite (Table 1). Mutation of either Pro247 or Pro263 to alanine did not significantly abolish binding of ITK SH3 to TSAD in pulldown experiments (Fig. 2, C and D), indicating that more than one amino acid in these regions is involved in binding to the ITK SH3 domain. Consistent with the presence of two ITK SH3 interaction sites on TSAD within aa 239–273 (Fig. 2, A and B), we found that a deletion mutant of TSAD lacking both the Pro247 and the Pro263 motifs (TSAD Δ239–274) displayed minimal binding to ITK SH3 (Fig. 2, E (lane 3) and F) as has been shown previously (10). In contrast, separate deletion constructs encompassing either Pro247 or Pro263, TSAD Δ239–256 and TSAD Δ257–274, respectively, both retained some binding to ITK SH3 (Figs. 1A and 2E (lanes 4 and 5)). The deletion construct lacking Pro263 (TSAD Δ257–274) showed a significantly greater reduction in binding ITK SH3 compared with TSAD Δ239–256 (Fig. 2F).
Table 1.
Verified and putative ITK SH3 motifs
The observation that neither of the two shorter deletions completely eliminated ITK SH3 interaction indicated that the continued presence of the remaining Pro247 or Pro263 motifs were sufficient for ITK SH3 interaction. Mutation of Pro263 to Ala263 in TSAD Δ239–256 resulted in significantly reduced interaction of ITK SH3 with TSAD. Also, the corresponding Pro247 to Ala247 mutation in TSAD Δ257–274 conferred reduced interaction with ITK SH3; however, the difference failed to reach statistical significance (Fig. 2, G and H). Taken together, we therefore hypothesized that the interaction of TSAD Pro263 with ITK SH3 was the most likely to be of biological relevance and hence set out to analyze this interaction in further detail using NMR spectroscopy.
The human ITK SH3 domain binds to TSAD aa 262–269 in a class I orientation
We determined the NMR resonance assignments (deposited as BMRB entry 18119) and three-dimensional structure of the human ITK SH3 domain (PDB entry 2LMJ) using standard methods (24). The structures are shown in Fig. 3, and the structure statistics are shown in Table 2. The obtained structure showed an SH3 structure similar to that of murine ITK SH3 (PDB entry 2RNA) (25). Similarly, no major differences are observed between this structure and the NMR structure of human ITK SH3 determined by the RIKEN structural genomics initiative (PDB entry 2YUQ).
Figure 3.
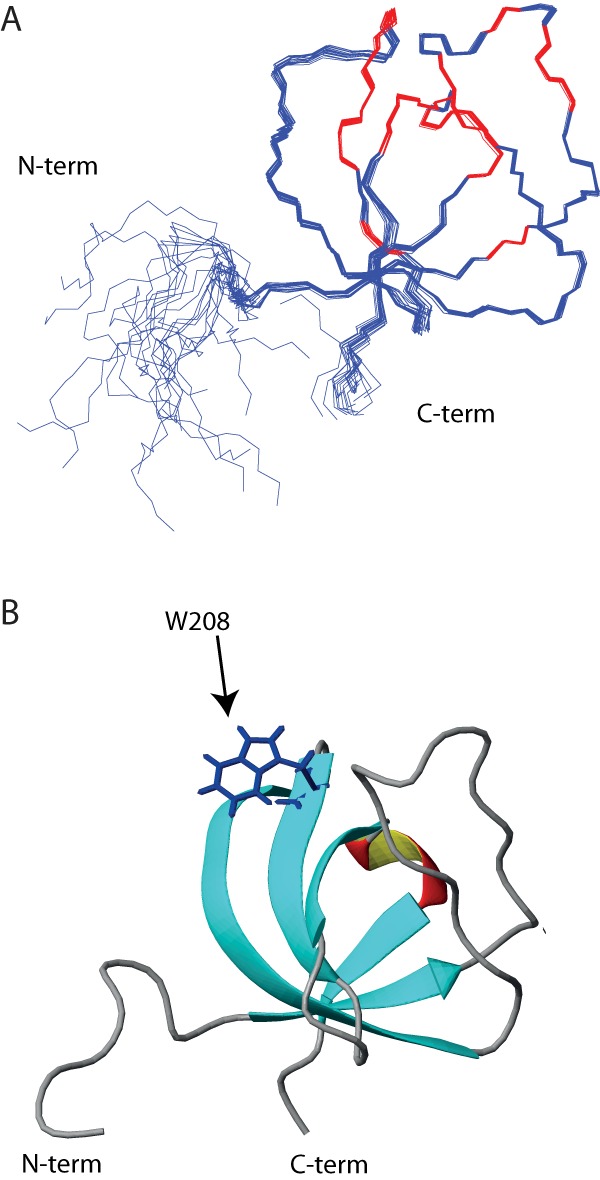
The human ITK SH3 domain solution structure. A, backbone trace of the 20 structures comprising the lowest-energy NMR ensemble is shown. Red color indicates residues affected by ligand binding. B, ribbon representation of one of the ITK SH3 domain structures shown in A. The tryptophan at position 208, which is critical for polyproline ligand binding, is indicated.
Table 2.
Constraints and structural statistics of ITK-SH3 NMR structure
| Parameter | Value |
|---|---|
| Constraints | |
| NOE constraints | 1,267 |
| Intraresidue | 267 |
| Sequential (ji − jj = 1) | 337 |
| Medium range (1 < ji − jj < 5) | 139 |
| Long-range (ji − jj > 5) | 524 |
| Hydrogen bonds/constraints | 17/34 |
| Dihedral angle constraints | 80 |
| Total no. of constraints | 1,381 |
| Structural statistics | |
| Constraint violations | |
| Distance constraints (Å) | 0.0063 ± 0.0008 |
| Dihedral angle constraints (degrees) | 0.57 ± 0.04 |
| Maximum distance constraint violation (Å) | <0.01 |
| Maximum dihedral constraint violation (degrees) | <4 |
| r.m.s. difference to mean structure residues 8–65 | |
| Backbone (Å) | 0.29 ± 0.09 |
| Heavy atoms (Å) | 0.98 ± 0.15 |
| Ramachandran statistics (%) | |
| Core regions | 84.1 |
| Allowed regions | 15.9 |
| Generous regions | 0 |
| Disallowed regions | 0 |
| CYANA (Å) | |
| Total | 0.54 ± 0.0616 |
A series of 2D 1H-15N HSQC experiments were recorded after titration of increasing amounts of TSAD peptide. The peptide *IPVPRHRPAPRPKPSNP# (aa 262–278) was chosen for analysis because it contains both a partial class II motif including Pro263 and a class I motif (15) corresponding to the consensus sequences PXXPX(R/K) and (R/K)XXPXXP, respectively. N-terminal and C-terminally truncated versions of the peptide (aa 262–269 (*IPVPRHRP#) containing the partial class II motif and aa 271–278 (*PRPKPSNP#) containing the class I motif) were also analyzed. The chemical shift changes that occurred upon ligand binding to the N-terminal peptide aa 262–269 (Fig. 4A) indicate SH3 residues that were affected by ligand binding. The longer aa 262–278 peptide gave similar shifts as the aa 262–269 peptide, whereas the aa 271–278 peptide did not induce chemical shift changes (data not shown). The chemical shift changes induced upon binding of TSAD aa 262–269 as well as TSAD aa 262–278 mapped within the conventional binding surface of SH3 domains (13) onto the structure of the human ITK SH3 (data not shown). These data did not, however, reveal information about the orientation of the peptide binding to ITK SH3.
Figure 4.

The human ITK SH3 domain binds to TSAD aa 262–269 in a class I orientation. A, 1H-15N HSQC of human 20 μm ITK SH3 domain without (blue) and with increasing amounts (200 μm to 1 mm, light blue to green-red) of TSAD aa 262–269 (*IPVPRHRP#) added. Chemical shift changes are observed indicating binding of the peptide to the ITK SH3 domain. B, TOAC experiment. HSQC experiments using TSAD aa 262–269 with an N-terminal TOAC aa were performed with and without a reducing agent (ascorbate) added to the solution to remove the effect of the TOAC aa. TOAC aa causes broadening of peaks in the NMR spectrum representing neighboring aa. The graph shows the percentage reduction in peak volume of the four most affected aa (Leu201, Trp208, Arg214, and Val176) in addition to three aa (Trp209, Val221 and Ser223) that the titration experiments had identified to be affected by peptide binding. C, three-dimensional structure of human ITK SH3 domain with the TOAC-labeled TSAD peptide aa 262–269 (light gray) docked onto the SH3 domain using constraints given by the aa most affected by the peptide titration. The N and C termini of the TSAD peptide are labeled N-term and C-term, respectively. The location of Leu201, Trp208, Arg214, and Val176 (resonances most affected by TOAC) are shown on the SH3 structure.
To map the binding orientation of the peptide, we used a custom made spin-labeled peptide containing a helicogenic nitroxyl aa (i.e. 2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4-carboxyl acid (TOAC)) at its N terminus. The TOAC aa in the bound peptide will influence the chemical environment of the ITK SH3 domain upon peptide binding and cause broadening of peaks in the NMR spectrum corresponding to aa in proximity of the spin label. Comparison of 1H-15N HSQC spectra of 500 μm 15N-labeled recombinant human ITK SH3 domain alone and in complex with 1 mm TOAC-peptide revealed that the TOAC label most strongly affected the peaks corresponding to Leu201, Trp208, Arg214, and Val176, with a 25–35% reduction in peak volumes (Fig. 4B). These data indicate that binding of the TSAD peptide to ITK SH3 brings the N-terminal TOAC moiety close to these aa, which all are located in the vicinity of the specificity pocket of human ITK SH3 domain. Based on our data, we thus modeled the TSAD peptide bound to ITK SH3 with the N-terminal pointing toward the specificity pocket by application of HADDOCK (26). The model of the TOAC-labeled TSAD peptide bound to ITK SH3 is shown in Fig. 4C. Taken together, our data indicate that the TSAD peptide aa 262–269 on human ITK SH3 is in a class I binding orientation (15) with the N-terminal end of the peptide ligand pointing toward the specificity pocket.
Both LCK SH3 and ITK SH3 may bind simultaneously to the TSAD PRR
Because LCK SH3 and ITK SH3 bind to overlapping peptides in the TSAD PRR (Fig. 2A) and the ITK SH3 domain binds to the TSAD aa 262–269 peptide (Fig. 4), we directly assessed whether LCK SH3 and ITK SH3 may bind to adjacent sites on the same TSAD peptide. We first performed pulldown of TSAD deletion mutants using GST-LCK SH3 as a bait (Fig. 5, A–C). The results showed that whereas TSAD Δ239–274 essentially abolished interaction of LCK SH3 with TSAD, both of the two shorter deletions (TSAD Δ239–256 and Δ257–274) retained some binding to TSAD (Fig. 5, A and C). Mutation of Pro263 to Ala263 in TSAD Δ239–256 significantly reduced interaction of LCK SH3 further, whereas the mutation Pro247 to Ala247 in TSAD Δ257–274 did not significantly reduce interaction with LCK SH3 (Fig. 5, B and C).
Figure 5.
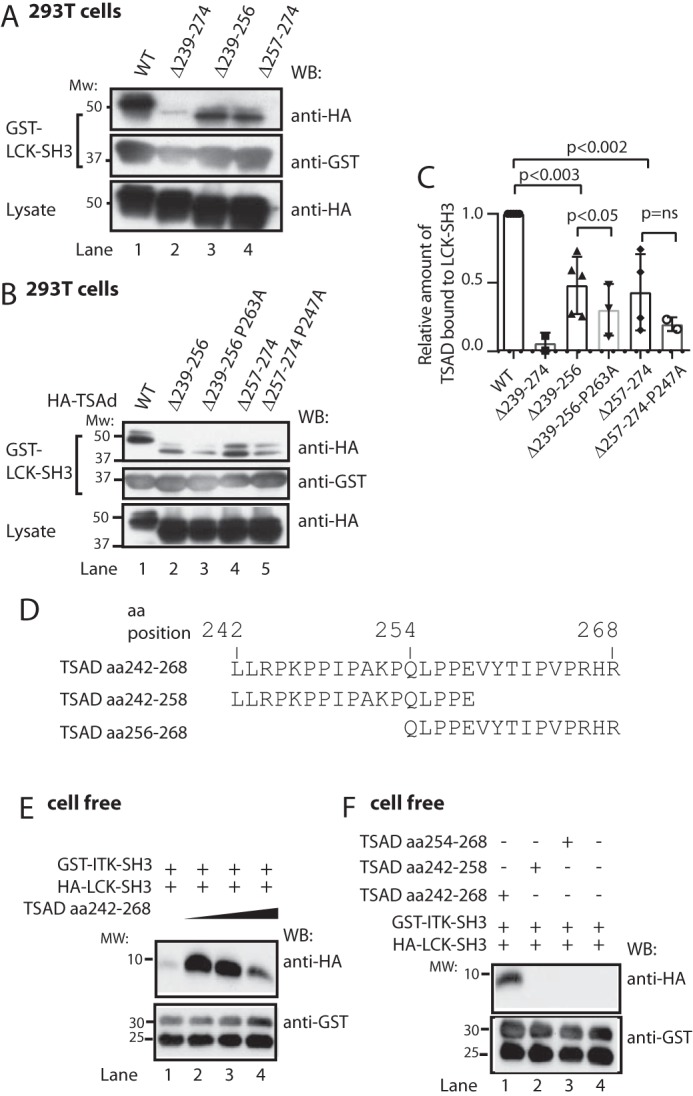
LCK SH3 and ITK SH3 may bind simultaneously to TSAD aa 242–268. A–C, LCK SH3 interacts with both TSAD aa 239–256 and aa 257–274. Additional mutation of Pro247 or Pro263 further reduces binding. A and B, pulldown experiment in 293T cells using the same TSAD constructs as in Fig. 2, E and G, respectively. Pulled down proteins and lysates were probed with the indicated antibodies. C, the graph represents ImageJ analysis of data shown in A and B (n = 3, mean ± S.D. (error bars)). D, sequences of custom synthesized TSAD peptides aa 242–268, 242–256, and 254–268. E, in vitro pulldown experiment using a 10 μm concentration of each SH3 domain and increasing amounts of TSAD aa 242–268 (10, 20, and 40 μm) in a total volume of 100 μl. The GST-ITK SH3 domain was added while attached to glutathione SepharoseTM beads. To eliminate bead loss, a 4-fold amount of GST-glutathione SepharoseTM beads was added to the mixture, as is evidenced from the bottom panel. F, in vitro pulldown experiment performed as in D, using a 10 μm concentration of each SH3 domain, and a 10 μm concentration of the indicated TSAD peptides. WB, Western blot.
To further substantiate that LCK SH3 and ITK SH3 both bind to adjacent sites in the TSAD PRR, a 27-amino acid peptide representing TSAD aa 242–268 (*LLRPKPPIPAKPQLPPEVYTIPVPRHR#), and two shorter peptides derived from TSAD aa 242–258 and aa 254–268 were synthesized (Figs. 1A and 5D). An in vitro pulldown experiment using an equimolar mixture of GST-ITK SH3 (bound to GSH SepharoseTM beads) with soluble HA-tagged LCK SH3 in the absence or the presence of increasing amounts of the TSAD aa 242–268 peptide was then performed. The result showed that GST-ITK SH3 associated with HA-LCK SH3 only in the presence of TSAD aa 242–268 (Fig. 5E). Moreover, increasing the concentration of TSAD aa 242–268 in the pulldown assay resulted in decreased association of HA-LCK SH3 with GST-ITK SH3 (Fig. 5E). This latter finding suggests that at higher concentrations of TSAD peptide, binary SH3/peptide complexes are favored over a ternary complex containing TSAD and two SH3 domains. We also found that only the long peptide (TSAD aa 242–268) facilitates association between the ITK and LCK SH3 domains, as identical pulldown experiments using the two shorter TSAD peptides (aa 242–258 and aa 254–268) resulted in no detectable association between GST-ITK SH3 and HA-LCK SH3 (Fig. 5F).
ITK SH3 competes with LCK SH3 for binding to TSAD PRR
To further assess the association between TSAD and the ITK and LCK SH3 domains, we introduced both SH3 domains into the same NMR experiment. The 15N-labeled LCK SH3 domain was first titrated with TSAD aa 242–268 (Fig. 6A). A subset of resonances showed chemical shift perturbations consistent with binding of the TSAD peptide to the LCK SH3 domain. Next, the unlabeled ITK SH3 domain was added to the 1:2 LCK SH3/TSAD peptide mixture in the NMR tube. For all LCK SH3 resonances that shifted upon the addition of TSAD aa 242–268, we observed resonance changes in the direction of unbound LCK SH3 (Fig. 6, A and B). Moreover, we did not detect chemical shift changes in the LCK SH3 domain outside of the peptide-binding pocket upon the addition of ITK SH3, suggesting a lack of interaction between the LCK and ITK SH3 domains themselves (data not shown). When we performed the experiment in the reverse order, using 15N-labeled ITK SH3 domain titrated with TSAD aa 242–268, and added unlabeled LCK SH3 domain to the 1:2 ITK SH3/TSAD peptide mixture in the NMR tube, similar results were obtained (data not shown).
Figure 6.

ITK SH3 competes with LCK SH3 for binding to TSAD aa 242–268. A, chemical shift deviations of selected residues in HSQC titration experiments performed with 15N-labeled LCK SH3 (initially 0.25 mm) in the presence of increasing concentration of TSAD aa 242–268 (0.25 mm (red) and 0.5 mm (green)) followed by increasing amounts of nonlabeled ITK SH3 (orange, yellow). B, chemical shift deviations of selected LCK residues from the HSQC experiment depicted in A (at 1:2:0.25, concentrations of LCK SH3, TSAD peptide, and ITK SH3 were 0.2, 0.4, and 0.04 mm respectively, whereas at 1:2:1, the corresponding concentrations were 0.1, 0.2, and 0.1 mm). C, chemical shift deviations of selected residues in HSQC titration experiments performed with 15N-labeled LCK SH3 and ITK SH3 added to the same NMR tube in the presence of increasing concentration of TSAD aa 242–258 or TSAD aa 254–268 peptide. D, titration curves for the indicated peptides based on HSQC shifts of selected amino acid signals from the indicated SH3 domains.
Taken together, our data presented in Figs. 2 and 5 suggest that ITK SH3 as well as LCK SH3 may each have two adjacent binding sites on TSAD. To further examine this notion, using NMR spectroscopy, we titrated an equimolar mixture of the two 15N-labeled isolated SH3 domains with either of the two shorter TSAD-derived peptides aa 242–258 and aa 254–268. Both SH3 domains displayed resonances that shifted upon the addition of both peptides, representing amino acids located to the peptide-binding surface of the SH3 domain (Fig. 6C). Each of the LCK SH3 or ITK SH3 resonances that shifted upon the addition of the peptides shifted in different directions, depending on the identity of the peptide (Fig. 6C), indicating that the chemical environments provided by the two peptides were indeed different. These results show that both SH3 domains may bind to two adjacent ligands contained within TSAD aa 242–268.
Finally, we characterized the binding of ITK SH3 and LCK SH3 with the TSAD PRR. Using the NMR titration data shown in Fig. 6C, where both SH3 domains were present in equimolar amounts, as well as similar titrations using TSAD aa 242–268 (not shown), we assessed the relative affinities of the different regions of the TSAD PRR for the ITK and LCK SH3 domains. TSAD aa 242–268, spanning the PRR, bound equally well in vitro to both ITK and LCK SH3 domains (Table 3 and Fig. 6D). TSAD aa 242–258, the N-terminal region of the PRR, also bound to both ITK and LCK SH3 domains (Table 3 and Fig. 6D). The dissociation constants for TSAD aa 242–268 and the N-terminal half of the same peptide, TSAD aa 242–256, were similar (Table 3), suggesting that most of the in vitro binding between the SH3 domains and the TSAD peptide is mediated by the aa stretch between residues 242 and 256. This was further supported by titrations using TSAD aa 254–268, representing the C-terminal half of the TSAD peptide. Chemical shift changes that resulted from titration of this peptide did not approach saturation within the range of concentrations tested (Fig. 6D). Thus, in this NMR titration experiment, the N-terminal half of the TSAD PRR appears to contain the preferred binding site for both ITK and LCK SH3 domains.
Table 3.
Kd values (mm) estimated from HSQC titration experiments
| Peptide | SH3 domain |
|
|---|---|---|
| ITK | LCK | |
| TSAD aa 242–256 | 150 ± 12 | 69 ± 3 |
| TSAD aa 242–268 | 123 ± 22 | 161 ± 75 |
| TSAD aa 254–268 | NDa | ND |
a ND, not determined.
The TSAD aa 239–274 sequence is required for LCK-mediated phosphorylation of ITK
Taken together, our data point to a model for the LCK, TSAD, ITK signaling system that involves binding of both kinases to the TSAD scaffold, to facilitate LCK-mediated activation of ITK. Both the peptide array data (Fig. 2A) and the mutagenesis and deletions within the TSAD sequence (Figs. 2 (E and F) and 5 (A–C)) as well as the NMR titration experiments suggest that the longer stretch of TSAD that includes Pro263 (TSAD aa 242–268) is involved in association with LCK and ITK.
To directly assess whether the longer stretch of TSAD is required for LCK-mediated phosphorylation of ITK, we transfected plasmids encoding the intact and truncated versions of TSAD together with LCK and Myc-tagged ITK into 293T cells and assessed the amount of ITK tyrosine phosphorylation. The results showed that in the absence of the PRR encoded by the aa 239–274, TSAD does not promote phosphorylation of ITK (Fig. 7A). However, when either aa 239–256 or aa 257–274 is present in TSAD, a 50% reduction in the phosphorylation of ITK is observed compared with that observed in the presence of intact TSAD (Fig. 7, A and B). This result indicates that both SH3-binding sites on TSAD contribute to the adapter function of TSAD, although the presence of both is not strictly required for TSAD to fulfill its role as a scaffold for LCK and ITK interaction.
Figure 7.

Association followed by dissociation of ITK to TSAD PRR is required for maximal phosphorylation of ITK in the presence of LCK. A, immunoprecipitation experiment showing phosphorylation of ITK in the absence or presence of intact or mutated TSAD molecules. 293T cells were transfected with the indicated cDNA plasmids. Myc-tagged ITK proteins were immunoprecipitated from the cell lysates, and the level of phosphorylation was assessed by immunoblotting. The result is one representative of two experiments. B, the graph shows quantitation of signal densities using ImageJ of the experiment shown in A (n = 4, mean ± S.D. (error bars)). C and D, immunoprecipitation experiments as in A, including also a Myc-ITK-Y180F mutant (n = 3, mean ± S.D.). E–G, immunoprecipitation experiments in 293T cells expressing the indicated plasmids immunoblotted with the indicated antibodies. F, graph shows quantitation of signal densities using ImageJ of the experiment shown in E and shows the relative amount of pTyr511 signal where the pTyr511 signal in ITK is set to 1 (n = 5, mean ± S.D.). H, the graph shows quantitation of signal densities using ImageJ of the experiment shown in G and shows the relative amount of ITK co-immunoprecipitated with TSAD (n = 5, mean ± S.D.).
ITK Tyr180 affects phosphorylation of ITK-Tyr511 as well as ITK association with TSAD
We then assessed the role of the autophosphorylation site ITK Tyr180 for LCK-mediated phosphorylation of ITK. As we had found that the Y180E mutant ITK SH3 domain mimicking ITK-pTyr180 displayed reduced interaction with TSAD (Fig. 1C), we speculated that failure to phosphorylate ITK on Tyr180 would influence the dynamics of the TSAD-ITK interaction and thus phosphorylation of ITK by LCK on Tyr511 followed by autophosphorylation on Tyr180. To assess to what extent the ITK-pTyr signal we observed in Fig. 7A represented either of these two tyrosines, we repeated the experiment shown in Fig. 7A (lanes 1 and 2) with a Myc-ITK-Y180F mutant. The results showed that Myc-ITK-Y180F displayed significantly reduced phosphorylation of ITK (Fig. 7, C and D). This could be explained by much of the ITK phospho-Tyr signal observed using a pan-pTyr antibody as being contributed by ITK pTyr180. Some of the reduced signal could also be attributed to reduced interaction of the ITK-Y180F mutant with TSAD (Figs. 1 (C and D) and 7C).
To distinguish between these two possibilities, we immunoprecipitated ITK from cells expressing Myc-ITK or Myc-ITK-Y180F in the presence of LCK and TSAD and directly assessed the total amount of ITK pTyr511 using a phosphosite-specific antibody. The result showed that in the absence of the autophosphorylation site Tyr180, ITK was also less phosphorylated on Tyr511 (Fig. 7, E and F). This indicates that in the presence of TSAD, autophosphorylation of ITK Tyr180 is required for maximal phosphorylation of ITK Tyr511. Reduced binding ability of ITK pTyr180 to TSAD PRR, as indicated by our initial pulldown experiments using the phosphotyrosine mimic ITK Y180E SH3 domain (Fig. 1, C and D), may ensure that only nonphosphorylated ITK associates with TSAD once the TSAD PRR is accessible for binding. An additional consequence of this model could be that ITK that cannot be phosphorylated on Tyr180 will occupy TSAD PRR to a larger extent than WT ITK, because an ITK molecule that is already bound to TSAD may immediately reassociate once it dissociates from the TSAD PRR. To test this assumption, we therefore immunoprecipitated TSAD from cells expressing either Myc-ITK or Myc-ITK-Y180F in the presence of TSAD and LCK and probed for co-immunoprecipitation of ITK. This experiment revealed that more ITK-Y180F than WT ITK was associated with TSAD (Fig. 7, G and H). Taken together, these data show that ITK Tyr180 regulates association of ITK to TSAD and thereby also the phosphorylation of ITK Tyr511 by LCK.
Discussion
The adapter TSAD has multiple interaction sites and thus provides a platform for complex multimodular protein-protein interactions within the T cell. We have previously mapped in detail how TSAD interacts with LCK (16) and have provided evidence that TSAD promotes LCK-mediated phosphorylation of ITK on Tyr511 (10) in the activation loop of the kinase domain, a phosphorylation event that is critical for activation of ITK. In our present study, we have begun to delineate the mode of interaction between TSAD and ITK. We show that TSAD may serve as a scaffold whereby ITK and LCK are brought into the vicinity of one another in the T cell. We propose that this LCK-ITK adapter function of TSAD promotes signaling downstream of ITK. A schematic overview of our model for how LCK and ITK interact with TSAD is shown in Fig. 8.
Figure 8.
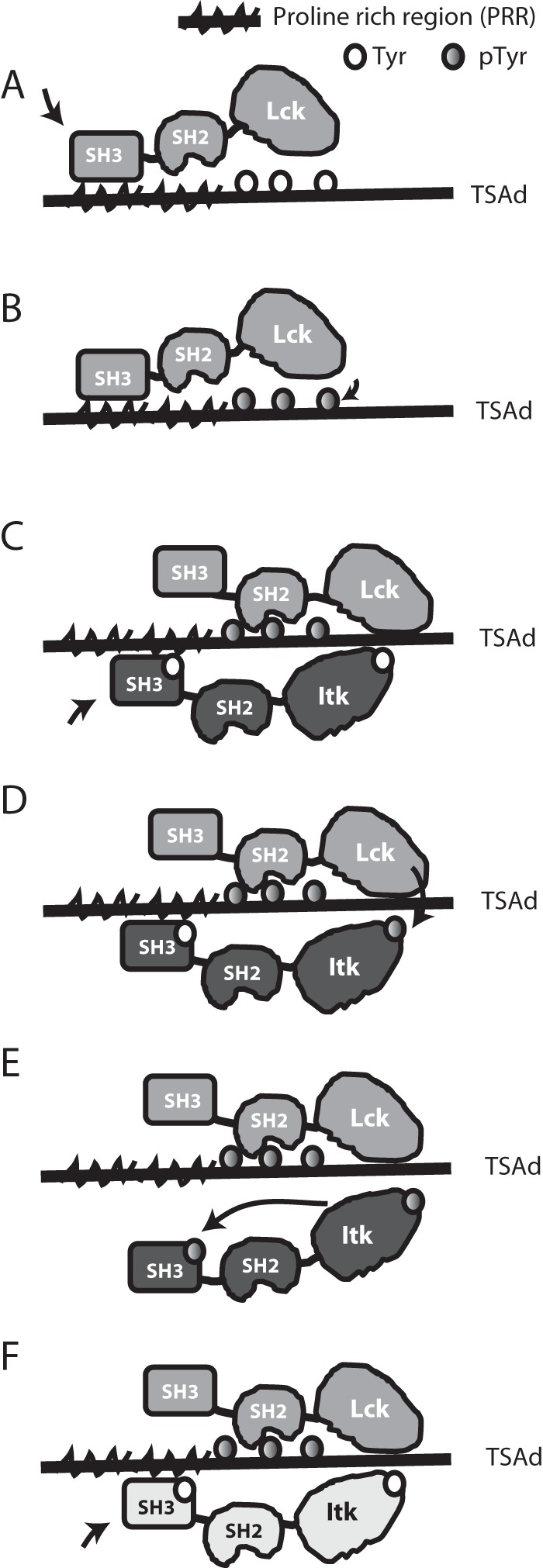
Schematic representation of TSAD-ITK-LCK interactions and their putative sequence. A, open LCK binds to TSAD PRR and becomes activated. B, active LCK phosphorylates the three TSAD C-terminal tyrosines. C, the LCK SH2 domain binds to TSAD pTyr, allowing LCK SH3 to detach from TSAD PRR. ITK binds to TSAD PRR. D, active LCK phosphorylates ITK Tyr511. E, active ITK pTyr511 autophosphorylates ITK Tyr180. ITK pTyr180 does not bind to TSAD PRR. F, active LCK remains bound to TSAD pTyr. Next, the ITK molecule (indicated in a different color) binds to TSAD PRR, and the process starts over from D.
In the current study, the biochemical analysis of the TSAD ITK LCK interactions was done exclusively in a heterologous cell system. The human embryonic kidney 293 (known as HEK293) cells, also expressing the SV40 large T antigen (known as 293T cells (27)) are commonly used for dissection of molecular mechanisms by exogenous expression of proteins. These cells have several advantages. Whereas LCK is constitutively expressed in T cells, both ITK and TSAD are expressed only at low to moderate levels in resting or nonstimulated T cells and T-cell lines. An additional advantage in using 293T cells for protein expression in eukaryotic cells is that these cells do not express other T cell–specific molecules that might interfere with the intermolecular interactions studied here. However, the stoichiometry and/or concentration of the interaction partners studied in 293T cells may differ from the situation in actual T cells. For instance, whereas the interactions between TSAD, LCK, and ITK appear to be independent of external, activating signals in 293T cells, this may not be the case at physiological conditions in T cells.
Our main finding in this study is that the ITK and LCK SH3 domains bind to adjacent sites on TSAD. Although they may both bind to TSAD at the same time, they also may compete for binding to the same site(s). LCK can bind to the three C-terminal TSAD tyrosines Tyr280, Tyr290, and Tyr305 via its SH2 domain. The TSAD PRR interacting with LCK SH3 strongly promotes phosphorylation of these tyrosines (16). Thus, it is possible that LCK and ITK interact with TSAD sequentially, with LCK binding to TSAD first through its SH3 domain (Fig. 8A) and subsequently—after having phosphorylated the three C-terminal TSAD tyrosines—via its SH2 domain (Fig. 8B), leaving the PRR free to bind for ITK SH3 (Fig. 8C). This mechanism is consistent with the observations from NMR indicating direct competition between the SH3 domains for binding to the two shorter peptides representing the N- and the C-terminal parts of the TSAD PRR.
An increasing body of evidence suggests that within the cell, signaling molecules may form large complexes with multiple copies of each interacting partner (i.e. signalosomes) (28, 29). The stoichiometric arrangement of ITK, LCK, and TSAD in the cell may therefore be quite complex, as has been suggested for the Grb2/LAT/SOS1 signaling system (30, 31). The cell interior is virtually packed with molecules, probably resulting in localized regions with comparably high concentrations of given molecules (32). It is increasingly being recognized that phase separation of signaling molecules may be functionally relevant (33). Thus, the relatively high dissociation constants measured in the NMR tube with both SH3 domains present in high and equimolar concentrations may still reflect physiologically meaningful molecular associations in the immediate vicinity of TSAD within the cell.
Once ITK is activated by LCK through phosphorylation of the Tyr511 (Fig. 8D), ITK autophosphorylates Tyr180 in its own SH3 domain in cis (19, 20). Moreover, autophosphorylation of ITK Tyr180 is required for full activity of ITK (19). Our findings suggest that autophosphorylation of the ITK SH3 domain in cis results in the release of ITK from the TSAD scaffold (Fig. 8E). This creates an open binding site on TSAD for recruiting and activation of another ITK molecule by LCK (Fig. 8F). In accordance with this notion, we observed that ITK Tyr180 was required for maximal phosphorylation of ITK Tyr511. Thus, our data strongly indicate that autophosphorylation of Tyr180 promotes phosphorylation of Tyr511. In addition, the inhibition of reassociation of ITK pTyr180 to TSAD upon activation of ITK by LCK, has the added advantage of leaving the ITK SH3 domain free to recruit other targets to the activated kinase.
We have recently found that once activated by LCK, ITK will phosphorylate LCK Tyr192 in the LCK SH2 domain, which increases the binding of LCK to some of its ligands, including TSAD and ITK (8). Importantly, LCK SH2 domain mutated to mimic Tyr(P)192 (i.e. Y192E) displays increased binding affinity to phosphopeptides representing ITK-pTyr180 and ITK-pTyr511 (8). It has been reported that one potential ligand for the ITK SH2 domain is indeed the LCK-pTyr192 (34). It is therefore possible that once the interaction between ITK and LCK has been promoted via their mutual interaction with TSAD through their SH3 domains, TSAD, ITK, and LCK are dynamically altering their interaction modes, depending on the phosphorylation status of the three interacting partners.
At the start of the current study, the solution structure of the human ITK SH3 domain was yet to be determined. We thus determined the ITK SH3 domain solution structure using NMR spectroscopy and found that it is similar to the structure of the mouse ITK SH3 domain (25). SH3 domains typically interact with peptides adopting a polyproline II helix, and the canonical PXXP motif commonly found in SH3 ligands ensures the formation of such a regular helix, with exactly three aa per turn (35). The flat binding surface of SH3 domains with two hydrophobic grooves lined mainly by aromatic residues is ideally suited to accommodate the XP dipeptides that form the base of the polyproline helix (X most often being a hydrophobic amino acid). However, some SH3 domains also bind to nonclassical motifs, like the GRB2 SH3 binding to XXXRXXKP (36), the SKAP55 SH3 domain binding to RKXXYXXY (37), the PEX5P SH3 domain binding to WXXXFXXLE (38), the EPS8 SH3 domain binding to PXXDY (39), and the GADS C-terminal SH3 domain binding to the RXXK motif of SLP-76 (40, 41).
Our initial NMR work involved the C-terminal ITK binding TSAD peptide aa 262–269 (IPVPRHRP) based on the results obtained upon mutagenesis of Pro263. TSAD aa 262–269 lacks a PXXP motif, so the interaction of ITK SH3 with TSAD at this site can be added to the list of nonconventional SH3-binding motifs. Moreover, in contrast to the classic type I or II ligands of SH3 domains, where a terminal basic residue determines the binding orientation of the peptide on the SH3-binding surface (15), our data suggest that in this TSAD peptide, the hydrophobic aa Ile262 occupies this anchoring position. Thus, although the TSAD aa 262–269 peptide binding to ITK SH3 is a partial class II ligand with positively charged aa in the C terminus, our data suggest that its binding orientation on ITK SH3 is similar to that of class I ligands.
In conclusion, we have used biochemical analyses as well as NMR spectroscopy to analyze ITK SH3 and LCK SH3 interactions with TSAD. Our data indicate that LCK and ITK binding to TSAD is complex and may be characterized by simultaneously binding of the ITK and LCK SH3 domains to the TSAD PRR under some conditions and sequential binding of the ITK and LCK SH3 domains under other conditions. Further molecular mapping of the interaction between TSAD, ITK, and LCK is required to understand the temporal dynamics of this trimolecular complex. In particular, it will be important to characterize formation of this protein complex using larger fragments or full-length proteins because multivalency may play a significant role in organizing the architecture of the functional TSAD-based signaling complex.
Experimental procedures
Cells
Human embryonal kidney 293T cells were cultured in RPMI 1640 supplemented with l-glutamine, 5–10% fetal calf serum, 1 mm sodium pyruvate, 1 mm nonessential amino acids, 100 units/ml penicillin, and 100 μg/ml streptomycin (all from GIBCOBRL®, Thermo Fisher Scientific).
Plasmids
pGex-2T with an insert encoding intact or mutated tryptophan to lysine at position 208 (W208K), murine ITK SH3-domain GSH-S-transferase (GST) fusion protein (aa 171–230 of ITK), and the pGex-6P-1 construct encoding human LCK SH3 were described previously (17, 18, 42). The pEF-Myc-ITK was kindly provided by Professor Leslie Berg. The pEF-LCK was kindly provided by Dr. Tomas Mustelin. LCK SH3 with a C-terminal hemagglutinin (HA) tag was constructed by amplifying the LCK SH3 sequence with a C-terminal primer encoding for this epitope, and the PCR product was cloned into the pGex-6P1 vector. The resulting construct yields a GST-tagged and HA-tagged LCK SH3 domain. The human ITK SH3 domain (aa 168–232) was isolated from a human cDNA library (4) using PCR and subcloned into pGex-6P-1 to generate the GST fusion protein expression construct. TSAD cDNA encoding TSAD WT sequence or TSAD containing a deletion of aa 239–274, 239–256, or 257–274 cloned into the expression vector pEF-HA (17) was used for transient expression of HA-tagged TSAD in eukaryote cells. Mutations of TSAD-P247A and -P263A in the pEF-HA-TSAD expression vectors, of ITK-Y180F and ITK-Y180E in ITK SH3 in the pGex-6P-1 vector as well as the pEF-Myc-ITK vector, and of W208K in the pEF-Myc-ITK vector were performed using QuikChange mutagenesis and appropriate custom-made primers. The sequences of all constructs were verified by sequencing.
Protein preparation
GST-SH3 domains were expressed in Escherichia coli BL21(DE3). Cells were grown at 25 °C until optical density (measured at 600 nm) reached 0.6. Isopropyl β-d-thiogalactopyranoside (IPTG) was then added to a final concentration of 0.2 mm to induce protein expression. In Luria-Bertani broth, the culture was incubated with IPTG for 5 hours, whereas in minimal medium (6 g/liter glucose, 5 g/liter Na2PO4, 3 g/liter KH2PO4, 1 g/liter 15NH4Cl, 0.5 g/liter NaCl, 0.12 g/liter MgSO4, 10 mg/liter CaCl2) incubation with IPTG was carried out overnight (∼15 h) at 15 °C. Minimal medium was used when incorporation of 15N and 13C was needed. The bacteria were then pelleted and resuspended in phosphate buffer (75 mm KH2PO4, 50 mm NaCl, 0.1 mm DTT, pH 7.4) containing lysozyme (1 mm) and frozen at −80 °C. After thawing, phenylmethylsulfonyl fluoride (1 mm) and DNase (5 μg/ml) were added, and the lysate was gently mixed at 4 °C for 8–16 h. Upon completed lysis, the debris was removed by centrifugation (20,000 × g, 4 °C, 15 min). Fusion protein was purified by GSH SepharoseTM 4B beads (Amersham Biosciences) in batch mode as described by the manufacturer. Briefly, lysate and GSH SepharoseTM 4B beads were mixed and incubated with gentle agitation at 4 °C overnight. Beads were subsequently washed extensively in phosphate buffer (75 mm KH2PO4, 50 mm NaCl, 0.1 mm DTT, pH 7.4) prior to cleavage of the GST fusion protein with the appropriate protease (thrombin (Amersham Biosciences) or PreScissionTM Protease (Amersham Biosciences)). The purity and integrity of the purified proteins was analyzed by SDS-PAGE followed by Coomassie Brilliant Blue (Bio-Rad) staining. Protein concentration was measured by NanoDrop spectrophotometry (Thermo Scientific, Wilmington, DE) at a wavelength of 280 nm and a theoretically obtained extinction coefficient (ProtParam). Purity of NMR samples typically exceeded 90%. If necessary, further concentration of the protein samples was performed using Amicon Ultra-15 centrifugal filter units (Millipore). NMR samples were supplemented with 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS) (Larodan Fine Chemicals AB, Malmö, Sweden) to a final concentration of 0.2 mm for internal chemical shift referencing. 5% D2O was added to the samples to provide lock signal. Structure determination was performed for protein dissolved in 20 mm phosphate buffer, pH 6.5.
Peptides
Peptides derived from the human TSAD PRR aa 248–268 (*LLRPKPPIPAKPQLPPEVYTIPVPRHR#), aa 242–258 (*LLRPKPPIPAKPQLPPE#), aa 254–268 (*QLPPEVYTIPVPRHR#), aa 262–269 (*IPVPRHRP#), aa 262–278 (*IPVPRHRPAPRPKPSNP#), and aa 271–278 (*PRPKPSNP#) were synthesized with blocked ends (* and #) by GeneScript or Pepscan Systems to more than 99% purity as estimated by HPLC. The IPVPRHRP peptide was also custom-synthesized with an N-terminal TOAC label by Anaspec (San Jose, CA).
Peptide array
Peptide arrays were synthesized on nitrocellulose membranes using a MultiPep automated peptide synthesizer (INTAVIS Bioanalytical Instruments AG, Germany) as described (43, 44). Peptide array membranes were probed with GST-tagged SH3 domains (100 μg/ml) and blotted with mouse anti-GST antibody (Santa Cruz Biotechnology, Inc.) and anti-mouse HRP secondary antibody followed by chemiluminescent detection.
Transfection of cells
Transient transfections of 293T cells took place for 5–6 h with 1–2 μg of DNA in 25 μl of Lipofectin (GIBCO-BRL) or 16,125 μg of polyethyleneimine (Polysciences Inc., Warrington, PA). Transfected cells were further propagated for 16–24 h in the presence of fetal calf serum (5% final concentration).
Cell lysis and pulldown experiments
293T cells were transfected with different HA-tagged constructs of TSAD cDNA. Cells were lysed in buffer containing 1% Igepal CA-630 (Sigma), 25 mm Tris (pH 7.5), 100 mm NaCl, 20 mm NaF, 1 mm NaVO3, and 10 μg/ml antipain, chymostatin, leupeptin, and pepstatin A (Sigma-Aldrich). Lysates representing 2 × 106 to 4 × 106 transfected cells were precleared three times for 30 min to 1 h with a 2:1 GST-GSH 4B SepharoseTM beads/4B SepharoseTM beads mixture at 4 °C. Precleared lysates were added to aliquots of GST-ITK SH3 or GST-LCK SH3 bound to GSH 4B SepharoseTM beads and gently mixed at 4 °C for 1 h. Finally, the beads were washed three times using lysis buffer and boiled in SDS loading buffer containing mercaptoethanol before separation of proteins on SDS-PAGE. In vitro pulldown experiments using isolated HA-tagged LCK SH3 domains and GST-tagged ITK SH3 domains bound to GSH 4B SepharoseTM bead SH3 domains were performed in buffer containing 1% Igepal CA-630, 25 mm Tris (pH 7.5), 100 mm NaCl, 2% BSA, and a 10 μm concentration of each SH3 domain and the indicated amount of TSAD peptides. Prior to use in the in vitro pulldown assay, the GST-ITK SH3 GSH 4B SepharoseTM beads were mixed 1:2 (bead volume) with GST GSH 4B SepharoseTM beads.
Immunoprecipitation experiments
293 T cells were transfected with HA-tagged TSAD constructs along with LCK and Myc-tagged ITK WT or ITK W208K. The cells were lysed in buffer containing 0.1% LDS, 0.5–1% Triton X-100, 50 mm HEPES, 0.05 m lithium chloride, 0.5 mm phenylmethylsulfonyl fluoride, 2.5 mm EDTA (pH 8.0), 1 mm NaVO3, 10 μg/ml antipain, chymostatin, leupeptin, and pepstatin A (Sigma-Aldrich), and 1 mg/ml DNase for 45 min. The lysate was sonicated briefly to fragment the DNA. The lysate was preabsorbed twice at 4 °C for 30 min using Dynabeads® Protein G (Life Technologies). The preabsorbed lysate was added to the Dynabeads® Protein G coated with anti-Myc antibody (clone 9E10, Sigma) or anti-TSAD antibody (4) for 1 h in a rotating wheel at 4 °C. The beads were washed three times using lysis buffer and boiled in loading buffer containing 2-mercaptoethanol before separation of proteins on SDS-PAGE. Proteins were detected by using the following primary antibodies: anti-ITK antibody (Médimabs, clone 2F12), anti-LCK (Santa Cruz Biotechnology, clone 3A5), anti-HA1.1 (Bio Site, clone 16B12), anti-pTyr (clone 4G10), or anti-pTyr511 ITK (clone 24a/Btk, BD Biosciences) in Tris-buffered saline (pH 7.4) with 0.1% Tween (Sigma-Aldrich) plus 3% BSA (Biotest) or 3% skimmed milk (Sigma-Aldrich). Signals were detected by horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG or goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories) and Super Signal® West Pico Stable Peroxide Solution (Pierce) (5). Images of scanned WB films were analyzed using ImageJ software (45).
NMR spectroscopy
The following NMR experiments were performed to assign the backbone chemical shifts and determine structural restraints: 15N HSQC, 13C HSQC, 15N NOESYHSQC, 13C NOESYHSQC, HNHA, HNCA, CBCACONH, CBCANH, HNCO, HNCACO, HNCOCA, (H)CCCONH, HCCCONH, HBHACONH, HBHANH HCCH-TOCSY (46). 1H-15N NOESY with a mixing time of 3 s was performed to investigate the relaxation properties of the protein (47). Experiments for structure determination were run on a 600-MHz Bruker Avance II spectrometer with four channels and a 5-mm TCI cryoprobe at 25 °C. 1H-15N HSQC titration experiments were recorded at 25 °C on Varian INOVA spectrometers at 600 MHz or a Bruker Avance II spectrometer at 600 or 700 MHz. All instruments were equipped with cryoprobes.
Data processing
Data were processed using Topspin 2.1 (Bruker Biospin). DSS was used as a chemical shift standard, and 13C and 15N data were referenced using frequency ratios as described by Wishart et al. (48). Time-domain to frequency-domain processing was carried out using NMRPipe (49) or TopSpin software, respectively, and subsequent analysis of titration experiments was performed using Sparky.
Assignment
For visualization and assignment the computer program CARA (50) was used. The spectra were assigned using standard methods (46). The chemical shifts are deposited in the Biological Magnetic Resonance Data Bank (BMRB) with the access code 18119.
Structure calculation and docking of TSAD peptide to the ITK SH3 structure
The 15N and 13C NOESY-HSQC spectra were peak picked using CARA (50). NOESY upper distance constraints were generated by the CANDID routine in CYANA 3.0 (51). Torsion angle constraints were determined from the chemical shifts by the application of TALOS (52). Temperature dependence more positive than −4.5 ppb/K for the amide proton was taken as a proof of the existence of a hydrogen bond (53). Hydrogen bond and torsion angle restraints were introduced in the final stage of structure determination. 100 structures were calculated using CYANA 3.0, and the 20 structures with the lowest energy were kept in the structure ensemble.2
NMR titration
The NMR samples consisted of a uniformly 15N-labeled recombinant ITK SH3 or LCK SH3 protein of human origin dissolved in phosphate buffer (75 mm KH2PO4, 50 mm NaCl, 0.1 mm DTT, pH 7.4) at concentrations ranging from 40 to 300 μm, containing 5% D2O. Titration was done by the stepwise addition of a defined amount of TSAD peptide or unlabeled SH3 domain, dissolved in the same phosphate buffer, to the LCK SH3 and/or ITK SH3 NMR samples. At each step, a 15N HSQC spectrum was recorded. 1H and 15N chemical shifts were referenced to DSS. The chemical shift deviations were quantified using the formula, Δav = ((ΔHN)2 + (0.2ΔN)2)½ (54), where ΔHN and ΔN correspond to the amide proton and nitrogen chemical shift changes upon peptide binding, respectively. The dissociation constants were derived from binding curves generated using the Matlab (version 5.3.1, The Mathworks Inc.) suite of programs by plotting Δave versus ligand concentration.
TOAC experiment
A 15N HSQC NMR spectrum of uniformly 15N-labeled, recombinant, ITK SH3 dissolved in phosphate buffer (as described above) at a concentration of ∼500 μm was recorded after adding N-terminally TOAC-labeled peptide, TOAC-IPVPRHRP, to a final concentration of 1 mm. In addition, a 15N-HSQC NMR spectrum of the same solution, adjusted to 400 μm sodium ascorbate, was recorded. The addition of the reducing agent sodium ascorbate abolishes the effect the TOAC label has on the spectrum.
Docking model of IPVPRHRP to ITK SH3
The peptide structure of IPVPRHRP was generated by mutating the polypeptide of 1VZJ using PyMOL. Docking of the peptide to ITK SH3 was performed using the HADDOCK server (26). Ambiguous restraints where defined between residues Thr184, Asp186, Glu189, Leu192, His207, Trp208, Trp209, Val211, Tyr220, Val221, Ser223, and Tyr225 and the peptide IPVPRHRP.
Author contributions
T. C. B. A., P. E. K., Z. H., D. B. F., B. G. K., A. H. A., and A. S. data curation; T. C. B. A., P. E. K., Z. H., M. U. J., R. P. G., H. K., V. S.-G., S. G., M. S., D. B. F., B. G. K., A. H. A., and A. S. investigation; T. C. B. A., P. E. K., Z. H., R. P. G., H. K., S. B., P. H. B., B. G. K., A. H. A., and A. S. visualization; T. C. B. A., A. H. A., and A. S. writing-original draft; P. E. K., V. S.-G., B. G. K., A. H. A., and A. S. conceptualization; P. E. K., M. S., D. B. F., and A. H. A. resources; P. E. K., M. U. J., S. B., P. H. B., D. B. F., B. G. K., and A. H. A. software; P. E. K., S. B., and A. H. A. formal analysis; P. E. K., M. U. J., V. S.-G., S. G., D. B. F., B. G. K., A. H. A., and A. S. supervision; P. E. K. and A. S. funding acquisition; P. E. K., S. B., M. S., P. H. B., D. B. F., B. G. K., and A. H. A. methodology; P. E. K., Z. H., V. S.-G., and A. S. writing-review and editing; A. S. project administration.
Acknowledgments
We thank Leslie Berg and Tomas Mustelin for providing valuable reagents. The technical assistance of Bjørg Simonsen and Lise Koll is highly appreciated. We acknowledge the Research Council of Norway for investment in the 600-MHz NMR spectrometer at the University of Oslo.
This work was supported by grants from the University of Oslo, Norwegian Research Council Grants 196386 and 214202, Norwegian Cancer Society Grant 18200, 18201, 163359, and 163361, Novo Nordisk Fonden, Unifor, Anders Jahres fond til vitenskapens fremme, and the University of Oslo. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 2LMJ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Chemical shift data have been deposited in the Biological Magnetic Resonance Data Bank (BMRB) with the access code 18119.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PTK
- protein-tyrosine kinase
- aa
- amino acid(s)
- ITK
- inducible Tec kinase
- LCK
- lymphocyte-specific kinase
- HA
- hemagglutinin
- PRR
- proline-rich region
- SH2 and SH3
- SRC homology 2 and 3, respectively
- TCR
- T-cell receptor
- 2D
- two-dimensional
- TOAC
- 2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4-carboxylic acid
- TSAD
- T cell–specific adapter protein
- HSQC
- heteronuclear single quantum coherence
- PDB
- Protein Data Bank
- GST
- glutathione S-transferase
- IPTG
- isopropyl β-d-thiogalactopyranoside
- DSS
- 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt
- HRP
- horseradish peroxidase.
References
- 1. Koretzky G. A., and Myung P. S. (2001) Positive and negative regulation of T-cell activation by adaptor proteins. Nat. Rev. Immunol. 1, 95–107 10.1038/35100523 [DOI] [PubMed] [Google Scholar]
- 2. Mustelin T., and Taskén K. (2003) Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem. J. 371, 15–27 10.1042/bj20021637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Felices M., Falk M., Kosaka Y., and Berg L. J. (2007) Tec kinases in T cell and mast cell signaling. Adv. Immunol. 93, 145–184 10.1016/S0065-2776(06)93004-1 [DOI] [PubMed] [Google Scholar]
- 4. Spurkland A., Brinchmann J. E., Markussen G., Pedeutour F., Munthe E., Lea T., Vartdal F., and Aasheim H. C. (1998) Molecular cloning of a T cell-specific adapter protein (TSAD) containing an Src homology (SH) 2 domain and putative SH3 and phosphotyrosine binding sites. J. Biol. Chem. 273, 4539–4546 10.1074/jbc.273.8.4539 [DOI] [PubMed] [Google Scholar]
- 5. Granum S., Sundvold-Gjerstad V., Dai K. Z., Kolltveit K. M., Hildebrand K., Huitfeldt H. S., Lea T., and Spurkland A. (2006) Structure function analysis of SH2D2A isoforms expressed in T cells reveals a crucial role for the proline rich region encoded by SH2D2A exon 7. BMC. Immunol. 7, 15 10.1186/1471-2172-7-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choi Y. B., Kim C. K., and Yun Y. (1999) Lad, an adapter protein interacting with the SH2 domain of p56lck, is required for T cell activation. J. Immunol. 163, 5242–5249 [PubMed] [Google Scholar]
- 7. Rajagopal K., Sommers C. L., Decker D. C., Mitchell E. O., Korthauer U., Sperling A. I., Kozak C. A., Love P. E., and Bluestone J. A. (1999) RIBP, a novel Rlk/Txk- and itk-binding adaptor protein that regulates T cell activation. J. Exp. Med. 190, 1657–1668 10.1084/jem.190.11.1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Granum S., Sundvold-Gjerstad V., Gopalakrishnan R. P., Abrahamsen G., Berge T., Koll L., Sørlie M., and Spurkland A. (2014) Itk and the adapter TSAD co-operate to promote phosphorylation dependent altered specificity of Lck. Sci. Signal. 7, ra118 10.1126/scisignal.2005384 [DOI] [PubMed] [Google Scholar]
- 9. Sundvold V., Torgersen K. M., Post N. H., Marti F., King P. D., Røttingen J. A., Spurkland A., and Lea T. (2000) T cell-specific adapter protein inhibits T cell activation by modulating Lck activity. J. Immunol. 165, 2927–2931 10.4049/jimmunol.165.6.2927 [DOI] [PubMed] [Google Scholar]
- 10. Berge T., Sundvold-Gjerstad V., Granum S., Andersen T. C., Holthe G. B., Claesson-Welsh L., Andreotti A. H., Inngjerdingen M., and Spurkland A. (2010) T cell specific adapter protein (TSAD) interacts with Tec kinase ITK to promote CXCL12 induced migration of human and murine T cells. PLoS One 5, e9761 10.1371/journal.pone.0009761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Drappa J., Kamen L. A., Chan E., Georgiev M., Ashany D., Marti F., and King P. D. (2003) Impaired T cell death and lupus-like autoimmunity in T cell-specific adapter protein-deficient mice. J. Exp. Med. 198, 809–821 10.1084/jem.20021358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith C. I., Islam T. C., Mattsson P. T., Mohamed A. J., Nore B. F., and Vihinen M. (2001) The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays 23, 436–446 10.1002/bies.1062 [DOI] [PubMed] [Google Scholar]
- 13. Kuriyan J., and Cowburn D. (1997) Modular peptide recognition domains in eukaryotic signaling. Annu. Rev. Biophys. Biomol. Struct. 26, 259–288 10.1146/annurev.biophys.26.1.259 [DOI] [PubMed] [Google Scholar]
- 14. Tran T., Hoffmann S., Wiesehan K., Jonas E., Luge C., Aladag A., and Willbold D. (2005) Insights into human Lck SH3 domain binding specificity: different binding modes of artificial and native ligands. Biochemistry 44, 15042–15052 10.1021/bi051403k [DOI] [PubMed] [Google Scholar]
- 15. Feng S., Chen J. K., Yu H., Simon J. A., and Schreiber S. L. (1994) Two binding orientations for peptides to the Src SH3 domain: development of a general model for SH3-ligand interactions. Science 266, 1241–1247 10.1126/science.7526465 [DOI] [PubMed] [Google Scholar]
- 16. Granum S., Andersen T. C., Sørlie M., Jørgensen M., Koll L., Berge T., Lea T., Fleckenstein B., Spurkland A., and Sundvold-Gjerstad V. (2008) Modulation of Lck function through multisite docking to T cell-specific adapter protein. J. Biol. Chem. 283, 21909–21919 10.1074/jbc.M800871200 [DOI] [PubMed] [Google Scholar]
- 17. Sundvold-Gjerstad V., Granum S., Mustelin T., Andersen T. C., Berge T., Shapiro M. J., Shapiro V. S., Spurkland A., and Lea T. (2005) The C terminus of T cell-specific adapter protein (TSAD) is necessary for TSAD-mediated inhibition of Lck activity. Eur. J. Immunol. 35, 1612–1620 10.1002/eji.200425638 [DOI] [PubMed] [Google Scholar]
- 18. Bunnell S. C., Henry P. A., Kolluri R., Kirchhausen T., Rickles R. J., and Berg L. J. (1996) Identification of Itk/Tsk Src homology 3 domain ligands. J. Biol. Chem. 271, 25646–25656 10.1074/jbc.271.41.25646 [DOI] [PubMed] [Google Scholar]
- 19. Wilcox H. M., and Berg L. J. (2003) Itk phosphorylation sites are required for functional activity in primary T cells. J. Biol. Chem. 278, 37112–37121 10.1074/jbc.M304811200 [DOI] [PubMed] [Google Scholar]
- 20. Joseph R. E., Fulton D. B., and Andreotti A. H. (2007) Mechanism and functional significance of Itk autophosphorylation. J. Mol. Biol. 373, 1281–1292 10.1016/j.jmb.2007.08.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Obenauer J. C., Cantley L. C., and Yaffe M. B. (2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 10.1093/nar/gkg584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cory G. O., MacCarthy-Morrogh L., Banin S., Gout I., Brickell P. M., Levinsky R. J., Kinnon C., and Lovering R. C. (1996) Evidence that the Wiskott-Aldrich syndrome protein may be involved in lymphoid cell signaling pathways. J. Immunol. 157, 3791–3795 [PubMed] [Google Scholar]
- 23. Bunnell S. C., Diehn M., Yaffe M. B., Findell P. R., Cantley L. C., and Berg L. J. (2000) Biochemical interactions integrating Itk with the T cell receptor-initiated signaling cascade. J. Biol. Chem. 275, 2219–2230 10.1074/jbc.275.3.2219 [DOI] [PubMed] [Google Scholar]
- 24. Cavanagh J., Fairbrother W. J., Palmer I. I. I., A. G., Rance M., and Skelton N. J. (2007) Principles and Practice of Protein NMR Spectroscopy, 2nd Ed., Elsevier Academic Press, Burlington, MA [Google Scholar]
- 25. Severin A., Fulton D. B., and Andreotti A. H. (2008) Murine Itk SH3 domain. J Biomol. NMR 40, 285–290 10.1007/s10858-008-9231-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Vries S. J., van Dijk M., Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 10.1038/nprot.2010.32 [DOI] [PubMed] [Google Scholar]
- 27. Pear W. S., Nolan G. P., Scott M. L., and Baltimore D. (1993) Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U.S.A. 90, 8392–8396 10.1073/pnas.90.18.8392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kagan J. C., Magupalli V. G., and Wu H. (2014) SMOCs: supramolecular organizing centres that control innate immunity. Nat. Rev. Immunol. 14, 821–826 10.1038/nri3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li P., Banjade S., Cheng H. C., Kim S., Chen B., Guo L., Llaguno M., Hollingsworth J. V., King D. S., Banani S. F., Russo P. S., Jiang Q. X., Nixon B. T., and Rosen M. K. (2012) Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 10.1038/nature10879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Houtman J. C., Yamaguchi H., Barda-Saad M., Braiman A., Bowden B., Appella E., Schuck P., and Samelson L. E. (2006) Oligomerization of signaling complexes by the multipoint binding of GRB2 to both LAT and SOS1. Nat. Struct. Mol. Biol. 13, 798–805 10.1038/nsmb1133 [DOI] [PubMed] [Google Scholar]
- 31. Kortum R. L., Balagopalan L., Alexander C. P., Garcia J., Pinski J. M., Merrill R. K., Nguyen P. H., Li W., Agarwal I., Akpan I. O., Sommers C. L., and Samelson L. E. (2013) The ability of Sos1 to oligomerize the adaptor protein LAT is separable from its guanine nucleotide exchange activity in vivo. Sci. Signal. 6, ra99 10.1126/scisignal.2004494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Phillip Y., and Schreiber G. (2013) Formation of protein complexes in crowded environments–from in vitro to in vivo. FEBS Lett. 587, 1046–1052 10.1016/j.febslet.2013.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boeynaems S., Alberti S., Fawzi N. L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L., Tompa P., and Fuxreiter M. (2018) Protein phase separation: a new phase in cell biology. Trends Cell Biol. 28, 420–435 10.1016/j.tcb.2018.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koytiger G., Kaushansky A., Gordus A., Rush J., Sorger P. K., and MacBeath G. (2013) Phosphotyrosine signaling proteins that drive oncogenesis tend to be highly interconnected. Mol. Cell Proteomics 12, 1204–1213 10.1074/mcp.M112.025858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li S. S. (2005) Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem. J. 390, 641–653 10.1042/BJ20050411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lewitzky M., Kardinal C., Gehring N. H., Schmidt E. K., Konkol B., Eulitz M., Birchmeier W., Schaeper U., and Feller S. M. (2001) The C-terminal SH3 domain of the adapter protein Grb2 binds with high affinity to sequences in Gab1 and SLP-76 which lack the SH3-typical P-x-x-P core motif. Oncogene 20, 1052–1062 10.1038/sj.onc.1204202 [DOI] [PubMed] [Google Scholar]
- 37. Kang H., Freund C., Duke-Cohan J. S., Musacchio A., Wagner G., and Rudd C. E. (2000) SH3 domain recognition of a proline-independent tyrosine-based RKxxYxxY motif in immune cell adaptor SKAP55. EMBO J. 19, 2889–2899 10.1093/emboj/19.12.2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barnett P., Bottger G., Klein A. T., Tabak H. F., and Distel B. (2000) The peroxisomal membrane protein Pex13p shows a novel mode of SH3 interaction. EMBO J. 19, 6382–6391 10.1093/emboj/19.23.6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mongioví A. M., Romano P. R., Panni S., Mendoza M., Wong W. T., Musacchio A., Cesareni G., and Di Fiore P. P. (1999) A novel peptide-SH3 interaction. EMBO J. 18, 5300–5309 10.1093/emboj/18.19.5300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berry D. M., Nash P., Liu S. K., Pawson T., and McGlade C. J. (2002) A high-affinity Arg-X-X-Lys SH3 binding motif confers specificity for the interaction between Gads and SLP-76 in T cell signaling. Curr. Biol. 12, 1336–1341 10.1016/S0960-9822(02)01038-2 [DOI] [PubMed] [Google Scholar]
- 41. Liu Q., Berry D., Nash P., Pawson T., McGlade C. J., and Li S. S. (2003) Structural basis for specific binding of the Gads SH3 domain to an RxxK motif-containing SLP-76 peptide: a novel mode of peptide recognition. Mol. Cell 11, 471–481 10.1016/S1097-2765(03)00046-7 [DOI] [PubMed] [Google Scholar]
- 42. Andreotti A. H., Bunnell S. C., Feng S., Berg L. J., and Schreiber S. L. (1997) Regulatory intramolecular association in a tyrosine kinase of the Tec family. Nature 385, 93–97 10.1038/385093a0 [DOI] [PubMed] [Google Scholar]
- 43. Kramer A., and Schneider-Mergener J. (1998) Synthesis and screening of peptide libraries on continuous cellulose membrane supports. Methods Mol. Biol. 87, 25–39 10.1385/0-89603-392-9:25 [DOI] [PubMed] [Google Scholar]
- 44. Frank R. (2002) High-density synthetic peptide microarrays: emerging tools for functional genomics and proteomics. Comb. Chem. High Throughput Screen. 5, 429–440 10.2174/1386207023330165 [DOI] [PubMed] [Google Scholar]
- 45. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sattler M., Schleucher J., and Griesinger C. (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectr. 34, 93–158 10.1016/S0079-6565(98)00025-9 [DOI] [Google Scholar]
- 47. Renner C., Schleicher M., Moroder L., and Holak T. A. (2002) Practical aspects of the 2D 15N-[1H]-NOE experiment. J. Biomol. NMR 23, 23–33 10.1023/A:1015385910220 [DOI] [PubMed] [Google Scholar]
- 48. Wishart D. S., Bigam C. G., Yao J., Abildgaard F., Dyson H. J., Oldfield E., Markley J. L., and Sykes B. D. (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6, 135–140 10.1007/BF00211777 [DOI] [PubMed] [Google Scholar]
- 49. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 50. Keller R. L. J. (2004) Optimizing the process of nuclear magnetic resonance spectrum analysis and computer aided resonance assignment, Ph.D. thesis, Thesis 15947, ETH Zurich, Switzerland [Google Scholar]
- 51. Herrmann T., Güntert P., and Wüthrich K. (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 10.1016/S0022-2836(02)00241-3 [DOI] [PubMed] [Google Scholar]
- 52. Cornilescu G., Delaglio F., and Bax A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302 10.1023/A:1008392405740 [DOI] [PubMed] [Google Scholar]
- 53. Baxter N. J., and Williamson M. P. (1997) Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 9, 359–369 10.1023/A:1018334207887 [DOI] [PubMed] [Google Scholar]
- 54. Radhakrishnan I., Pérez-Alvarado G. C., Parker D., Dyson H. J., Montminy M. R., and Wright P. E. (1999) Structural analyses of CREB-CBP transcriptional activator-coactivator complexes by NMR spectroscopy: implications for mapping the boundaries of structural domains. J. Mol. Biol. 287, 859–865 10.1006/jmbi.1999.2658 [DOI] [PubMed] [Google Scholar]
- 55. Marengère L. E., Okkenhaug K., Clavreul A., Couez D., Gibson S., Mills G. B., Mak T. W., and Rottapel R. (1997) The SH3 domain of Itk/Emt binds to proline-rich sequences in the cytoplasmic domain of the T cell costimulatory receptor CD28. J. Immunol. 159, 3220–3229 [PubMed] [Google Scholar]
- 56. Perez-Villar J. J., O'Day K., Hewgill D. H., Nadler S. G., and Kanner S. B. (2001) Nuclear localization of the tyrosine kinase Itk and interaction of its SH3 domain with karyopherin α (Rch1α). Int. Immunol. 13, 1265–1274 10.1093/intimm/13.10.1265 [DOI] [PubMed] [Google Scholar]
- 57. Hansson H., Okoh M. P., Smith C. I., Vihinen M., and Härd T. (2001) Intermolecular interactions between the SH3 domain and the proline-rich TH region of Bruton's tyrosine kinase. FEBS Lett. 489, 67–70 10.1016/S0014-5793(00)02438-8 [DOI] [PubMed] [Google Scholar]