

Abstract
During enteroviral infections, the canonical translation factor eukaryotic translation initiation factor 4 γ I (eIF4GI) is cleaved by viral protease 2A. The resulting C-terminal fragment is recruited by the viral internal ribosome entry site (IRES) for efficient translation of the viral RNA. However, the 2A protease is not present in the viral capsid and is synthesized only after the initial round of translation. This presents the conundrum of how the initial round of translation occurs in the absence of the C-terminal eIF4GI fragment. Interestingly, the host protein DAP5 (also known as p97, eIF4GIII, and eIF4G2), an isoform of eIF4GI, closely resembles the eIF4GI C-terminal fragment produced after 2A protease–mediated cleavage. Using the Coxsackievirus B3 (CVB3) IRES as a model system, here we demonstrate that DAP5, but not the full-length eIF4GI, is required for CVB3 IRES activity for translation of input viral RNA. Additionally, we show that DAP5 is specifically required by type I IRES but not by type II or type III IRES, in which cleavage of eIF4GI has not been observed. We observed that both DAP5 and C-terminal eIF4GI interact with CVB3 IRES in the same region, but DAP5 exhibits a lower affinity for CVB3 IRES compared with the C-terminal eIF4GI fragment. It appears that DAP5 is required for the initial round of viral RNA translation by sustaining a basal level of CVB3 IRES activity. This activity leads to expression of 2A protease and consequent robust CVB3 IRES-mediated translation by the C-terminal eIF4GI fragment.
Keywords: translation, RNA virus, eukaryotic translation initiation factor 4G (eIF4G), eukaryotic translation initiation, viral protease, Coxsackievirus B3, DAP5, internal initiation of translation, IRES mediated translation
Introduction
Enteroviruses such as EV71, poliovirus, and Coxsackievirus possess a type I IRES5 in their genomes, which is characterized by its conserved structure and requirement of all canonical translation initiation factors except eIF4E (1). Other IRESs, such as the Hepatitis C Virus (HCV) IRES, which is a type III IRES, are completely independent of the eIF4F complex (consisting of eIF4G, eIF4A, and eIF4E) (1). Interestingly, upon infection by enteroviruses, viral protease 2A cleaves the scaffolding protein eIF4GI into two fragments (2–4). The C-terminal eIF4GI fragment directly interacts with stem loop V in type I IRES via its MIF4G domain, and this interaction is critical for recruitment of the ribosome (5). The mutations in the 2A protease that inhibit cleavage of eIF4GI lead to abrogation of the poliovirus life cycle (6). The aforementioned reports clearly suggest that 2A protease–mediated cleavage of eIF4GI is essential for the successful life cycle of the virus and signify the importance of the interaction between the C-terminal eIF4GI and IRES in viral RNA translation.
Upon entry of enteroviruses into cells, only the viral genomic RNA is released into the cells. This genomic RNA has to undergo its first round of translation in the absence of 2A protease and, hence, in the absence of C-terminal eIF4GI. This suggests that the initial rounds of translation occur by an alternate mechanism in which the C-terminal eIF4GI is not required. It has been suggested that the full-length eIF4GI may interact with type I IRES and mediate translation; however, no experimental evidence has been provided (7). In this study, Coxsackievirus B3 (CVB3) was used as a model system, and the focus was on the functional relevance of the full-length eIF4GI and its cellular isoforms on IRES-mediated translation of Coxsackievirus B3 genomic RNA. Interestingly, it was found that the host protein death-associated protein 5 (DAP5), an N-terminal truncated isoform of eIF4GI, resembles C-terminal eIF4GI with respect to domain organization. It contains an MIF4G domain and eIF4A-interacting domain, similar to the C-terminal eIF4GI that is produced after cleavage by 2A protease. DAP5 has been studied extensively with respect to its role in IRES-mediated translation of cellular mRNAs (8, 9). It has been shown that DAP5 undergoes caspase-mediated cleavage during apoptosis, which leads to production of an 86-kDa truncated protein that acts as a stimulator for cellular IRES-containing mRNA like c-Myc, DAP5, and XIAP (X-linked inhibitor of apoptosis) (10, 11). Unlike its isoform eIF4GI, DAP5 does not have an eIF4E-interacting domain, and it has been shown to be dispensable for cap-dependent translation (8, 12). However, it retains the regions involved in the interaction with eIF3 and eIF4A, which are essential for ribosome recruitment on the target RNA.
Unlike many nuclear resident host RNA–binding proteins that act as IRES trans-acting factors (ITAFs) for viral IRES (13–20), DAP5 is a resident of the cytoplasm, making it readily available for incoming viral RNA. Considering the cytoplasmic localization and its domain organization, similar to C-terminal eIF4GI, we studied the role of DAP5 and full-length eIF4GI in the initial rounds of translation of viral IRES. Our study suggests that DAP5, but not the full-length eIF4GI, is required for the initial round of translation during CVB3 infection. A comparative study was carried out with type I, type II, and type III IRES, and it was found that only type I IRES requires DAP5. This is interesting because only after infection with type I IRES-containing viruses, eIF4GI is cleaved by 2A protease but not in the closely related Encephalomyocarditis Virus (EMCV) virus, which contains type II IRES. The results suggest that DAP5 directly interacts with the CVB3 IRES but not the HCV IRES. DAP5 also interacts with stem loop V, similar to the C-terminal eIF4GI, but with a lower affinity. Based on these results, we propose a model in which the viral RNA undergoes two different stages of translation. The first stage is the basal level of translation during the initial stages (before cleavage of eIF4GI), which is mediated by DAP5. Consequently, viral proteins are produced, leading to 2A protease–mediated cleavage of eIF4GI. The C-terminal eIF4GI is then utilized by IRES to carry out robust protein synthesis. Our study provides new insights into the mechanism of translation initiation at the initial stages of virus infection and provides a model to explain how input viral RNA could be translated in the milieu of limited resources (host factors) in the cytoplasm, with better availability of ITAFs pending.
Results
DAP5 is required for the initial round of CVB3 IRES–mediated translation
As discussed above, eIF4G and its isoforms (Fig. 1A) are known to play a central role in both cap-dependent and cap-independent internal initiation of translation. To understand the mechanism of the initial round of translation in CVB3 genomic RNA, the possible role of the isoforms of eIF4G was explored. DAP5 is an N-terminally truncated isoform of eIF4G that resembles the C-terminal part of eIF4GI and eIF4GII, which is formed after cleavage by 2A protease. Interestingly, DAP5 contains the MIF4G domain, which is known to interact with stem loop V of the CVB3 5′ UTR and also retains the eIF3- and eIF4A-interacting regions. The N terminus region that interacts with eIF4E is absent in DAP5.
Figure 1.

DAP5 is required for CVB3 IRES mediated translation. A, schematic of isoforms of eIF4G in mammalian cells. Different domains in individual proteins, the length of the proteins, and the 2A protease cleavage site are indicated. Poly-A binding protein (PABP) interacting domain and Nuclear Localisation Signal (NLS) are indicated in the N-Terminus of the schematic. B, effect of partial silencing of DAP5 on the CVB3 life cycle. Top panel, schematic of CVB3 replicon RNA. F Luc indicated the Firefly Luciferase gene in the CVB3 replicon. CVB3 replicon RNA was transfected in cells transfected previously with the indicated siRNAs, and cells were processed for luciferase activity 8 h after replicon RNA transfection. Center panel, luciferase activity. Bottom panel, Western blot indicating the DAP5 and β-actin protein levels. The band intensities were quantified, and silencing efficiency is represented normalized to β-actin. C, effect of knockdown of DAP5 or eIF4GI or both on the early stage of the CVB3 life cycle. Top panel, luciferase activity indicating translation of the CVB3 IRES. Bottom panel, Western blot showing the DAP5 and β-Actin protein levels upon treatment of siDAP5, sieIF4GI, or both. D, schematic of WT and 2A protease mutant CVB3 replicon RNAs. The G122E mutation in the 2A protease region of the CVB3 replicon RNA is indicated. E, effect of partial silencing of DAP5 on WT and mutant CVB3 replicon RNA translation in early stages. Left panel, luciferase activity indicating translation of the CVB3 IRES. Right panel, Western blotting showing the DAP5 and β-actin protein levels upon siDAP5 treatment. F, effect of partial silencing of DAP5 and eIF4GI on CVB3 IRES activity 1.5 h after CVB3 infection. The CVB3-RLuc virus was used for this experiment, and the top panel represents percentage luciferase activity. All graphs represent the average of three or more independent experiments. Error bars represent standard deviation. *, p ≤ 0.05; **, p ≤ 0.01. Band intensity normalized to β-actin is indicated below the western blots. Similar sets of blots were used to indicate the molecular weight position in the western blots.
To study the role of DAP5 in the CVB3 life cycle, CVB3 replicon RNA was used, wherein a firefly luciferase gene substitutes the region encoding for structural proteins (Fig. 1B) (21). The in vitro transcribed RNA was directly transfected in cells, and the efficiency of translation and replication was quantified by measuring the luciferase activity. Initially, siRNA-mediated knockdown of DAP5 was carried out in a dose-dependent manner, followed by transfection of CVB3 replicon RNA. The luciferase activity from replicon RNA–transfected cells was measured 8 h after transfection. Compared with nonspecific siRNA–transfected cells, the luciferase activity was found to be decreased in a dose-dependent manner in siDAP5-transfected cells, suggesting that DAP5 is important for the CVB3 life cycle (Fig. 1B). To study the role of full-length eIF4GI and DAP5 in the initial round of translation, the cells were harvested 1.5 h after CVB3 replicon RNA transfection. At this early stage, luciferase activity is a measure of translation alone, as replication only begins 2 h after transfection of the CVB3 replicon RNA (Fig. 1C). Partial silencing of DAP5 resulted in a reduction of luciferase activity, suggesting that DAP5 regulates CVB3 RNA translation during the early stages of the viral life cycle (Fig. 1C). Interestingly, partial silencing of full-length eIF4GI caused a significant increase in translation of CVB3 RNA, suggesting that full-length eIF4GI does not mediate CVB3 IRES–mediated translation at the earlier stages (Fig. 1C). Partial knockdown of both DAP5 and eIF4GI rescued the translation that was inhibited by siDAP5 treatment (Fig. 1C).
To eliminate the possibility of a low level of eIF4GI cleavage at the earlier stages of infection that could have been missed during detection by western blotting, an alternate strategy was adopted. A CVB3 replicon RNA harboring a mutation in the 2A protease region was generated; this is known to inhibit eIF4GI cleavage (Fig. 1D) (22). Partial knockdown of DAP5 led to a reduction in CVB3 IRES–mediated translation 1.5 h after transfection in the WT as well as the mutant replicon (Fig. 1E), suggesting that DAP5 mediates CVB3 IRES–mediated translation independent of eIF4GI cleavage. Of note, it was confirmed that the siRNAs targeting DAP5 and eIF4GI were specific to their targets (Fig. S1, Table S1, and Table S2). To rule out nonspecific effects because of transfection of the CVB3 replicon RNA, the same assay was performed using a CVB3 virus containing a Renilla luciferase gene in the genome (as described by Lanke et al. (21)). Partial silencing of DAP5 led to a dramatic reduction in CVB3 IRES–mediated translation 1.5 h after infection. As expected, partial silencing of full-length eIF4GI did not reduce CVB3 IRES–mediated translation (Fig. 1F). To rule out differences in luciferase activity because of differential cell physiology as a result of siRNA treatment, an MTT assay was performed. As shown in Fig. S2, no significant change in cell proliferation was observed because of siDAP5 or sieIF4GI treatment. These results suggest that, during the initial stages of the viral life cycle, DAP5, but not full-length eIF4GI, is required for CVB3 IRES–mediated translation.
Role of DAP5 and eIF4GI in viral IRES–mediated translation
To study the role of full-length eIF4GI and DAP5 proteins in CVB3 RNA translation, a bicistronic reporter was utilized. In the reporter construct, Renilla luciferase activity represents cap-dependent translation, and firefly luciferase activity represents CVB3 IRES–mediated translation (Fig. S3). Upon expression of 2A protease, CVB3 IRES activity was stimulated, whereas cap-dependent translation was reduced drastically (Fig. S3). This suggests that the IRES activity measured from the bicistronic construct in the absence of 2A protease represents a basal level of IRES activity and is a good system to study the initial round of translation that happens in the absence of the viral proteins. siRNA-mediated knockdown of DAP5 and full-length eIF4GI was performed in a dose-dependent manner, followed by transfection of capped CVB3 bicistronic RNAs. Cells were processed for measuring luciferase activity 10 h after transfection (Fig. 2A). Partial silencing of DAP5 reduced CVB3 IRES activity in a dose-dependent manner, but cap-dependent translation was unaffected (Fig. 2A). Consistent with observations using the CVB3 replicon, partial silencing of eIF4GI led to a significant increase in CVB3 IRES activity while reducing cap-dependent translation (Fig. 2A).
Figure 2.

Role of DAP5 in modulating functions of different IRES elements. A, role of DAP5 and eIF4GI in CVB3 IRES–mediated translation. Top panel, schematic of bicistronic RNAs. B, role of DAP5 and eIF4GI in HCV IRES–mediated translation. C, role of DAP5 and eIF4GI in EMCV IRES–mediated translation. F luc activity represents IRES-mediated translation, and R luc activity represents cap-dependent RNA translation throughout. *, p < 0.05; **, p < 0.01. D and E, Western blots indicating silencing of DAP5 (D) and eIF4GI (E). Band intensity normalized to β-actin is indicated below the western blots. Similar set of blots were used to indicate the molecular weight position in the western blots. F Luc, Firefly luciferase; R Luc, Renilla Luciferase.
Both type I (CVB3 IRES) and type II IRES (e.g. EMCV IRES) require all canonical translation initiation factors except eIF4E. However, in the case of EMCV virus infection, eIF4GI cleavage has not been observed. Unlike type I and type II IRESs, type III IRES (e.g. HCV IRES) do not require the eIF4F complex (eIF4E, eIF4G, eIF4A, and eIF4B) for translation initiation. The study was extended to different IRES to understand the role of DAP5 in IRES-mediated translation. Partial silencing of DAP5 had no effect on EMCV or HCV IRES–mediated translation, suggesting that DAP5 is only required for type I IRES but not for type II and type III IRES (Fig. 2, B and C, respectively). Interestingly, silencing of eIF4GI increased the IRES activity of both EMCV and HCV IRES, similar to CVB3 IRES (Fig. 2, B and C, respectively). It was surprising to observe the inhibitory role of full-length eIF4GI for all IRES. We reason that this could be due to the increased availability of other initiation factors (associated with eIF4GI) to viral IRES as a consequence of silencing of eIF4GI.
DAP5 interacts with CVB3 IRES
From earlier studies, it is known that C-terminal eIF4GI directly interacts with stem loop V in the CVB3 5′ UTR via its MIF4G domain (5). The crystal structures of the MIF4G domain of eIF4GI and DAP5 have been solved previously (PDB codes 4IUL and 1HU3, respectively) (23, 24). Overlaying these structures revealed a remarkable similarity in overall folding of the MIF4G domains in DAP5 and eIF4GI (Fig. S4). The folding of the RNA-interacting region in the MIF4G domain of C-terminal eIF4GI and the corresponding region in DAP5 was similar, with minor differences in the composition of amino acids (Fig. S4). We tested whether DAP5 also interacts with the CVB3 5′ UTR, similar to C-terminal eIF4GI. The UV cross-linking experiment was performed using radiolabeled CVB3 5′ UTR RNA and recombinant DAP5 protein. The unlabeled CVB3 5′ UTR and HCV IRES RNAs were used as competitors in this experiment. As observed in Fig. 3A, DAP5 was able to specifically interact with the CVB3 5′ UTR (lanes 2 and 3) but not with HCV IRES (lanes 4 and 5). The bar graph represents the band intensities from three independent experiments (Fig. 3A). This suggests that DAP5 specifically interacts with the CVB3 IRES but not with the HCV IRES, which is independent of the eIF4F complex for its function. This result is also consistent with the experiments illustrated in Fig. 2B, which suggests that DAP5 is not required for HCV IRES.
Figure 3.

Interaction of DAP5 with CVB3 IRES. A, UV cross-linking experiment carried out with the radiolabeled CVB3 5′ UTR and recombinant DAP5 protein in the presence of unlabeled CVB3 5′ UTR RNA (lanes 2 and 3) or HCV IRES RNA (lanes 4 and 5) as competitor RNAs. NC, no competition. B, schematic of stem loop V in the CVB3 IRES. The binding site of eIF4GI is indicated in the box, and this site is mutated from CCG to AAA. C, UV cross-linking experiment carried out using a radiolabeled CVB3 RNA probe with recombinant DAP5 protein in the presence of the WT unlabeled CVB3 5′ UTR and CCG→AAA mutant CVB3 5′ UTR RNA as competitor RNAs. Normalized band intensities are indicated under individual lanes. D, RNA immunoprecipitation experiment carried out 1.5 h after CVB3 replicon RNA transfection. Top panel, CVB3 positive-strand RNA after immunoprecipitation. Bottom panel, protein levels by Western blotting (WB). All graphs indicate the average of normalized band intensities from three independent experiments. Error bars represent standard deviation. *, p ≤ 0.05; **, p ≤ 0.01.
A mutation in stem loop V at the 541 nt position has been reported to inhibit the C-terminal 4GI interaction with the CVB3 IRES (Fig. 3B) (25). We investigated whether the same mutation could also inhibit the interaction of DAP5 with the CVB3 IRES. The UV cross-linking experiment was carried out using the radiolabeled CVB3 5′ UTR and recombinant DAP5 protein in the presence of unlabeled WT CVB3 5′ UTR or 541 CCG→AAA mutant 5′ UTR competitor RNAs (Fig. 3C; the bar represents the normalized band intensities in three independent experiments). The mutant CVB3 RNA was also able to compete with the probe, but to a much lesser extent, suggesting that the mutation in stem loop V could partially inhibit the interaction of DAP5 with the CVB3 IRES. The minor difference in the amino acid sequence in the MIF4G domain of DAP5 and eIF4G1 could account for this difference in the DAP5 and eIF4G1 interactions. These results demonstrate that both DAP5 and C-terminal eIF4GI interact with the CVB3 IRES at the stem loop V region via their MIF4G domain. Of note, when purifying the recombinant proteins from Escherichia coli for carrying our UV cross-linking assays, contaminant proteins were also copurified (Fig. S5). However, these proteins did not interact with the CVB3 IRES in the assay; hence, we believe that the presence of the contaminants does not affect the conclusions.
To confirm whether DAP5 interacts with the CVB3 IRES in the early stages of the virus life cycle, Myc-tagged DAP5 was overexpressed, immunoprecipitation of DAP5 was performed 1.5 h after CVB3 replicon RNA transfection, and its association with CVB3 genomic RNA was checked. As shown in Fig. 3D, CVB3 genomic RNA was found to be associated with DAP5 in earlier stages in cells.
Significance of DAP5 in CVB3 IRES–mediated translation
The results suggest that DAP5 interacts with the CVB3 IRES and helps in the initial round of translation during the virus life cycle. However, previous studies have indicated that inhibition of eIF4GI cleavage by a mutation in 2A protease leads to an abrogated life cycle, suggesting that DAP5 alone is unable to sustain the entire life cycle and that cleavage of eIF4GI is required. We hypothesized that this could be because DAP5 is less efficient in translation compared with C-terminal eIF4GI. To address this question, surface plasmon resonance experiments were carried out with recombinant DAP5 and C-terminal eIF4GI proteins to measure their affinity for the CVB3 5′ UTR. The affinity of DAP5 was found to be weaker compared with C-terminal eIF4GI for the CVB3 IRES (Fig. 4, A and B). It is known that the affinity of DAP5 for eIF4A is lower compared with the affinity of eIF4GI for eIF4A (24), suggesting that DAP5 is weak in recruiting the downstream factors required for IRES-mediated translation. Together, these results highlight the relatively weak IRES trans-activating potential of DAP5 compared with C-terminal eIF4GI because of its lower affinity with both the CVB3 IRES as well as eIF4A. We propose a model in which DAP5 interacts with the CVB3 IRES and mediates its basal level of translation during the early stages of the CVB3 life cycle. This leads to synthesis of 2A protease and, hence, cleavage of eIF4GI and the subsequent activated mode of translation that is mediated by the cleaved C-terminal part of eIF4GI (Fig. 5).
Figure 4.
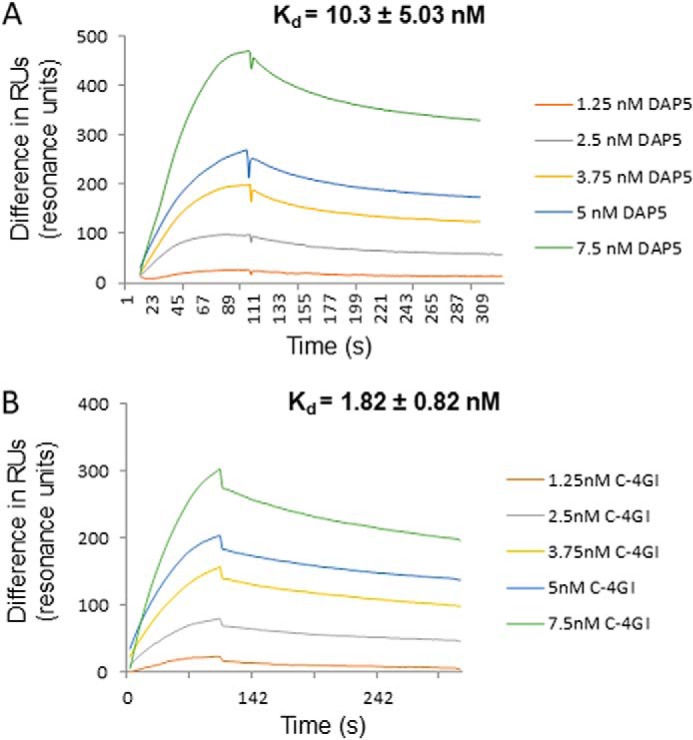
Affinity of DAP5 and C-terminal eIF4GI proteins for binding CVB3 IRES. A and B, sensorgrams representing the interaction of DAP5 (A) or C-terminal eIF4GI (B) with CVB3 5′ UTR RNA at the indicated protein concentrations. The y axis represents the change in RUs during association and dissociation phases. The average Kd is indicated in the inset.
Figure 5.

Role of DAP5 in the initial round of translation. Shown is the proposed model for the initial round of translation in type I IRES. Upon infection, DAP5 interacts with CVB3 IRES and leads to the basal level of translation. This produces viral protein 2A protease, which can now cleave eIF4GI. The C-terminal eIF4GI now interacts with CVB3 IRES and leads to robust activation of translation, which is essential for rapid progression of the viral life cycle.
Discussion
The enterovirus life cycle takes up to 8–10 h in cell culture. To achieve this, viral RNA must undergo robust protein synthesis. Enteroviruses manage to accomplish this task by hijacking the cellular translation machinery for its own protein synthesis and inhibiting host cell translation. A well-studied example is cleavage of eIF4GI and eIF4GII by viral protease 2A, which benefits the virus in two ways. It leads to inhibition of host cell translation, making ribosomes and other initiation factors available for viral IRES–mediated translation (3, 4). The cleaved C-terminal part of eIF4GI directly interacts with IRES, bridging the ribosomes to viral RNA (5). Apart from the canonical translation initiation factors, several host RNA-binding proteins are recruited to viral IRES to facilitate efficient translation (13–20). These RNA-binding proteins are predominantly nuclear residents, and upon viral infection, they relocalize to the cytoplasm. However, relocalization of these proteins is induced only 3–4 h after viral infection (20, 26). Hence, the intracellular host factors that are required for viral IRES–mediated translation are limiting during the initial stages of viral infection because of the absence of viral protease and limited amounts of ITAFs in the cytoplasm. In light of these observations, DAP5, which is located in the cytoplasm and resembles cleaved eIF4GI, is a vital host factor for viral IRES–mediated translation in the initial stages.
A recent report suggested that eIF4E is required for translation initiation by the picornavirus IRES, implying that eIF4GI could also be required. However, this possibility has not been studied directly (7). One possible explanation could be that loss of eIF4E could increase the ability of eIF4G to interact with the other members of eIF4F (eIF4A and eIF4B), ultimately leading to reduced availability of the factors that are required for type I IRES–mediated translation. Another report mentioned that overexpression of eIF4G1 enabled rescue of translation of the mutant poliovirus (27). This is not surprising, as eIF4G1 would also increase the abundance of C-terminal eIF4G1 and, hence, increase translation. However, because of the partial silencing of eIF4GI achieved, our study does not completely rule out that full-length eIF4GI can also function in input viral RNA translation in the first and subsequent rounds. Further studies involving IRES–eIF4GI interaction are required to understand the role of full-length eIF4GI in type I IRES–mediated translation. The results regarding the role of DAP5 in the initial round of translation were consistent in all systems used in this study: CVB3 replicon RNA, bicistronic reporter RNA, as well as CVB3 Renilla luciferase virus infection.
The type I (CVB3) and type II (EMCV) IRES require all canonical initiation factors except eIF4E, whereas the type III IRES (HCV) functions independent of the eIF4F complex. The results suggest that DAP5 does not interact with the HCV IRES and is not required for its activity. Interestingly, it was found that full-length eIF4GI is dispensable for EMCV IRES–mediated translation. The EMCV virus harbors a type II IRES for its translation, and eIF4GI cleavage has not been observed during EMCV infection. It was found in this study that DAP5 is dispensable for EMCV IRES–mediated translation. Interestingly, partial silencing of intact eIF4GI led to an increase in EMCV IRES activity. These results are in contrast with earlier reports that suggest that eIF4GI interaction with the J–K loop in the EMCV IRES is required for ribosome recruitment (28–30). However, more recently it has been shown that the EMCV IRES could directly recruit the 40S ribosome subunit in the absence of eIF4G, suggesting that alternative mechanisms of translation initiation may exist (31). In another study, depletion of the full-length eIF4GI by expression of CVB3 2A protease during EMCV infection led to an increase in EMCV IRES activity, pointing to the fact that intact eIF4GI may not be required for EMCV IRES activity (32). We propose that down-regulation of eIF4GI by siRNAs led to an increase in the available amount of eIF4A and eIF3 for viral IRESs and, hence, facilitates ribosome recruitment. This claim is supported by evidence showing that eIF4GI is predominantly present in the monosomal and polysomal fractions in the cell rather than the ribosome-free fractions, suggesting tight associations with initiation factors and ribosomes (33).
DAP5 has been studied previously with respect to CVB3 infection; however, its role in viral IRES–mediated translation has not been studied. It was found that DAP5 is also cleaved by viral 2A protease into N- and C-terminal parts so that the N-terminal fragment retains the MIF4G domain (34). Interestingly, it was shown that the N-terminal fragment of DAP5 containing the MIF4G domain translocated to the nucleus. Translocation of N-DAP5 to the nucleus after 2A protease–mediated cleavage could favor interaction with the C-terminal eIF4GI, as both proteins share a common binding site on the CVB3 IRES, as observed in this study. The sequence of stem loop V seems to be conserved in type I IRES–containing viruses (Fig. S6), suggesting that the role of DAP5 could be conserved in all viruses containing type I IRES.
Interaction of DAP5 with the CVB3 IRES is a key step in translation initiation in the early stages. Our study found that DAP5 has a lower affinity for the CVB3 IRES compared with C-terminal eIF4GI. However, interaction with RNA alone is not sufficient for translation initiation. Recruitment of downstream initiation factors like eIF4A by DAP5 is also required for IRES function. It has been observed that DAP5 has a 10 times lower affinity for eIF4A compared with eIF4GI (24). These observations suggest that DAP5 is not as potent as C-terminal eIF4GI in ribosome recruitment on the CVB3 IRES and explains why DAP5 alone is unable to sustain the entire life cycle. We propose a model in which a basal level of translation is carried out by DAP5 in the initial stages, followed by a robust mode of translation mediated by C-terminal eIF4GI in the later stages. In the subsequent stages, many ITAFs also relocalize from the nucleus to the cytoplasm, and these proteins could also contribute to a robust mode of translation. This study provides novel insights into a previously ignored but essential stage of the virus life cycle.
Experimental procedures
Cell line, transfections, and luciferase assay
For all cell culture experiments in this study, HeLa cells were used. CVB3 replicon RNA used for transfections was prepared from the pRib-T7/luc plasmid (a kind gift from Prof. Frank van Kuppeveld, Utrecht University) as described earlier (20). CVB3 replicon RNA was transfected in HeLa cells, and at the indicated time points, cells were processed for the luciferase assay using luciferase assay reagent (Promega) according to the manufacturer's protocol. siDAP5 and sieIF4GI were acquired from Integrated DNA Technologies and transfected at 50 nm or 100 nm concentration. All transfections were carried out using Lipofectamine 2000 reagent using the manufacturer's protocol (Invitrogen).
pCDNA3-CVB3 bicis, pCDNA3-HCV bicis, and pCDNA3-EMCV bicis plasmids were used to prepared various capped bicistronic RNAs. These plasmids were linearized with the PmeI restriction enzyme, and the linear plasmids were used as a template in in vitro transcription reactions. The luciferase assays for bicistronic RNAs were performed using Dual-Luciferase reagent kit (Promega) according to the manufacturer's protocol.
Virus preparation and infection
The pRLuc-CVB3 plasmid (a kind gift from Prof. Frank Van Kuppeveld) was used to prepare the CVB3 virus containing the Renilla luciferase gene. In vitro transcribed CVB3 Renilla luciferase RNA was transfected in HeLa cells, and the virus was purified from the cell culture supernatant. Virus titer was calculated by performing a plaque assay in Vero cells, and plaque-forming units per milliliter were estimated. For experiments, HeLa cells were infected with a multiplicity of infection of 15.
In vitro transcriptions
CVB3 5′ UTR RNA was prepared from the pCDNA3-CVB3 5′ UTR plasmid as described earlier (20). α-32P was included in in vitro transcription reactions carried out to prepare radiolabeled RNAs. In vitro transcription reactions were carried out using T7 RNA polymerase (Thermo Scientific) according to the manufacturer's protocol. To prepare biotin-labeled CVB3 5′ UTR RNA, biotin-11–UTP was included in in vitro transcription reactions carried out using the Ribomax Kit (Promega).
Capped RNAs were prepared by adding a cap analog in in vitro transcription reactions carried out using the Ribomax kit (Promega) according to the manufacturer's protocol. These RNAs were transfected in HeLa cells, and 10 h after transfection, cells were processed for the luciferase assay. Dual-Luciferase assays were carried out using the DLR kit (Promega) according to the manufacturer's protocol.
Plasmids and protein purification
Recombinant DAP5 protein was purified using the pET28a-DAP5 plasmid (a kind gift from Prof. Adi Kimchi, Weizmann Institute) as described earlier (12). C-terminal eIF4GI was cloned into the pET28a vector from the pCDNA3-eIF4GI-HA plasmid (a kind gift from Prof. Nahum Sonenberg, McGill University). The pET28a–C-terminal eIF4GI plasmid was transformed in the E. coli BL21 strain, and protein expression was induced at 0.6 optical density by 0.6 mm isopropyl 1-thio-β-d-galactopyranoside for 3 h, followed by purification of His-tagged protein using nickel-nitrilotriacetic acid beads. Myc-tagged DAP5 was cloned in the pCDNA3 vector for expression in HeLa cells.
Western blotting
Cells were lysed using radioimmune precipitation assay buffer, and the protein was quantified using Bradford reagent. Equal amounts of proteins were resolved on SDS-PAGE, followed by transfer of proteins over a PVDF membrane (Millipore). Primary antibodies used in this study were as follows: anti-DAP5 antibody (Imgenex), anti-eIF4GI antibody (Cell Signaling Technology), and HRP-conjugated β-actin antibody (Sigma). Primary antibodies were incubated with the blots for 12 h, followed by washing with TBST buffer (20 mm Tris (pH 7.5), 137 mm NaCl, and 0.1% Tween 20) and subsequent incubation with secondary antibodies (anti-rabbit HRP-conjugated (Sigma) or anti-mouse HRP-conjugated (Sigma)). Blots were developed by chemiluminescence using WesternBright (Advansta).
UV-induced cross-linking of RNA–protein complexes
UV cross-linking assays were carried out essentially as described earlier (20). Briefly, recombinant DAP5 or C-terminal eIF4GI protein was incubated with radiolabeled CVB3 5′ UTR RNA at 30 °C for 30 min in presence of RNA binding buffer (25 mm HEPES (pH 7.6), 1.25 mm ATP and 2 mm MgCl2, 19% glycerol, 0.5 mm EDTA, 1.25 mm ATP, and 2 mm GTP). The reaction also contained 10 μg of yeast tRNAs as nonspecific RNAs. The RNA–protein complexes were then cross-linked using UV light for 20 min, followed by RNase A treatment for 45 min at 37 °C. Laemmli's buffer was added, the samples were boiled for 5 min, and proteins were resolved on SDS-PAGE. The RNA-bound proteins were visualized by autoradiography. For competition UV cross-linking assays, the indicated amounts of unlabeled RNAs were additionally included in the reaction mixture. Radiolabeled RNAs were prepared by carrying out in vitro transcription in the presence of [α-32P]UTP (BRIT, Hyderabad, India); hence, the RNAs were U-labeled.
RNA immunoprecipitation
RNA immunoprecipitation was essentially performed as described previously (20). Briefly, cells were lysed in immunoprecipitation buffer containing 100 mm KCl, 5 mm MgCl2, 10 mm HEPES (pH 7.0), 0.5% NP-40, 1 mm DTT, and 100 units/ml RNase inhibitor. Protein G beads were saturated with antibodies (anti-Myc or IgG control) and incubated with an equal amount of lysate at 4 °C overnight. The beads were washed with wash buffer twice and resuspended in 100 μl of immunoprecipitation buffer. To analyze the proteins in the pulldown, Western blotting was performed using a 10% aliquot of the pulldown sample. To check the RNAs associated with the pulled-down proteins, the sample was subjected to proteinase K treatment, and subsequently RNA was isolated using TRIzol. The association of CVB3 genomic RNA was tested by semiquantitative PCR using CVB3-specific primers: CVB3 forward, 5′-GAATGCGGCTAATCCTAACTGC-3′; CVB3 reverse, 5′-GCTCTATTAGTCACCGGATGGC-3′.
Surface plasmon resonance experiments
Surface plasmon resonance spectroscopy was performed using a BIAcore3000 optical biosensor (GE Healthcare Lifescience) to study the binding kinetics of DAP5 and C-terminal eIF4GI proteins with CVB3 5′ UTR RNA. Biotin-labeled CVB3 RNA was immobilized on streptavidin-coated sensor chips (GE Healthcare Lifescience) to a final concentration of ≈1000 Resonance Units (RU)/flow cell. RNA–protein interactions were carried out in a continuous flow of Tris buffer (25 mm Tris (pH 7.5), 100 mm KCl, 7 mm β-mercaptoethanol, and 10% glycerol) at 25 °C at a flow rate of 10 μl/min. Increasing concentrations of DAP5 or C-terminal eIF4GI protein were loaded on the biosensor chip for 100 s (characterized as the association phase), followed by a dissociation phase of 300 s with buffer alone. A blank surface without any RNA was used to determine the background nonspecific interaction by simultaneous injections of the sample during the experiment. The on rate, kon (M−1 s−1), and off rate, koff (s−1), were determined using BIAevaluation software (version 3.0) by using a 1:1 Langmuir binding model. The Kd was determined using the following equation: Kd = koff/kon.
MTT assay
To study the effect of siDAP5 and sieIF4GI on cell proliferation and growth, a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed as described previously (35).
Author contributions
P. D., B. G., and S. D. conceptualization; P. D. and S. D. resources; P. D., B. G., H. R., P. R., P. B., and S. D. data curation; P. D., B. G., H. R., P. R., P. B., and S. D. formal analysis; P. D. and S. D. supervision; P. D. and S. D. funding acquisition; P. D., B. G., H. R., P. R., P. B., and S. D. investigation; P. D., B. G., H. R., P. R., P. B., and S. D. methodology; P. D. and B. G. writing-original draft; P. D., B. G., H. R., P. R., P. B., and S. D. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Prof. Nahum Sonenberg, Prof. Frank Van Kuppeveld, and Prof. Adi Kimchi for sharing plasmid constructs. We also thank Prof. D. N. Rao and the surface plasmon resonance facility at the Indian Institute of Science for help.
This work was supported by the Fund for Improvement of Science and Technology Infrastructure level II infrastructure and the University Grants Commission Centre of Advanced Studies. This work was also supported by the Department of Biotechnology, Ministry of Science and Technology, Government of India DBT-IISc (Department of Biotechnology - Indian Institute of Science) Partnership Program. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6 and Tables S1 and S2.
- IRES
- internal ribosomal entry site(s)
- CVB3
- Coxsackievirus B3
- ITAF
- IRES trans-acting factor
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- bicis
- bicistronic
- EMCV
- encephalomyocarditis virus.
References
- 1. Jackson R. J., Hellen C. U., and Pestova T. V. (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11, 113 10.1038/nrm2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Etchison D., Milburn S. C., Edery I., Sonenberg N., and Hershey J. W. (1982) Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J. Biol. Chem. 257, 14806–14810 [PubMed] [Google Scholar]
- 3. Kräusslich H. G., Nicklin M. J., Toyoda H., Etchison D., and Wimmer E. (1987) Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J. Virol. 61, 2711–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gradi A., Svitkin Y. V., Imataka H., and Sonenberg N. (1998) Proteolysis of human eukaryotic translation initiation factor eIF4GII, but not eIF4GI, coincides with the shutoff of host protein synthesis after poliovirus infection. Proc. Natl. Acad. Sci. U.S.A. 95, 11089–11094 10.1073/pnas.95.19.11089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Breyne S., Yu Y., Unbehaun A., Pestova T. V., and Hellen C. U. (2009) Direct functional interaction of initiation factor eIF4G with type 1 internal ribosomal entry sites. Proc. Natl. Acad. Sci. U.S.A. 106, 9197–9202 10.1073/pnas.0900153106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu S. F., Benton P., Bovee M., Sessions J., and Lloyd R. E. (1995) Defective RNA replication by poliovirus mutants deficient in 2A protease cleavage activity. J. Virol. 69, 247–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Avanzino B. C., Fuchs G., and Fraser C. S. (2017) Cellular cap-binding protein, eIF4E, promotes picornavirus genome restructuring and translation. Proc. Natl. Acad. Sci. U.S.A. 114, 9611–9616 10.1073/pnas.1704390114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liberman N., Gandin V., Svitkin Y. V., David M., Virgili G., Jaramillo M., Holcik M., Nagar B., Kimchi A., and Sonenberg N. (2015) DAP5 associates with eIF2β and eIF4AI to promote internal ribosome entry site-driven translation. Nucleic Acids Res. 43, 3764–3775 10.1093/nar/gkv205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoffe Y., David M., Kalaora R., Povodovski L., Friedlander G., Feldmesser E., Ainbinder E., Saada A., Bialik S., and Kimchi A. (2016) Cap-independent translation by DAP5 controls cell fate decisions in human embryonic stem cells. Genes Dev. 30, 1991–2004 10.1101/gad.285239.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Henis-Korenblit S., Strumpf N. L., Goldstaub D., and Kimchi A. (2000) A novel form of DAP5 protein accumulates in apoptotic cells as a result of caspase cleavage and internal ribosome entry site-mediated translation. Mol. Cell. Biol. 20, 496–506 10.1128/MCB.20.2.496-506.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Henis-Korenblit S., Shani G., Sines T., Marash L., Shohat G., and Kimchi A. (2002) The caspase-cleaved DAP5 protein supports internal ribosome entry site-mediated translation of death proteins. Proc. Natl. Acad. Sci. U.S.A. 99, 5400–5405 10.1073/pnas.082102499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weingarten-Gabbay S., Khan D., Liberman N., Yoffe Y., Bialik S., Das S., Oren M., and Kimchi A. (2014) The translation initiation factor DAP5 promotes IRES-driven translation of p53 mRNA. Oncogene 33, 611–618 [DOI] [PubMed] [Google Scholar]
- 13. Huang P.-N., Lin J.-Y., Locker N., Kung Y.-A., Hung C.-T., Lin J.-Y., Huang H.-I., Li M.-L., and Shih S.-R. (2011) Far upstream element binding protein 1 binds the internal ribosomal entry site of enterovirus 71 and enhances viral translation and viral growth. Nucleic Acids Res. 39, 9633–9648 10.1093/nar/gkr682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fitzgerald K. D., and Semler B. L. (2011) Re-localization of cellular protein SRp20 during poliovirus infection: bridging a viral IRES to the host cell translation apparatus. PLOS Pathog. 7, e1002127 10.1371/journal.ppat.1002127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verma B., Bhattacharyya S., and Das S. (2010) Polypyrimidine tract-binding protein interacts with coxsackievirus B3 RNA and influences its translation. J. Gen. Virol. 91, 1245–1255 10.1099/vir.0.018507-0 [DOI] [PubMed] [Google Scholar]
- 16. Kafasla P., Morgner N., Robinson C. V., and Jackson R. J. (2010) Polypyrimidine tract-binding protein stimulates the poliovirus IRES by modulating eIF4G binding. EMBO J. 29, 3710–3722 10.1038/emboj.2010.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kempf B. J., and Barton D. J. (2008) Poly(rC) binding proteins and the 5′ cloverleaf of uncapped poliovirus mRNA function during de novo assembly of polysomes. J. Virol. 82, 5835–5846 10.1128/JVI.01513-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ray P. S., and Das S. (2002) La autoantigen is required for the internal ribosome entry site-mediated translation of coxsackievirus B3 RNA. Nucleic Acids Res. 30, 4500–4508 10.1093/nar/gkf583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blyn L. B., Towner J. S., Semler B. L., and Ehrenfeld E. (1997) Requirement of poly(rC) binding protein 2 for translation of poliovirus RNA. J. Virol. 71, 6243–6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dave P., George B., Sharma D. K., and Das S. (2017) Polypyrimidine tract-binding protein (PTB) and PTB-associated splicing factor in CVB3 infection: an ITAF for an ITAF. Nucleic Acids Res. 45, 9068–9084 10.1093/nar/gkx519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lanke K. H., van der Schaar H. M., Belov G. A., Feng Q., Duijsings D., Jackson C. L., Ehrenfeld E., and van Kuppeveld F. J. (2009) GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J. Virol. 83, 11940–11949 10.1128/JVI.01244-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu S., Wang Y., Lin L., Si X., Wang T., Zhong X., Tong L., Luan Y., Chen Y., Li X., Zhang F., Zhao W., and Zhong Z. (2014) Protease 2A induces stress granule formation during coxsackievirus B3 and enterovirus 71 infections. Virol J 11, 192 10.1186/s12985-014-0192-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marcotrigiano J., Lomakin I. B., Sonenberg N., Pestova T. V., Hellen C. U., and Burley S. K. (2001) A conserved HEAT domain within eIF4G directs assembly of the translation initiation machinery. Mol. Cell 7, 193–203 10.1016/S1097-2765(01)00167-8 [DOI] [PubMed] [Google Scholar]
- 24. Virgili G., Frank F., Feoktistova K., Sawicki M., Sonenberg N., Fraser C. S., and Nagar B. (2013) Structural analysis of the DAP5 MIF4G domain and its interaction with eIF4A. Structure 21, 517–527 10.1016/j.str.2013.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sweeney T. R., Abaeva I. S., Pestova T. V., and Hellen C. U. (2014) The mechanism of translation initiation on type 1 picornavirus IRESs. EMBO J. 33, 76–92 10.1002/embj.201386124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fitzgerald K. D., Chase A. J., Cathcart A. L., Tran G. P., and Semler B. L. (2013) Viral proteinase requirements for the nucleocytoplasmic relocalization of cellular splicing factor SRp20 during picornavirus infections. J. Virol. 87, 2390–2400 10.1128/JVI.02396-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Avanzino B. C., Jue H., Miller C. M., Cheung E., Fuchs G., and Fraser C. S. (2018) Molecular mechanism of poliovirus Sabin vaccine strain attenuation. J. Biol. Chem. 293, 15471–15482 10.1074/jbc.RA118.004913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kolupaeva V. G., Pestova T. V., Hellen C. U., and Shatsky I. N. (1998) Translation eukaryotic initiation factor 4G recognizes a specific structural element within the internal ribosome entry site of encephalomyocarditis virus RNA. J. Biol. Chem. 273, 18599–18604 10.1074/jbc.273.29.18599 [DOI] [PubMed] [Google Scholar]
- 29. Kolupaeva V. G., Lomakin I. B., Pestova T. V., and Hellen C. U. (2003) Eukaryotic initiation factors 4G and 4A mediate conformational changes downstream of the initiation codon of the encephalomyocarditis virus internal ribosomal entry site. Mol. Cell. Biol. 23, 687–698 10.1128/MCB.23.2.687-698.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jackson R. J. (2013) The current status of vertebrate cellular mRNA IRESs. Cold Spring Harb. Perspect. Biol. 5, a011569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chamond N., Deforges J., Ulryck N., and Sargueil B. (2014) 40S recruitment in the absence of eIF4G/4A by EMCV IRES refines the model for translation initiation on the archetype of type II IRESs. Nucleic Acids Res. 42, 10373–10384 10.1093/nar/gku720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Song Q.-Q., Lu M.-Z., Song J., Chi M.-M., Sheng L.-J., Yu J., Luo X.-N., Zhang L., Yao H.-L., and Han J. (2015) Coxsackievirus B3 2A protease promotes encephalomyocarditis virus replication. Virus Res. 208, 22–29 10.1016/j.virusres.2015.05.020 [DOI] [PubMed] [Google Scholar]
- 33. Nousch M., Reed V., Bryson-Richardson R. J., Currie P. D., and Preiss T. (2007) The eIF4G homolog p97 can activate translation independent of caspase cleavage. RNA 13, 374–384 10.1261/rna.372307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hanson P. J., Ye X., Qiu Y., Zhang H. M., Hemida M. G., Wang F., Lim T., Gu A., Cho B., Kim H., Fung G., Granville D. J., and Yang D. (2016) Cleavage of DAP5 by coxsackievirus B3 2A protease facilitates viral replication and enhances apoptosis by altering translation of IRES-containing genes. Cell Death Differ. 23, 828–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ciofani G., Danti S., D'Alessandro D., Moscato S., and Menciassi A. (2010) Assessing cytotoxicity of boron nitride nanotubes: interference with the MTT assay. Biochem. Biophys. Res. Commun. 394, 405–411 10.1016/j.bbrc.2010.03.035 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.