

Abstract
Unlike many other well-characterized bacteria, the opportunistic human pathogen Pseudomonas aeruginosa relies exclusively on the Entner-Doudoroff pathway (EDP) for glycolysis. Pyruvate kinase (PK) is the main “pacemaker” of the EDP, and its activity is also relevant for P. aeruginosa virulence. Two distinct isozymes of bacterial PK have been recognized, PykA and PykF. Here, using growth and expression analyses of relevant PK mutants, we show that PykA is the dominant isoform in P. aeruginosa. Enzyme kinetics assays revealed that PykA displays potent K-type allosteric activation by glucose 6-phosphate and by intermediates from the pentose phosphate pathway. Unexpectedly, the X-ray structure of PykA at 2.4 Å resolution revealed that glucose 6-phosphate binds in a pocket that is distinct from the binding site reported for this metabolite in the PK from Mycobacterium tuberculosis (the only other available bacterial PK structure containing bound glucose 6-phosphate). We propose a mechanism by which glucose 6-phosphate binding at the allosteric site communicates with the PykA active site. Taken together, our findings indicate remarkable evolutionary plasticity in the mechanism(s) by which PK senses and responds to allosteric signals.
Keywords: glycolysis, pentose phosphate pathway (PPP), bacterial metabolism, pyruvate kinase, X-ray crystallography, Pseudomonas, allostery, Entner-Doudoroff Pathway, Pseudomonas aeruginosa, PykA
Introduction
Pyruvate kinase (EC 2.7.1.40) is a key enzyme in glycolysis, and a major site of pathway regulation. The enzyme catalyzes the conversion of phosphoenolpyruvate and ADP into pyruvate and ATP (1). Given its central position in metabolism, pyruvate kinase (PK)2 activity can have a major impact on the carbon economy of the cell (2, 3). More recently, it has been shown that PK activity can also influence pathogencity (4). Bacterial PK is encoded by two isozymes, pykA and pykF (5–7). Most organisms encode either pykA or pykF, although a few species also encode both isozymes. Among the organisms encoding both pykA and pykF that have been characterized to date, PykF is usually the dominant isoform. The PykA and PykF isozymes have been especially well-studied in the Enterobacteriaceae, and are distinguished by being differentially regulated and cladistically distinct (at a sequence level). For example, PykF isozymes are typically activated by fructose 1,6-bisphosphate (8), whereas PykA is activated by adenosine 5′-monophosphate (AMP) (9). Several crystal structures are available for PykF isozymes, although no structures have been solved for PykA. Recently, the structure of Mycobacterium tuberculosis PK bound to AMP and glucose 6-phosphate was solved (the first structure to be solved for a bacterial PK with bound allosteric regulators). The regulators were found to bind at distinct sites in the allosteric cleft and were proposed to communicate with the active site through a series of domain re-orientations denoted the “rock-shape-lock” mechanism.
Pseudomonas aeruginosa is an opportunistic pathogen that commonly causes pulmonary (10), urinary tract (11), and soft tissue-associated infections in hospitalized patients. The pathogen is intrinsically resistant to most clinically-used antibiotics (12) and was recently designated by the World Health Organization (WHO) as a “critical priority pathogen.” Unlike the Enterobacteriaceae, which mainly employ the Embden-Meyerhof-Parnas (EMP) pathway for glucose catabolism, P. aeruginosa lacks phosphofructokinase and utilizes the Entner-Doudoroff pathway (EDP) instead (13, 14). As a consequence, PK is likely to be the major site of glycolytic flux regulation in this organism. P. aeruginosa encodes two uncharacterized PK isozymes, annotated as pykA (PA4329) and pykF (PA1498). Here, we show that in contrast with most organisms encoding both PK isozymes, in P. aeruginosa, PykA plays the dominant physiological role. We also present the first structure of a PykA isozyme. Surprisingly, this structure revealed that glucose 6-phosphate binds in a site distinct to that observed for the M. tuberculosis enzyme.
Results
PykA is the dominant pyruvate kinase isozyme in P. aeruginosa
P. aeruginosa encodes two PK isozymes: PykA (PA4329, 58% amino acid identity with Escherichia coli PykA) and PykF (PA1498, 37% amino acid identity with E. coli PykF). An alignment of selected relevant PykA and PykF sequences is shown in Fig. S1. Phylogenetic classification of P. aeruginosa PykA and PykF (Fig. S2) indicates that the isozymes belong to two distinct subclasses of PK. To establish which isozyme is dominant in P. aeruginosa, we measured the PK activity in cell extracts of mutants defective in pykA, pykF, or pykA and pykF together (pykA pykF). Following growth on minimal medium containing glucose, glycerol, or acetate as a sole carbon source, pyruvate kinase activity was >90% lower in the pykA mutant and in the pykA pykF mutant than it was in the WT control (Fig. 1). However, pyruvate kinase activity in the pykF mutant remained high on these substrates. These data indicate that PykA contributes most to PK activity in P. aeruginosa. Consistent with this, Western analyses using antibodies raised against purified P. aeruginosa PykA or PykF confirmed that the latter is essentially undetectable during growth on the carbon sources tested unless overexpressed in trans from a plasmid (Fig. 1). Note that the antibodies were preabsorbed against a pykA mutant (for the anti-PykA antibodies) or a pykF mutant (for the PykF antibodies). The absence of PykF expression in a pykA mutant led to weak cross-reactivity of the anti-PykA antibodies against shared PykA/PykF epitopes in PykF, although this was only apparent when PykF was overexpressed. We conclude that pykA is expressed at higher levels in these conditions than pykF.
Figure 1.

PykA is the dominant PK in P. aeruginosa. Upper panel, PK activity of the WT and indicated mutants grown in M9 minimal media supplemented with 20 mm glucose, 30 mm glycerol, or 40 mm acetate. Where indicated, the pykA mutant, pykF mutant, or pykA pykF double mutant were complemented with a plasmid expressing pykA (denoted ppykA+) or pykF (denoted ppykF+). Cell cultures were collected for measurement of enzyme activity after 10 h of growth. Middle panel, Western blots showing expression of PykA and PykF (as indicated) in the different carbon sources. Isocitrate dehydrogenase (ICD) was probed using anti-ICD antibodies as a loading control in each blot. The doublet observed when anti-PykA antibodies were used to probe cell extracts of the pykF mutant expressing pykF in trans indicates weak cross-reactivity of the anti-PykA antibodies with PykF (the lower of the bands in the doublet being PykF). No such cross-reactivity was apparent with the anti-PykF antibodies. Lower panel, growth of the indicated PK mutants (± complementation) in different carbon sources. Data represent mean ± S.D. for three independent biological replicates.
To explore the phenotypic consequences of pykA and pykF mutation, we assessed the growth characteristics of each mutant on different carbon sources (Fig. 1). The pykA mutant and the pykA pykF mutant grew slowly compared with the WT and the pykF mutant in minimal media containing glucose or glycerol as a sole carbon source. That there was any observable growth of these mutants at all on glucose is presumably due to the pyruvate produced at the 2-keto-3-deoxy-6-phosphogluconate (KDPG) aldolase-catalyzed step of the EDP. Similarly, the slow, but nonnegligible growth in glycerol (which feeds in to the EDP after the KDPG aldolase-catalyzed step) can be rationalized by invoking operation of the recently-defined EDEMP cycle (15). The EDEMP cycle allows triose phosphates such as those derived from glycerol to be recycled through the KDPG aldolase step, thereby yielding pyruvate. Growth on glucose or glycerol was restored in the pykA mutant and the pykA pykF mutant by expression of either pykA or pykF in trans, suggesting that both isozymes have the potential to complement the pykA mutant growth defect. These data also indicate that the low PK activity in the pykA mutant is likely due to low expression of PykF, rather than low intrinsic activity of the isozyme. Consistent with this, pyruvate kinase activity increased and PykF expression became detectable during growth on glucose when pykF was expressed in trans in the pykF mutant (Fig. 1). (PykF is predicted to be part of a bicistronic operon alongside a putative glycerate kinase, PA1499. The ppykF+ complementation plasmid encoded both PA1499 and pykF, see “Materials and Methods”.) In contrast, during growth on acetate, the pykA, pykF, and pykA pykF mutants all grew at essentially the same rate as the WT. This suggests that PykA plays an important role in catabolizing substrates that feed into the EDP such as glucose and glycerol, but contributes less to growth on acetate (which does not require flux through the PK-catalyzed reaction).
Kinetic properties and regulation of PykA
Purified PykA was an ∼200 kDa tetramer in solution based on analytical ultracentrifugation data (Figs. S5 and S6). At saturating phosphoenolpyruvate (PEP) concentration, the enzyme displayed Michaelis-Menten kinetics with respect to [ADP], with a Km value of 0.07 mm (Fig. 2, Table 1). In contrast, at saturating ADP concentration, the PEP-dependence was sigmoidal, with an s0.5 value of 0.67 mm (Fig. 2, Table 1) and a Hill coefficient of 2.14, indicative of positive cooperativity.
Figure 2.

Kinetic characterization of PykA. A and B, PykA kinetics with respect to PEP and ADP. The PEP titration was carried out using a saturating concentration of ADP (2 mm), whereas the ADP titration was carried out using a saturating concentration of PEP (5 mm). C, effects of monovalent and divalent cations on PykA activity. MgCl2 and KCl were added at 10 and 100 mm concentrations, respectively. PEP and ADP were at saturating initial concentrations (5 and 2 mm, respectively). D, the effect of putative metabolic regulators on PykA activity at low [PEP] (0.3 mm). [ADP] was fixed at 2 mm. The rationale here was to identify potential activatory molecules. Potential regulators were added at a final concentration of 1 mm, except for F6P, R5P, and X5P, which were added at 0.2, 0.15, and 0.5 mm concentration, respectively. Data of A–D represent the mean ± S.D. of three independent experiments. E, Lineweaver-Burk plots demonstrating that the indicated metabolic molecules primarily act to decrease s0.5 of PykA compared with control (without regulators).
Table 1.
The effect of metabolic regulators and monovalent ions on PykA activity
Kinetic parameters were calculated using GraphPad Prism from best-fit nonlinear regression analysis of the data. The allosteric sigmoid function was used for the PEP titration and the Michaelis-Menten function was used for ADP titration.
| No additive | 0.2 mm F6P | 0.15 mm R5P | 1 mm G3P | 1 mm G6P | 1 mm KDPG | 0.5 mm X5P | 1 mm RL5P | 100 mm KCl | 100 mm NH4Cl | 100 mm NaCl | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PEP titration | |||||||||||
| s0.5 (mm) | 0.670 ± 0.058 | 0.159 ± 0.021 | 0.195 ± 0.017 | 0.297 ± 0.017 | 0.290 ± 0.026 | 0.432 ± 0.036 | 0.209 ± 0.011 | 0.269 ± 0.012 | 1.187 ± 0.086 | 1.402 ± 0.084 | 1.492 ± 0.116 |
| Hill coefficient (h) | 2.14 ± 0.34 | 1.45 ± 0.31 | 1.65 ± 0.25 | 1.89 ± 0.21 | 1.78 ± 0.32 | 1.52 ± 0.18 | 1.87 ± 0.22 | 1.61 ± 0.12 | 2.36 ± 0.28 | 2.31 ± 0.22 | 2.42 ± 0.32 |
| Vmax (mm/min) | 0.099 ± 0.004 | 0.096 ± 0.004 | 0.092 ± 0.003 | 0.092 ± 0.002 | 0.100 ± 0.003 | 0.100 ± 0.004 | 0.110 ± 0.002 | 0.110 ± 0.002 | 0.087 ± 0.002 | 0.130 ± 0.003 | 0.120 ± 0.004 |
| kcat(s−1) | 431.9 | 418.8 | 401.4 | 401.4 | 436.3 | 436.3 | 479.9 | 479.9 | 379.6 | 567.2 | 523.6 |
| kcat/s0.5(s−1 mm−1) | 644.1 | 2626 | 2055 | 1348 | 1504 | 1008 | 2286 | 1778 | 319.8 | 404.6 | 350.9 |
| ADP titration | |||||||||||
| Km (mm) | 0.070 ± 0.015 | NAa | NA | NA | NA | NA | NA | NA | 0.147 ± 0.020 | 0.155 ± 0.026 | 0.146 ± 0.033 |
| Vmax (mm/min) | 0.080 ± 0.005 | NA | NA | NA | NA | NA | NA | NA | 0.089 ± 0.004 | 0.067 ± 0.003 | 0.078 ± 0.005 |
| kcat (s−1) | 349.0 | NA | NA | NA | NA | NA | NA | NA | 388.3 | 292.3 | 340.3 |
| kcat/Km (s−1 mm−1) | 4952 | NA | NA | NA | NA | NA | NA | NA | 2633 | 1876 | 2321 |
a NA, not applicable (the regulator had no significant effect on PykA kinetics compared with the control (no additive present)). The values of kcat were calculated using [Et] = [PK monomer].
All PK enzymes require divalent cations for activity (mostly Mg2+), and some have also been reported to require K+ to achieve maximum catalysis (16). The presence of these ions is important to facilitate binding of the substrate, transfer of the phosphoryl group from PEP to ADP, and possibly also acquisition of an active enzyme conformation (17, 18). We found that the activity of P. aeruginosa PykA was independent of K+ concentration (Fig. 2). Indeed, addition of monovalent cations (K+, NH4+ or Na+) even somewhat decreased the overall catalytic activity of the enzyme (Fig. S7, Table 1). The K+-mediated activation of pyruvate kinase activity is known to be dependent on the nature of the residue immediately preceding strand Bβ1 in the structure (19); in K+-dependent enzymes, this residue is a glutamate, whereas in K+-independent enzymes, it is often a lysine. In P. aeruginosa PykA, the corresponding residue is a lysine (Fig. S1).
Pyruvate kinase is a major site for the regulation of glycolytic flux in many species, and most of the known regulators in other species are associated with the EMP. However, P. aeruginosa relies solely on the EDP for glycolysis. To investigate the regulation of P. aeruginosa PykA further, we screened a wide range of metabolic intermediates, guided by a knowledge of the pathways feeding in and out of the EDP, for their ability to activate or inhibit PykA in vitro. Putative regulatory molecules were screened at low PEP concentration (0.3 mm) to identify activating ligands, and high PEP concentration (3 mm) to identify inhibitory ligands (Fig. 2 and Fig. S8, respectively). More detailed kinetic analyses (s0.5, kcat) were carried out on the “hits” identified this way (Fig. 2, Fig. S9, and Table 1). No inhibitors were identified. However, PykA was strongly activated by glucose 6-phosphate (G6P) from the EDP and by intermediates from the reductive PPP (fructose 6-phosphate (F6P), glyceraldehyde 3-phosphate (G3P), ribose 5-phosphate (R5P), ribulose 5-phosphate (RL5P), and xylulose 5-phosphate (X5P)) (Table 1). The main effect of these regulators was to decrease s0.5, suggestive of K-type allosteric regulation, and to change the substrate dependence kinetics on PEP from a sigmoidal to a hyperbolic (i.e. Michaelis-Menten) profile. Although these regulators had a large impact on the PEP-dependence of the reaction, they had little or no effect on its ADP-dependence. Interestingly, AMP and fructose 1,6-bisphosphate (two well-established regulators of PK in many other species) had almost no effect on PykA activity.
Structure of PykA
Currently, there is no structure available for any PykA isozyme. We were therefore fortunate to obtain crystals of PykA containing a substrate analogue (malonate) and Mg2+ bound in the active site, and a bound allosteric regulator (G6P). The crystals diffracted X-rays to 2.4 Å resolution. The asymmetric unit of PykA contained 12 polypeptides assembled into three complete homotetramers. This quaternary structure is consistent with analytical ultracentrifugation analyses data indicating that PykA is a tetramer in solution (Fig. S6). Refinement statistics are shown in Table 2.
Table 2.
Crystallographic data collection and refinement statistics of PykA
Values in parentheses are for the highest resolution shell.
| PDB code | 6QXL |
|---|---|
| Synchrotron/X-ray source | Diamond Light Source |
| Beamline | IO4-1 |
| Data collection | |
| Wavelength (Å) | 0.9159 |
| Resolution range (Å) | 405.44–2.43 (2.49–2.43) |
| Space group | P31 2 1 |
| Unit cell | |
| a, b, c, (Å) | 182.48, 182.48, 405.04 |
| a, b, g (°) | 90, 90, 120 |
| Total reflections | 4,318,494 (226,510) |
| Unique reflections | 292,996 (21,477) |
| Multiplicity | 14.7 (10.5) |
| Completeness (%) | 100.0 (100.0) |
| Mean I/σ(I) | 11.1 (1.1) |
| Resolution (Å) at mean I/σ(I) > 2 | 2.65 Å |
| Wilson B-factor | 55.5 |
| Rmerge | 0.143 (2.210) |
| Rmeas | 0.148 (2.324) |
| CC1/2 | 0.999 (0.642) |
| Refinement | |
| Resolution range (high resolution) (Å) | 158.03–2.43 (2.49–2.43) |
| Reflections used in refinement | 289,310 (21,209) |
| Reflections used for Rfree | 14,331 (1,071) |
| Rwork | 0.227 (0.298) |
| Rfree | 0.251 (0.315) |
| Number of molecules in the ASU | 12 |
| Number of non-hydrogen atoms | |
| Macromolecules | 43,331 |
| Ligands | 306 |
| Solvent | 2,268 |
| Protein residues | |
| RMS (bonds) (Å) | 0.014 |
| RMS (angles) (°) | 1.64 |
| Ramachandran favored (%) | 96.98 |
| Ramachandran allowed (%) | 2.8 |
| Ramachandran outliers (%) | 0.23 |
| Average B-factor | |
| Macromolecules | 55.65 |
| Ligands | 62.51 |
| Solvent | 50.59 |
Each protomer of PykA had the typical tripartite domain organization associated with PKs, with the A and C domains of one protomer interacting with the A and C domains, respectively, from the adjacent protomers (Fig. 3, A and B). The A domain is comprised of 8 α/β structures assembled into a TIM barrel motif. Helices Aα6 and Aα8 are further subdivided into shorter segments forming Aα6′ and Aα8′. The Aα6′helix contains part of a highly conserved segment (245MVARGDLGVE254), which plays a key role in catalysis (Fig. 3C). The active site is located in the cleft between domains A and B (Fig. 3B). Relative to the A domain, the spatial disposition of the β-rich B domain varied somewhat between the different protomers, indicating that the domain is configurationally flexible as a unit. The αβ-rich C domain contains the bound allosteric regulator, G6P.
Figure 3.

X-ray crystal structure of PykA. A, the PykA homotetramer. The A-A and C-C interfaces are shown. Substrate and G6P-binding sites are indicated in the top left protomer. B, domain organization of a PykA protomer showing malonate bound in the active site and G6P bound in the allosteric site. C, close-up of the PykA active site. The network of interactions involved in holding the malonate-Mg2+ (orange mesh, Fo − Fc map contoured at 3σ) and water molecules are shown. D, superposition of the side chains involved in malonate binding in PykA (green) and pyruvate binding in rabbit muscle PK (PDB 1F3W, light blue). E, superposition of the side chains involved in malonate binding in P. aeruginosa PykA (green) with side chains present in the active site of E. coli PykF (PDB 1PKY, yellow).
The PykA active site contains a bound substrate analogue, malonate (present in the crystallization buffer), and Mg2+ (Fig. 3C). The malonate is bound by hydrogen bonds from both the polypeptide backbone (residues Gly249 (2.8 Å) and Asp250 (3.1 Å)) and the side chains of Lys221 (2.9 Å), Thr282 (2.6 Å), and Arg34 (3.3 Å). In addition, the active site Mg2+ also contributes to malonate binding through electrostatic interactions. The Mg2+ is octahedrally coordinated through interactions with the side chains of Glu223 and Asp250, and also with two water molecules. These active site residues are absolutely conserved in all bacterial PKs (Fig. S1), as is their spatial disposition, even when comparing malonate-bound PykA with pyruvate-bound PK from rabbit muscle (PDB 1F3W) (Fig. 3D). In contrast, superposition of the PykA active site with that of E. coli apo-PykF (PDB 1PKY) reveals that almost all the residues just discussed undergo significant spatial re-orientation in the presence of the substrate, indicative of induced fit (Fig. 3E).
An alignment of PykA sequences reveals that all PykA orthologues contain 3 residues not present in any of the PykF orthologues, corresponding to Ala229, Asp230, and Asp231 in P. aeruginosa PykA (Fig. S1). The presence of these three additional residues lengthens the loop connecting Aα5-Aβ5 resulting in the displacement of helix Aα5 away from the core of the enzyme (Fig. S10). The functional significance of this is not yet clear, although it is worth noting that the same loop of structure contributes two key residues to the active site: Glu223 and Lys221 (Fig. S10).
Kinetic analysis indicated that G6P is an activator of PykA, increasing the apparent catalytic efficiency (kcat/s0.5) of the enzyme around 3-fold (Table 1, Fig. 2, and Fig. S9). Inspection of the PykA structure revealed a molecule of G6P buried in each C-domain, far from the active site, indicating that G6P is an allosteric regulator (Fig. 3B). The structure of only one other bacterial PK with bound regulator has been solved, and that is the PykF family enzyme from M. tuberculosis (Mtb). A comparison of the allosteric binding site(s) observed in the Mtb enzyme reveals AMP bound in the same location as the G6P in P. aeruginosa PykA. The Mtb enzyme does bind G6P, but the ligand is located in an entirely different position in the allosteric cleft (PDB 5WSB) (Fig. 4A).
Figure 4.

The G6P-binding site in PykA. A, superposition of the allosteric site in PykA from P. aeruginosa (light green) and PK from M. tuberculosis (PDB 5WSB, blue). The G6P in PykA is shown in coral, whereas the AMP and G6P in Mtb PK are shown in pink and yellow, respectively. B, superposition of the allosteric pocket in G6P-bound PykA (light green) and unbound PykF from E. coli (yellow). The G6P is shown surrounded by an electron density map (orange mesh, Fo− Fc map contoured at 3σ). C, schematic of the interactions involved in binding G6P to PykA. D, interaction of the phosphate loop (light green) and ring loop (blue) in the G6P-binding site of PykA. The G6P is shown as magenta sticks.
The G6P-binding pocket in PykA is comprised of two loops: a “phosphate loop” and a “ring loop.” The phosphate loop (Cβ1-Cα2, residues 383–388) and partly the Cα2 helix (Thr389) binds the phosphate group of the ligand, whereas the ring loop (Cβ4-Cβ5, residues 460–471) anchors the hexose ring moiety. Superposition of the G6P-binding site in PykA with the PykF apo-enzyme (PDB 1PKY) from E. coli reveals that in the presence of G6P, the ring loop of PykA is pulled tightly over the allosteric pocket (Fig. 4B), whereas the phosphate loop shares a similar disposition in both structures. The movement of the ring loop in the presence of G6P causes a shift and partial unwinding of the Cα4 helix in PykA.
The bound G6P interacts with the surrounding residues through a network of hydrogen bonds involving both the phosphate group and the sugar ring hydroxyls (Fig. 4C). In addition, occupation of the allosteric site also appears to promote secondary interactions between the phosphate loop and the ring loop of PykA; Ser386 and Thr389 on the phosphate loop form hydrogen bonds with Gln467 and Thr470 on the ring loop, respectively. Interestingly, Lys460 and Tyr464 on the ring loop also interact, tightening up closure of the binding pocket (Fig. 4D). This may be unique to the PK subclass represented by P. aeruginosa PykA, because its ring loop is less well-conserved than the ring loop in other PykA enzymes (Fig. S1). Furthermore, half of the residues that mediate the interaction between the phosphate and the ring loops are unique to P. aeruginosa PykA (Lys460, Tyr464, and Gln467).
Comparison of the G6P-bound PykA structure with the E. coli apo-PykF structure (PDB 1PKY) revealed differences in the disposition of residues at the protomer interfaces. The PykA tetramer contains four inter-protomer interfaces, two of which are between adjacent A-domains (A-A interface) and two between adjacent C-domains (C-C interface) (Fig. 3A). The A-A interface (∼1450 Å2) is formed through pairing of Aα6, Aα6′, Aα7, Aα8, loop Aα6-Aα6′, loop Aα7-Aβ7, and loop Aα8-Cα1 from one protomer with the same secondary structural units (albeit, with inverted symmetry) on the second protomer (Fig. S11). By contrast, the apo-structure of PykF does not include Aα6′ or loop Aα8-Cα1 as parts of the interface and instead, there is an Aα7-Aα7 interaction that is absent in PykA (Fig. 5A). The A-A interface in PykA is very close to the active site, so structural changes at the interface affect the disposition of active site residues including Arg248, Gly249, Glu283, Asp250, and Thr282 (Fig. S12). Indeed, the main chain amide nitrogen of Gly249 is directly involved in binding the substrate analogue, whereas the amide carbonyl moiety of this residue forms a hydrogen bond with the side chain of Arg296 on helix Aα7 on the adjacent protomer (Fig. 5C). The A-A interface is stabilized by just two salt bridges (Fig. S13), which contrasts with the situation seen in E. coli apo-PykF, where most of the interactions at the A-A involve salt bridges.
Figure 5.
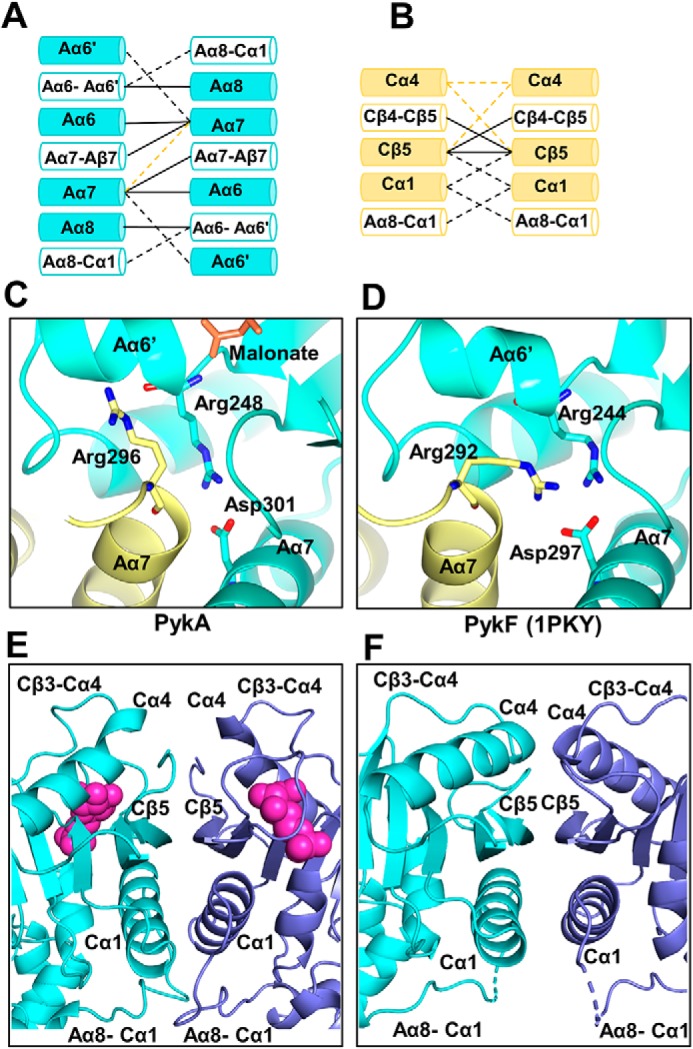
The inter-protomer interfaces in PykA. Comparison between (A) the A-A interface and (B) the C-C interface in PykA and E. coli PykF. Interactions present in both PykA and PykF are shown as solid black lines. Interactions unique to PykA only are shown as black dashed lines. Interactions unique to PykF only are shown as yellow dashed lines. C, close-up view of the A-A interface in PykA showing Arg296 oriented toward the active site. D, close-up view of the A-A interface in E. coli PykF showing Arg292 (equivalent to Arg296 in PykA) oriented away from the active site. E, the C-C interface in PykA. G6P is depicted as large magenta spheres. F, the C-C interface in E. coli PykF.
Comparison of G6P-bound PykA with apo-PykF (PDB 1PKY) also reveals major differences at the C-C interface (Fig. 5B). The C-C interface in PykF is comprised of Cα4, Cβ5, and the ring loop between Cβ4 and Cβ5. By contrast, the C-C interface in PykA is comprised of Cβ5, the ring loop, Cα1, and the long, flexible loop between Aα8-Cα1. Essentially, the bound G6P in PykA displaces the ring loop and Cα4, resulting in an outward movement of Cα1. This drags the Aα8-Cα1 loop region closer toward the C-C interface (Fig. 5, E and F). This movement of the Aα8-Cα1 loop could, in turn, communicate the conformational change from the G6P-binding site to the A-A interface and the active site, providing a plausible mechanism by which the allosteric regulator elicits its effect.
Discussion
Consistent with its role as the major PK isozyme in P. aeruginosa, a pykA mutant was defective in growth on glucose and glycerol. These data raise the question of why P. aeruginosa encodes two distinct PK isozymes? This is intriguing, especially given that the gene encoding one of the isozymes (pykF) appeared to show negligible expression under any of the growth conditions tested. One likely solution is that pykF appears to be part of a larger cluster of genes (PA1498–PA1502) that may be involved in the catabolism of compounds such as ethylene glycol (20). Therefore, although artificially expressed pykF is capable of rescuing the growth defect associated with the pykA mutant (Fig. 1), its expression is likely to be induced only under very select conditions.
P. aeruginosa lacks phosphofructokinase (encoded by pfk) and is therefore incapable of carrying out the EMP pathway of glycolysis (13). Instead, and in common with around 13% of species for which a genome sequence is available, it is exclusively reliant on the EDP for glucose oxidation (21). In this pathway, pyruvate kinase is the major regulatory enzyme. PykA activity was activated primarily by G6P, F6P, G3P, and by intermediates of the reductive PPP. In many organisms, the upper arm of the reductive PPP is “fed” by the conversion of 6-phosphogluconate into ribulose 5-phosphate; a reaction catalyzed by 6-phosphogluconate dehydrogenase (gnd) (22–24). However, and like many other bacteria that exclusively utilize the EDP, P. aeruginosa does not encode a homologue of gnd (13, 25). As a consequence, carbon can only flow into the PPP from G3P or from gluconeogenesis-derived F6P (Fig. 6) (26). Not surprisingly, given the lack of redundancy in input pathways, many of the PPP-encoding genes are essential in P. aeruginosa (26). It is therefore logical that P. aeruginosa has evolved to coordinate glucose oxidation with the supply of biosynthetic precursors by titrating both gluconeogenic and PPP intermediates. These two anabolic pathways are tightly inter-twined; as shown in Fig. 6, EDP-derived G3P, and F6P derived from gluconeogenesis also play a central role in the cyclical series of sugar phosphate interconversions that comprise the reductive PPP. High levels of these, and other key PPP intermediates (R5P, X5P, and RL5P) are presumably physiological indicators that biosynthetic precursors are abundant (27), and that carbon flux can be redirected toward energy production via PykA activation. Similarly, high levels of G6P indicate sufficiency of either glucose supply or of gluconeogenic flux, so it makes good metabolic sense that this too should feed forward to stimulate PykA, and hence, carbon catabolism.
Figure 6.

PykA is primarily regulated by PPP metabolites. Pathways of glucose metabolism in P. aeruginosa are indicated by black (EDP), blue (PPP), and yellow (EMP) arrows, respectively. Identified PykA regulators are boxed and shown in green with the respective s0.5 indicated below each regulator. Fructose 6-phosphate and glyceraldehyde 3-phosphate are depicted as part of the PPP to emphasize the importance they play in this series of sugar interconversions. The s0.5 of PykA without regulators is shown in a violet box. RpiA, ribose 5-phosphate isomerase; Rpe, ribulose phosphate 3-epimerase; TktA, transketolase; Tal, transaldolase; Fda, fructose 1,6-bisphosphate aldolase; Fbp, fructose 1,6-bisphosphatase.
The only other bacterial PK structure containing bound regulatory ligands is the PK from M. tuberculosis (28). That work revealed that AMP binds in the heart of the C domain (Fig. 4A), and that G6P binds at the interface between the A and C domains. However, P. aeruginosa PykA was insensitive to AMP, and G6P was found to be bound to the same site as that occupied by AMP in the Mtb structure. This indicates that although the allosteric binding pocket is conserved as a cavity in PykA, there is considerable evolutionary plasticity in regulator binding. That different enzymes from the same family (albeit, from different species) have evolved to bind the same allosteric regulator (G6P) in different sites is remarkable, especially from the perspective of allosteric transitions. Moreover, and given the radically different structures of ring-sugar phosphates such as G6P and F6P compared with “configurationally locked” straight-chain forms such as RL5P and X5P, this suggests that the latter may well bind PykA at other, yet to be characterized sites.
Unfortunately, we were unable to obtain crystals of apo-PykA that diffracted beyond about 4 Å. We therefore based our structural comparisons on the PykF apoenzyme from E. coli, which does not contain bound substrate or regulatory ligands. In the PykF structure, the allosteric pocket is open, and is uncapped by the ring loop (29). This unbound configuration allows the Cα4 helices to form prominent interactions across the C-C interface. However, when the allosteric site becomes occupied by G6P, the ring loop is pulled down toward the ligand. This partially displaces Cα4 and firmly closes the allosteric pocket through interactions with both the ligand and the phosphate loop. The new configuration of the allosteric site seems to loosen up the interactions from Cα4, concomitantly recruiting other structures to the interface, most notably Cα1 and the Aα8-Cα1 loop (Fig. 5). Thus, in PykA, the Aα8-Cα1 loop contacts two protomers simultaneously via the A-A interface and the C-C interface. Given the proximity of the A-A interface with the active site, this suggests a likely mechanism by which G6P binding may influence catalysis. Fig. 7 illustrates this proposed mechanism. G6P binding to the allosteric pocket leads to movement of the ring loop, which displaces Cα4 and allows outward movement of Cα1. This, in turn, leads to a re-structuring of the Aα8-Cα1 loop, which directly or indirectly affects the A-A interface, especially Aα6′ (which forms part of the active site). This proposed mechanism contrasts with the rigid-body rock-shape-lock mechanism proposed for Mtb PK (28).
Figure 7.

Spatial disposition of key secondary structural elements in P. aeruginosa PykA and E. coli PykF. Note how the movement of the ring loop in PykA leads to disruption of the Cα4-Cα4 interaction and concomitant outward movement of Cα1. This, in turn, tugs on the Aα8-Cα1 loop, engaging it with the C-C interface. The resulting movement of Aα8 directly or indirectly affects the A-A interface by forming new interactions with Aα6-Aα6′. Given that Aα6′ forms part of the active site, this set of interactions provides a plausible mechanism by which G6P binding to the allosteric pocket in the structure can lead to altered catalysis.
Materials and methods
For primers and bacterial strains, see Table S1.
Construction of mutants
The pykA and pykF mutants used were from the Washington Mutant Library (identities PW8308 (pykA-B04::ISlacZ/hah, which carries a Tn insertion at position 903/1452) and PW3705 (pykF-C02::ISphoA/hah, which carries a Tn insertion at position 213/1434), respectively). The insertion site of the transposon in each mutant was confirmed by PCR using the primers recommended by the library curators. To generate the pykA pykF double mutant, we first had to remove the TetR marker from the pykA mutant. This was done by introducing plasmid pFLP2-cre into the pykA mutant by electroporation (to enable site-specific cre-mediated excision of the transposon) followed by plasmid curing on LBA plates containing 5% sucrose (pFLP2-cre carries sacB as a counter-selectable marker). Site-specific excision of the TetR marker leaves behind a 63-codon scar within the target gene, and this was confirmed using PCR (Fig. S3). The TetS pykA mutant was then used as a recipient for the TetR marker from a pykF donor, introduced by generalized transduction with φPA3 as a vehicle (30). The pykA pykF double mutant was confirmed using (i) PCR to demonstrate the presence of the 63 codon scar in the pykA ORF (Fig. S3), (ii) the absence of a PCR-amplifiable pykF fragment in the transductants (Fig. S4), and (iii) the TetR phenotype of the transductants.
Construction of complementing plasmids
To complement the pykA mutant, a region encompassing the entire pykA ORF and 500 bp upstream of this were PCR-amplified using primers pykA-pLP170-FD and pykA-pET19m-RV. The amplified DNA fragment was digested with EcoRI and BamHI and introduced into similarly digested pUCP20, yielding ppykA+. To complement the pykF mutant, we used a different strategy. The pykF ORF is predicted to form an operon with an adjacent glycerate kinase-encoding gene, PA1499. Therefore, we PCR amplified the entire region spanning from upstream of PA1499 to the 3′ end of pykF (using primers PA1499-pLP170-FD and pykF-pET19m-RV primers). The amplified DNA fragment was digested with EcoRI and HindIII and introduced into similarly digested pUCP20, yielding ppykF+.
Bacterial growth
The indicated strains/mutants were grown in LB overnight and washed twice in PBS. Cultures were inoculated to an initial A600 of 0.05 in 50 ml of minimal media containing the indicated carbon sources and/or antibiotics. The cultures were grown at 37 °C with vigorous aeration. Samples were taken every hour for optical density measurement. Each growth curve was carried out in triplicate.
Enzyme assays
Cultures were grown in 50 ml of minimal media containing the indicated carbon sources and/or antibiotics. The cultures were grown at 37 °C with vigorous aeration. After the indicated period of growth, the cells were harvested by centrifugation (3220 × g, 15 min, 4 °C) and resuspended in lysis buffer (50 mm Tris-HCl, 0.1 m NaCl, pH 7.5) containing a protease inhibitor mixture tablet (Roche, 1 tablet per 50 ml of buffer). The cells were lysed by sonication on ice and the lysates were clarified by centrifugation. Protein was determined using the Bradford assay. PK activity was measured using a lactate dehydrogenase (LDH)-coupled assay. The reaction mixtures contained 50 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 units of rabbit muscle LDH, 0.2 mm NADH, 5 mm PEP, and 2 mm ADP. Reactions were initiated by the addition of an equal volume of cell lysate (normalized for protein concentration) and allowed to proceed at 37 °C. NADH consumption was monitored at 340 nm. Each measurement was carried out in triplicate.
Kinetics of purified PykA
Pyruvate kinase activity was measured at 37 °C in 1-ml reactions containing 50 mm Tris-HCl, 10 mm MgCl2, 10 units of rabbit muscle LDH, 0.2 mm NADH (pH 7.5). For the PEP titrations, [ADP] was kept constant at 2 mm. For the ADP titrations, [PEP] was kept constant at 5 mm. Reactions were initiated by addition of purified PykA to a final concentration of 0.2 μg/ml. Reaction progress was monitored at 340 nm. Regulator screens were carried out using 1 mm final concentration of the indicated regulator candidates and a fixed concentration of either 2 mm ADP and 0.3 mm PEP (when screening for activators), or 2 mm ADP and 2 mm PEP (when screening for inhibitors). The effect of the hits identified this way on the detailed kinetics were re-measured with 1 mm of each potential regulator and variable concentrations of PEP and ADP, as above. The only exception to this was that F6P, R5P, and X5P were added at 0.2, 0.15, and 0.5 mm final concentrations, respectively, in all experiments. GraphPad Prism 7 was used to analyze the data and to extract the kinetic constants. All experiments were carried out in triplicate.
Western blotting
Anti-PykA and anti-PykF antibodies were raised against purified PykA and PykF proteins, respectively (Biogenes.De). The PykA and PykF antisera were pre-absorbed onto an acetone extract of whole lysate protein from a pykA mutant or from a pykF mutant, respectively. The primary anti-PykA and anti-PykF antibodies were used at 1:2,000 and 1:3,000 dilution, respectively. Horseradish peroxidase-conjugated goat anti-rabbit (1:10,000) was used as a secondary antibody. The isocitrate dehydrogenase isozyme, ICD, was used as a loading control. Antibodies raised against ICD has been described previously (31).
Cloning, expression, and purification of PykA and PykF
The ORFs encoding PykA (PA4329) and PykF (PA1498) were PCR-amplified from P. aeruginosa genomic DNA using primers pykA-pET19m-FD and pykA-pET19m-RV (for pykA amplification) and pykF-pET19m-FD and pykF-pET19m-RV (for pykF amplification). The amplified ORFs were cloned into pET-19m, generating pET-19m (pykA) and pET-19m (pykF), respectively. Expression from pET-19m yields a tobacco etch virus protease-cleavable N terminally His6-tagged protein product. For protein expression, E. coli BL21 (DE3) containing pET-19m (pykA) or pET-19m (pykF), as appropriate, was grown in 1 liter of LB containing 50 μg/ml of carbenicillin and 34 μg/ml of chloramphenicol at 37 °C until the A600 reached 0.5. The temperature was then decreased to 20 °C and protein expression was induced by adding isopropyl β-d-thiogalactopyranoside to a final concentration of 1 mm. Following overnight growth, the cells were harvested by centrifugation (4,000 × g, 30 min, 4 °C) and re-suspended in 50 ml of ice-cold lysis buffer (50 mm Tris-HCl, 200 mm NaCl, 10% (v/v) glycerol, 10 mm imidazole (pH 8.0)) containing a dissolved EDTA-free protease inhibitor mixture tablet (Roche). The resuspended cells were lysed by sonication on ice and the sample was then clarified by sedimentation (10,000 × g, 30 min, 4 °C). The clear supernatant was loaded onto an nickel-nitrilotriacetic acid column that had been previously pre-equilibrated with lysis buffer. The column was washed overnight with lysis buffer. The bound protein was eluted in 50 mm Tris-HCl, 200 mm NaCl, 10% (v/v) glycerol, 250 mm imidazole (pH 8.0). The eluted protein was mixed with His6-tagged tobacco etch virus protease (1 mg/20 mg of target protein) and loaded into a dialysis bag (10,000 MWCO). The sample was dialyzed overnight against 20 mm Tris-HCl, 100 mm NaCl, 5% (v/v) glycerol, 1 mm DTT, 0.1 mm EDTA (pH 7.5). After dialysis, the protein was transferred into a Falcon tube and incubated end-over-end for 2 h at 4 °C with a slurry of nickel-nitrilotriacetic acid that had been previously pre-equilibrated in dialysis buffer. Unbound protein was concentrated by ultrafiltration (Vivaspin column, 30,000 MWCO) then aliquoted and flash frozen in liquid nitrogen. The protein concentration was determined by spectrophotometry using a calculated molar extinction coefficient of 24,410 m−1 cm−1 for PykA and 25,440 m−1cm−1 for PykF.
Analytical ultracentrifugation
Purified PykA was dialyzed against 20 mm Tris-HCl, 100 mm NaCl, and 0.1 mm EDTA (pH 7.5) for glycerol removal. The centerpieces of the Epon double-sector of a Beckman Optima XL-I (AN-60 Ti rotor) were filled with 400 μl of purified protein or blank buffer. The sample was sedimented at 29,160 × g, for 24 h at 20 °C. Absorbance data (280 nm) were acquired every 2 min with interference scans collected every minute. Data analysis and the calculations of buffer viscosity, protein partial specific volumes, and frictional rations were performed using SEDFIT and SEDNTERP (32, 33).
Amino acid sequence analysis
Amino acid sequences were extracted from UniProt in FASTA format, aligned by Clustal Omega (34, 35), and displayed by ESPript (36).
Crystallization of PykA
PykA was crystallized using the sitting drop vapor diffusion method. PykA (22 mg/ml in 20 mm Tris-HCl, 100 mm NaCl, 5% (v/v) glycerol, 1 mm DTT, 0.1 mm EDTA, 20 mm MgCl2, 2 mm G6P, 2 mm PEP (pH 7.5)) was mixed 1:1 with the reservoir buffer (200 nl each), which contained 20% (w/v) PEG3350, 0.1 m BIS-TRIS propane, and 0.2 m disodium malonate (pH 7.5). Crystals grew within 1 week. The crystals were mounted on nylon loops and cryoprotected in mother liquor supplemented with 40% (v/v) glycerol before being flash frozen in liquid N2.
X-ray diffraction and structure refinement
Diffraction data were collected at the Diamond Light Source (Didcot, UK) on beamline IO4 (MX14043-47). The PykA structure was obtained from molecular replacement with Phaser (37) using the pyruvate kinase of Trypanosoma brucei (PDB 4HYV) as a structural template. Coot (38) was used for model building, and refinement was performed by BUSTER (39). Table 2 shows the data collection and refinement statistics. Structural coordinates were deposited in the PDB with accession code PDB 6QXL. PDBePISA (40) was used for analysis of the tetramer and ligand interfaces. Figures were generated using CCP4mg and PyMOL (41, 42).
Author contributions
Y. A. and M. W. conceptualization; Y. A., P. B., J. G., X. C., T. R., and M. W. formal analysis; Y. A., T. R., and M. W. supervision; Y. A. and M. W. funding acquisition; Y. A., P. B., J. G., X. C., and T. R. investigation; Y. A., P. B., and M. W. methodology; Y. A., P. B., and M. W. writing-original draft; Y. A. and M. W. project administration; Y. A., P. B., J. G., X. C., T. R., and M. W. writing-review and editing; P. B. and J. G. resources.
Supplementary Material
This work was supported by a Ph.D. studentship from the Yousef Jameel Foundation (to Y. A.), a BBSRC studentship (to J. G.), and BBSRC Grant BB/M019411/1. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S13 and Table S1.
The atomic coordinates and structure factors (code 6QXL) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PK
- pyruvate kinase
- KDPG
- 2-keto-3-deoxy-6-phosphogluconate
- EDP
- Entner-Doudoroff pathway
- PEP
- phosphoenolpyruvate
- F6P
- fructose 6-phosphate
- G6P
- glucose 6-phosphate
- R5P
- ribose 5-phosphate
- RL5P
- ribulose 5-phosphate
- X5P
- xylulose 5-phosphate
- LDH
- lactate dehydrogenase
- BIS-TRIS propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- PDB
- Protein Data Bank
- RMS
- root mean square
- G3P
- glyceraldehyde 3-phosphate.
References
- 1. Kayne F. J. (1973) 11 Pyruvate kinase. Enzymes 8, 353–382 10.1016/S1874-6047(08)60071-2 [DOI] [Google Scholar]
- 2. Al Zaid Siddiquee K., Arauzo-Bravo M. J., and Shimizu K. (2004) Metabolic flux analysis of pykF gene knockout Escherichia coli based on 13C-labeling experiments together with measurements of enzyme activities and intracellular metabolite concentrations. Appl. Microbiol. Biotechnol. 63, 407–417 10.1007/s00253-003-1357-9 [DOI] [PubMed] [Google Scholar]
- 3. Sawada K., Zen-in S., Wada M., and Yokota A. (2010) Metabolic changes in a pyruvate kinase gene deletion mutant of Corynebacterium glutamicum ATCC 13032. Metab. Eng. 12, 401–407 10.1016/j.ymben.2010.01.004 [DOI] [PubMed] [Google Scholar]
- 4. Bücker R., Heroven A. K., Becker J., Dersch P., and Wittmann C. (2014) The pyruvate-tricarboxylic acid cycle node: a focal point of virulence control in the enteric pathogen Yersinia pseudotuberculosis. J. Biol. Chem. 289, 30114–30132 10.1074/jbc.M114.581348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ponce E., Flores N., Martinez A., Valle F., and Bolívar F. (1995) Cloning of the two pyruvate kinase isoenzyme structural genes from Escherichia coli: the relative roles of these enzymes in pyruvate biosynthesis. J. Bacteriol. 177, 5719–5722 10.1128/jb.177.19.5719-5722.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garcia-Olalla C., and Garrido-Pertierra A. (1987) Purification and kinetic properties of pyruvate kinase isoenzymes of Salmonella typhimurium. Biochem. J. 241, 573–581 10.1042/bj2410573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hofmann J., Heider C., Li W., Krausze J., Roessle M., and Wilharm G. (2013) Recombinant production of Yersinia enterocolitica pyruvate kinase isoenzymes PykA and PykF. Protein Expr. Purif. 88, 243–247 10.1016/j.pep.2013.01.010 [DOI] [PubMed] [Google Scholar]
- 8. Waygood E. B., Mort J. S., and Sanwal B. D. (1976) The control of pyruvate kinase of Escherichia coli: binding of substrate and allosteric effectors to the enzyme activated by fructose 1,6-bisphosphate. Biochemistry 15, 277–282 10.1021/bi00647a006 [DOI] [PubMed] [Google Scholar]
- 9. Waygood E. B., Rayman M. K., and Sanwal B. D. (1975) The control of pyruvate kinases of Escherichia coli: II. effectors and regulatory properties of the enzyme activated by ribose 5-phosphate. Can. J. Biochem. 53, 444–454 10.1139/o75-061 [DOI] [PubMed] [Google Scholar]
- 10. Martínez-Solano L., Macia M. D., Fajardo A., Oliver A., and Martinez J. L. (2008) Chronic Pseudomonas aeruginosa infection in chronic obstructive pulmonary disease. Clin. Infect. Dis. 47, 1526–1533 10.1086/593186 [DOI] [PubMed] [Google Scholar]
- 11. Mittal R., Aggarwal S., Sharma S., Chhibber S., and Harjai K. (2009) Urinary tract infections caused by Pseudomonas aeruginosa: a minireview. J. Infect. Public Health 2, 101–111 10.1016/j.jiph.2009.08.003 [DOI] [PubMed] [Google Scholar]
- 12. Williamson K. S., Richards L. A., Perez-Osorio A. C., Pitts B., McInnerney K., Stewart P. S., and Franklin M. J. (2012) Heterogeneity in Pseudomonas aeruginosa biofilms includes expression of ribosome hibernation factors in the antibiotic-tolerant subpopulation and hypoxia-induced stress response in the metabolically active population. J. Bacteriol. 194, 2062–2073 10.1128/JB.00022-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lessie T. G., and Phibbs P. V. (1984) Alternative pathways of carbohydrate utilization in Pseudomonads. Annu. Rev. Microbiol. 38, 359–388 10.1146/annurev.mi.38.100184.002043 [DOI] [PubMed] [Google Scholar]
- 14. Kersters K., and De Ley J. (1968) The occurrence of the Entner-Doudoroff pathway in bacteria. Antonie Van Leeuwenhoek 34, 393–408 10.1007/BF02046462 [DOI] [PubMed] [Google Scholar]
- 15. Nikel P. I., Chavarría M., Fuhrer T., Sauer U., and de Lorenzo V. (2015) Pseudomonas putida KT2440 strain metabolizes glucose through a cycle formed by enzymes of the Entner-Doudoroff, Embden-Meyerhof-Parnas, and pentose phosphate pathways J. Biol. Chem. 290, 25920–25932 10.1074/jbc.M115.687749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kachmar J. F., and Boyer P. D. (1953) Kinetic analysis of enzyme reactions: II. the potassium activation and calcium inhibition of pyruvic phosphoferase. J. Biol. Chem. 200, 669–682 [PubMed] [Google Scholar]
- 17. Larsen T. M., Benning M. M., Wesenberg G. E., Rayment I., and Reed G. H. (1997) Ligand-induced domain movement in pyruvate kinase: structure of the enzyme from rabbit muscle with Mg2+, K+, and l-phospholactate at 2.7 Å resolution Arch. Biochem. Biophys. 345, 199–206 10.1006/abbi.1997.0257 [DOI] [PubMed] [Google Scholar]
- 18. Oria-Hernández J., Cabrera N., Pérez-Montfort R., and Ramírez-Silva L. (2005) Pyruvate kinase revisited: the activating effect of K+. J. Biol. Chem. 280, 37924–37929 10.1074/jbc.M508490200 [DOI] [PubMed] [Google Scholar]
- 19. Laughlin L. T., and Reed G. H. (1997) The monovalent cation requirement of rabbit muscle pyruvate kinase is eliminated by substitution of lysine for glutamate 117. Arch. Biochem. Biophys. 348, 262–267 10.1006/abbi.1997.0448 [DOI] [PubMed] [Google Scholar]
- 20. Franden M. A., Jayakody L. N., Li W.-J., Wagner N. J., Cleveland N. S., Michener W. E., Hauer B., Blank L. M., Wierckx N., Klebensberger J., and Beckham G. T. (2018) Engineering Pseudomonas putida KT2440 for efficient ethylene glycol utilization. Metab. Eng. 48, 197–207 10.1016/j.ymben.2018.06.003 [DOI] [PubMed] [Google Scholar]
- 21. Flamholz A., Noor E., Bar-Even A., Liebermeister W., and Milo R. (2013) Glycolytic strategy as a tradeoff between energy yield and protein cost. Proc. Natl. Acad. Sci. U.S.A. 110, 10039–10044 10.1073/pnas.1215283110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scott D. B. M., and Cohen S. S. (1953) The oxidative pathway of carbohydrate metabolism in Escherichia coli: 1. the isolation and properties of glucose 6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase. Biochem. J. 55, 23–33 10.1042/bj0550023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scott D. B., and Cohen S. S. (1957) The oxidative pathway of carbohydrate metabolism in Escherichia coli: V. isolation and identification of ribulose phosphate produced from 6-phosphogluconate by the dehydrogenase of E. coli. Biochem. J. 65, 686–689 10.1042/bj0650686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McNair Scott D. B. (1956) The oxidative pathway of carbohydrate metabolism in Escherichia coli: III. glucose 6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase in cells grown under different conditions. Biochem. J. 63, 587–593 10.1042/bj0630587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mithani A., Hein J., and Preston G. M. (2011) Comparative analysis of metabolic networks provides insight into the evolution of plant pathogenic and nonpathogenic lifestyles in Pseudomonas. Mol. Biol. Evol. 28, 483–499 10.1093/molbev/msq213 [DOI] [PubMed] [Google Scholar]
- 26. Lee S. A., Gallagher L. A., Thongdee M., Staudinger B. J., Lippman S., Singh P. K., and Manoil C. (2015) General and condition-specific essential functions of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 112, 5189–5194 10.1073/pnas.1422186112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stincone A., Prigione A., Cramer T., Wamelink M. M., Campbell K., Cheung E., Olin-Sandoval V., Grüning N.-M., Krüger A., Tauqeer Alam M., Keller M. A., Breitenbach M., Brindle K. M., Rabinowitz J. D., and Ralser M. (2015) The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 90, 927–963 10.1111/brv.12140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhong W., Cui L., Goh B. C., Cai Q., Ho P., Chionh Y. H., Yuan M., Sahili A. E., Fothergill-Gilmore L. A., Walkinshaw M. D., Lescar J., and Dedon P. C. (2017) Allosteric pyruvate kinase-based “logic gate” synergistically senses energy and sugar levels in Mycobacterium tuberculosis. Nat. Commun. 8, 1986 10.1038/s41467-017-02086-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mattevi A., Valentini G., Rizzi M., Speranza M. L., Bolognesi M., and Coda A. (1995) Crystal structure of Escherichia coli pyruvate kinase type I: molecular basis of the allosteric transition. Structure 3, 729–741 10.1016/S0969-2126(01)00207-6 [DOI] [PubMed] [Google Scholar]
- 30. Monson R., Foulds I., Foweraker J., Welch M., and Salmond G. P. (2011) The Pseudomonas aeruginosa generalized transducing phage PA3 is a new member of the KZ-like group of “jumbo” phages, and infects model laboratory strains and clinical isolates from cystic fibrosis patients. Microbiology 157, 859–867 10.1099/mic.0.044701-0 [DOI] [PubMed] [Google Scholar]
- 31. Crousilles A., Dolan S. K., Brear P., Chirgadze D. Y., and Welch M. (2018) Gluconeogenic precursor availability regulates flux through the glyoxylate shunt in Pseudomonas aeruginosa. J. Biol. Chem. 293, 14260–14269 10.1074/jbc.RA118.004514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hayes D., Laue T., and Philo J. (1995) Program Sednterp: Sedimentation Interpretation Program. Alliance Protein Laboratories, Thousand Oaks, CA [Google Scholar]
- 34. Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., and Lopez R. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699 10.1093/nar/gkq313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., and Bricogne G. (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER–TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 10.1107/S0907444904016427 [DOI] [PubMed] [Google Scholar]
- 40. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 41. McNicholas S., Potterton E., Wilson K. S., and Noble M. E. M. (2011) Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 67, 386–394 10.1107/S0907444911007281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. DeLano W. L. (2012) The PyMOL Molecular Graphics System, Version 2.0, Schrödinger, LLC, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.