

Abstract
The CD38 molecule (CD38) catalyzes biogenesis of the calcium-mobilizing messenger cyclic ADP-ribose (cADPR). CD38 has dual membrane orientations, and type III CD38, with its catalytic domain facing the cytosol, has low abundance but is efficient in cyclizing cytosolic NAD to produce cADPR. The role of cell surface type II CD38 in cellular cADPR production is unknown. Here we modulated type II CD38 expression and assessed the effects of this modulation on cADPR levels. We developed a photoactivatable cross-linking probe based on a CD38 nanobody, and, combining it with MS analysis, we discovered that cell surface CD38 interacts with CD71. CD71 knockdown increased CD38 levels, and CD38 knockout reciprocally increased CD71, and both could be cocapped and coimmunoprecipitated. We constructed a chimera comprising the N-terminal segment of CD71 and a CD38 nanobody to mimic CD71's ligand property. Overexpression of this chimera induced a dramatically large decrease in CD38 via lysosomes. Remarkably, cellular cADPR levels did not decrease correspondingly. Bafilomycin-mediated blockade of lysosomal degradation greatly elevated active type II CD38 by trapping it in the lysosomes but also did not increase cADPR levels. Retention of type II CD38 in the endoplasmic reticulum (ER) by expressing an ER construct that prevented its transport to the cell surface likewise did not change cADPR levels. These results provide first and direct evidence that cADPR biogenesis occurs in the cytosol and is catalyzed mainly by type III CD38 and that type II CD38, compartmentalized in the ER or lysosomes or on the cell surface, contributes only minimally to cADPR biogenesis.
Keywords: calcium intracellular release, CD38, cell compartmentalization, cell surface enzyme, cyclic ADP Ribose (cADPR), membrane topology
Introduction
CD38 is a signaling enzyme with multiple forms and functions. It regulates various physiopathological processes, including insulin secretion (1), HIV infection (2), malignancies like leukemia (3) and myeloma (4), as well as dysfunction of tumor-infiltrating CD8+ T cells (5).
CD38 was first identified on the surface of lymphocytes by monoclonal typing (6). It has long been accepted that it is a type II membrane protein with its carboxyl domain (C-domain)3 facing out. It has been considered to function only as a surface antigen. In T lymphocytes, it has been suggested to interact with CD4 and, in doing so, protect the cells from infection by HIV (2). In monocytes, surface CD38 has been reported to associate with proteins of the major histocompatibility complexes (MHCs) (7) and has also been shown to act as the receptor for ligands such as CD31 (8).
Since the subsequent discovery that CD38 is an NAD-utilizing enzyme responsible for producing a second messenger for calcium mobilization, cyclic ADP-ribose (cADPR), more than two decades ago (9), its membrane topology has been an unresolved paradox. It is puzzling how a type II protein, with its catalytic domain facing out, can use cytosolic NAD to produce cADPR, which targets the calcium stores in the endoplasmic reticulum (ER). Recently, we showed that it also naturally exists in an opposite orientation as a type III membrane protein, with its catalytic domain facing the cytosol (10, 11). This form of CD38 is highly efficient in using cytosolic NAD as a substrate and producing cellular cADPR (11). The activity is regulated by a cytosolic protein, CIB1, that binds to the catalytic domain of CD38 (11).
The question that remains unanswered is the role of type II CD38 in the production of cellular cADPR. In this study, we aimed to modulate type II CD38 in cells and assess the effects on cellular cADPR production. We first developed a versatile technique to identify and map out the interacting partners of surface CD38 in myeloma cells, LP-1, which express high levels of CD38 in both type II and type III orientations (11). We designed and synthesized a nanobody-based photoaffinity cross-linker (NPC). Using it, we identified CD71 as a so far unknown and specific interaction partner of surface type II CD38. We further elucidated the functional consequences of the interaction for expression of CD38 and intracellular cADPR levels.
Results
Development of the nanobody-based photoaffinity labeling approach
A common approach for mapping the interacting molecules of a surface protein is to immunopurify its associated complex after solubilization, followed by identification with MS. This approach normally generates a large number of candidates. The majority of these may not interact directly with the protein of interest but are only secondarily and peripherally associated in the complex. For proteins that are known to be in lipid rafts, highly stringent solubilization can disrupt weaker interactions and lead to false negative results. Indeed, CD38 has been found in lipid rafts in various cells, such as T cells (12), monocytes (7), and B cells (13). A better profiling method is needed.
Here we designed and developed a nanobody-based photoaffinity labeling (NPL) approach to identify the interacting membrane proteins of CD38. We recently produced a series of nanobodies targeting three different epitopes on the C-domain of CD38, which is also its catalytic domain (14). The design of the NPC is shown in Fig. 1A. Its cross-linker portion (sulfo-SBED) has three functional groups. The NHS group (Fig. 1A, light green square) is used to conjugate the probe to the primary amine groups of the four lysines at the surface of the CD38 nanobody, Nb-1053 (Fig. 1A, red) (14). The UV-activatable phenyl azide group (Fig. 1A, blue square) is used to cross-link to the interacting candidates. The biotin group (Fig. 1A, light red square) is for purification of the cross-linked candidates using streptavidin (Strep) beads. The disulfide linkage in the middle (Fig. 1A, yellow square) allows release of Nb-1053 from the construct by reduction.
Figure 1.

Design of NPL. A, the chemical structure of sulfo-SBED and the partners of the three functional groups. The NHS group (green box) is for covalent linkage with nanobodies (Nb-1053, surface structure). The p-azido benzamido group (blue box) is for photosensitive linkage with the candidate interaction proteins (IPs), and biotin (red box) is for affinity purification with streptavidin beads. The distance between the succinimidyl group and azido group is around 14.3 Å. In the crystal structure of Nb-1053 (PDB code 5F1K), the CDRs are colored yellow, and the four lysines are colored red. B, protocol and schematic of the NPL approach. The CD38/Nb-1053 complex is the actual crystal structure (PDB code 5F1K) we solved previously. The four red stars represent the biotins in sulfo-SBED, which were transferred from Nb-1053 to CD38, and its interaction proteins.
The purity of Nb-1053 used in the construct is shown in Fig. S1A. The resulting NPC was validated by SDS-PAGE and Western blots, which showed that NPC could bind streptavidin (Fig. S1B). The analysis procedure is diagrammed in Fig. 1B. NPC (Fig. 1B, green and yellow, cartoon mode) is incubated with live cells, and its Nb-1053 directs specific binding of the probe to the surface type II CD38 (Fig. 1B, beige, in surface mode). UV irradiation activates the azido groups (Fig. 1B, red stars) into highly reactive nitrenes (Fig. 1B, red lines), which then cross-link NPC to CD38 as well as its interacting proteins (Fig. 1B, light blue, labeled with a question mark). Subsequent treatment with the reductive reagent DTT releases Nb-1053 from CD38 and the candidate, which are linked with biotin (Fig. 1A, red square) and can be pulled down by Strep beads for Western blots or MS identification (Fig. 1B). The release of the nanobody is to avoid its interference in subsequent MS analysis. The theoretical distance between Nb-1053 and the azido group is around 14.3 Å. The allosteric constraint of NPC ensures that it can only cross-link to candidates that bind directly and closely to CD38. Using the crystal structure of the CD38/Nb-1053 complex (PDB code 5F1K, Fig. 1B), we determined that the four lysines of Nb-1053 in the NPC should preferentially cross-link to candidates that bind to the opposite side of the catalytic pocket of CD38.
The methodology was first validated in vitro on recombinant CD38 (reCD38) (15) using Nb-5512 as a model CD38-binding protein and a positive control. BSA was used as a negative control. Nb-5512 (the purity is shown in Fig. S1A) is a tandem dimer of another nanobody of CD38, Nb-551, which binds to CD38 at an epitope distinct from that of Nb-1053, and they can form a triplex (16). The size of Nb-5512 is twice that of Nb-1053, allowing its easy identification in gels and blots. As shown in Fig. 2A, top panel (visualized with HRP-conjugated streptavidin), the biotin labeling could be detected in reCD38 and Nb-5512 but not in BSA, even though 2-fold more BSA than reCD38 was used (Fig. 2A, bottom panel, Coomassie Blue staining), indicating that the labeling was highly specific. It should be noted that the biotin signal of Nb-5512 was much stronger than that of reCD38, indicating that the NPC probe preferentially cross-linked to the CD38-binding proteins than to CD38 itself. This is expected because, from the crystal structure of the CD38/Nb-1053 complex, only one of the NPC-conjugating lysines on Nb-1053 is close to the CD38 binding interface (Fig. 1B, Nb-1053, yellow).
Figure 2.

NPC efficiently captures the interaction proteins of CD38 in vitro and in cellulo. A, the proteins, including Nb-5512, a known CD38 binder serving as a positive control, and BSA, a negative control, were used to test the efficiency and specificity of NPL in vitro. The indicated proteins were incubated at 4 °C for 1 h, UV-irradiated for 8 min on ice, treated with 100 mm DTT for 15 min, and analyzed by Western blotting and Coomassie Blue staining. The molar ratio of NPC probe:reCD38:Nb-5512:BSA is 5:1:1:2. All protein markers are indicated on the left of the gels or blots in kilodaltons. B, the indicated proteins were incubated with LP-1 cells on ice for 1 h, followed by exposure to UV light for 20 min on ice. The lysates were treated with 100 mm DTT, and the biotinylated proteins were precipitated by the Strep beads and analyzed by Western blotting. IB, immunoblot. C, schematic of the semiquantitative chemical proteomics workflow. D, dot plots of the enriched proteins in NPC-labeled LP-1 cells versus those in CD38-KO LP-1 cells identified by MS. The numbers (≥2) of unique peptides of candidate proteins were plotted. CD71, MHCs, and CD38 are highlighted in red, purple, and green, respectively. The MS experiments were done twice.
Profiling the CD38-interacting membrane proteins
We next applied the NPC to a multiple myeloma cell line, LP-1, which expresses high levels of surface type II CD38. A CD38 knockout LP-1 cell line (CD38-KO) (11) was used as a negative control to test CD38 dependence. As shown in Fig. 2B, CD38 was detected in the input of LP-1 samples, but it could be pulled down by Strep beads only when NPC was applied, which indicated that NPC could label CD38 in LP-1 cells. Nb-5512 was added as a reference binding protein. The biotin labeling of Nb-5512 was much stronger than CD38 itself (Fig. 2B, third blot with Strep-HRP, the indicated bands in the second lane versus the third lane) and was CD38-dependent because CD38-KO cells did not develop any signal. Addition of Nb-5512 competed the signal of CD38, indicating preferential cross-linking of NPC on CD38-binding proteins than CD38 itself and was consistent with what was seen with reCD38 described above.
To further verify that NPC can bind to and label surface CD38 in LP-1 cells, the experiments were repeated with the following modifications. First, to further demonstrate specificity, 1053-biotin, the biotin-conjugated Nb-1053, was included, which should bind to surface CD38 but not transfer biotin to CD38 or its partners. Second, the probe-associated complexes were precipitated by anti-FLAG beads without DTT pretreatment. As shown in Fig. S2A, CD38 could be pulled down by incubation of either NPC or 1053-biotin (second blot, lanes 3 and 4). On the contrary, biotinylation of CD38, Nb-5512, or the complex was only detected in the NPC-treated sample (Fig. S2A, third blot, lane 3), which was consistent with the results in Fig. 2B.
The Strep-HRP blot (Fig. 2B, first lane) shows that there were abundant endogenous biotinylated proteins, obscuring the very low amounts of the CD38 binding partners. We then turned to high-resolution MS to identify unique peptides in the NPC-labeled pulldowns prepared from LP-1 cells. Samples were prepared as depicted in the scheme in Fig. 2C from NPC-labeled LP-1 cells and three controls, including nontreated LP-1 cells, NPC-labeled CD38-KO/LP-1 cells, and 1053-biotin incubated LP-1 cells. The numbers of unique peptides identified by MS of the samples from LP-1 and CD38-KO LP-1 cells are plotted in Fig. 2D; peptides found in both samples fall along the dotted diagonal line (black dots). Peptides corresponding to CD38 (Fig. 2D, green dot) were seen only in the LP-1 samples. Likewise, peptides corresponding to CD71 (Fig. 2D, red dot) and six MHC proteins (Fig. 2D, purple dots) were also enriched in NPC-treated WT LP-1 cells but not in CD38-KO LP-1 cells. These peptides were also absent in the other controls shown in Fig. S2B, plotting peptides in NPC-labeled LP-1 (NPC/LP-1) versus LP-1 cells not treated with NPC (vehicle/LP-1), nor were they found in LP-1 cells incubated with 1053-biotin (Fig. S2C, 1053-biotin/LP-1).
The MS results indicated that CD71 and MHCs are CD38 binding partners on the surface of LP-1 cells. MHCs have been reported previously to be associated with CD38 (7). We confirmed that NPC could label one of the MHCs, HLA-A, by Western blotting. As shown in Fig. S3A, HLA-A and CD38 were present in the immunoprecipitants either by anti-FLAG (on NPC) beads (fifth blot) or by Strep beads (eighth blot) in NPC-treated LP-1 cells (lane 3) but not in the controls (lanes 1, 2, and 4).
CD71 interacted with CD38 and regulated its protein levels
The MS results above documented that at least six peptides corresponding to CD71 were present only in samples from NPC-cross-linked LP-1 cells, more than that of MHCs, a known CD38-associated protein (7). This strongly suggests that CD71 is a more abundant CD38 binding partner than MHCs in LP-1 cells, which has never been reported before. We next validated the MS result by probing the Western blots of Strep pulldown samples from NPC-cross-linked LP-1 cells using anti-CD71 instead of the Strep-HRP in Fig. 2B. Indeed, both CD38 (Fig. 3A, third blot) and CD71 (Fig. 3A, fourth blot) were detected in the precipitates by Strep beads in NPC-treated LP-1 cells but not in the two controls, NPC-treated CD38-KO (Fig. 3A, lane 1, 3rd and 4th blots) or nontreated LP-1 cells (Fig. 3A, second lane, third and fourth blots). The inputs shown also in Fig. 3A, first and second blots, indicated equal loading for all samples. The Western blot results confirm that CD71 is naturally associated with endogenous CD38 in LP-1 cells.
Figure 3.

CD71 interacts with CD38 and regulates its protein level and cellular cADPR production. A, the interaction between CD71 and CD38 was validated by NPL-immunoprecipitation-Western blotting. Sample preparation is shown in Fig. 2B. The proteins CD71 and CD38 in the lysate (Input) or immunoprecipitants were analyzed by anti-CD71 and anti-CD38, respectively. B, WT (center panel) or CD38-KO (top panel) LP-1 cells were incubated with anti-CD38 at 4 °C for 1 h, followed by 37 °C for 20 min to induce capping of CD38. Cocapping (arrows) of CD71 was observed by immunostaining with anti-CD71. Green, CD38; purple, CD71. Negative DAPI staining was used to ensure cell integrity. The low-temperature control (bottom panel) was done as above, except that the cells were not moved to 37 °C but maintained in 4 °C for 20 min. C, the lysates of WT and CD38-KO/LP-1 cells were subjected to Western blotting (top panel). Bottom panel, the band intensities were analyzed by ImageLab (Bio-Rad), normalized by those of WT LP-1, and plotted with GraphPad software. D, the same experiments as in C were done with LP-1 cells stably knocking down CD71 with lentiviral vectors. E, the same experiments as in D were done with HEK293 cells transiently knocking down CD71 with siRNAs. F, the total activities of CD38 were measured by ϵNAD assay with the lysates from D and E. G, the cellular cADPR contents were measured by cycling assay. All experiments were repeated at least three times (mean ± SD; n = 3; Student's t test; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001).
Because both CD38 and CD71 are present on the cell surface, a cocapping assay was used to further substantiate the interaction. As shown in Fig. 3B, the antibody against CD38 could induce capping of surface CD38 and formation of the clusters, as we have shown before (10). Fig. 3B shows that the capping of CD38 (second row, green, indicated by arrows) in WT LP-1 cells and CD71 appeared in the same clusters (Fig. 3B, second row, purple, indicated by arrows). The same treatment did not induce clustering of CD71 in CD38-KO LP-1 cells (Fig. 3B, first row) or in controls incubated on ice to inhibit capping (Fig. 3B, third row). All of these data indicate that CD71 is strongly associated with CD38, either naturally in LP-1 cells or when transfected into HEK293 cells.
We next determined the functional consequences of the association between CD38 and CD71. Fig. 3C shows that LP-1 cells with CD38 knocked out expressed significant higher levels of CD71 compared with normal LP-1 cells (top panel, representative blots; bottom panel, statistical analysis of band intensities from three independent experiments). Because knockout of CD71 severely impaired cell viability, we used knockdown with shRNA or siRNA instead in LP-1 cells (Fig. 3D) or in HEK293 cells overexpressing CD38 (Fig. 3E, CD38/HEK-293), respectively. Both showed that knocking down CD71 could significantly increase the protein levels of CD38. The increased CD38 was active, as measured by its NADase activities in the cell lysates, which showed a corresponding increase in enzymatic activity after CD71 knockdown (Fig. 3F).
Surprisingly, CD71 knockdown in LP-1 and CD38/HEK293, causing increases in both total protein and activity of CD38 (Fig. 3, C–F), did not result in changing the cellular cADPR levels (Fig. 3G). In fact, slight decreases were actually seen in both HEK293 and LP-1 cells. This was the first indication that cellular levels of surface type II CD38 (cf. Fig. 3F) do not correlate with cADPR.
Chimeric CD71N-551 decreased CD38 levels but did not alter cellular cADPR
It is generally known that ligand binding to surface proteins can induce endocytic degradation of the proteins. This mechanism underlies the ligand-induced desensitization of many surface receptors. The effects of CD71 on CD38 levels suggest that it could function as a natural ligand for surface CD38.
To test this hypothesis, we constructed a binding chimera to see whether it can mimic the effects of CD71. The construct was composed with the N terminus of CD71 (amino acids 1–96) fused to a CD38 nanobody (Fig. 4A), Nb-551 (CD71N-551). Nb-551 was used because it is a good model ligand for type II CD38 (cf. Fig. 2, A and B). Indeed, we have shown previously that surface CD38 possesses an intrinsic endocytic mechanism that can be potently activated by binding to its nanobody as an exogenous ligand (17). CD71 may well be a natural ligand for activating this endocytic mechanism.
Figure 4.

The chimeric CD71N-551 significantly decreased CD38 protein levels but did not decrease intracellular cADPR production. A, schematic of the construction of chimeric CD71N-551, with CD71N-NbGFP and CD71-NbEGFR as the CD38-unrelated controls. B, the expression and subcellular localization of CD38 and the chimeric Nbs. HEK293 cells stably expressing CD38 (CD38/HEK-293) were infected with a lentivirus carrying the expression cassettes for the chimeric Nbs. The cells were stained with anti-CD38 and anti-FLAG (for the chimeric Nbs) with the protocol described under “Experimental procedures.” Green, CD38; purple, chimeric Nbs; blue, DAPI for the nucleus. C, the levels of CD38/CD71N-Nbs and cellular cADPR in HEK293 cells coexpressing CD38 and the indicated CD71-Nbs were analyzed by Western blotting (center panel, representative blots; top panel, quantification of band intensities) and cycling assay (bottom panel). D, the cyclase activities of the above lysates were analyzed by NGD assay. E and F, HEK293/CD38 cells were infected with a lentivirus carrying the inducible expression cassettes for the chimeric Nbs. The proteins (E) and cADPR (F) levels were analyzed at the indicated time points with treatment of 2 μg/ml doxycycline. G, the levels of mutCD38/CD71N-Nbs in HEK293 cells coexpressing mutCD38 and the indicated CD71-Nbs were analyzed by Western blotting (bottom panel, representative blots; top panel, quantification of band intensities). All experiments were repeated at least three times (mean ± SD; n = 3; Student's t test; *, p < 0.05; **, p < 0.01; ****, p < 0.0001; ns, not significant).
As nonbinder controls, chimeras CD71N-NbGFP with an Nb against GFP (18) and CD71N-NbEGFR with an Nb against epidermal growth factor receptor (19), were also prepared. All Nbs were also FLAG-tagged.
Fig. 4B shows that CD38/HEK-293 cells expressed CD38 mainly in the cell membrane (CD38, control, green). Coexpression with CD71N-551 showed similar membrane localization as CD38 (Fig. 4B, anti-FLAG, CD71N-551, purple) and dramatically reduced CD38 expression (Fig. 4B, CD38, CD71N-551). Controls cotransfected with a nonbinder, CD71N-NbGFP or CD71N-NbEGFR (Fig. 4B, third and fourth rows, respectively), showed no reduction in CD38, and both were colocalized on the cell surface.
Western blot analyses confirmed the results from the immunostaining experiments. Expression of the chimeras was verified by anti-FLAG blots (Fig. 4C, center panel, second blot). The CD38 blot (Fig. 4C, center panel, first blot) shows that CD71N-551 could induce a dramatic reduction in CD38 levels by 80% ± 5% (Fig. 4C, top panel and center panel, first blot). Nonbinding chimeras (Fig. 4C, top panel and center panel, fourth and fifth lanes) did not alter CD38 levels, which remained similar to the control without the chimera (Fig. 4C, top panel and center panel, third lane). The results show that the binding chimeras can reproduce the reducing effect of CD71 on CD38 levels and are consistent with the hypothesis that binding to surface CD38 can activate its intrinsic mechanism of endocytic degradation. In fact, the CD38 reduction induced by the binding chimeras was even more dramatic than that of CD71, which may be related to the higher affinity of Nb-551 for CD38 (14). Consistently, the association of the MHCs is much weaker than that of CD71, as indicated by the MS results (Fig. 2D), and knockdown of HLA A produced no change in CD38 levels (Fig. S3B).
Strikingly, the cellular levels of cADPR showed no decrease (Fig. 4C, bottom panel, last column), even with the dramatic decrease of the CD38 protein induced by the CD38 targeting chimera. Measurements of the cyclase activity of CD38 in cell lysates (Fig. 4D) showed a reduction similar to the protein levels.
Because CD38 is responsible for synthesizing cellular cADPR, and the great majority is type II on the cell surface, that the large reduction in its level is not reflected in cADPR content is a big surprise. It does, however, strongly suggest that the type II CD38 is not contributing to cellular cADPR production. We have documented previously that CD38 naturally coexists in cells in two opposite membrane orientations (10, 11). The great majority of CD38 is type II CD38, expressed on the cell surface, whereas type III, a very minor portion, is present intracellularly, with its catalytic carboxyl domain facing the cytosol. We have also shown that type III CD38 is enzymatically active and can elevate cellular cADPR levels using cytosolic NAD as a substrate (10, 20). We have further documented a direct correlation of type III CD38 levels with cADPR content even though type III CD38 is of very low abundance compared with total CD38 (11), which is in direct contrast with the noncorrelation of type II CD38 levels described above.
This truly remarkable dichotomy of the type II CD38 and cADPR levels affected by CD71 and the binding chimeras was further substantiated by constructing tetracycline-responsive vectors to control the expression of the chimeras. As shown in Fig. 4E, treatment with doxycycline gradually induced time-dependent expression of the chimeras in CD38/HEK293 cells (center blot in each set). Correlating with expression of the binding chimera CD71N-551, the CD38 levels (Fig. 4E, first blot in the first set) showed a concomitant decrease. Induction of nonbinder chimeras produced no such change. The time courses in Fig. 4F show that the cellular cADPR levels remained essentially constant, and no decreases were observed.
In striking contrast, CD71N-551 had no effect on the expression level of type III CD38, as shown in Fig. 4G. We have shown previously that type II CD38 can be switched to type III by mutating the positively charged residues in the N-terminal tail (10). Type III CD38 expression in cells expressing CD71N-551 remained the same as that of those expressing the nonbinder or no chimeras (Fig. 4G). This was fully expected because the epitope in the C-domain of type III CD38 is facing the cytosol and is separated from the Nb-551 of the binding chimera.
Type II CD38 has its catalytic domain facing out, and it is unclear whether it can produce cellular cADPR; the results described above strongly suggest that it cannot. The binding chimera thus unexpectedly provides a way to resolve the long-standing and unanswered question of whether type II contributes to the production of intracellular cADPR.
The degradation of CD38 was partially through lysosomes, and the lysosomal inhibitor did not alter cADPR production
If the chimera effect is indeed due to the reduction of type II CD38, then it is expected to be via endocytic degradation through lysosomes. To test this, we treated CD38/HEK293 cells coexpressing CD38 and CD71N-551 with Bafilomycin A1 (Baf-A1) or MG132, inhibitors of lysosomes or proteasomes, respectively, the two best-known protein degradation machineries. As shown in Fig. 5A, Baf-A1 significantly increased the levels of CD38 but MG132 did not, consistent with CD71N-551 inducing degradation of CD38 through lysosomes. The increased CD38 was active, as measured by its ADP-ribosyl cyclase activity in cell lysates that showed increases correlating with the protein levels (Fig. 5B).
Figure 5.

CD38 was degraded in lysosomes, and lysosomal CD38 did not contribute to cADPR production. A, HEK293 cells coexpressing CD38 and CD71N-551 were treated with 5 μm MG132 for 6 h or 0.4 μm Baf-A1 for 15 h, and the cell lysates were analyzed by Western blotting with the indicated antibodies. The band intensities of CD38 were normalized by those of tubulin, plotted, and statistically analyzed by GraphPad software. B, the same cells as in A were treated with 0.4 μm Baf-A1 or DMSO as a vehicle control for 15 h, and the cell lysates were subjected to an NGD assay. The activities were normalized with those from the DMSO group. C, the same cells as in A were treated with Baf-A1 or DMSO as a control and stained with the indicated antibodies, followed by confocal imaging. Green, CD38; red, LAMP1 as an indicator of lysosomes; purple, anti-FLAG for CD71N-551; blue, DAPI for the nucleus. D, PCCs were calculated by overlapping the signals of CD38 with those of CD71N-551 (blue dots) or LAMP1 (red squares) in the presence of Baf-A1 from the experiments in C. E, the cellular cADPR contents were measured after Baf-A1 treatment. All experiments were repeated at least three times (mean ± SD; n = 3; Student's t test; ***, p < 0.001; ****, p < 0.0001; ns, not significant).
The immunostaining and confocal images confirmed the expression increase of CD38 following the Baf-A1 treatment (Fig. 5C, CD38, DMSO, and Baf-A1). Also shown is colocalization of CD71N-551 (Fig. 5C, purple) with lysosomes (Fig. 5C, LAMP1, red). Fig. 5D shows that the quantitative Pearson correlation coefficient (PCC) of CD38 overlapping CD71N-551 was around 0.6, indicating that most of the CD38 and CD71N-1053 formed complexes, whereas the PCC of CD38 overlapping LAMP1 was also around 0.6, which statistically reflected the significant localization of CD38 in lysosomes. The lower PCC value is due to a portion of CD38 remaining in the plasma membrane, as shown in Fig. 5C.
Although the amount of CD38 was increased more than 4-fold after Baf-A1 treatment (Fig. 5A), cellular cADPR did not change at all (Fig. 5E). These results provide strong evidence that type II CD38, either on the surface or endocytically delivered to the lysosomes (cf. Fig. 5C) contributes minimally to intracellular cADPR production, as proposed above.
CD38 retained in the ER could not elevate cellular cADPR levels
As a surface protein, type II CD38 is expected to be translated in the ER with its catalytic carboxyl domain facing the lumen. It is topologically equivalent to type II CD38 endocytically delivered to lysosomes, as described above. It is thus similarly expected not to contribute to cellular cADPR production. To test this expectation, we made an ER retention construct (erNbGFP) consisting of NbGFP fused with the signal peptide and ER retention signal of calreticulin (21) (Fig. 6A). erNbGFP was expressed in HEK293 cells stably expressing CD38 with an EGFP tag in its C terminus (see “Experimental procedures”).
Figure 6.

CD38, retained in the ER, could not elevate cellular cADPR levels. A, schematic of erNbGFP, the ER-retained NbGFP with a His6 tag. SP, signal peptide of calreticulin; KEDL, an ER retention signal motif. B, CD38-EGFP/HEK293 cells were infected with a lentivirus encoding erNbGFP. The resulting cell line was stained with anti-GRP78, an ER marker, anti-His6 for erNbGFP, and DAPI. The fluorescence developed by antibody and dyes, together with that from the EGFP tag of CD38, was imaged under a Nikon A1 confocal microscope. Green, CD38; red, GRP78; purple, erNbGFP; blue, DAPI for the nucleus. C, PCCs were calculated by overlapping the signals of CD38 or erNbGFP with those of GRP78 from the experiment in B. D, the total expression levels of CD38 were analyzed by Western blotting, together with erNbGFP, blotted by anti-CD38 and anti-His6, respectively. E, the ADP-ribosyl cyclase activities of the lysates were measured with an NGD assay. F, the cellular cADPR levels were measured by a cycling assay. All experiments were repeated at least three times (mean ± SD; n = 3; Student's t test; **, p < 0.01, 0.0001; ns, not significant).
As shown in Fig. 6B, the expression of erNbGFP (purple) induced retention of CD38 to the ER (green for CD38, red for the ER marker) instead of expression in the plasma membrane. As shown in Fig. 6C, erNbGFP or CD38 overlapping with the ER marker, GRP-78, showed a PCC around 0.8 compared with 0.3 in control cells, indicating ER retention of CD38 by erNbGFP as expected. Concomitant with the expression of erNbGFP (Fig. 6D, second blot), the levels of CD38 (Fig. 6D, first blot and top panel) and total cyclase activity in the cell lysates (Fig. 6E) increased significantly, indicating that the ER-retained CD38 remained active. Again, the cellular cADPR levels did not change (Fig. 6F). The results document that type II CD38 compartmented in the ER does not contribute to the production of cellular cADPR, which indicates that there might not be an available NAD pool in the ER lumen.
Discussion
In this study, we developed a new methodology, NPL, to profile the extracellular interactome of the plasma membrane protein CD38. The key innovation was the use of the nanobody of CD38, which was coupled with a cross-linker, sulfo-SBED. The high specificity of the nanobody allows direct targeting of surface CD38 in live cells. The small size of the nanobody also ensures that only proteins that bind closely and directly to CD38 are cross-linked. The three functional groups of sulfo-SBED (Fig. 1) provide easy attachment to the nanobody and can transfer a biotin group to the cross-linked proteins, facilitating subsequent purification. These advantages of NPL resolve the clear shortcomings of the conventional immunoprecipitation approach, which cannot distinguish secondary and nonspecific binders even with elaborate solubilization procedures.
Adopting high-resolution MS to identify unique peptides that were present only in the Strep pulldown samples but not in the CD38 knockout controls, we documented CD71 and MHCs as specific binding partners of the endogenous CD38 on the surface of LP-1 cells. MHCs are known to associate with CD38 and serve to validate the NPL. CD71, a receptor for transferrin (22) and several viruses (23, 24), has not been reported as a specific ligand for CD38. We documented the specific association of CD38 and CD71 by immunoprecipitation and cocapping.
Elucidation of the ligand properties of CD71 unexpectedly revealed valuable clues regarding the mechanism of CD38 signaling and prompted us to develop probes to address a long-standing and unresolved question regarding the functional role of type II CD38 in cellular cADPR synthesis. We then engineered a binding chimera to simulate the effects of CD71 on CD38, and the results, summarized in Fig. 7, provide the first direct evidence documenting the distinct roles of type II and type III CD38 in cellular cADPR production.
Figure 7.
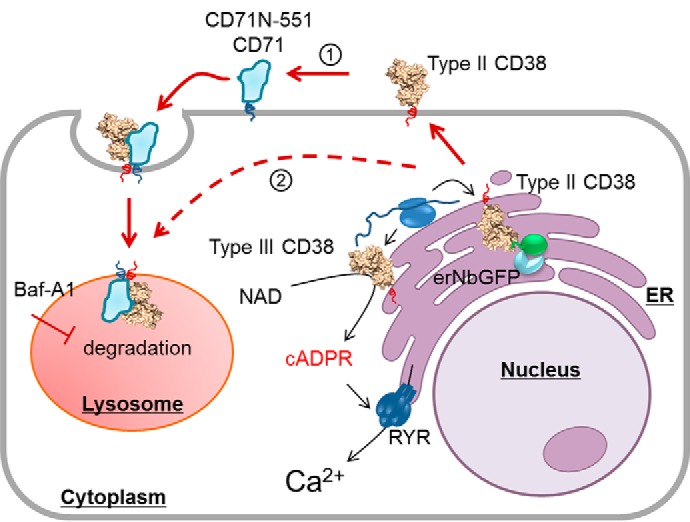
A working model of CD38 signaling. Type II CD38 is synthesized in the ER and sorted to the plasma membrane, where it interacts with the endogenous or chimeric CD71 (1). The interaction induces endocytosis, and more than 80% of CD38 is degraded in the lysosomes. Possible aberrant protein sorting (2) may also deliver type II CD38 directly to the lysosomes. The cellular levels of cADPR, however, remains unchanged. Retention of type II CD38, either in the lysosomes via Baf-A1 treatment or in the ER by expressing an ER retention protein for CD38, erNbGFP, greatly increases the type II CD38 levels in the cells but does not increase cellular cADPR levels. The results indicate that type II CD38 contributes minimally to cellular cADPR levels. This is consistent with our previously published findings documenting that it is mainly type III CD38, and not type II, that catalyzes the biosynthesis of cADPR and mediates Ca2+ signaling.
As depicted in Fig. 7, type II CD38 is translated in the ER (ribosome, blue double ovals) and transported to the cell surface, similarly as CD71 and the chimera, CD71N-551, as both are also expressed on cell surface (Figs. 3B and 4B). Binding of the chimera to surface CD38 activates endocytosis and subsequent lysosomal degradation (Fig. 5), resulting in a dramatic decrease in CD38 content (Fig. 4, C and E). No change in cellular cADPR (Fig. 4, C and F) was seen, indicating that surface type II CD38 does not contribute to cADPR synthesis. Blocking lysosomal degradation led to a large accumulation of CD38 in lysosomes (Fig. 5), and, again, no change in cADPR was observed (Fig. 5E). Likewise, retaining the type II CD38 in the ER using the erNb construct does not elevate cADPR content (Fig. 6). It is clear that type II CD38 compartmented in the ER, on the surface, and in lysosomes contributes minimally to cellular cADPR production.
Type III CD38 is thus mainly responsible for cellular cADPR. Indeed, we have shown previously that it is efficient in doing so (11). Type III CD38 is also translated in the ER (Fig. 7), and the membrane orientation is determined by the charges in the N-terminal tail of CD38, according to the “positive-inside rule” (25). For CD38, the two sides of the transmembrane segment differ only by one positive charge (10), making type II more favorable, but type III is also translated. This is consistent with our observation that transfection of CD38 to cells leads to production of type II predominantly but type III as well (11). Type III CD38, with its C-domain facing the cytosol, was not affected by the chimera CD71N-551 and erNb. This is because the Nb epitopes are on the C-domain, and, for type III CD38, they are topologically separated from the Nb constructs.
It is also possible that CD71 or the chimera CD71N-551 can interact with type II CD38 in the ER before they reach the plasma membrane. The binding could trigger direct delivery to lysosomes for degradation (Fig. 7, dashed lines, pathway 2), resulting in a decrease in CD38.
The type II CD38 in the extracytosolic compartment is enzymatically active (Figs. 5B and 6E). That it is inhibited from producing cADPR is likely due to the acidic environment of the endolysosomal compartment as well as the absence of substrate. Indeed, treatments that import exogenous substrate, such as nicotinic acid, can result in production of the base exchange product NAADP, another calcium messenger produced by CD38 (26). This is, in fact, the main evidence for the substrate-limited signaling mechanism of CD38 we proposed previously (26).
In summary, we developed a new NPL method in this study and identified CD71 as an endogenous interaction protein of CD38. Our investigation of the ligand properties of CD71 provide the first direct evidence of the topological compartmentation of type II and type III CD38 and their cADPR production and completes a comprehensive view of the signaling mechanism of CD38. In a broader view, the NPL method can be easily applied to profile the extracellular interactome of other membrane proteins. The strategy of using a membrane-bound nanobody chimera is a tool for selective knockdown of type II membrane proteins, which is especially important for membrane proteins with dual orientations.
Experimental procedures
Chemicals
FBS, DMEM, Iscove's modified Dulbecco's medium, penicillin/streptomycin solution, trypsin, sulfo-SBED (33033), and Alexa Fluor–conjugated donkey anti-mouse or anti-rabbit IgG were purchased from Thermo Fisher Scientific. MG132, Baf-A1, 4′,6-diamidino-2-phenylindole (DAPI), anti-FLAG M2 magnetic beads, 3×FLAG peptide, anti-FLAG M2 (F3165), and reagents for the cycling assay and εNAD assay were obtained from Sigma-Aldrich. Anti-LAMP1 (9091) was obtained from Cell Signaling Technology. Anti-CD71 (66180-1-lg) and anti-HLA A (66013-1-lg) were obtained from Proteintech. Anti-CD38 (AF2404) was obtained from R&D Systems. Anti-GRP 78 (C-20) was obtained from Santa Cruz Biotechnology.
Cell culture
HEK293 and HEK293T cells were purchased from the American Type Culture Collection, and the myeloma cell line LP-1 was kindly provided by Annie An (Peking University, Beijing, China). Cell line authentication was achieved by genetic profiling using polymorphic short tandem repeat loci. The cells were cultured in DMEM (HEK293 and HEK293T) or Iscove's modified Dulbecco's medium (LP-1 and CD38-KO/LP-1) supplemented with 10% FBS and 1% penicillin/streptomycin solution. The cultures were maintained at 37 °C in a humidified atmosphere with 5% CO2.
Expression and purification of the recombinant proteins
ReCD38 and nanobodies (Nb-1053 and Nb-551) were expressed and purified as described previously (14). To make the expression vector for Nb-5512, one copy of the DNA sequence encoding Nb-551 without a stop codon was first subcloned to pET-28a (+) by EcoRI and HindIII, resulting in pET-28a-551, and the sequence encoding a 10-amino acid flexible polypeptide linker (Gly-Gly-Gly-Gly-Ser)2 followed by another copy of Nb-551, with a stop codon, was subcloned to pET-28a-551 by HindIII and NotI. The recombinant protein Nb-5512 was expressed and purified as Nb-551 (14).
Plasmids for CD71N-Nb chimeras
To construct the constitutive expression vectors for CD71N-Nb chimeras, the genes encoding Nb-551/EGFR/GFP were fused to the C terminus of the CD71 fragment (amino acids 1–96) by overlapping PCR (27). The resulted genes with a FLAG tag in the C terminus were subcloned to pCDH-neo. To make the inducible expression vector for the CD71N-Nb chimeras, the corresponding genes were subcloned to the lentivector pInducer20 (Addgene, 44012).
Plasmids for erNbGFP and CD38-EGFP
The DNA encoding erNbGFP was synthesized and contained a signal peptide of calreticulin in the N terminus and subsequent NbGFP and KDEL motif (21), which was subcloned to pLenti-puro to make the expression vector. To make the expression vector for CD38-EGFP, pEGFPN1-CD38 (28) was used as a template and subcloned into pCDH-neo by NheI and NotI.
Transfection and infection
The siRNAs were transfected to CD38/HEK293 cells using Lipofectamine RNAi MAX (Thermo Fisher Scientific). Lentiviral particles were prepared by transfecting HEK293T cells with pLenti or pCDH lentivectors, pMD2.G and psPAX2, with Lipofectamine 2000 (Thermo Fisher Scientific). Cell infection was performed as described previously (28), followed by G418 or puromycin selection.
siRNAs were synthesized in GenePharma. The sequences were 5′-UUCUCCGAACGUGUCACGUtt-3′ (for scramble), 5′-AGGAGACACGGAAUGUGAAtt-3′ (for HLA A), and 5′-CUCCUGUGAAUGGAUCUAUtt-3′ (for CD71).
Preparation and in vitro characterization of NPC
Recombinant Nb-1053 was labeled with sulfo-SBED according to the manufacturer's instructions. Briefly, 200 μg of Nb-1053 was mixed with a 5-fold molar excess of sulfo-SBED, incubated at 4 °C for 3 h, and stopped by hydroxylammonium chloride. The resulting NPC was desalted and stored in aliquots at −80 °C, protected from light.
To test cross-linking of NPC with reCD38 or Nb-5512, NPC was mixed with reCD38 with or without Nb-5512 or BSA (negative control) and incubated on ice for 1 h, followed by exposure to UV irradiation in the UVP CL-1000L UV Cross-linker (254 nm) at a distance of ∼8 cm on ice for 8 min. The mixtures were then treated with 100 mm DTT and applied to SDS-PAGE.
Cross-linking and pulldown of CD38 and its interaction proteins on the LP-1 cell surface
NPC (2 μg) or the control probes were incubated with 1 × 107 LP-1 cells for 1 h at 4 °C with end-over-end rotation. Then the cells were subjected to UV irradiation in the UVP CL-1000 UV Cross-linker (254 nm) at a distance of ∼8 cm on ice for 20 min. The cells were lysed at room temperature in 1 ml of lysis buffer (buffer A: 50 mm Tris (pH 7.4), 500 mm NaCl, 0.4% SDS, 5 mm EDTA, and 1 mm DTT; buffer B: 50 mm Tris (pH 7.4) and 4% Triton X-100; buffer A and B were mixed at equal volumes before use) containing a protease inhibitor mixture (EDTA-Free Complete, Roche). The lysates were applied to sonication (two 5-s pulses, 8-s pause in between). After centrifugation at 13,000 rpm at 4 °C, the supernatants were incubated with 25 μl of Dynabeads (MyOne Streptavidin C1, Thermo) overnight. At room temperature, the beads were collected and washed twice for 8 min in 1 ml of wash buffer I (2% SDS in distilled H2O). This was repeated once with wash buffer II (0.1% deoxycholate, 1% Triton X-100, 500 mm NaCl, 1 mm EDTA, and 50 mm Hepes (pH 7.5)) and twice with wash buffer III (50 mm Tris (pH 7.4), and 50 mm NaCl). One fraction (10%) of the samples was applied to Western blots, and the remaining samples were separated on SDS-PAGE, followed by MS-compatible Coomassie Brilliant Blue staining, in-gel digestion by trypsin, and MS analysis.
MS analysis
MS data acquisition was performed by LC-MS/MS using a nanoLC.2D (Eksigent Technologies) coupled with a TripleTOF 5600+ system (AB Sciex, Concord, ON, Canada). The details were similar to a previous protocol (29). Briefly, samples were separated in a nanoLC column with an acetonitrile gradient ((v/v) 2%–80%) in 0.1% (v/v) formic acid. MS1 spectra were collected in the range of 350–1,500 m/z for 250 ms. The 50 most intense precursors with charge state 2–5 were selected for fragmentation, and MS2 spectra were collected in the range of 100–2,000 m/z for 100 ms; precursor ions were excluded from reselection for 15 s. The original MS data were analyzed by ProteinPilot software (version 4.5, AB Sciex) as described previously (29). The endogenous biotinylated proteins were excluded before plotting the unique peptide numbers of the candidate proteins from NPC-treated LP-1 samples versus those from control samples by GraphPad software.
Cocapping assay
Cocapping experiments were conducted in a 1.5-ml eppendorf tube. 1 × 106 LP-1 or CD38 KO cells were incubated with 2 μg of homemade rabbit polyclonal antibody (anti-CD38) in 200 μl of PBS with 1% BSA for 1 h on ice. Then cells were divided into two aliquots. One aliquot (I) was incubated at 37 °C for 40 min to induce capping of CD38. Another aliquot (II) was incubated on ice. Both aliquots were stained with Alexa FluorTM 488–conjugated anti-rabbit and Allophycocyanin-conjugated anti-CD71 in PBS with 0.5% BSA on ice for 1 h. The cells were stained with DAPI for 5 min, fixed with 4% paraformaldehyde on ice for 15 min, mounted on a coverslip, and visualized by confocal microscopy. Cells were washed twice with ice-cold PBS between steps.
SDS-PAGE and Western blots
The expression levels of CD38 and CD71 in different cell lines were measured by Western blotting as described previously (29). The primary antibodies used in this study were anti-CD38 (homemade), anti-CD71(Proteintech), anti-FLAG (Sigma), and anti-tubulin (Transgen). Images were acquired by Chemidoc MP (Bio-Rad) and analyzed by ImageLab (Bio-Rad).
Measurement of cellular cADPR
The cells were lysed with 0.6 m perchloric acid. After centrifugation, cADPR in the supernatant was measured by a cycling assay (30). The protein pellets were redissolved in 1 m NaOH and quantified by Bradford assay (Quick Start Bradford Kit, Bio-Rad). The results were presented as picomoles of cADPR per milligram of total protein or further normalized to control samples, as indicated in the main text.
Measurement of the activities of CD38
The cells were lysed with ice-cold lysis buffer (50 mm Tris (pH 8.0), 150 mm NaCl, 1 mm EDTA, and 0.5% Triton X-100) containing a protease inhibitor mixture (EDTA-Free Complete, Roche). The activities of CD38 in the lysates were measured by fluorimetric reactions (excitation 300 nm/emission 410 nm), utilizing εNAD as the substrate for NADase activity or NGD for ADP-ribosyl cyclase activity (31). The activities were calculated as nanomoles of εNAD or cGDPR per minute per microgram of lysate and normalized by the control samples.
Statistical analysis
Quantification of the Western blots was performed with ImageLab software. NIS Elements software was used for colocalization analysis of the confocal signals. Statistical analysis was done with GraphPad Prism software, and the significance of differences was analyzed by unpaired Student's t test. The data shown in the figures are mean ± SD from at least three repeats.
Author contributions
Q. W. D., T. L., H. C. L., and Y. J. Z. conceptualization; Q. W. D., J. Z., T. L., W. M. H., L. F., H. C. L., and Y. J. Z. formal analysis; Q. W. D., T. L., H. C. L., and Y. J. Z. supervision; Q. W. D., J. Z., T. L., W. M. H., L. F., H. C. L., and Y. J. Z. investigation; Q. W. D., J. Z., T. L., W. M. H., L. F., H. C. L., and Y. J. Z. methodology; Q. W. D., T. L., H. C. L., and Y. J. Z. writing-original draft; Q. W. D., T. L., H. C. L., and Y. J. Z. writing-review and editing; Y. J. Z. project administration.
Supplementary Material
This study was supported by National Science Foundation of China grants (31571438, 31871403, and 31671463). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
- C-domain
- carboxyl domain
- cADPR
- cyclic ADP-ribose
- MHC
- major histocompatibility complex
- ER
- endoplasmic reticulum
- NPC
- nanobody-based photoaffinity crosslinker
- NPL
- nanobody-based photoaffinity labeling
- Strep
- streptavidin
- Nb
- nanobody
- Baf-A1
- Bafilomycin A1
- PCC
- Pearson correlation coefficient
- EGFP
- enhanced GFP
- DAPI
- 4′,6-diamidino-2-phenylindole
- sulfo-SBED
- Sulfo-N-hydroxysuccinimidyl-2-(6-[biotinamido]-2-(p-azido benzamido)-hexanoamido) ethyl-1,3′-dithioproprionate
- NGD
- Nicotinamide Guanine Dinucleotide.
References
- 1. Kato I., Yamamoto Y., Fujimura M., Noguchi N., Takasawa S., and Okamoto H. (1999) CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J. Biol. Chem. 274, 1869–1872 10.1074/jbc.274.4.1869 [DOI] [PubMed] [Google Scholar]
- 2. Giorgi J. V., Ho H. N., Hirji K., Chou C. C., Hultin L. E., O'Rourke S., Park L., Margolick J. B., Ferbas J., and Phair J. P. (1994) CD8+ lymphocyte activation at human immunodeficiency virus type 1 seroconversion: development of HLA-DR+ CD38− CD8+ cells is associated with subsequent stable CD4+ cell levels: the Multicenter AIDS Cohort Study Group. J. Infect. Dis. 170, 775–781 10.1093/infdis/170.4.775 [DOI] [PubMed] [Google Scholar]
- 3. Dürig J., Naschar M., Schmücker U., Renzing-Köhler K., Hölter T., Hüttmann A., and Dührsen U. (2002) CD38 expression is an important prognostic marker in chronic lymphocytic leukaemia. Leukemia 16, 30–35 10.1038/sj.leu.2402339 [DOI] [PubMed] [Google Scholar]
- 4. Ruiz-Argüelles G. J., and San Miguel J. F. (1994) Cell surface markers in multiple myeloma. Mayo Clin. Proc. 69, 684–690 10.1016/S0025-6196(12)61350-0 [DOI] [PubMed] [Google Scholar]
- 5. Philip M., Fairchild L., Sun L., Horste E. L., Camara S., Shakiba M., Scott A. C., Viale A., Lauer P., Merghoub T., Hellmann M. D., Wolchok J. D., Leslie C. S., and Schietinger A. (2017) Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 10.1038/nature22367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. States D. J., Walseth T. F., and Lee H. C. (1992) Similarities in amino acid sequences of Aplysia ADP-ribosyl cyclase and human lymphocyte antigen CD38. Trends Biochem. Sci. 17, 495 [DOI] [PubMed] [Google Scholar]
- 7. Zilber M. T., Setterblad N., Vasselon T., Doliger C., Charron D., Mooney N., and Gelin C. (2005) MHC class II/CD38/CD9: a lipid-raft-dependent signaling complex in human monocytes. Blood 106, 3074–3081 10.1182/blood-2004-10-4094 [DOI] [PubMed] [Google Scholar]
- 8. Deaglio S., Morra M., Mallone R., Ausiello C. M., Prager E., Garbarino G., Dianzani U., Stockinger H., and Malavasi F. (1998) Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 160, 395–402 [PubMed] [Google Scholar]
- 9. Lee H. C., Walseth T. F., Bratt G. T., Hayes R. N., and Clapper D. L. (1989) Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 264, 1608–1615 [PubMed] [Google Scholar]
- 10. Zhao Y. J., Lam C. M., and Lee H. C. (2012) The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal. 5, ra67 [DOI] [PubMed] [Google Scholar]
- 11. Liu J., Zhao Y. J., Li W. H., Hou Y. N., Li T., Zhao Z. Y., Fang C., Li S. L., and Lee H. C. (2017) Cytosolic interaction of type III human CD38 with CIB1 modulates cellular cyclic ADP-ribose levels. Proc. Natl. Acad. Sci. U.S.A. 10.1073/pnas.1703718114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zubiaur M., Fernández O., Ferrero E., Salmerón J., Malissen B., Malavasi F., and Sancho J. (2002) CD38 is associated with lipid rafts and upon receptor stimulation leads to Akt/protein kinase B and Erk activation in the absence of the CD3-ζ immune receptor tyrosine-based activation motifs. J. Biol. Chem. 277, 13–22 10.1074/jbc.M107474200 [DOI] [PubMed] [Google Scholar]
- 13. Deaglio S., Vaisitti T., Billington R., Bergui L., Omede' P., Genazzani A. A., and Malavasi F. (2007) CD38/CD19: a lipid raft-dependent signaling complex in human B cells. Blood 109, 5390–5398 10.1182/blood-2006-12-061812 [DOI] [PubMed] [Google Scholar]
- 14. Li T., Qi S., Unger M., Hou Y. N., Deng Q. W., Liu J., Lam C. M. C., Wang X. W., Xin D., Zhang P., Koch-Nolte F., Hao Q., Zhang H., Lee H. C., and Zhao Y. J. (2016) Immuno-targeting the multifunctional CD38 using nanobody. Sci. Rep. 6, 27055 10.1038/srep27055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Graeff R., Munshi C., Aarhus R., Johns M., and Lee H. C. (2001) A single residue at the active site of CD38 determines its NAD cyclizing and hydrolyzing activities. J. Biol. Chem. 276, 12169–12173 10.1074/jbc.M011299200 [DOI] [PubMed] [Google Scholar]
- 16. Li T., Li S. L., Fang C., Hou Y. N., Zhang Q., Du X., Lee H. C., and Zhao Y. J. (2018) Nanobody-based dual epitopes protein identification (DepID) assay for measuring soluble CD38 in plasma of multiple myeloma patients. Anal. Chim. Acta 1029, 65–71 10.1016/j.aca.2018.04.061 [DOI] [PubMed] [Google Scholar]
- 17. An N., Hou Y. N., Zhang Q. X., Li T., Zhang Q. L., Fang C., Chen H., Lee H. C., Zhao Y. J., and Du X. (2018) Anti-multiple myeloma activity of nanobody-based anti-CD38 chimeric antigen receptor T cells. Mol. Pharm. 15, 4577–4588 10.1021/acs.molpharmaceut.8b00584 [DOI] [PubMed] [Google Scholar]
- 18. Kubala M. H., Kovtun O., Alexandrov K., and Collins B. M. (2010) Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci. 19, 2389–2401 10.1002/pro.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heukers R., van Bergen en Henegouwen P. M., and Oliveira S. (2014) Nanobody-photosensitizer conjugates for targeted photodynamic therapy. Nanomedicine 10, 1441–1451 10.1016/j.nano.2013.12.007 [DOI] [PubMed] [Google Scholar]
- 20. Zhao Y. J., Zhu W. J., Wang X. W., Zhang L. H., and Lee H. C. (2015) Determinants of the membrane orientation of a calcium signaling enzyme CD38. Biochim. Biophys. Acta 1853, 2095–2103 10.1016/j.bbamcr.2014.10.028 [DOI] [PubMed] [Google Scholar]
- 21. Kendall J. M., Badminton M. N., Dormer R. L., and Campbell A. K. (1994) Changes in free calcium in the endoplasmic reticulum of living cells detected using targeted aequorin. Anal. Biochem. 221, 173–181 10.1006/abio.1994.1394 [DOI] [PubMed] [Google Scholar]
- 22. Sutherland R., Delia D., Schneider C., Newman R., Kemshead J., and Greaves M. (1981) Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proc. Natl. Acad. Sci. U.S.A. 78, 4515–4519 10.1073/pnas.78.7.4515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin D. N., and Uprichard S. L. (2013) Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. U.S.A. 110, 10777–10782 10.1073/pnas.1301764110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Radoshitzky S. R., Abraham J., Spiropoulou C. F., Kuhn J. H., Nguyen D., Li W., Nagel J., Schmidt P. J., Nunberg J. H., Andrews N. C., Farzan M., and Choe H. (2007) Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446, 92–96 10.1038/nature05539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Higy M., Junne T., and Spiess M. (2004) Topogenesis of membrane proteins at the endoplasmic reticulum. Biochemistry 43, 12716–12722 10.1021/bi048368m [DOI] [PubMed] [Google Scholar]
- 26. Fang C., Li T., Li Y., Xu G. J., Deng Q. W., Chen Y. J., Hou Y. N., Lee H. C., and Zhao Y. J. (2018) CD38 produces nicotinic acid adenosine dinucleotide phosphate in the lysosome. J. Biol. Chem. 293, 8151–8160 10.1074/jbc.RA118.002113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bryksin A. V., and Matsumura I. (2010) Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. BioTechniques 48, 463–465 10.2144/000113418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Y. J., Zhang H. M., Lam C. M., Hao Q., and Lee H. C. (2011) Cytosolic CD38 protein forms intact disulfides and is active in elevating intracellular cyclic ADP-ribose. J. Biol. Chem. 286, 22170–22177 10.1074/jbc.M111.228379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu Y., Zhang J., Fang L., Lee H. C., and Zhao Y. J. (2019) A cytosolic chaperone complex controls folding and degradation of type III CD38. J. Biol. Chem. 294, 4247–4258 10.1074/jbc.RA118.005844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Graeff R., and Lee H. C. (2002) A novel cycling assay for cellular cADP-ribose with nanomolar sensitivity. Biochem. J. 361, 379–384 10.1042/bj3610379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Graeff R. M., Walseth T. F., Fryxell K., Branton W. D., and Lee H. C. (1994) Enzymatic synthesis and characterizations of cyclic GDP-ribose: a procedure for distinguishing enzymes with ADP-ribosyl cyclase activity. J. Biol. Chem. 269, 30260–30267 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.