

Abstract
Learning is a process of plastic adaptation through which a neural circuit generates a more preferable outcome; however, at a microscopic level, little is known about how synaptic activity is patterned into a desired configuration. Here, we report that animals can generate a specific form of synaptic activity in a given neuron in the hippocampus. In awake, head-restricted mice, we applied electrical stimulation to the lateral hypothalamus, a reward-associated brain region, when whole-cell patch-clamped CA1 neurons exhibited spontaneous synaptic activity that met preset criteria. Within 15 min, the mice learned to generate frequently the excitatory synaptic input pattern that satisfied the criteria. This reinforcement learning of synaptic activity was not observed for inhibitory input patterns. When a burst unit activity pattern was conditioned in paired and nonpaired paradigms, the frequency of burst-spiking events increased and decreased, respectively. The burst reinforcement occurred in the conditioned neuron but not in other adjacent neurons; however, ripple field oscillations were concomitantly reinforced. Neural conditioning depended on activation of NMDA receptors and dopamine D1 receptors. Acutely stressed mice and depression model mice that were subjected to forced swimming failed to exhibit the neural conditioning. This learning deficit was rescued by repetitive treatment with fluoxetine, an antidepressant. Therefore, internally motivated animals are capable of routing an ongoing action potential series into a specific neural pathway of the hippocampal network.
Keywords: awake mouse, depression, hippocampus, motivation, operant conditioning, synaptic activity
Introduction
Learning is believed to rely on the persistent modification of action potential flow in neuronal networks. Because a neuron has a membrane potential threshold for generating action potentials, it does not fire unless it receives highly synchronized spikes from a presynaptic neuron population. Therefore, learning can be described as a plastic process by which neurons become responsive to specific presynaptic cell assemblies (Hebb, 1949). Cell assembly dynamics have been well documented in coordinated activity of the hippocampus (Harris et al., 2003; Buzsáki, 2010), a brain region involved in certain forms of learning and memory. For example, cell assemblies that appeared during learning are often spontaneously reactivated as synchronous barrages during quiet awake states or slow-wave sleep (Buzsáki et al., 1983; Wilson and McNaughton, 1994; Lee and Wilson, 2002). Therefore, barrage activities of hippocampal neurons serve as an experimental model with which to investigate neural processes underlying learning and memory.
One major form of learning is operant conditioning, in which the frequency of voluntary behavior increases or decreases after reward or punishment (Domjan and Grau, 2003). Half a century ago, Fetz (1969) demonstrated a modified form of the operant conditioning; that is, by the use of neurofeedback training, motor cortical activity in monkeys was operantly reinforced in a given single neuron. More recently, Cerf et al. (2010) reported that humans can voluntarily control the firing rate of a specific neuron in the medial temporal lobe. However, no studies have addressed the synaptic dynamics that mediates the control of a single neuron's firing. Moreover, in these pioneer studies, the activity of the controlled neuron might be correlated with some feature in the real world, including behavior, cognition, attention, and extrinsic rewards such as food or money (Basmajian, 1963; Fetz, 1969; Fetz and Finocchio, 1975; Fetz, 2007; Cerf et al., 2010; Kobayashi et al., 2010; Schafer and Moore, 2011; Sakurai and Takahashi, 2013).
In the present work, we used in vivo patch-clamp techniques in the mouse hippocampus and an internal neural process-based paradigm with direct electrical stimulation of the reward system, which did not require an external reward, an explicit cognitive task, or a specific pretraining for the operant conditioning. Using reward stimulation through a real-time neurofeedback system, we attempted to reinforce the event frequencies of the predefined patterns of spontaneous EPSCs (sEPSCs), spontaneous IPSC (sIPSCs), or spontaneous unit activity. In addition, we examined how this neural reinforcement learning was modulated by anesthesia or animal's state. As a result, we demonstrated that mice could rapidly learn to receive more reward stimuli by increasing or decreasing the frequency of the predefined activity patterns in a single hippocampal CA1 pyramidal cell. This reinforcement learning did not occur in less motivated animals.
Materials and Methods
Animals.
Experiments were performed with the approval of the animal experiment ethics committee at the University of Tokyo (approval number P21-6) and according to the University of Tokyo guidelines for the care and use of laboratory animals. Male C57BL/6J mice (21–27 d old) were housed in cages under standard laboratory conditions (12 h light/dark cycle, ad libitum access to food and water). All efforts were made to minimize the animals' suffering and the number of animals used.
Drug.
MK801 was applied intraperitoneally (0.1 mg/kg) or locally into the CA1 region. Local application was performed using a pressure injection (16 psi for 20 s) of 300 μm MK801 in artificial CSF (aCSF) consisting of the following (in mm): 127 NaCl, 3.5 KCl, 1.24 KH2PO4, 1.2 MgSO4, 2.0 CaCl2, 26 NaHCO3, and 10 d-glucose through glass pipettes (1–3 MΩ). SCH23390 (50 μg/kg) and fluoxetine (20 mg/kg) were applied intraperitoneally.
Habituation.
Mice were anesthetized with ketamine (50 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). Anesthesia was confirmed by the lack of paw withdrawal, whisker movement, and eye blink reflexes. The head skin was then removed and the animal was implanted with a metal head-holding plate. After 2 d of recovery, the head-fixation training on a custom-made stereotaxic fixture was repeated for 1–3 h per day until the implanted animal learned to remain quiet. During and after each session, the animal was rewarded with ad libitum access to sucrose-containing water. During the last three sessions, sham experiments were conducted to habituate the animal to experimental conditions and noise. On the last 2–3 d, the animal was kept virtually immobile—quiet but awake—for >2 h.
Electrophysiology.
Unanesthetized and head-restrained mice were used unless otherwise specified. After full habituation, the animal was anesthetized with a ketamine/xylazine mixture. A craniotomy (1 × 1 mm2), centered at 2.2 mm posterior and 2.0 mm lateral to the bregma, was performed and the dura was surgically removed. The exposed cortical surface was covered with 1.7% agar. Throughout the experiments, a heating pad maintained the rectal temperature at 37°C and the surgical region was analgesized with 0.2% lidocaine. After recovery from anesthesia, patch-clamp recordings were obtained using borosilicate glass electrodes (4–7 MΩ). Neurons in the CA1 pyramidal cell layer of the hippocampus or the layer II/III of the primary somatosensory cortex were targeted for patch clamping. In the experiments shown in Figure 3, D and E, and Figure 5, I and J, urethane (2.0–2.2 g/kg) was intraperitoneally injected before surgery and electrophysiology was conducted in a manner similar to that for the unanesthetized mice. For voltage-clamped recordings, the intrapipette solution consisted of the following (in mm): 140 Cs-methanesulfonate, 5 HEPES, 10 TEA-Cl, 1 EGTA, 10 2Na-Phosphocreatine, and 1 Mg-ATP, pH 7.2, containing 0.2% biocytin. sEPSCs and sIPSCs were obtained by maintaining the membrane potential at −70 and 0 mV, respectively. For cell-attached recordings, the patch pipettes were filled with aCSF. The signals were amplified and digitized at a sampling rate of 20 kHz using a MultiClamp 700B amplifier and a Digidata 1440A digitizer that were controlled by pCLAMP 10.3 software (Molecular Devices). Data were analyzed offline using custom MATLAB (The MathWorks) routines. Experiments in which the series resistance exceeded 70 MΩ or changed by >15% during the entire recording session were discarded. Hippocampal pyramidal cells were electrophysiologically identified by their characteristic spiking pattern of high-frequency bursts (HFBs; 100–300 Hz, 3–6 spikes) (Kandel and Spencer, 1961; Ranck, 1973) and further confirmed by post hoc histological analysis. Data obtained from mice that received conditioning for >20 min under these criteria were used for analysis. Neurons that emitted HFBs at <0.033 Hz (twice per minute) during the preconditioning session were not used. Local field potentials (LFPs) were recorded from CA1 stratum pyramidale using borosilicate glass pipettes (1–2 MΩ) filled with aCSF. The signals were amplified (1000×) and digitized at 20 kHz. LFPs were band-pass filtered between 120 and 250 Hz and the root-mean-square power was calculated by sliding a 10 ms time window at a step interval of 10 ms. Successive time windows during which the ripple power continued to be >3–7× SD were defined as a single ripple epoch. Events shorter than 20 ms were discarded. The coincidence ratio was calculated by dividing the total number of events in which HFBs and ripples occurred simultaneously by the total number of events in which both or either of them occurred.
Figure 3.

Experimental conditions for sEPSCsupra reinforcement. A, Raw data (dots) and the boxplot represent the distribution of baseline frequencies of HFBs in control and MK801-treated mice during the period 0–5 min before the onset of conditioning. The initial frequencies did not differ between two groups. B, Time courses of the percent changes in the sEPSCsupra frequency in mice treated with MK801 (n = 5), mice with pseudorandom (yoked) conditioning (n = 5), and mice that received stimulation of the ventromedial (VM) thalamus (n = 5). Error bars indicate SEMs. C, Mean changes in the frequencies of sEPSCsupras (left) and sEPSCsubs (right) during the period between 15 and 20 min after the onset of conditioning. **p = 0.0091 versus preconditioning baseline, paired t test; #p = 0.021 versus reinforcement, Bonferroni's/Dunn's test. D, Time courses of the percent changes in the sEPSCsupra frequency of during the 25 min of conditioning in urethane-anesthetized mice. Error bars indicate the SEMs of five neurons. E, Mean changes in the sEPSCsupra frequency during the 15–20 min period after the onset of conditioning. F, Time courses of the percent changes in the sEPSCsupra frequency of layer II/III pyramidal cells in the somatosensory cortex during the 25 min of conditioning. Error bars indicate the SEMs of five neurons. G, Mean changes in the sEPSCsupra frequency during the 15–20 min after the onset of conditioning. ns, p > 0.05 versus preconditioning baseline of each group, paired t test.
Figure 5.

Selective reinforcement of HFBs in unanesthetized and anesthetized mice. A, Unit activity recorded from a representative neuron at −5, 0, and 20 min relative to the onset of conditioning. Colored bars indicate LH stimulation periods. B, Time courses of the percent changes in the frequency of HFB events during the 25 min of conditioning. Error bars indicate the SEMs of 30 neurons (reinforcement). Of 30 neurons, 21 exhibited significant changes in the HFB frequency. C, Time courses of the percent changes in the HFB frequency in mice treated with intraperitoneal MK801 (n = 5) and in yoked control mice (n = 9). D, Representative image of locally injected SR101 (red) counterstained by Hoechst 33342. E, Time courses of the percent changes in the frequency of HFBs during the 25 min of conditioning in mice injected with MK801 into the CA1 region. F, Mean changes in the HFB frequency during the period between 15 and 20 min after the onset of conditioning. **p = 8.2 × 10−7, ns, p > 0.05 versus preconditioning baseline, paired t test; #p = 0.032, ##p = 0.0014, ns, p > 0.05 versus reinforcement, Bonferroni's/Dunn's test. G, Time courses of the percent changes in the frequency of single spikes during the 25 min of conditioning. H, Mean changes in the single-spike frequency during the period between 15 and 20 min after the onset of conditioning. ns, p > 0.05 versus preconditioning baseline, paired t test. I, Time courses of the percent changes in the frequency of HFB events during the 25 min of conditioning in urethane-anesthetized mice. Error bars indicate the SEMs of six neurons. J, Mean changes in the HFB frequency during the period between 15 and 20 min after the onset of conditioning. ns, p > 0.05 versus preconditioning baseline, paired t test.
Real-time feedback delivery of lateral hypothalamus stimulation.
After a 5 min session of baseline recording, the distribution of synaptic charges was obtained using a custom-built MATLAB routine. First, synaptic events with amplitudes >7 pA were defined as sEPSCs. This program calculated the difference between the successive local maximums and minimums after local filtering; every event with a peak amplitude larger than 7 pA was regarded as a putative sEPSC (or sIPSC). The onset time, peak time, and offset time of the synaptic event at a threshold of 20% of the peak amplitude above baseline noise were then calculated. Any event with a rise time larger than its decay time was considered a false positive and was discarded from subsequent analyses. Second, the instantaneous amplitude of a synaptic event was integrated from the onset time to the offset time and was defined as a synaptic charge. This procedure was repeated for all detected sEPSCs (or sIPSCs) and the distribution of baseline synaptic charges (i.e., prior distribution) was obtained. Finally, the suprathreshold for detecting sEPSCsupras (or sIPSCsupras) was set as the top 1% of the distribution. This preset threshold was embedded in a LabVIEW (National Instruments) routine for triggering lateral hypothalamus (LH) stimulation. For cell-attached recordings, HFBs, defined herein as events that included more than two spikes at >100 Hz, were used as a preset criterion for triggering LH stimulation. During operant conditioning, neuronal activities were monitored at a rate of 20 kHz by a custom-made program designed in Linux or a Windows 7 environment. The preset criteria were detected in every 200 and 20 ms segment for voltage-clamped recordings and cell-attached recordings, respectively. When sEPSCsupras, sIPSCsupras, and HFBs were detected, TTL signals were emitted from a PCI-6024E data acquisition board (National Instruments) to a stimulator and electrical stimulation was immediately delivered to the LH (a reward-related region; AP −1.3 mm; LM −1.0 mm; DV: −5.0 mm) or the ventromedial thalamus (a reward-irrelevant region; AP −1.3 mm; LM −0.8 mm; DV: −3.7 mm). In yoked control experiments, LH stimulation was randomly delivered independently of the preset criteria. Each electrical stimulus consisted of 50 0.1-ms-duration pulses at 100 Hz. The event frequency of the targeted activities (sEPSCsupras, sIPSCsupras, HFBs, or ripples) was analyzed every 1 min. The frequency was normalized by the baseline mean value during the preconditioning period and their percent changes were expressed as the “%Δ” unit and plotted as a function of time.
Electromyogram.
Electromyogram (EMG) electrodes were implanted on the left forelimbs of the mice. EMG signals were amplified, filtered, and digitized at a sampling rate of 20 kHz. All processing was performed post hoc using a custom-made MATLAB routine. EMG signals were filtered at 0.01–2 kHz and resampled at 10 kHz. They were then rectified and smoothed using a Gaussian kernel (SD = 25 ms). The power spectrum of 15–300 Hz was calculated every 2 s using the average discrete Fourier transform coefficients.
Whisker movement measurement.
Whisker movements were monitored at 1000 Hz using a high-speed CMOS camera. A commercially available image analysis program running in the ImageJ environment was used to measure the whisker angle. Whiskers were considered to have moved when the whisker deflection was 2× SD. The majority of the whisking occurred spontaneously in the absence of overt external events. The whisker angle was integrated over 1 min to assess the whisker movement.
Forced swimming test.
Individual mice were forced to swim inside a vertical Plexiglas cylinder (inner ϕ = 12 cm). The water temperature was 22 ± 1°C, the depth was 15 cm, and the above-water wall height was 8 cm. The mice were kept in the water for 15 min until they spent 60% of their time in immobility. The immobility time was evaluated using a video-based automatic detection system. After mice were removed from the cylinder, they were dried and used for experiments. In acute-stressed mice, the operant conditioning was started 1–2 h after the forced swimming test for 15 min. In depression-model mice, the operant conditioning was started 1–2 h after the forced swimming test for 5 min on the second day following the same 15 min test on the first day. Saline or fluoxetine (20 mg/kg) was intraperitoneally injected 3 times 1, 19, and 23 h after the forced swimming test on the first day test. The water was changed for each mouse.
Histology.
After each experiment, the region of electrical stimulation was histologically confirmed. A direct current (50 μA for 20 s) was administered through the stimulating electrode to mark the position of the electrode tip. Mice were perfused transcardially with 4% paraformaldehyde. The brains were coronally sectioned at a thickness of 100 μm using a vibratome. The lesioned area was located in serial Nissl-stained sessions. Mice in which the intended region was confirmed to be stimulated were used in the data analyses. For biocytin reconstruction, sections were incubated with 0.3% H2O2 for 30 min and permeabilized with 0.2% Triton X-100 for 1 h. The sections were processed with ABC reagent (PK-6100; Vector Laboratories) containing 0.2% Triton X-100 at 4°C overnight and developed with 0.0003% H2O2, 0.02% diaminobenzidine, and 10 mm (NH4)2Ni(SO4)2. To estimate the spatial diffusion of locally applied MK801, we injected 0.1 mm sulforhodamine 101 (SR101), a fluorescent dye, simultaneously with MK801. After experiments, the animals were perfused transcardially with 0.01 m PBS and then 4% paraformaldehyde. The brains were removed carefully, postfixed overnight at 4°C in 4% paraformaldehyde, and coronally sectioned at a thickness of 100 μm using a DTK-1500 vibratome (Dosaka). The sections were incubated with 0.5 μg/ml Hoechst 33342 for 5 min. The dye-injected area was visualized using a macrozoom fluorescence microscope (MVX10 MacroView; Olympus).
Results
Reinforcement of synaptic activity
We used a closed-loop system in which ongoing activity of a patch-clamped neuron in a mouse was analyzed online at an update rate of 20 kHz and applied feedback stimulation to the brain of the same animal (Fig. 1A). In an awake, head-fixed mouse, a single dorsal hippocampal CA1 pyramidal neuron was voltage clamped at −70 mV to monitor sEPSCs (Fig. 1B). A tungsten-stimulating electrode was implanted in the ipsilateral LH, one of the reward-related regions of the brain (Margules and Olds, 1962). First, we collected sEPSCs for 5 min and obtained the “prior” distribution of the area above the curves of individual sEPSC events (sEPSC charges). We then defined the preset threshold as the top 1% of sEPSC charges in this prior distribution and started operant conditioning (Fig. 1C).
Figure 1.
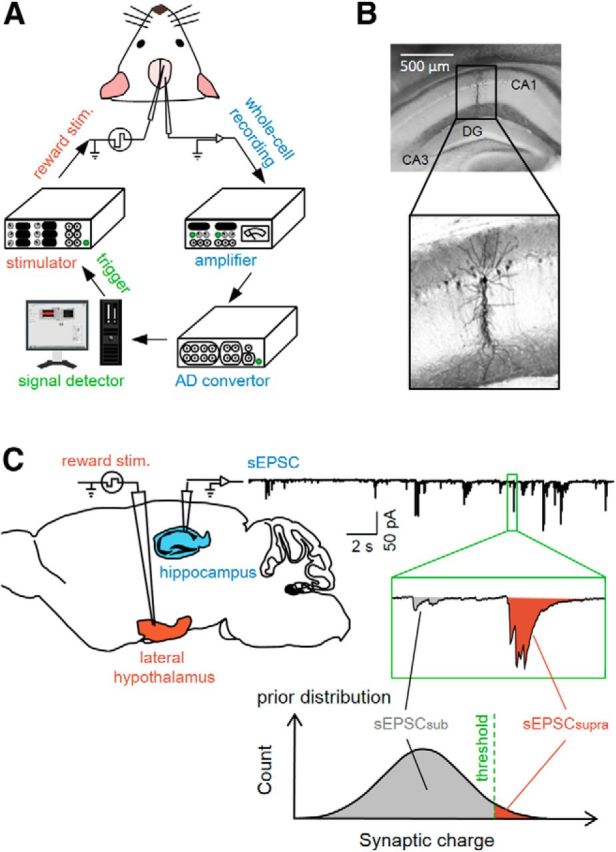
Experimental paradigm for a closed-loop neurofeedback system. A, Whole-cell recordings of sEPSCs were obtained from CA1 pyramidal cells in awake mice and were digitized at 20 kHz after amplification. The largest 1% of sEPSCs detected were used to trigger electrical reward stimulation. B, Representative biocytin reconstruction of a whole-cell-recorded CA1 neuron. C, The prior distribution of sEPSC charges was obtained during a baseline period of 5 min and the top 1% was defined as a preset threshold between sEPSCsupras and sEPSCsubs. Reward stimuli were applied to the LH immediately after the occurrence of individual sEPSCsupras.
During the conditioning session, we continued to monitor sEPSCs for at least 20 min (Fig. 2). When a sEPSC larger than the preset threshold (sEPSCsupra) was recorded, an electrical rewarding stimulus (50 pulses at 100 Hz; pulse width of 0.1 ms; pulse amplitude of 30–40 μA) was immediately delivered to the LH (Fig. 2A) in <50 ms delay. The mean frequency of sEPSCsupras was 0.19 ± 0.03 Hz during the preconditioning baseline period (mean ± SEM of 19 cells from 19 mice; Fig. 3A). The sEPSCsupra frequency started to increase ∼5 min after the onset of the conditioning and reached a steady state at an increased level of 126.6 ± 45.2% (mean ± SEM of 9 cells, p = 0.0091, t8 = 3.41, paired t test; Fig. 2B, closed circles, Fig. 3C, left). This increment was observed regardless of the initial sEPSCsupra frequency (Fig. 2C) and persisted for at least 10 min after termination of the LH stimulation (Fig. 2B). In a single whole-cell recording, we routinely measured the membrane properties during the preconditioning and postconditioning sessions and did not find that the neuron exhibited a change in the intrinsic excitability (data not shown), suggesting that the increase in sEPSCsupras was unlikely to be a result of the overall trend of neuronal excitation. Consistent with this idea, the overall mean frequency of subthreshold sEPSCs (sEPSCsubs) was not changed by the conditioning (Fig. 3C, right), although the top 10% of sEPSCsubs were reinforced (Fig. 2D). The conditioning did not alter the mean charges of sEPSCsupras (p = 0.38, t8 = 0.92, paired t test; Fig. 2B, open circles). In three trained mice, we also monitored the electromyogram power of their forelimbs and the movement of their whiskers, but we did not observe apparent changes in the body motion level during the conditioning (Fig. 2E). LH stimulation did not reinforce the sEPSCsupra frequency in urethane-anesthetized mice (p = 0.99, t4 = 0.013, paired t test, n = 5 cells; Fig. 3D,E).
Figure 2.

Neurofeedback-mediated reinforcement of sEPSCsupras in hippocampal neurons. A, sEPSC traces recorded from a representative neuron at −5, 0, and 20 min relative to the onset of conditioning. Colored bars indicate LH stimulation periods. B, Time courses of the percent changes in the mean frequency (closed circles) and the mean charge (open circles) of sEPSCsupras during the 25 min of conditioning. Error bars indicate the SEMs of 9 neurons from 9 mice; 6 of 9 neurons exhibited a significant change in the sEPSCsupra frequency. C, The sEPSCsupra frequency 15–20 min after the onset of LH stimulation was plotted against that measured 0–5 min before the reinforcement. Each data point represents the mean ± SEM of a single neuron. D, Percent changes in the frequency distribution of sEPSC charges during the postconditioning session relative to the preconditioning baseline. The sEPSC charges were rank ordered and normalized in each experiment. E, During operant conditioning, EMGs were recorded from forelimbs (top) and whisker movement was monitored (bottom). Neither the EMG power nor the whisker movement integral changed in the course of the development of sEPSCsupra reinforcement. Error bars indicate the SEMs of three mice.
To examine the involvement of NMDA receptors, which are known to be involved in the induction of synaptic plasticity in hippocampal excitatory transmission, we treated mice with intraperitoneal administration of 0.1 mg/kg MK801, an NMDA receptor antagonist, 15–30 min before conditioning. MK801 prevented neural operant conditioning (p = 0.53, t4 = 0.69, paired t test, n = 5 cells; p = 0.021, Bonferonni's/Dunn's test vs reinforcement; Fig. 3B,C, left), whereas MK801 per se did not alter the sEPSCsupra frequency during the preconditioning period (p = 0.92, t22 = 0.10, Student's t test; Fig. 3A).
As a control, we used a random trigger generator and arbitrarily applied LH stimulation regardless of the sEPSCsupra timing. In this yoked conditioning, the timing of LH stimulation was randomly generated at a mean frequency of 0.1 Hz, which was determined based on the mean stimulation frequency during the reinforcement experiments shown in Figure 2. The randomly timed rewarding electrical stimulation did not induce an increase in sEPSCsupra frequency (p = 0.48, t4 = 0.78, paired t test, n = 5 cells; Fig. 3B,C, left).
To examine whether sEPSCsupra reinforcement is driven by reward stimulation or by contingency of electrical stimulation of the brain, we applied contingent electrical stimulation to the ventromedial thalamus, a reward-unrelated thalamic nucleus that is located close to the LH and projects to the hippocampus (Su and Bentivoglio, 1990; Varela et al., 2013). Stimulation to the ventromedial thalamus was not shown to increase the sEPSCsupra frequency, at least during our 25 min observation period (p = 0.11, t4 = 2.04, paired t test, n = 5 cells; Fig. 3B,C, left). Therefore, the increased sEPSCsupras represent a contingent reward-driven, NMDA-receptor-dependent reinforcement of synaptic activity.
In layer II/III pyramidal cells of the primary somatosensory cortex, which is located immediately above the dorsal hippocampus, LH stimulation did not reinforce the sEPSCsupra frequency during our observation period (p = 0.65, t4 = 0.49, paired t test, n = 5 cells; Fig. 3F,G). Therefore, hippocampal neurons appeared to be more plastic than somatosensory cortical neurons.
Next, hippocampal CA1 pyramidal neurons were voltage clamped at 0 mV, the reversal potential of glutamatergic receptor-mediated currents, to monitor sIPSCs (Fig. 4). We applied a similar conditioning protocol to sIPSCs; the LH was stimulated with an IPSC larger than the top 1% of the prior distribution of sIPSC charges (sIPSCsupra) in the patched CA1 neurons (Fig. 4A). The mean frequency of sIPSCsupra before the conditioning was 0.14 ± 0.02 Hz (mean ± SEM of 5 cells from 5 mice). In the LH reinforcement and yoked control groups, neither the frequencies of sIPSCsupras nor those of sIPSCsubs changed (sIPSCsupra: p = 0.87, t4 = 0.18, paired t test, n = 5 cells for the reinforcement group; p = 0.79, t2 = 0.31, n = 3 cells for the yoked control; sIPSCsub: p = 0.12, t4 = 1.99, paired t test for the reinforcement group; p = 0.40, t2 = 1.05 for the yoked control; Fig. 4B,C).
Figure 4.

sIPSCsupras are not reinforced during the conditioning. A, sIPSC traces recorded from a representative neuron at −5, 0, and 20 min relative to the onset of conditioning. Colored bars indicate LH stimulation periods. B, Time courses of the percent changes in the frequency of sIPSCsupras during the conditioning. Error bars indicate the SEMs of five (reinforcement) and three neurons (yoked control). C, Mean changes in the frequencies of sIPSCsupras (left) and sEPSCsubs (right) during the period between 15 and 20 min after the onset of conditioning. ns, p > 0.05 versus preconditioning baseline of each group, paired t test.
Reinforcement of spiking activity
We sought to link the plasticity of sEPSCsupras to a change in spike output. We patch clamped CA1 neurons in the cell-attached configuration and monitored spontaneous spike series. The neurons exhibited two types of spiking patterns, single spikes and transient HFBs of spikes (Kandel and Spencer, 1961; Ranck, 1973). We defined an HFB as an event in which at least three spikes occurred at >100 Hz. The neurons that emitted HFBs at >0.033 Hz (twice per minute) during the preconditioning session were selected, so that mice were sufficiently given reward stimuli while the patch-clamp configuration was maintained. Based on this criterion, approximately 80% of the recorded cells were discarded. In the remaining datasets, HFBs occurred at a mean frequency of 0.12 ± 0.02 Hz (mean ± SEM of 39 cells from 39 mice; see Fig. 9B). We then delivered LH stimulation upon HFB occurrence (Fig. 5A). HFBs were reinforced in a time course similar to the sEPSCsupra conditioning (p = 8.2 × 10−7, t29 = 6.24, paired t test vs the preconditioning baseline, n = 30 cells; Fig. 5B,F), whereas the frequency of single spikes was not changed (p = 0.30, t29 = 1.07; Fig. 5G,H). The reinforcement did not occur in MK801-treated mice (p = 0.85, t4 = 0.20, paired t test, n = 5 cells; P = 0.032, Bonferroni's/Dunn's test vs reinforcement' Fig. 5C,F), yoked control mice (p = 0.34, t8 = 1.02, n = 9 cells; p = 0.0014, Bonferroni's/Dunn's test vs reinforcement; Fig. 5C,F), or urethane-anesthetized mice (p = 0.35, t5 = 1.02, n = 6 cells; Fig. 5I,J), even though MK801 did not alter the HFB frequency during the preconditioning period (0.15 ± 0.03 Hz, mean ± SEM; p = 0.70, Bonferroni's/Dunn's test vs reinforcement). To examine the involvement of NMDA receptors in the CA1 region, we injected 300 μm MK801 parenchymally into the CA1 region. The diffusion of MK801 was estimated by SR101 injection (Fig. 5D), because MK801 and SR101 are both water-soluble small molecules (MK801: 337 Da; SR101: 601 Da). After the pipette used for local MK801 injection was carefully pulled out from the brain, neurons in MK801-injected areas were patch clamped and the conditioning started within 30 min. Local application of MK801 attenuated the HFB reinforcement; MK801-treated neurons did not exhibit a significant increase in HFB frequency (p = 0.16, t7 = 1.59, paired t test vs baselines, n = 8 cells; p = 0.18, Bonferroni's/Dunn's test vs reinforcement; Fig. 5E,F), whereas local application of MK801 did not alter the HFB frequency during the preconditioning period (0.12 ± 0.04 Hz, mean ± SEM; p = 0.99, Bonferroni's/Dunn's test vs reinforcement).
Figure 9.

A lack of HFB reinforcement in stressed or depression-model mice. A, Time courses of the percent changes in the HFB frequency during the conditioning in SCH23390-treated mice (n = 10). B, Raw data (dots) and the boxplot represent the distribution of baseline frequencies of HFBs in control, SCH23390-treated, and acutely stressed mice during the period 0–5 min before the onset of conditioning. There were no statistical differences in the initial frequency across the groups versus reinforcement, Bonferroni's/Dunn's test. C, The percentage of time spent for immobility during every 1 min period (day 1) or during the total 5 min (day 2) in a water-filled cylinder in vehicle-treated and fluoxetine-treated mice. The horizontal line in the right panel indicates the mean value of the first 5 min in naive mice in the day 1 test. Fluoxetine was administered 3 times between the day 1 and day 2 tests. **p = 0.0003 versus vehicle group, Student's t test. D, Time courses of the percent changes in the HFB frequency during the conditioning in acutely stressed mice that experienced the day 1 test (n = 10). E, Time courses of the HFB frequency in mice treated with saline (n = 10) or fluoxetine (n = 8) after the day 2 test. F, Mean changes in the HFB frequency during the period between 15 and 20 min after the onset of conditioning in acutely stressed mice (day 1) and in stressed mice treated with vehicle or fluoxetine (day 2). #p = 0.021 versus vehicle group, Student's t test; *p = 0.028; ns, p > 0.05 versus preconditioning baseline of each group, paired t test.
To examine the circuit specificity of HFB reinforcement, we conducted cell-attached recordings simultaneously from two CA1 neurons that were located ∼200 μm apart from one another. The LH was stimulated upon HFBs in one neuron but not in the other neuron. The conditioned neuron was randomly assigned in each pair. The HFB frequency increased in the conditioned neurons (p = 0.042, t4 = 2.95, paired t test, n = 5 cells; Fig. 6), but not in the nonconditioned ones (p = 0.37, t4 = 1.02, paired t test, n = 5 cells; p = 0.015 for conditioned vs nonconditioned, t8 = 3.09, Student's t test), suggesting pathway specificity of the operant conditioning.
Figure 6.

Selective reinforcement of HFBs in a pathway-specific manner. A, Time courses of the HFB frequencies of two simultaneously recorded neurons during the conditioning of one neuron (n = 5 pairs). Error bars indicate the SEMs of five pairs. B, Mean changes in the HFB frequencies during the period between 15 and 20 min after the onset of conditioning. *p = 0.042; ns, p > 0.05 versus preconditioning baseline of each group, paired t test; #p = 0.015, Student's t test.
We also tested explicitly nonpaired controls; LH stimulation was applied when HFBs did not occur for a given period. The “endurance” period was set at 6.3 ± 1.2 s such that the initial frequency of LH stimulation was ∼0.1 Hz (Fig. 9B). With the nonpaired reinforcement, HFBs decreased within 15 min (p = 0.011, t5 = 3.92, paired t test, n = 6 cells; Fig. 7A,B) without a change in the frequency of single spikes (p = 0.96, t5 = 0.05; Fig. 7C,D).
Figure 7.
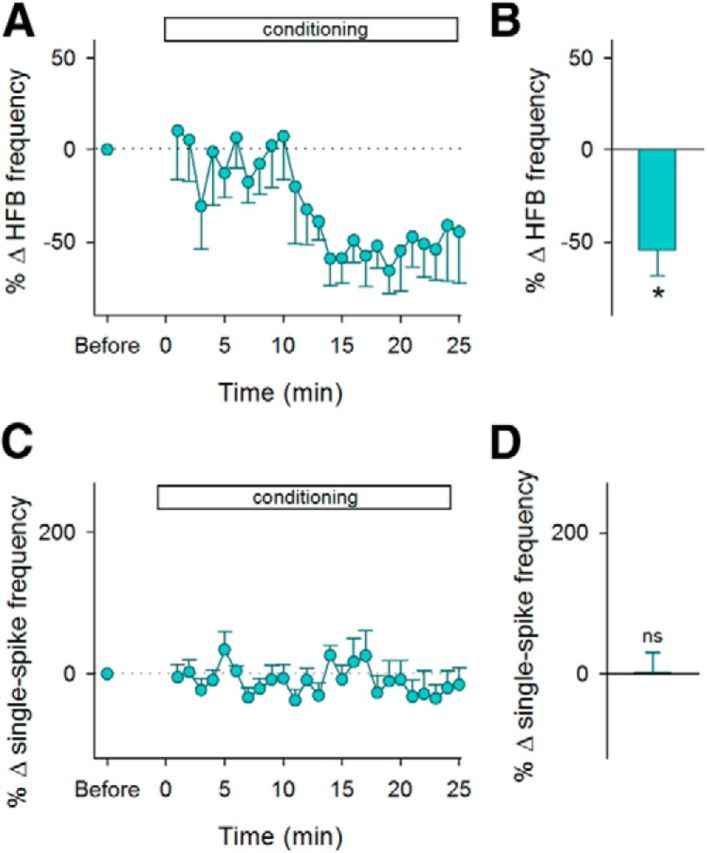
Reinforcement for the disappearance of HFBs. A, Time courses of the HFB frequency during the reinforcement for the disappearance of HFBs. Error bars indicate the SEMs of six neurons. B, Mean changes in the HFB frequency during the period between 15 and 20 min after the onset of conditioning. *p = 0.011 versus preconditioning baseline, paired t test. C, Time courses of the percent changes in the frequency of single spikes during the 25 min of conditioning. D, Mean changes in the single-spike frequency during the period between 15 and 20 min after the onset of conditioning. ns, p > 0.05 versus preconditioning baseline, paired t test.
Ripple reinforcement
To relate the HFB reinforcement to hippocampal network activity, we recorded LFPs from the CA1 stratum pyramidale and compared the timings of HFBs and ripple events, transient high-frequency oscillations in the hippocampus (Buzsáki et al., 1992) (Fig. 8A). During the preconditioning period, the event frequency of ripples was 0.20 ± 0.03 Hz (mean ± SEM of 5 mice) and was significantly higher than that of HFBs (p = 0.024, t4 = 3.55, paired t test, n = 5 mice). The coincidence ratio of HFBs and ripples was as low as 0.017 ± 0.003 (mean ± SEM of 5 mice). Nonetheless, HFBs and ripples were both reinforced when HFBs were conditioned regardless of ripple occurrence (p = 0.0017, t4 = 7.51, paired t test, n = 5 cells from 5 mice for HFBs; p = 0.0018, t4 = 7.41, paired t test, n = 5 mice for ripples; Fig. 8B,C). This concomitant reinforcement was not dependent on the initial coincidence ratios of HFBs and ripples (Fig. 8D). In 4 of 5 mice, the conditioning increased the coincidence ratios (Fig. 8E). We also found that ripple events per se could be reinforced: when ripple event timings were targeted for reward stimulation, the frequency of ripples started to increase ∼5–10 min after the onset of the conditioning and reached an increased level of 151.7 ± 54.3% after 20–25 min (p = 0.038, t5 = 2.79, paired t test, n = 6 mice; Fig. 8F).
Figure 8.

Effect of HFB conditioning on ripples. A, Representative traces of cell-attached unit recording and LFP recording. A ripple event is magnified in the inset. B, Time courses of the percent changes in the frequency of HFBs and ripples during the 25 min of conditioning to HFBs. Error bars indicate the SEMs of five neurons from five mice. C, Mean changes in the frequency of HFBs and ripples 15–20 min after the onset of conditioning. **p < 0.005 versus preconditioning baseline of each group, paired t test. D, The mean changes in the frequency of HFBs (closed circles) and ripples (open circles) 15–20 min after the conditioning were plotted against the coincidence ratios of HFBs and ripples 0–5 min before the reinforcement. Each data point represents the mean of a single neuron. Simultaneously recorded data are linked with vertical lines. E, The coincidence ratios 0–5 min before and 15–20 min after the reinforcement was plotted. Each symbol represents the mean of a single neuron and their means ± SEM are shown by lines. F, Time courses of the percent changes in the frequency of ripples during the 25 min of conditioning to ripples. Error bars are SEMs of six mice.
Effect of motivation on HFB reinforcement
The operant conditioning of HFBs did not occur in mice that received intraperitoneal administration of 50 μg/kg SCH23390, a selective dopamine D1 receptor antagonist, 15–30 min before the conditioning (p = 0.71, t9 = 0.50, paired t test, n = 10 cells; Fig. 9A), whereas SCH23390 did not alter the baseline frequency of HFBs before the conditioning (p = 0.18, Bonferroni's/Dunn's test vs reinforcement; Fig. 9B). Dopamine is believed to be associated with reward and internal motivation (Salamone and Correa, 2012). Therefore, to examine whether mice under conditions of depression and anxiety can acquire HFB conditioning, we conducted a forced swimming test, a behavioral despair model (Porsolt et al., 1977), to burden mice with acute stress. This acute stress is known to cause various aberrations of the morphology and electrophysiological properties of hippocampal neurons (Howland and Wang, 2008; Maras and Baram, 2012). Mice were placed in a water-filled cylinder for 15 min until they spent approximately 60% of their time in immobility (Fig. 9C, day 1). In these acutely stressed mice, the baseline frequency of HFBs was not different from that of control mice (p = 0.23, Bonferroni's/Dunn's test vs reinforcement; Fig. 9B), but LH stimulation did not reinforce the HFB frequency (p = 0.40, t9 = 0.89, paired t test, n = 10 cells; Fig. 9D,F).
As another series of experiments, the stressed mice were again placed in the same water-filled cylinder for 5 min on the next day. They spent 51.8 ± 3.6% of their time in immobility (mean ± SEM of 10 vehicle-treated mice; Fig. 9C). This immobility period was reduced to 29.5 ± 3.0% in stressed mice that had been repeatedly administered with 20 mg/kg fluoxetine between the day 1 and day 2 tests (mean ± SEM of 8 fluoxetine-treated mice; p = 0.0003, t16 = 4.62, Student's t test; Fig. 9C). After the day 2 test, the stressed mice failed to exhibit HFB reinforcement (p = 0.61, t9 = 0.52, paired t test, n = 10 cells; Fig. 9E,F); however, this depressive effect was rescued in fluoxetine-treated mice (p = 0.028, t7 = 2.77, paired t test, n = 8 cells, p = 0.021, t16 = 2.78, Student's t test; Fig. 9E,F). Neither day 2 forced swimming stress nor fluoxetine treatment changed the baseline HFB frequency during the preconditioning period (p = 0.67, Bonferroni's/Dunn's test for day 2 forced swimming stress group vs reinforcement; p = 1.00, Bonferroni's/Dunn's test for fluoxetine-treated group vs reinforcement; Fig. 9B).
Discussion
In this study, we demonstrated that some form of hippocampal neuronal activity could be rapidly and selectively reinforced by contingent rewards through a neurofeedback operant conditioning paradigm without any pretraining session. The findings include the following: (1) this reinforcement learning emerged as an NMDA receptor-dependent internal neural process without apparent body movement; (2) the reinforcement did not occur under anesthesia, in marked contrast to involuntary learning that can occur under anesthesia, such as fear conditioning (Weinberger et al., 1984; Rosenkranz and Grace, 2002); and (3) LH stimulation did not work as a motivational reward in mice under stressed or depressed conditions.
Previous studies on operant conditioning of single-cell activity have emphasized the firing rates of neocortical neurons in the motor cortex (Fetz, 1969; Fetz and Finocchio, 1975), prefrontal (Kobayashi et al., 2010; Schafer and Moore, 2011), and temporal cortex (Cerf et al., 2010) rather than hippocampal neurons in primates including humans. The subjects had to be trained or pretested in neural decoding for hours or days, combined with external reward and motor/perceptual modulation, before they could control cortical unit activity (Fetz, 2007). Therefore, our experimental approach was unique in the following ways: (1) naive, untrained mice were used, (2) the internal reward was applied electrophysiologically, (3) neurons of the hippocampus were conditioned, and (4) subthreshold synaptic activity was monitored. Under these novel conditions, we succeeded in reinforcing sEPSCs within 15 min. Because mice are useful in molecular biology and genetics studies, our work will help bridge the gap between a behavioral operant and the underlying molecular mechanisms.
Spike rates of pyramidal cells in CA1 are reported to conform to a lognormal distribution with the median HFB event rate being approximately 0.01 Hz (Mizuseki and Buzsáki, 2013). The firing rates of hippocampal neurons recorded in our experiments were higher than those reported previously. This difference is mainly because we did not use ∼80% neurons, which emitted HFBs at <0.033 Hz. It is also possible that cell-attached recordings per se affected the excitability of the recorded neurons (Alcami et al., 2012). Whether neurons with lower firing rates can also be reinforced remains to be addressed.
The reinforcement required NMDA receptor activity. NMDA receptors are known to be involved in the induction of synaptic plasticity in hippocampal excitatory transmission. Indeed, the HFB conditioning efficiency was reduced by local injection of an NMDA receptor antagonist into the CA1 region. Therefore, a simple explanation for the neural operant is that CA3-to-CA1 synapses in the recorded CA1 neurons exhibited long-term potentiation during the reinforcement. However, we do not believe that this scenario can completely account for our results, because local MK801 injection only partially prevented the HFB reinforcement. Moreover, the sEPSCsupra reinforcement increased their frequencies, but not their charges. This result suggests that the conditioned CA1 pyramidal neuron came to receive synaptic inputs more frequently from specific assemblies of CA3 presynaptic neurons, rather than to receive stronger synaptic inputs from these CA3 neurons or to recruit more numbers of CA3 neurons. This notion seems consistent with two observations: first, the sEPSCsupra or HFB conditioning did not alter the frequency of sEPSCsubs or single spikes and, second, the HFB conditioning of one neuron did not alter the HFB frequency of the unconditioned neuron in a simultaneously recorded neuron pair. We consider the involvement of NMDA receptors to be outside of the CA1 region (perhaps CA3), because the preventing effect of systemic MK801 on the HFB conditioning was stronger than that of local MK801. The reinforcement may emerge from CA3 network reorganization through NMDA-receptor-dependent synaptic plasticity so that specific activities of CA3 ensembles are more frequently sent to the targeted CA1 neurons. Another possible involvement of NMDA receptors is the modulation of dopaminergic neurons of the ventral tegmental area. Phasic firings of these neurons are impaired in mice lacking the NR1 subunit of NMDA receptors in the dopaminergic neurons (Zweifel et al., 2009), which projects to the hippocampus (Oades and Halliday, 1987). Therefore, NMDA receptor blockade may decrease the efficiency of the LH stimulation by altering the firing properties of ventral tegmental area neurons. To confirm this idea, local injection with SCH23390 is required because dopamine D1 receptors are widely expressed in both reward-related and reward-unrelated brain regions, including the ventral tegmental area, the medial prefrontal cortex, and the hippocampus.
Barrage activities of hippocampal CA1 neurons, such as sEPSCsupras and HFBs, originate in the CA3 network and are most likely to be associated with ripple (or sharp-wave) events in LFPs (Ylinen et al., 1995; Maier et al., 2011). One of the simplest mechanisms for the reinforcement of sEPSCsupras and HFBs is that animals learned merely to increase the ripple event frequency. However, this mechanism is unlikely. Although we found that the HFB reinforcement was accompanied by an increase of ripple events, the mean frequency of HFBs was lower than that of ripple events, which is consistent with previous reports showing that CA1 pyramidal cells sparsely discharge burst spikes during ripple oscillations (Ylinen et al., 1995; Csicsvari et al., 1999). Moreover, the coincidence ratio between HFBs and ripples was always <0.1. Interestingly, this ratio increased after the HFB conditioning. Therefore, there is no doubt that the conditioning of single neuron activity induced a change in local network activity. Conversely, our dual cell-attached recordings (n = 5) revealed that conditioning of one neuron did not affect the activity of the other neurons. In the recordings, HFBs were rarely coincident between two neurons. Therefore, these neurons were unlikely to be involved in the identical cell assemblies that constitute ripples. This may be a reason why the activities of two neurons could be modulated independently. Consistent with this idea, King et al. (1999) has reported that, when depolarizing currents are injected into a CA1 pyramidal cell simultaneously with spontaneous ripples, its synapses are selectively potentiated.
Inhibitory interneurons are also known to shape ripples/sharp waves (Klausberger et al., 2003), but we failed to reinforce sIPSCsupras during our experimental periods. This may be because inhibitory interneurons are less plastic than pyramidal cells and require additional time for reinforcement or because they are connected with pyramidal cells in a less selective manner (Kerlin et al., 2010; Fino and Yuste, 2011; Packer and Yuste, 2011) and cannot be reinforced by operant conditioning of a specific neural pathway. However, we do not rule out the involvement of inhibitory interneurons in the reinforcement. For example, CA3 axo-axonic cells are reported to reduce their firing rates by septum-driven inhibition during ripples and may trigger ripple events (Viney et al., 2013). It is possible that the modulation of their activity patterns contributes to the conditioning of CA1 neuronal activity.
Footnotes
This work was supported by Grants-in-Aid for Science Research on Innovative Areas, “Mesoscopic Neurocircuitry” (Grants 22115003 and 23115101) and “Mental Time” (Grant 25119004) from the Japan Society for the Promotion of Science through the Funding Program for Next Generation World-Leading Researchers (NEXT Program) initiated by the Council for Science and Technology Policy (LS023) and Strategic Research Program for Brain Sciences.
The authors declare no competing financial interests.
References
- Alcami P, Franconville R, Llano I, Marty A. Measuring the firing rate of high-resistance neurons with cell-attached recording. J Neurosci. 2012;32:3118–3130. doi: 10.1523/JNEUROSCI.5371-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basmajian JV. Control and training of individual motor units. Science. 1963;141:440–441. doi: 10.1126/science.141.3579.440. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Neural syntax: cell assemblies, synapsembles, and readers. Neuron. 2010;68:362–385. doi: 10.1016/j.neuron.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Leung LW, Vanderwolf CH. Cellular bases of hippocampal EEG in the behaving rat. Brain Res. 1983;287:139–171. doi: 10.1016/0165-0173(83)90037-1. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Horváth Z, Urioste R, Hetke J, Wise K. High-frequency network oscillation in the hippocampus. Science. 1992;256:1025–1027. doi: 10.1126/science.1589772. [DOI] [PubMed] [Google Scholar]
- Cerf M, Thiruvengadam N, Mormann F, Kraskov A, Quiroga RQ, Koch C, Fried I. On-line, voluntary control of human temporal lobe neurons. Nature. 2010;467:1104–1108. doi: 10.1038/nature09510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurkó A, Mamiya A, Buzsáki G. Fast network oscillations in the hippocampal CA1 region of the behaving rat. J Neurosci. 1999;19:RC20. doi: 10.1523/JNEUROSCI.19-16-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domjan M, Grau JW. The principles of learning and behavior. Ed 5. Australia, California: Thomson/Wadsworth; 2003. [Google Scholar]
- Fetz EE. Operant conditioning of cortical unit activity. Science. 1969;163:955–958. doi: 10.1126/science.163.3870.955. [DOI] [PubMed] [Google Scholar]
- Fetz EE. Volitional control of neural activity: implications for brain-computer interfaces. J Physiol. 2007;579:571–579. doi: 10.1113/jphysiol.2006.127142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetz EE, Finocchio DV. Correlations between activity of motor cortex cells and arm muscles during operantly conditioned response patterns. Exp Brain Res. 1975;23:217–240. doi: 10.1007/BF00239736. [DOI] [PubMed] [Google Scholar]
- Fino E, Yuste R. Dense inhibitory connectivity in neocortex. Neuron. 2011;69:1188–1203. doi: 10.1016/j.neuron.2011.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, Csicsvari J, Hirase H, Dragoi G, Buzsáki G. Organization of cell assemblies in the hippocampus. Nature. 2003;424:552–556. doi: 10.1038/nature01834. [DOI] [PubMed] [Google Scholar]
- Hebb DO. The organization of behavior; a neuropsychological theory. New York: Wiley; 1949. [Google Scholar]
- Howland JG, Wang YT. Synaptic plasticity in learning and memory: stress effects in the hippocampus. Prog Brain Res. 2008;169:145–158. doi: 10.1016/S0079-6123(07)00008-8. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Spencer WA. Electrophysiology of hippocampal neurons. II. After-potentials and repetitive firing. J Neurophysiol. 1961;24:243–259. doi: 10.1152/jn.1961.24.3.243. [DOI] [PubMed] [Google Scholar]
- Kerlin AM, Andermann ML, Berezovskii VK, Reid RC. Broadly tuned response properties of diverse inhibitory neuron subtypes in mouse visual cortex. Neuron. 2010;67:858–871. doi: 10.1016/j.neuron.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C, Henze DA, Leinekugel X, Buzsáki G. Hebbian modification of a hippocampal population pattern in the rat. J Physiol. 1999;521:159–167. doi: 10.1111/j.1469-7793.1999.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Magill PJ, Márton LF, Roberts JD, Cobden PM, Buzsáki G, Somogyi P. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature. 2003;421:844–848. doi: 10.1038/nature01374. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Schultz W, Sakagami M. Operant conditioning of primate prefrontal neurons. J Neurophysiol. 2010;103:1843–1855. doi: 10.1152/jn.00173.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AK, Wilson MA. Memory of sequential experience in the hippocampus during slow wave sleep. Neuron. 2002;36:1183–1194. doi: 10.1016/S0896-6273(02)01096-6. [DOI] [PubMed] [Google Scholar]
- Maier N, Tejero-Cantero A, Dorrn AL, Winterer J, Beed PS, Morris G, Kempter R, Poulet JF, Leibold C, Schmitz D. Coherent phasic excitation during hippocampal ripples. Neuron. 2011;72:137–152. doi: 10.1016/j.neuron.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Maras PM, Baram TZ. Sculpting the hippocampus from within: stress, spines, and CRH. Trends Neurosci. 2012;35:315–324. doi: 10.1016/j.tins.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margules DL, Olds J. Identical “feeding” and “rewarding” systems in the lateral hypothalamus of rats. Science. 1962;135:374–375. doi: 10.1126/science.135.3501.374. [DOI] [PubMed] [Google Scholar]
- Mizuseki K, Buzsáki G. Preconfigured, skewed distribution of firing rates in the hippocampus and entorhinal cortex. Cell reports. 2013;4:1010–1021. doi: 10.1016/j.celrep.2013.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oades RD, Halliday GM. Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res. 1987;434:117–165. doi: 10.1016/0165-0173(87)90011-7. [DOI] [PubMed] [Google Scholar]
- Packer AM, Yuste R. Dense, unspecific connectivity of neocortical parvalbumin-positive interneurons: a canonical microcircuit for inhibition? J Neurosci. 2011;31:13260–13271. doi: 10.1523/JNEUROSCI.3131-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- Ranck JB., Jr Studies on single neurons in dorsal hippocampal formation and septum in unrestrained rats. I. Behavioral correlates and firing repertoires. Exp Neurol. 1973;41:461–531. doi: 10.1016/0014-4886(73)90290-2. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA. Dopamine-mediated modulation of odour-evoked amygdala potentials during Pavlovian conditioning. Nature. 2002;417:282–287. doi: 10.1038/417282a. [DOI] [PubMed] [Google Scholar]
- Sakurai Y, Takahashi S. Conditioned enhancement of firing rates and synchrony of hippocampal neurons and firing rates of motor cortical neurons in rats. Eur J Neurosci. 2013;37:623–639. doi: 10.1111/ejn.12070. [DOI] [PubMed] [Google Scholar]
- Salamone JD, Correa M. The mysterious motivational functions of mesolimbic dopamine. Neuron. 2012;76:470–485. doi: 10.1016/j.neuron.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer RJ, Moore T. Selective attention from voluntary control of neurons in prefrontal cortex. Science. 2011;332:1568–1571. doi: 10.1126/science.1199892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su HS, Bentivoglio M. Thalamic midline cell populations projecting to the nucleus accumbens, amygdala, and hippocampus in the rat. J Comp Neurol. 1990;297:582–593. doi: 10.1002/cne.902970410. [DOI] [PubMed] [Google Scholar]
- Varela C, Kumar S, Yang JY, Wilson MA. Anatomical substrates for direct interactions between hippocampus, medial prefrontal cortex, and the thalamic nucleus reuniens. Brain Struct Funct. 2013 doi: 10.1007/s00429-013-0543-5. doi: 10.1007/s00429-013-0543-5. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viney TJ, Lasztoczi B, Katona L, Crump MG, Tukker JJ, Klausberger T, Somogyi P. Network state-dependent inhibition of identified hippocampal CA3 axo-axonic cells in vivo. Nat Neurosci. 2013;16:1802–1811. doi: 10.1038/nn.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger NM, Gold PE, Sternberg DB. Epinephrine enables Pavlovian fear conditioning under anesthesia. Science. 1984;223:605–607. doi: 10.1126/science.6695173. [DOI] [PubMed] [Google Scholar]
- Wilson MA, McNaughton BL. Reactivation of hippocampal ensemble memories during sleep. Science. 1994;265:676–679. doi: 10.1126/science.8036517. [DOI] [PubMed] [Google Scholar]
- Ylinen A, Bragin A, Nádasdy Z, Jandó G, Szabó I, Sik A, Buzsáki G. Sharp wave-associated high-frequency oscillation (200 Hz) in the intact hippocampus: network and intracellular mechanisms. J Neurosci. 1995;15:30–46. doi: 10.1523/JNEUROSCI.15-01-00030.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA, Phillips PE, Palmiter RD. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci U S A. 2009;106:7281–7288. doi: 10.1073/pnas.0813415106. [DOI] [PMC free article] [PubMed] [Google Scholar]