

Abstract
The intestinal microbiota has several effects on host physiology. Previous work from our laboratory demonstrated that the microbiota influences systemic iron homeostasis in mouse colitis models by altering inflammation-induced expression of the iron-regulating hormone hepcidin. In the present study, we examined the impact of the gut commensal bacterium Bacteroides fragilis on the expression of the iron exporter ferroportin, the target of hepcidin action, in macrophages, the cell type that plays a pivotal role in iron recycling. Mouse bone marrow-derived macrophages were exposed to B. fragilis and were analyzed by quantitative RT-PCR and western blotting. We found that B. fragilis down-regulated ferroportin transcription independently of bacterial viability. Medium conditioned by the bacteria also reduced ferroportin expression, indicating the involvement of soluble factors, possibly Toll-like receptor ligands. Consistent with this idea, several of these ligands were able to down-regulate ferroportin. The B. fragilis-induced decrease in ferroportin was functionally important since it produced a significant increase in intracellular iron concentrations that prevented the effects of the iron chelator deferoxamine on Salmonella-induced IL-6 and IL-1β production. Our results thus reveal that B. fragilis can influence macrophage iron handling and inflammatory responses by modulating ferroportin expression.
Keywords: macrophage, iron metabolism, microbiome, Bacteroides fragilis, metal homeostasis
Summary sentence:
Soluble molecules released by Bacteroides fragilis decrease transcription of the macrophage iron exporter ferroportin, thereby increasing intracellular iron concentrations and altering inflammatory function.
Graphical ABSTRACT
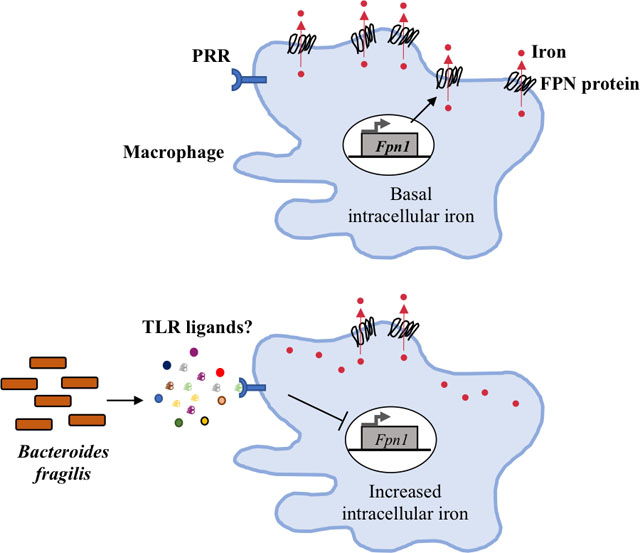
Soluble molecules released by Bacteroides fragilis, possibly redundantly-acting Toll-like receptor (TLR) ligands, down-regulate transcription of the macrophage iron exporter ferroportin (FPN), with a consequent increase in intracellular iron concentration.
INTRODUCTION
Iron, with its ability to undergo oxidation/reduction reactions, is critical for cell survival, growth and maturation. It is required in oxygen transport and as a cofactor for proteins that play roles in a number of basic cellular processes including metabolism, development, the immune response and cognition (1,2). Iron can be toxic when present in excess or under certain pathologic circumstances because of its ability to participate in the generation of free radicals (3). Thus, its levels in biological fluids are under stringent regulation. Disorders of iron homeostasis can lead to clinical problems such as anemia or hemochromatosis that affect over a billion people worldwide (4,5). Elucidation of the mechanisms that regulate iron metabolism will provide insights into the pathogenesis of these conditions and may aid in the development of ways to prevent or treat them.
Mammalian iron metabolism consists of uptake, storage and transport (6). The enterocytes of the duodenum are the primary sites of uptake of dietary iron (1–2 mg/day). Most of the body’s requirement (~20 mg/day) however is met by recycling of iron from aged or damaged erythrocytes by resident macrophages of the spleen, liver and bone marrow (7). In both cases, the dietary or recycled iron is transported into the bloodstream via ferroportin (FPN, SLC40A1), a multi-pass plasma membrane protein that is the sole exporter of iron. Iron released into the blood is transported to other cells bound to a plasma protein called transferrin (Tf) and is taken up via receptor mediated endocytosis through Tf receptor 1 (TfR1). Tf-bound iron is released in endosomes and transported into the cytosol via the transporter Nramp2 (DMT-1) to add to the cytosolic labile iron pool (LIP) for utilization by the cell. Excess iron is stored in a cage-like structure made up of dimers of ferritin heavy and light chain proteins or may also be exported via FPN.
Export of iron into the circulation by FPN is rigorously regulated in order to maintain iron homeostasis (8). This regulation is achieved by modulating FPN expression at multiple levels. A major mechanism involves the hepatocyte-derived hormone hepcidin, the expression of which is regulated in response to changes in systemic iron status and requirements, and which acts post-translationally by binding to FPN and inducing its degradation (9–12). In addition, intracellular iron, in the form of the LIP, controls the translation of FPN mRNA by influencing the binding of iron regulatory proteins (IRPs) to the iron responsive element (IRE) in the 5’ untranslated region (13,14). Transcriptional regulation of FPN expression has also been described. The FPN promoter contains several regulatory elements that are able to bind transcription factors responsive to oxidative stress, hypoxia, and metals such as zinc and cadmium (15–17). FPN can also be down-regulated at the transcriptional level in macrophages by lipopolysaccharide (LPS) and by pro-inflammatory cytokines such as interferon γ but the mechanisms involved are not well understood (18–20).
The gastrointestinal tract harbors a resident community of microorganisms, including bacteria, viruses, fungi and protozoa, that is referred to as the microbiota. This microbial community is present in a state of dynamic, two-way interaction with the host and has profound effects on immunity, physiology and metabolism (21,22). Bacteria represent an important component of the intestinal microbiota, with members of Bacteroidetes being the most prominent Gram-negative bacteria (23,24). Several studies have shown that Bacteroidetes are important for the development of the gastrointestinal tract and the immune system, as well as for protection against colonization by pathogens (24). Previous work from our lab has demonstrated that the commensal microbiota impacts iron metabolism in mouse models of inflammatory bowel disease (IBD) by influencing hepcidin expression (25). The goal of the present study was to test the hypothesis that commensal bacteria can affect iron metabolism by directly altering macrophage FPN expression.
MATERIALS AND METHODS
Cell culture
Primary murine bone marrow derived macrophages (BMDMs) from wild-type C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME, USA), as well as from mice deficient in TLR2 (kindly provided by Dr. Nitya Jain, Massachusetts General Hospital, Boston, MA, USA), were prepared and cultured in 24-well tissue culture plates (2–4 × 106/well) as described previously (26,27). The cells were washed and placed in antibiotic-free Dulbecco’s Modified Eagle medium (DMEM, Invitrogen, Waltham, MA, USA) with 10% heat-inactivated fetal bovine serum (FBS, Genesee Scientific, San Diego, CA, USA) prior to experiments.
Anaerobic culturing and processing of commensal bacteria
Bacteroides fragilis (strain NCTC 9343), Bifidobacter infantis, and Lactobacillus plantarum were obtained originally from the American Type Culture Collection and were provided by Dr. Deepak Vijaykumar, Massachusetts General Hospital. The bacteria were cultured anaerobically at 37°C in an anaerobic chamber containing 5% carbon dioxide, 5% hydrogen and 90% nitrogen for 20–22 hours in appropriate medium: liquid brain heart infusion (BHI, Becton-Dickinson, Franklin Lakes, NJ, USA) medium for B. fragilis and De Mann Rogosa Shapre broth for B. infantis and L. plantarum. Stationary phase cultures were pelleted at 3500 g for 15 mins at 4°C and resuspended in phosphate-buffered saline (PBS, Invitrogen, Waltham, MA, USA) at a density of 2.5 × 108 colony forming units (cfu)/mL. The bacteria were either used directly as live organisms or heat-killed by boiling in a water bath for 10 minutes. B. fragilis conditioned medium (BCM) was prepared by adding bacterial cells (live or heat-killed) corresponding to 5 × 107 cfu/mL to serum-containing, antibiotic-free DMEM and incubating at 37°C for 1 hour. The bacteria were subsequently pelleted and the supernatant passed through a 0.22 micron filter to generate BCM.
Infection of BMDM with B. fragilis and treatment with BCM
The BMDMs were exposed to live or heat-killed B. fragilis corresponding to 2.5 × 107 cfu per well of a 24-well plate for one hour at 37°C and 5% CO2. The bacteria were then washed off with PBS and the cells were incubated in antibiotic-free, serum-containing DMEM overnight. Similarly, cells were incubated overnight with BCM prepared as indicated above. The following day, the supernatants were collected for analysis and the cells washed in PBS followed by lysis in TRIzol reagent (Life Technologies, Grand Island, NY, USA) for RNA isolation, or in hypotonic or RIPA lysis buffer (Sigma-Aldrich, St Louis, MO, USA) containing HALT protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, USA) for western blotting.
RNA isolation and quantitative real-time PCR
Total RNA was isolated using the TRIzol extraction method according to manufacturer’s recommendations, then subsequently treated with DNase. One to 2 micrograms of DNase treated-RNA were used to synthesize first strand cDNA after priming with random hexamers (Invitrogen, Waltham, MA, USA) using MuLV reverse transcriptase (Life Technologies, Grand Island, NY, USA) or using a cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). Quantitative real-time-PCR (qRT-PCR) was carried out as described previously (28) using SYBR Green master mix (Bio-Rad, Hercules, CA, USA) and transcript-specific primers that have been described previously (25,27,29–32). Levels of the mRNA of interest were normalized to the levels of the mRNA for 36B4 (acidic ribosomal phosphoprotein P0) in the same sample. Relative expression was calculated as the difference (ΔCt) between the Ct (threshold cycle) values of the target gene and of 36B4, and expressed as 2−ΔCt.
Actinomycin D treatment and measurement of FPN mRNA stability
Macrophages were exposed to BCM in the presence or absence of actinomycin D (1 μg/mL) for 2, 4 and 6 hrs. FPN expression was determined by qRT-PCR and plotted against time on a semi-logarithmic plot to obtain the slope of the line that best fitted the data, and which was used to calculate half-life (33).
Preparation of BMDM membrane fraction
Equivalent numbers of BMDMs were exposed to BCM as described above or left untreated as a control, following which the cells were washed in ice-cold PBS and incubated on ice for 10 minutes with hypotonic lysis buffer (3 mM Tris-HCl pH 8.0, 1 mM magnesium chloride and 1 mM sodium chloride) containing HALT protease inhibitor cocktail. The cells were then disrupted with a Dounce homogenizer. The homogenates were centrifuged at 2000 g for 10 minutes at 4°C to remove nuclei and unbroken cells. The post-nuclear supernatants were centrifuged at 100,000 g for 1 hour at 4°C to pellet the membrane fraction. The membrane pellets were solubilized directly in equal volumes of 1X Laemmli sample buffer.
Western blotting
Immunoblotting was carried out as previously described (27). The membrane was blocked and probed with antibodies to ferritin heavy chain (Cell Signaling Technology, Danvers, MA, USA), FPN (Alpha Diagnostics, San Antonio, TX, USA), sodium-potassium ATPase (Novus Biologicals, Littleton, CO, USA) or β-actin (Sigma-Aldrich, St Louis, MO, USA). The blots were developed with fluorescently-tagged anti-rabbit or anti-mouse secondary antibodies as appropriate and the signals were visualized with an Odyssey infra-red fluorescence imaging system (Li-Cor Biosciences, Lincoln, NE, USA).
Measurement of intracellular iron
The iron-sensitive fluorescent dye calcein acetoxymethyl ester (Calcein-AM, Molecular Probes, Eugene, OR, USA) was used to measure intracellular labile iron as described previously (31). Briefly, BMDMs that had either been treated or not with BCM overnight, were stained with 0.5 μM calcein for 15 minutes at 37°C followed by three washes with PBS. Cellular fluorescence was assessed with a Life Technologies Attune NxT flow cytometer and the results were analyzed using CellQuest software, with a decrease in calcein fluorescence indicative of a relative increase in intracellular labile iron.
Infection of BMDMs with Salmonella
BMDMs were plated and differentiated as described above. They were either pre-treated or not with BCM, 100 μM deferoxamine, or BCM plus deferoxamine, overnight. The following day the macrophages were infected with the wild-type Salmonella enterica serovar Typhimurium strain SL1344 in the presence of the corresponding pre-treatment reagents using the protocol described previously (34). Briefly, S. Typhimurium was cultivated aerobically overnight and used to infect macrophages at a multiplicity of infection of 10:1 for one hour at 37°C. Unbound bacteria were washed off and extracellular bacteria killed using 100 μg/mL of gentamicin for one hour followed by 10 μg/mL of gentamicin for the remaining incubation period. The infection was allowed to proceed for a total of 7 hours at which time supernatants were collected from each well for cytokine measurement by ELISA while the cells were washed in PBS and lysed in 1% TritonX-100, diluted and plated to ascertain intracellular bacterial counts.
IL-6 and IL-1β ELISAs
IL-6 and IL-1β levels in culture supernatants were measured as previously described using kits specific for the mouse cytokines (BD Pharmingen, San Diego, CA, USA and eBioscience, San Diego, CA, USA, respectively) (27).
Statistical analysis
Data has been shown as mean ± standard deviation (SD) of at least three independent biological replicates. For comparisons involving two groups, statistical analysis was carried out by two-tailed, unpaired student’s t test and for comparisons involving multiple groups, one-way ANOVA analysis followed by post-hoc tests were employed using SigmaPlot version 11. A p value < 0.05 was considered significant. The test used and the corresponding n and p values are specified in the legend of each figure.
RESULTS
Bacteroides fragilis down-regulates FPN in macrophages
Macrophages in the spleen, liver and other tissues play a central role in iron metabolism by virtue of their ability to recover iron from phagocytosed senescent erythrocytes and then export the metal via FPN (7). Changes in macrophage FPN expression thus influence how much iron enters the circulation from the recycling of red blood cells, a process that constitutes the major source of the body’s daily iron requirements (7,35,36). Macrophages in the gut and systemic tissues are exposed to soluble products of the microbiota (ligands for pattern recognition receptors, metabolites, other molecules) that are translocated across the intestinal epithelium under basal conditions and in the context of intestinal inflammation (21,22). In light of this exposure to microbiota-derived molecules, we wished to determine if commensal bacteria or their products might influence FPN expression on macrophages and thus alter iron handling. Although no single macrophage type is completely representative of all tissue macrophages, BMDMs are widely used in the field because they are easy to prepare in large numbers and have phenotypic and functional characteristics that reflect those seen in macrophage populations in vivo (26). Accordingly, we used these cells in our experiments.
We exposed BMDMs to Bacteroides fragilis, a Gram-negative commensal bacterium that is representative of the genus Bacteroides, an important component of the adult gut microbiota (24), at a multiplicity of ~10:1 and assessed FPN expression by qRT-PCR. We used a relatively low multiplicity of infection in order to strike a balance between mimicking the in vivo situation (where B. fragilis is only one of many organisms that macrophages are exposed to) and the need to use sufficient bacteria to obtain consistent results. The infection resulted in a significant down-regulation of FPN mRNA (Fig. 1A). Similar results were obtained when an equivalent number of heat-killed bacteria was used (Fig. 1B), indicating that bacterial viability was not required. The exposure to B. fragilis did not affect the viability of the macrophages as indicated by lactate dehydrogenase release (data not shown). Conditioned medium of live or heat-killed B. fragilis (BCM) also induced down-regulation of FPN mRNA in the macrophages (Fig. 1C, results with BCM from killed bacteria), indicating that a soluble molecule released from the bacteria was able to mediate the effect. The decrease in FPN mRNA was associated with a decrease in FPN protein detected in the membrane fraction of BMDMs treated with BCM (Fig. 2A). This decrease in FPN protein was likely to be mediated independently of hepcidin since there was no change in hepcidin mRNA expression in BMDMs exposed to BCM (Fig. 2B).
Fig. 1.

Bacteroides fragilis down-regulates FPN mRNA in bone marrow-derived macrophages. BMDMs were exposed to live (A) or heat-killed (B) B. fragilis for one hour followed by washing and incubation overnight at 37°C. Bacteroides conditioned medium (BCM) was prepared as described in the text and applied to differentiated macrophages overnight (C). FPN expression was assessed by qRT-PCR. Results are shown as mean ± S.D. from 7, 3 and 6 independent replicates per group respectively. *(A) p = 0.0044, (B) p = 0.013, (C) p =0.0185.
Fig. 2.

Bacteroides fragilis down-regulate FPN protein independently of hepcidin. (A) Control or BCM-treated BMDMs were processed for isolation of membrane fractions and then probed for FPN via western blotting. Sodium-potassium ATPase was used for normalization. The western blot is representative of three independent experiments. (B) BMDMs exposed to BCM overnight were analyzed for mRNA expression levels of hepcidin by qRT-PCR. Results are shown as mean ± S.D. from 5 independent replicates.
In order to understand if the down-regulation of FPN was caused by changes in transcription or mRNA stability, we analyzed FPN heterogeneous nuclear RNA (hnRNA), which represents the precursor to the mature transcript and reflects transcriptional activity (32). We found that when macrophages were exposed to BCM, a significant reduction in FPN hnRNA was observed, suggesting that the down-regulation was at the level of transcription (Fig. 3). We also measured the stability of the FPN mRNA following actinomycin D treatment and found that exposure to B. fragilis did not lead to a significant change in this parameter (FPN mRNA half-life in the presence of ActD – 4.35 ±1.12 hrs., half-life in the presence of ActD and B. fragilis – 4.24±2.2 hrs.).
Fig. 3.

BCM down-regulates ferroportin transcriptionally. BMDMs were exposed to BCM overnight. FPN hnRNA expression was assessed by qRT-PCR. Results are shown as mean ± S.D. from 7 independent replicates. * p = 0.02468.
Other intestinal bacteria also modulate macrophage FPN expression
We next assessed the ability of other intestinal bacteria to modulate FPN by exposing BMDMs to heat-killed Bifidobacter infantis, Lactobacillus plantarum, and Citrobacter rodentium. The commensals B. infantis and L. plantarum down-regulated FPN mRNA whereas the enteropathogen C. rodentium significantly increased it (Figs. 4A, 4B). B. infantis and L. plantarum are Gram positive bacteria and are not expected to activate TLR4, the receptor for LPS (37). B. fragilis is Gram negative, but it expresses an atypical LPS (i.e., it differs from the more typical enterobacterial LPS in being penta-acylated rather than hexa-acylated) that is believed to signal through TLR2 rather than TLR4 (38). C. rodentium expresses a typical LPS that activates TLR4 (39). In addition, studies by Guida et al. have implicated the TLR2/TLR6 pathway in the hepcidin-independent down-regulation of FPN (40). Given these observations, we considered the possibility that B. fragilis-induced down-regulation of FPN involved signaling through TLR2. However, BCM was able to induce a significant decrease in FPN mRNA in BMDMs from TLR2 knockout mice (Fig. 5A), indicating that the receptor was not essential for this process. It is likely that BCM consists of a complex mix of microbial molecules that may act redundantly via multiple pattern recognition receptors to decrease FPN expression. Consistent with this idea, we found that several TLR ligands – LPS (acting on TLR4), polyinosinic:polycytidylic acid (PolyI:C, acting on TLR3), and CpG oligodeoxynucleotide (ODN-1, acting on TLR9) – were able, individually, to decrease FPN expression in BMDMs (Fig. 5B).
Fig. 4.

Modulation of FPN mRNA by other intestinal bacteria. BMDMs were exposed overnight to the Gram-positive commensal bacteria B. infantis (A) or L. plantarum (B), or the Gram-negative intestinal pathogen Citrobacter rodentium (B). FPN expression was assessed by qRT-PCR. Results are shown as mean ± S.D. from at least 5 independent replicates. * p = 0.043.
Fig. 5.

Multiple TLR ligands can induce down-regulation of FPN mRNA. (A) TLR2 knockout BMDMs were exposed to BCM overnight. FPN expression was assessed by qRT-PCR. Results are shown as mean ± S.D. from 11 independent replicates. * p = 0.0008. (B) Wild-type BMDMs were exposed overnight to the indicated TLR ligands. FPN expression was assessed by qRT-PCR. Results are shown as mean ± S.D. from at least 3 independent replicates. . One way ANOVA test was performed on ranks. *p=0.022.
B. fragilis increases intracellular iron concentration in macrophages
Since FPN is the sole iron exporter, a reduction in its levels is likely to increase the intracellular iron pool. BMDMs exposed or not to BCM overnight were stained with the iron-sensitive fluorescent dye calcein-AM and the fluorescence measured by flow cytometry. As shown in Fig. 6A, BMDMs treated with BCM had appreciably lower calcein fluorescence than the control BMDMs. Since calcein fluorescence is quenched by free iron – preferentially by Fe3+ (41) – these results suggest that BCM treatment increased intracellular iron concentrations, which is consistent with the BCM-induced decrease in FPN expression. Given the formal possibility that the quenching of calcein fluorescence could reflect a shift in the relative concentrations of Fe2+ and Fe3+ rather than an increase in total free iron, we carried out additional assays to substantiate this result. For this purpose, we made use of well-documented observations indicating that an elevation in intracellular iron triggers compensatory mechanisms that lead to an increase in ferritin protein (promoting iron storage) and a decrease in transferrin receptor TfR1 mRNA (reducing iron import) (42). We found that macrophages exposed to BCM overnight showed a small but significant decrease in TfR1 mRNA (Fig 6B) and a clear increase in the ferritin heavy chain protein (FTH1, Fig. 6C). Although measurements of calcein fluorescence, TfR1 mRNA and ferritin protein are indirect indicators of intracellular iron, each with its own limitations, when the results of our analyses of these paramenters are taken together, they strongly suggest that the B. fragilis-induced down-regulation of FPN expression has the expected functional consequence of increasing the intracellular iron concentration. However, it must be noted that direct assessment of the effects of B. fragilis on FPN transport activity will require quantitation of iron uptake.
Fig. 6.

Bacteroides fragilis increases intracellular iron concentration within macrophages. (A) BMDMs were exposed to medium (Ctrl) or BCM (BCM) overnight followed by staining with the iron-sensitive fluorophore calcein-AM for 15 mins. The excess stain was washed off and the fluorescence intensities measured via flow cytometry. Results representative of three independent experiments are shown. (B) BMDMs exposed to BCM overnight were analyzed for mRNA expression levels of transferrin receptor by qRT-PCR. Results are shown as mean ± S.D. from 3 independent replicates. * p = 0.0067 (C) BMDMs were exposed to BCM overnight following which cells were lysed in RIPA buffer and probed for ferritin heavy chain via western blotting. Actin was used for normalization. The western blot is representative of three independent experiments.
We were interested in assessing whether the BCM-induced increase in intracellular iron affects macrophage responses. Accordingly, we measured the production of IL-6 by macrophages in response to infection with S. Typhimurium following pre-treatment with BCM, the iron chelator deferoxamine, or a combination of the 2 reagents. Pre-treatment of BMDMs with deferoxamine alone significantly reduced the levels of IL-6 released by macrophages in response to S. Typhimurium (Fig. 7A), consistent with our earlier observation that decreased intracellular iron attenuates macrophage inflammatory responses (29,31). BCM pre-treatment prevented the inhibition of IL-6 production by deferoxamine (Fig. 7A), suggesting that the BCM-induced down-regulation of FPN and consequent increase in intracellular iron counteracted the effect of the chelator. Very similar results were obtained when we analyzed the effects of deferoxamine and BCM on S. Typhimurium-induced IL-1β production (Fig. 7B), further substantiating the idea that commensals can influence macrophage inflammatory responses by modulating FPN expression. The effects of deferoxamine and BCM on Salmonella-induced cytokine production could not be explained by differences in intracellular bacterial burden. In fact, deferoxamine treatment increased intracellullar Salmonella numbers, an observation that has been documented previously and has been attributed to inhibition of the iron-dependent anti-microbial enzyme phagocyte oxidase (43), whereas BCM counteracted the effect of deferoxamine on intracellular bacterial burden (Fig. 7C). Thus, our results show that the down-regulation of FPN by B. fragilis has a clear influence on 2 important aspects of macrophage function, viz., inflammatory cytokine production and the ability to kill intracellular bacteria.
Fig. 7.

Exposure to BCM can overcome effects of iron chelation on Salmonella-induced cytokine secretion by macrophages. BMDMs were pre-treated with BCM or BCM plus deferoxamine overnight, followed by infection with S. Typhimurium in the presence of the corresponding pre-treatment reagent. Supernatants were collected 7 hrs after infection and ELISAs were carried out to measure IL-6 (A) or IL-1β (B), and the cells were lysed to quantitate intracellular bacterial burden (C). Results are shown as mean ± S.D. from 3 technical replicates of one experiment representative of three independent experiments. One way ANOVA test was performed on ranks. *p < 0.05.
DISCUSSION
The gut microbiota has the capacity to produce a diverse range of compounds that play a role in regulating the function of cells in distal tissues (21,22). Here, our results show that soluble molecules released by B. fragilis, a prominent member of the resident intestinal microbial community, are able to decrease FPN transcription in macrophages and induce a consequent increase in intracellular iron concentrations. These findings suggest that at least some gut commensal bacteria can modulate systemic iron homeostasis as a result of microbial molecules being translocated across the intestinal epithelium and acting to decrease FPN expression on macrophages independently of hepcidin. Hepcidin-independent down-regulation of FPN has been shown to lead to lowering of serum iron levels (40), so it is likely that the B. fragilis-induced decrease in macrophage FPN expression would have similar effects, especially since our results indicate that it is associated with intracellular iron sequestration. Translocation of FPN-inhibiting microbial molecules across the gut epithelium may occur under basal conditions but is likely to be particularly important in the context of intestinal inflammation, a situation in which gut epithelial barrier properties are compromised (22). Thus, the observations reported here complement our earlier findings indicating that the gut microbiota can influence intestinal inflammation-induced changes in hepcidin expression (25,27) and point to multiple mechanisms by which the commensal flora can affect iron metabolism.
It is worth noting that the B. fragilis-induced down-regulation of FPN may have effects on macrophages that extend beyond the role of these cells in iron homeostasis. Specifically, the results in Fig. 7 show that BCM exposure, and the consequent reduction of FPN expression and elevation of intracellular iron, prevented the decrease in Salmonella-induced IL-6 and IL-1β production caused by pre-treatment with the iron chelator deferoxamine. These findings are in keeping with our earlier observations that decreased intracellular iron levels inhibit macrophage inflammatory responses while increased intracellular iron promotes such responses (29,31). The data in Fig. 7 also demonstrate the effect of BCM-induced down-regulation of FPN on macrophage anti-microbial function. This effect is probably related to the fact that phagocyte oxidase, the enzyme that plays a key role in generating anti-microbial reactive oxygen species, is sensitive to iron chelation (43). Thus, the increase intracellular iron associated with the BCM-induced decrease in FPN helps to attenuate the inhibition of phagocyte oxidase activity by deferoxamine.
Taken together, the observations shown in Fig. 7 suggest that commensals such as B. fragilis that are able to decrease FPN expression could have beneficial effects by preserving iron-influenced and iron-dependent macrophage functions such as production of cytokines and reactive oxygen species in the face of factors that might otherwise decrease intracellular iron concentrations. An example of such a factor is systemic iron deficiency, which would have the effect of lowering hepcidin levels, thus allowing the amount of surface FPN protein to increase (6,7,9). The B. fragilis-induced decrease in FPN transcription would counteract this effect. Another example is provided by iron-binding siderophores produced by several pathogens during the course of infection. At least some of these siderophores have been shown to alter host cell physiology and responses as a result of decreased intracellular iron (44). Again, the B. fragilis-induced decrease in FPN expression could attenuate these siderphore-mediated actions. In addition to the cytokine and anti-microbial responses that we have examined, other iron-sensitive aspects of macrophage function, such as M1/M2 polarization and involvement in erythropoiesis, could also be influenced by commensal-induced modulation of FPN expression (45–48). Future studies, including in vivo experiments, will be required to delineate the range of macrophage functions that might be affected by commensal-induced modulation of FPN expression.
The identity of the FPN-inhibiting molecule(s) released by B. fragilis remains to be revealed. The fact that several TLR ligands, including LPS, are individually capable of down-regulating FPN raises the possibility that the relevant molecule belongs to this family. However, B. fragilis LPS is unlikely to be solely responsible for inhibiting FPN expression since BCM was able to bring about this effect in BMDMs deficient in TLR2, the receptor that recognizes this molecule (38). BCM is a crude preparation and so is likely to contain multiple microbial molecules acting redundantly to decrease FPN expression. Further work will be required to characterize and identify the relevant molecules. In this context, it is interesting that Citrobacter did not down-regulate FPN despite expressing a typical enterobacterial LPS that acts on TLR4 (39). In fact, Citrobacter increased FPN expression, similar to what has been reported for Salmonella (49–51). This finding suggests that bacterial factors other than LPS may also influence FPN expression and that the net effect may reflect the combined action of multiple molecules. The exact mechanism by which B. fragilis decreases FPN expression also remains to be elucidated. Our results indicate that it operates at the level of transcription. Identification of the FPN promoter elements that respond to B. fragilis, as well as the transcription factors that interact with these elements, will be the focus of future work.
A study by Deschemin et al. examined the effect of the microbiota on FPN expression and obtained results that are different from ours (52). They found that FPN expression was 2-fold lower in the duodenum of germ-free mice than in conventional animals and that introduction of a normal microbiota corrected this abnormality. These results indicate that the microbiota promotes FPN expression, contrasting with the B. fragilis-induced down-regulation of FPN seen in our study. This apparent discrepancy could be related to several differences in the experimental systems used in the Deschemin et al. investigation versus ours - duodenum versus macrophages, the effects of a complex microbiota versus a single organism, whole animal versus isolated macrophages. Nevertheless, the differences between the two sets of findings does highlight the important idea that different members of the microbiota community may have different effects on FPN expression and that these effects may vary with cell type. Thus, the ultimate effect of the microbiota on iron metabolism is likely to reflect the combined action of these factors.
The results of the experiments described here, as well as our earlier observations and the work of Deschemin et al. (25,27,52), indicate that the composition of the gut microbiota can have important effects on iron handling, both at baseline and during abnormal states such as intestinal inflammation. Thus, besides affecting the pathogenesis of the anemia that results from FPN down-regulation during inflammation (6), variations in the microbiota could contribute to the population-specific differences in basal iron status that have been observed in humans, e.g., the elevated iron stores in individuals of African heritage compared to Hispanics and Caucasians, or the increased iron absorption in Asian relative to Caucasian females (53,54). While some of this variation is undoubtedly attributable to genetic differences (53,55), alterations in the microbiota related to diet or other environmental factors could also play a role. This possibility deserves further investigation since it represents a potential opportunity for correcting abnormalities of iron metabolism by manipulating the microbiota. Further characterization of the molecular mechanisms involved in B. fragilis-induced FPN down-regulation will help in the development of such therapeutic strategies.
ACKNOWLEDGEMENTS
This work was supported by a grant to BJC from the National Institutes of Health (R01 AI089700). We are grateful to Dr. Deepak Vijaykumar, Massachusetts General Hospital for giving us stocks of the commensal bacterial strains used in this study. We are also grateful to Dr. Nitya Jain, Massachusetts General Hospital for providing TLR2 KO mice and to Ms Maria Teresa Ennamorati for her assistance with these animals.
ABBREVIATIONS
- AI
Anemia of inflammation
- BCM
Bacteroides fragilis conditioned medium
- BMDM
Bone marrow derived macrophages
- FPN
Ferroportin (SLC40A1)
- hnRNA
Heterogeneous nuclear RNA
- IBD
Inflammatory bowel disease
- IL-6
Interleukin-6
- IRE
Iron responsive element
- IRP
Iron regulatory protein
- LIP
Labile iron pool
- LPS
Lipopolysaccharide
- qRT-PCR
Quantitative real-time polymerase chain reaction
- SD
Standard deviation
- TfR1
Transferrin receptor 1
- TLR
Toll-like receptor
- 36B4
Acidic ribosomal phosphoprotein P0
Footnotes
CONFLICT OF INTEREST
The authors do not have any conflicts of interest relevant to the information in this manuscript.
REFERENCES
- 1.Evstatiev R, Gasche C, Scott BB. Iron sensing and signalling. Gut. 2012;61:933–952. [DOI] [PubMed] [Google Scholar]
- 2.Wessling-Resnick M Excess iron: considerations related to development and early growth. Am J Clin Nutr. 2017;106:1600S–1605S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kosman DJ. Redox cycling in iron uptake, efflux, and trafficking. J Biol Chem. 2010;285:26729–26735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wood MJ, Skoien R, Powell LW. The global burden of iron overload. Hepatol Int. 2009;3:434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kassebaum NJ, Jasrasaria R, Naghavi M, et al. A systematic analysis of global anemia burden from 1990 to 2010. Blood. 2014;123:615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verma S, Cherayil BJ. Iron and inflammation - the gut reaction. Metallomics. 2017;9:101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ganz T Macrophages and iron metabolism. Microbiol. Spectr 2016;4: doi: 10.1128/microbiolspec.MCHD-0037-2016. [DOI] [PubMed] [Google Scholar]
- 8.Drakesmith H, Nemeth E, Ganz T Ironing out ferroportin. Cell Metabolism. 2015;22:777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arezes J, Jung G, Gabayan V, et al. Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe. 2015;17:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefanova D, Raychev A, Arezes J, et al. Endogenous hepcidin and its agonist mediate resistance to selected infections by clearing non-transferrin-bound iron. Blood. 2017;130:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michels KR, Zhang Z, Bettina AM, et al. Hepcidin-mediated iron sequestration protects against bacterial dissemination during pneumonia. JCI Insight. 2017;2: e92002. doi: 10.1172/jci.insight.92002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stefanova D, Raychev A, Deville J, et al. Hepcidin protects against lethal Escherichia coli sepsis in mice inoculated with isolates from septic patients. Infect. Immun 2018;86: pii: e00253–18. doi: 10.1128/IAI.00253-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward DM, Kaplan J. Ferroportin-mediated iron transport: expression and regulation. Biochim Biophys Acta - Mol Cell Res. 2012;1823:1426–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilkinson N, Pantopoulos K. The IRP/IRE system in vivo: insights from mouse models. Front Pharmacol. 2014;5:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marro S, Chiabrando D, Messana E, et al. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position −7007 of the FPN1 promoter. Haematologica. 2010;95:1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiabrando D, Fiorito V, Marro S, et al. Cell-specific regulation of ferroportin transcription following experimentally-induced acute anemia in mice. Blood Cells, Mol Dis. 2013;50:25–30. [DOI] [PubMed] [Google Scholar]
- 17.Troadec M-B, Ward DM, Lo E, et al. Induction of FPN1 transcription by MTF-1 reveals a role for ferroportin in transition metal efflux. Blood. 2010;116:4657–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ludwiczek S, Aigner E, Theurl I, et al. Cytokine-mediated regulation of iron transport in human monocytic cells. Blood. 2003;101:4148–4154. [DOI] [PubMed] [Google Scholar]
- 19.Yang F, Liu X-B, Quinones M, et al. Regulation of reticuloendothelial iron transporter MTP1 (Slc11a3) by inflammation. J Biol Chem. 2002;277:39786–39791. [DOI] [PubMed] [Google Scholar]
- 20.Liu X-B, Nguyen N-BH, Marquess KD, et al. Regulation of hepcidin and ferroportin expression by lipopolysaccharide in splenic macrophages. Blood Cells, Mol Dis. 2005;35:47–56. [DOI] [PubMed] [Google Scholar]
- 21.Kundu P, Blacher E, Elinav E, Pettersson S. Our gut microbiome: the evolving inner self. Cell. 2017;171:1481–1493. [DOI] [PubMed] [Google Scholar]
- 22.Belkaid Y, Harrison OJ. Homeostatic immunity and the microbiota. Immunity. 2017;46:562–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zitomersky NL, Coyne MJ, Comstock LE. Longitudinal analysis of the prevalence, maintenance, and IgA response to species of the order Bacteroidales in the human gut. Infect Immun. 2011;79:2012–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wexler HM. Bacteroides: The good, the bad, and the nitty-gritty. Clin Microbiol Rev. 2007;20:593–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shanmugam NKN, Trebicka E, Fu L-L, et al. Intestinal inflammation modulates expression of the iron-regulating hormone hepcidin depending on erythropoietic activity and the commensal microbiota. J Immunol. 2014;193:1398–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc. 2008; 2008:pdb.prot5080. doi: 10.1101/pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- 27.Shanmugam NKN, Chen K, Cherayil BJ. Commensal bacteria-induced interleukin-1β (IL-1β) secreted by macrophages upregulates hepcidin expression in hepatocytes by activating the bone morphogenetic protein signaling pathway. J Biol Chem. 2015;290:30637–30647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen K, Shanmugam NKN, Pazos MA, et al. Commensal bacteria-induced inflammasome activation in mouse and human macrophages is dependent on potassium efflux but does not require phagocytosis or bacterial viability. PLoS One. 2016;11:e0160937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, Harrington L, Trebicka E, et al. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J Clin Invest. 2009;119:3322–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown EM, Wlodarska M, Willing BP, et al. Diet and specific microbial exposure trigger features of environmental enteropathy in a novel murine model. Nat Commun. 2015;6:7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, Johnson EE, Shi HN, et al. Attenuated inflammatory responses in hemochromatosis reveal a role for iron in the regulation of macrophage cytokine translation. J Immunol. 2008;181:2723–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aydemir F, Jenkitkasemwong S, Gulec S, et al. Iron loading increases ferroportin heterogeneous nuclear RNA and mRNA levels in murine J774 macrophages. J Nutr. 2009;139:434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen C-YA, Ezzeddine N, Shyu A-B. Messenger RNA half-life measurements in mammalian cells. Methods Enzymol. 2008; 448:335–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Q, Cherayil BJ. Role of Toll-like receptor 4 in macrophage activation and tolerance during Salmonella enterica serovar Typhimurium infection. Infect Immun. 2003;71:4873–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knutson MD, Oukka M, Koss LM, et al. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc. Natl. Acad. Sci. USA 2005;102:1324–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Z, Zhang F, An P, et al. Ferroportin 1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood. 2011;118:1912–1922. [DOI] [PubMed] [Google Scholar]
- 37.Odendall C, Kagan JC. Activation and pathogenic manipulation of sensors of the innate immune system. Microbes Infect. 2017;19:229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alhawi M, Stewart J, Erridge C, et al. Bacteroides fragilis signals through Toll-like receptor (TLR) 2 and not through TLR4. J Med Microbiol. 2009;58:1015–1022. [DOI] [PubMed] [Google Scholar]
- 39.Khan MA, Ma C, Knodler LA, et al. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect Immun. 2006;74:2522–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guida C, Altamura S, Klein FA, et al. A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood. 2015;125:2265–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas F, Serratrice G, Beguin C, et al. Calcein as fluorescent probe for ferric iron. Application to iron nutrition in plant cells. J Biol Chem. 1999;274:13375–13383. [DOI] [PubMed] [Google Scholar]
- 42.Theil EC. Regulation of ferritin and transferrin receptor mRNAs. J Biol Chem. 1990;265:4771–4774. [PubMed] [Google Scholar]
- 43.Collins HL, Kaufmann SH, Schaible UE. Iron chelation via deferoxamine exacerbates experimental salmonellosis via inhibition of the nicotinamide adenine dinucleotide phosphate oxidase-dependent respiratory burst. J Immunol. 2002;168:3458–3463. [DOI] [PubMed] [Google Scholar]
- 44.Holden VI, Bachman MA. Diverging roles of bacterial siderophores during infection. Metallomics. 2015;7:986–995. [DOI] [PubMed] [Google Scholar]
- 45.Kroner A, Greenhalgh AD, Zarruk JG, et al. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron. 2014;83:1098–1116. [DOI] [PubMed] [Google Scholar]
- 46.Handa P, Thomas S, Morgan-Stevenson V, et al. Iron alters macrophage polarization status and leads to steatohepatitis and fibrogenesis. J Leukoc Biol. 2019; doi: 10.1002/JLB.3A0318-108R. [DOI] [PubMed] [Google Scholar]
- 47.Alam MZ, Devalaraja S, Haldar M. The heme connection: linking erythrocytes and macrophage biology. Front Immunol. 2017;8:33. doi: 10.3389/fimmu.2017.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yeo JH, Colonne CK, Tasneem N, et al. The iron islands: erythroblastic islands and iron metabolism. Biochim Biophys Acta Gen Subj. 2019;1863:466–471. [DOI] [PubMed] [Google Scholar]
- 49.Nairz M, Schleicher U, Schroll A, et al. Nitric oxide-mediated regulation of ferroportin-1 controls macrophage iron homeostasis and immune function in Salmonella infection. J Exp Med. 2013;210:855–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nairz M, Fritsche G, Brunner P, et al. Interferonγ limits the availability of iron for intramacrophage Salmonella Typhimurium. Eur J Immunol. 2008;38:1923–1936. [DOI] [PubMed] [Google Scholar]
- 51.Nairz M, Theurl I, Ludwiczek S, et al. The co-ordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella Typhimurium. Cell Microbiol. 2007;9:2126–2140. [DOI] [PubMed] [Google Scholar]
- 52.Deschemin JC, Noordine ML, Remot A, et al. The microbiota shifts the iron sensing of intestinal cells. FASEB J. 2016;30:252–261. [DOI] [PubMed] [Google Scholar]
- 53.Ye K, Cao C, Lin X, et al. Natural selection on HFE in Asian populations contributes to enhanced non-heme iron absorption. BMC Genet. 2015;16:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zacharski LR, Ornstein DL, Woloshin S, et al. Association of age, sex, and race with body iron stores in adults: analysis of NHANES III data. Am Heart J. 2000;140:98–104. [DOI] [PubMed] [Google Scholar]
- 55.Moyo VM, Mandishona E, Hasstedt SJ, et al. Evidence of genetic transmission in African iron overload. Blood. 1998;91:1076–1082. [PubMed] [Google Scholar]