

Abstract
Superior to linear peptides in biological activities, cyclic peptides are considered to have great potentials used as therapeutic agents. In order to identify cyclic peptide ligands for therapeutic targets, selection from phage-displayed peptide libraries in which cysteines are conjugated covalently through either the disulfide bond or organic linkers has been widely adopted with great success. To resolve some technical drawbacks related to cysteine conjugation, we have invented a novel phage display technique in which its displayed peptides are cyclized through a proximity-driven Michael addition reaction between a cysteine and an amber codon-coded Nε-acryloyl-lysine (AcrK). Using a randomized 6-mer library in which peptides were cyclized at two ends through a cysteine-AcrK linker, we demonstrated the successful selection of potent ligands for TEV protease and HDAC8. All selected cyclic peptide ligands showed 4 to 6-fold stronger affinity to their protein targets than their linear counterparts. As a new addition to the phage display technique, we believe this novel approach will find broad applications in drug discovery.
Keywords: phage display, cyclic peptides, proximity-driven cyclization, HDAC8, Nε-acryloyl-lysine
Graphical Abstract
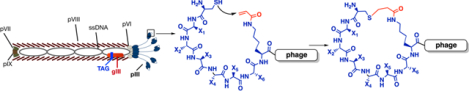
Using the amber suppression-based mutagenesis approach, Nε-acryloyl-lysine was genetically encoded in a phage-displayed peptide library for cyclization with a preinstalled cysteine. Selection from this novel phage display library afforded cyclic peptide ligands that bind TEV protease and HDAC8 much stronger than their linear counterparts
Introduction
Traditionally, therapeutic drugs have consisted of small molecules that are exquisite at binding their receptors. However, due to their small size, small molecules have achieved little success at targeting proteins that involve large, relative flat surfaces for interactions with other molecules. With the development of the recombinant protein expression technology, a new class of protein pharmaceuticals, dubbed as biologics, has emerged. Because of their larger sizes, biologics display far superior target affinity and selectivity compared to small molecules. However, their increased size and protein-based composition lead to poor tissue permeability and metabolic stability. With their intermediate size between small molecules and biologics, peptides offer a promising alternative to the two established classes of pharmaceutics. Being larger than small molecules, peptides offer increased potency and target selectivity while maintaining a potential for cell permeability and a lower cost of manufacturing than biologics.[1] Peptides are also extremely easy to screen. Using peptide display technologies,[2] such as phage display, which link the displayed peptide phenotype to the genotype, it is possible for a single researcher to screen a library of greater than 1010 unique peptides in a matter of days. Despite these advantages, peptide-based inhibitors have long been avoided for two reasons. First, peptides are generally unstructured in solution which leads to an entropic penalty upon binding to a target. Second, peptides are highly susceptible to proteolysis when applied in vivo.[3] It has long been known that macrocyclization can help to overcome some of the disadvantages of peptides.[4, 5] Macrocyclization imparts a degree of conformational rigidity to an unstructured peptide, which often increases the binding affinity of the peptide for its target.[6] Cyclic peptides are also significantly more resistant to proteolysis.[7] In several cases this has led to peptides so stable that they have been successfully used for oral delivery.[8]
Although peptide cyclization generally leads to better pharmacological properties, cyclizing a linear peptide identified through screening can have unknown consequences on the ability of the peptide to bind to a target protein.[9] For this reason, multiple binary coding systems have been developed for direct selection of cyclic peptide libraries.[10] These include phage display, bacterial display, yeast display, traditional mRNA display, RaPID, SICLOPPS, etc.[4, 11, 12] Screening one-bead-one-compound libraries is also an effective method for the identification of potent cyclic peptide ligands.[13] Phages natively display linear peptides. To cyclize phage-displayed linear peptides, several chemical approaches have been developed.[14] One involves the formation of a disulfide bond between two cysteine residues (Figure 1A).[11, 15] There are many examples about using this strategy to produce disulfide-cyclized peptides with higher affinity for a target protein than their linear counterparts.[16] While beneficial for some in vitro applications, peptides cyclized in this way cannot be used in vivo as they cannot withstand the reducing cellular environments. An alternative strategy relies on the reactivity of nucleophilic thiols towards small-molecule organic linkers to covalently connect two cysteines (Figure 1B).[17] This strategy has been successfully used for the formation of both mono- and bicyclic, phage-displayed peptide libraries and used to select ligands with inhibition constants as low as 2 nM (Figure 1C).[18] Although effective at forming cyclized peptide libraries, this method modifies native phage cysteines leading to low phage viability. Attempts have been made to construct phage strains with no surface cysteines.[19] However, these phages have low viability, limiting the phage production. Due to the non-selective nature while conjugating cysteines, most current organic linkers are symmetric and achiral for avoiding heterogeneity in the phage-displayed cyclic peptides that might pose significant challenges in the following synthesis and characterization of selected cyclic peptides. The requirement of using symmetric, achiral organic linkers limits the structural diversity of cyclized peptides. To resolve these limitations, we envisioned that an electrophilic noncanonical amino acid (ncAA) and a cysteine can be genetically installed in phage-displayed peptides in close proximity for peptide cyclization (Figure 1D). The incorporation of the ncAA into phages can be achieved by suppressing an amber mutation in the phage-displayed peptide coding region in E. coli cells that harbor a ncAA-specific aminoacyl-tRNA synthetase-amber suppressor tRNA pair and grow in the presence of the ncAA (Figure 1E).[20] Using this new method for the construction of a phage display library will afford a genetically encoded, phage-displayed cyclic peptide library whose spontaneous peptide cyclization requires neither the use of phage strains with no surface cysteines nor an organic linker for cyclization. Encouragingly, several cases featuring proximity-driven reactions using a genetically encoded ncAA and a cysteine or another amino acid has recently been demonstrated by Fasan, Suga, Wang, and their coworkers.[21, 22]
Figure 1.

Representative, existing and proposed cyclization strategies for phage-displayed peptides. (A) Cyclization through the disulfide bond between cysteines; (B) Cyclization through covalent conjugation of cysteines with organic linkers; (C) Representative organic linkers used for cysteine conjugation to generate mono- and bicyclic peptides; (D) A proposed proximity-driven cyclization between a cysteine and an electrophilic noncanonical amino acid (ncAA); (E) An amber suppression-based approach to link the phenotypic ncAA with the genotypic TAG mutation. The production of a phage with a TAG mutation at the coding region of its displayed peptide is produced in E. coli cells that harbour an evolved aminoacyl-tRNA synthetase and amber suppressing tRNA for the genetic incorporation of the designated ncAA.
Results and Discussion
Pyrrolysine (Pyl) is a naturally occurring 22nd proteinaceous amino acid that is genetically encoded by an amber codon.[23] Its incorporation is mediated by pyrrolysyl-tRNA synthetase (PylRS) and tRNA. In the past decade, a number of research groups including ours have evolved PylRS for the genetic incorporation of more than 100 ncAAs including both lysine and phenylalanine derivatives into proteins in E. coli. One of these ncAAs is Nε-acryloyl-lysine (AcrK), a Michael acceptor.[24] We previously demonstrated that AcrK reacts slowly with a thiol (the second-order rate constant is 0.004 M−1s−1) at physiological conditions but can be stably incorporated into proteins in E. coli using an evolved PylRS mutant (PrKRS) and . The slow reaction between AcrK and cysteine is desirable in that it avoids non-specific reactions with regular protein cysteines but allow rapid conjugation when AcrK and cysteine are located in close proximity in a peptide. By installing a cysteine and an AcrK at two ends of a phage displayed peptide, an automatic cyclization of the peptide is expected (Figure 2A). There is also an advantage to work with AcrK. Its acrylamide moiety undergoes Huisgen 1,3-cycloaddition reaction selectively with a non-fluorescent diphenylnitrilimine moeity to form an intense blue fluorescent final product.[25] Using HZC1 (Figure 2B) that undergoes rapid dehydrochloration in water to release a diphenylnitrile,[26] protein or phage with intact AcrK can be easily labeled and visualized. However, a proximity-driven Michael addition reaction between AcrK and cysteine in a peptide will annihilate the acrylamide moiety, leading to a cyclic peptide that cannot be labeled by HZC1.
Figure 2.

Cyclization of phage-displayed peptides through Michael addition between a cysteine and a genetically incorporated Nε-acryloyl-lysine (AcrK). (A) A diagram that illustrates proximity-driven peptide cyclization between a cysteine and an electron deficient ncAA; (B) The structure of AcrK and HZC1 whose dissociation product in water is a nitrilimine that reacts selectively with an acrylamide to show intense blue fluorescence; (C) Two superfolder green fluorescent protein (sfGFP) derivatives, one with a N-terminal CA5X peptide and the other with a N-terminal A6X peptide, and their fluorescent labeling with HZC1. X denotes AcrK. Proteins were denatured first and then analyzed by SDS-PAGE. In-gel fluorescence was recorded at the blue region with the excitation at 365 nm; (D) Two phage derivatives, one with a N-terminal CA5X peptide and the other with a N-terminal A6X peptide, and their titer results after reaction with the N-biotinyl-cysteamine probe and then capturing by streptavidin beads. Uncaptured phages were analyzed in the titer assay.
To demonstrate the proximity-driven cyclization between a genetically incorporated AcrK and an adjacent cysteine in a protein, we expressed superfolder green fluorescent protein (sfGFP) that had a N-terminal CA5X peptide (X denotes AcrK and is coded by an amber codon). To express this protein, we transformed E. coli BL21(DE3) cells with two plasmids. One was a previous described pEVOL-PrKRS plasmid that contained both PrKRS and genes and the other was a pETduet plasmid that contained a gene coding the CA5X-sfGFP protein. Growing the transformed cells in the presence of AcrK afforded CA5X-sfGFP. Labeling this protein with HZC1 led to no blue fluorescent product. However, a control sfGFP protein with an N-terminal A6X peptide, that we expressed similarly to CA5X-sfGFP and reacted with HZC1, provided an intense blue fluorescent protein band in a SDS-PAGE gel (Figure 2C). In parallel, we generated two phages, one with a CA5X peptide and the other with an A6X peptide at the N-terminus of the coating protein pIII. To construct the CA5X phage, we inserted a CA5X-coding DNA fragment between the PelB leader peptide-coding region and the phage pIII-coding (gIII) gene in the pADLg3 phagemid vector that we purchased from Antibody Design Labs. We used the afforded phagemid pADLg3-CA5X to transform E. coli Top10 cells that also harbored a plasmid pEVOL-PrKRS-CloDF and a mutant helper phage plasmid M13K07-g3TAA. pEVOL-PrKRS-CloDF was derived from pEVOL-PrKRS by switching the replication origin from p15a to CloDF for its compatible use with a helper phage plasmid that typically has a p15a replication origin. We constructed M13K07-g3TAA by introducing a deleterious ochre mutation at the Q350 coding site in the gIII gene of the M13K07 helper phage. Since M13K07-g3TAA had a non-functional gIII gene, its use together with PADLg3-CA5X drove the synthesis of a phage that contained pIII expressed only from the later plasmid. Growing the transformed cells in the presence of AcrK afforded the CA5X phage. We used the similar approach to produce the control A6X phage. Following their separation, we incubated them together with N-biotinyl-cysteamine, a probe that reacts with acrylamide. We used 60°C to speed up the relative slow Michael addition reaction. After reaction, we used streptavidin beads to capture all biotin-conjugated phages and tittered uncaptured phages. Our result (Figure 2D) showed that more than 90% A6X phage was captured but about 65% CA5X phage remained in the supernatant, supporting that the A6X phage had a free acrylamide moiety but the CA5X phage underwent a proximity-driven cyclization to eliminate its acrylamide. The loss of phages during substantial washing to remove residual N-biotinyl-cysteamine might contribute to the observation that only about 65% of the CA5X phage was recovered. To examine whether peptide chain lengths may significantly impact the proximity driven cyclization between a cysteine and a AcrK, we synthesized three Cys-containing peptides (CA5AcrK, CA6AcrK, and CA7AcrK) and their non-Cys control peptides (A6AcrK, CA7AcrK, and CA8AcrK). Labeling all six peptides with HZC1 in PBS buffer showed strong blue fluorescence for non-Cys peptides but low fluorescence for Cys-containing peptides indicating that all Cys-containing peptides cyclized (Supplementary Figure 4). Prolonging the peptide chain length did not significantly deter the cyclization process.
Encouraged by the in vitro results, we advanced to construct a phage-displayed 6-mer cyclic peptide library. To afford a phagemid library for the production of phages with displayed cyclic peptides, we inserted a 24 base-pair DNA fragment that encoded six randomized amino acids flanked by an N-terminal cysteine and a C-terminal AcrK between the PelB leader peptide-coding region and the gIII gene of the pADLg3 phagemid (Figure 3A). 20 clones from this library were sequenced to confirm the library diversity (Supplementary Figure 7). We used this phagemid library to transform E. coli Top10 cells that also contained pEVOL-PrKRS-CloDF and M13K07-g3TAA to afford close to 109 transformants and then grew the transformed cells in the presence of AcrK to produce phages. To demonstrate the viability of using this library to select cyclic peptide ligands for a protein target, we first tested it on a model protein, TEV protease that we conjugated with biotin for its loading onto streptavidin magnetic beads for undergoing selection. We carried out three rounds of affinity-based selection. Eluted phages were clearly enriched after each round (Supplementary Figure 8). After the third round, we sequenced 25 phage clones that converged to only three peptide sequences, CycTev1, CycTev2, and CycTev3 (Supplementary Figure 9). Using solid-phase peptide synthesis, we synthesized 5-FAM-conjugated CycTev1, CycTev2, as well as their linear counterparts and then measured their binding affinities to TEV protease using fluorescence polarization assays. Our results as shown in Figures 3B–E and Table 1 indicated that both CycTev1 and CycTev2 bind to TEV protease with a single digit μM dissociation constant and both cyclic peptides bind to TEV protease significantly better than their linear counterpart (>6-fold). These results established the feasibility of using our genetically encoded phage-displayed cyclic peptide library to identify potent ligands for protein targets and demonstrated that cyclization contributes to the binding.
Figure 3.

Selected TEV protease-binding cyclic peptides and their Kd measurements. (A) A diagram to show the phagemid structure for the production of a phage-displayed 6-mer cyclic peptide library; (B) The structure of 5FAM-CycTev1. CycTev1, highlighted in blue and red, was selected from phage display; (C) Fluorescence polarization analysis of 5FAM-CycTev1 binding to TEV protease. Data for a linear counterpart of 5FAM-CycTev1 with no linker is also included; (D) The structure of 5FAM-CycTev2; (E) Fluorescence polarization analysis of 5FAM-CycTev2 binding to TEV protease. Data for a linear counterpart of 5FAM-CycTev2 with no linker is also included.
Table 1.
Determined Kd and IC50 values of selected cyclic peptides and their linear counterparts when binding to their protein targets.
| Ligand | Sequence[a] | Protein target | Kd (μM) | IC50 (μM) |
|---|---|---|---|---|
| CycTev1 | CWRDYLIX | TEV protease | 8.2 ± 0.8 | |
| LinTev1 | CWRDYLIK | TEV protease | 50 ± 5 | |
| CycTev2 | CQWFSHRX | TEV protease | 6.9 ± 0.9 | |
| LinTev2 | CQWFSHRK | TEV protease | 39 ± 5 | |
| CycH8a | CQSLWMNX | HDAC8 | 7.1 ± 0.7 | 9.7 ± 0.7 |
| LinH8a | CQSLWMNK | HDAC8 | >50 | |
| LinH8a’ | CQSLWMNNIe | HDAC8 | 31 ± 7 |
X denotes AcrK.
HDAC8 is a Zn2+-dependent histone deacetylase that has been implicated as a therapeutic target in various diseases including cancer, X-linked intellectual disability, and parasitic infections.[27] Notable efforts have been made to identify potent HDAC8 inhibitors.[28] In order to identify novel cyclic peptide ligands for HDAC8, we carried out selection from our genetically encoded, phage-displayed 6-mer cyclic peptide library similar to that for TEV protease. Out of selected clones that we subsequently sequenced, the majority converged to a single sequence CycH8a (Table 1 and Supplementary Figure 10).
To determine the affinity of CycH8a to HDAC8, we synthesized 5-FAM-conjugated CycH8a (Figure 4A) and then characterized its binding to HDAC8 using the fluorescence polarization analysis. The result indicated a 7.1 μM dissociation constant (Figure 4B). We also synthesized 5-FAM-conjugated LinH8a, a linear counterpart of CycH8a and tested its binding to HDAC8. However, LinH8a bound very weakly to HDAC8. Due to the fact that HDAC8 aggregated at a concentration higher than 100 μM, we were not able to collect enough data to accurately determine Kd for LinH8a though it is estimated to be higher than 50 μM. LinH8a has a positively charged lysine residue; however, CycH8a has this charge neutralized. To resolve a concern that this charge difference might contribute to the binding disparity between the two peptides, we synthesized 5-FAM-conjugated LinH8a’ in which norleucine (Nle), a neutral amino acid spatially similar to lysine was installed at the lysine position in LinH8a and determined its dissociation constant toward HDAC8 as 31 μM (Table 1 and Supplementary Figure 12). This Kd value is four-fold higher than that of CycH8a, eliminating the possibility that theneutral charge of CycH8a contributes significantly to its strong binding to HDAC8. Therefore, the cyclization is critical to provide high potency to CycH8a for its binding to HDAC8. For a protein target, it doesn’t necessarily bind to the active site of the protein for direct inhibition. To test whether CycH8a can directly inhibit the deacetylation activity of HDAC8, we adopted a HDAC8 activity assay[29] as shown in Figure 4C and synthesized the substrate Boc-AcK-AMC. In this assay, HDAC8 catalyzed the deacetylation of Boc-AcK-AMC to afford Boc-K-AMC that reacted with the coupling enzyme trypsin to release the fluorescent AMC, a compound that we could easily track in a fluorescent plate reader. As shown in Figure 4D, providing CycH8a to the assay inhibited the deacetylation of Boc-AcK-AMC by HDAC8. The determined IC50 value in the conditions of 5 μM HDAC8 and 50 μM Boc-AcK-AMC is 9.7 μM, close to the determined Kd value. Given that IC50 is not a direct binding affinity indicator and influenced by the concentration of the substrate, its slightly higher value than Kd was expected. Collectively, results in Figure 4 demonstrated a successful application of using our genetically encoded, phage-displayed cyclic peptide library in identifying a potent cyclic peptide inhibitor for a therapeutic protein target.
Figure 4.

A selected cyclic peptide ligand CycH8a and its binding and inhibition of HDAC8. (A) The structure of 5FAM-CycH8a. The sequence of CycH8a, highlighted in blue and red, was selected from phage display; (B) Fluorescence polarization analysis of 5FAM-CycH8a binding to HDAC8. Data for a linear counterpart of 5FAM-CycH8a is also included; (C) A diagram to show a fluorogenic HDAC8 activity assay scheme; (D) The IC50 determination of 5FAM-CycH8a inhibition of HDAC8 using the assay shown in C.
To gain insight into how CycH8a might interact with HDAC8, we virtually docked CysH8a on HDAC8. HDAC8 naturally occurs in a dimeric form.[30] Therefore, we investigated both monomeric and dimeric HDAC8 as the receptor for docking. The docking results indicated that CycH8a binds weakly to monomeric HDAC8 but fits favorably in two grooves that are at the dimeric interface of HDAC8 and close to the two active sites (Figure 5). Several published crystal structures of HDAC8-substrate complexes have shown that the two dimer interface grooves are also part of channels for binding peptide substrates. The binding of CycH8a at the two grooves will block the entry of a peptide substrate to the two active sites, which provides an explanation for the inhibition of HDAC8 by CycH8a. This is different from most HDAC8 inhibitors that have been developed so far. Most HDAC8 inhibitors are hydroxamates or other metal binders that strongly chelate the active site Zn2+. They bind strongly to the active site tunnel of HDAC8 with IC50 values typically below 1 μM.[31] Our modeling results showed that CycH8a doesn’t directly interact with Zn2+ and other residues in the active site, explaining its relatively lower potency than hydroxamate inhibitors. However, CycH8a binds close to the active site tunnel of HDAC8. One possibility to develop more potent HDAC8 inhibitors is to conjugate an active site-targeting hydroxamate inhibitor and CycH8a to form a tight-binding, bidentate ligand. This is a research direction that we are actively exploring right now.
Figure 5.

The molecular docking results of CycH8a binding to the HDAC8 dimer. The top panel show different CycH8a conformers binding at two grooves of the HDAC8 dimer interface. The bottom panel presents the most favorable conformer of CycH8a binding at each groove.
Conclusion
In summary, we have developed a novel phage display technique that allows the construction of a genetically encoded, phage-displayed cyclic peptide library. The cyclization of phage displayed peptides are achieved by a proximity-driven Michael addition reaction between a cysteine and an AcrK that flank a randomized 6-mer peptide sequence. AcrK was encoded by an amber codon and its incorporation into phages was mediated by an evolved PylRS- pair in E. coli. Applying the developed library to selection against both TEV protease and HDAC8 afforded cyclic peptide ligands that bind to their protein targets with single digit μM Kd values and significantly better than their linear counterparts. As a proof of concept, the current study involved relatively small size peptides that randomized only 6 residues. It is expected that a library with much bigger randomized peptides will afford the selection of more potent ligands. Given that many electrophilic ncAAs have been incorporated into proteins using the amber suppression mutagenesis approach,[22, 32] they can all potentially be used to construct genetically encoded, phage-displayed cyclic peptide libraries. Since these ncAAs are structurally diverse, their use will impart different structural constraints to phage displayed cyclic peptides that will provide diverse structural diversity beneficial for selection. Unlike most other binary coding techniques for the construction of cyclic peptide libraries, the reported method leads to direct, irreversible, and simultaneous cyclization of displayed peptides right after their translation. There is no additional chemical intervention necessary. This feature, shared by the mRNA display-based RaPID technique, significantly simplifies the construction of cyclic peptide libraries. As a novel addition to the phage display technique, we anticipate that this developed technique will find broad applications in the identification of potent ligands for many surface receptors and strong inhibitors for enzymes and protein-protein/DNA/RNA binding interactions.
Experimental Section
Experimental details for the synthesis of all small molecules and peptides, protein expression, construction of plasmids, phagemids, and the phagemid library, selection, and characterization of selected peptides are presented in the supplementary information.
Supplementary Material
Acknowledgements
This work was supported in part by National Institute of Health (grant R01CA161158), Cancer Prevention and Research Institute of Texas (grant RP170797), and Welch Foundation (grant A-1715). We thank Sunshine Z. Leeuwon who proofread the manuscript.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Sato AK, Viswanathan M, Kent RB, Wood CR, Curr. Opin. Biotechnol 2006, 17, 638–642; [DOI] [PubMed] [Google Scholar]; Verdine GL, Hilinski GJ, Method. Enzymol 2012, 503, 3–33. [DOI] [PubMed] [Google Scholar]
- [2].Sternberg N, Hoess RH, Proceedings of the National Academy of Sciences of the United States of America 1995, 92, 1609–1613; [DOI] [PMC free article] [PubMed] [Google Scholar]; Li S, Millward S, Roberts R, J Am Chem Soc 2002, 124, 9972–9973; [DOI] [PubMed] [Google Scholar]; Bashiruddin NK, Suga H, Curr Opin Chem Biol 2015, 24, 131–138; [DOI] [PubMed] [Google Scholar]; Boder ET, Wittrup KD, Nat Biotechnol 1997, 15, 553–557; [DOI] [PubMed] [Google Scholar]; Yonezawa M, Doi N, Kawahashi Y, Higashinakagawa T, Yanagawa H, Nucleic Acids Res 2003, 31, e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Passioura T, Katoh T, Goto Y, Suga H, Annu. Rev. Biochem 2014, 83, 727–753. [DOI] [PubMed] [Google Scholar]
- [4].Passioura T, Suga H, Chem Commun (Camb) 2017, 53, 1931–1940. [DOI] [PubMed] [Google Scholar]
- [5].Kawamura A, Munzel M, Kojima T, Yapp C, Bhushan B, Goto Y, Tumber A, Katoh T, King ON, Passioura T, Walport LJ, Hatch SB, Madden S, Muller S, Brennan PE, Chowdhury R, Hopkinson RJ, Suga H, Schofield CJ, Nat Commun 2017, 8, 14773; [DOI] [PMC free article] [PubMed] [Google Scholar]; Morimoto J, Hayashi Y, Suga H, Angew Chem Int Ed Engl 2012, 51, 3423–3427; [DOI] [PubMed] [Google Scholar]; Zorzi A, Deyle K, Heinis C, Curr Opin Chem Biol 2017, 38, 24–29; [DOI] [PubMed] [Google Scholar]; Bionda N, Fasan R, Methods Mol Biol 2017, 1495, 57–76; [DOI] [PMC free article] [PubMed] [Google Scholar]; Owens AE, de Paola I, Hansen WA, Liu YW, Khare SD, Fasan R, J Am Chem Soc 2017, 139, 12559–12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen S, Rebollo IR, Buth SA, Morales-Sanfrutos J, Touti J, Leiman PG, Heinis C, J. Am. Chem. Soc 2013, 135, 6562–6569. [DOI] [PubMed] [Google Scholar]
- [7].Craik DJ, Science 2006, 311, 1563–1564. [DOI] [PubMed] [Google Scholar]
- [8].Craik DJ, Fairlie DP, Liras S, Price D, Chem. Biol. Drug Des. 2013, 81. [DOI] [PubMed] [Google Scholar]
- [9].Katasara M, Tselios T, Deraos S, Deraos G, Matsoukas MT, Lazoura E, Matsoukas J, Apostolopoulos V, Curr. Med. Chem 2006, 13, 2221–2232. [DOI] [PubMed] [Google Scholar]
- [10].Xiao W, Wang Y, Lau EY, Luo J, Yao N, Shi C, Meza L, Tseng H, Maeda Y, Kumaresan P, Liu R, Lightstone FC, Takada Y, Lam KS, Mol Cancer Ther 2010, 9, 2714–2723; [DOI] [PMC free article] [PubMed] [Google Scholar]; Hipolito CJ, Suga H, Curr Opin Chem Biol 2012, 16, 196–203; [DOI] [PubMed] [Google Scholar]; Vinogradov AA, Yin Y, Suga H, J Am Chem Soc 2019, 141, 4167–4181. [DOI] [PubMed] [Google Scholar]
- [11].Deyle K, Kong XD, Heinis C, Acc Chem Res 2017, 50, 1866–1874. [DOI] [PubMed] [Google Scholar]
- [12].Palei S, Becher KS, Nienberg C, Jose J, Mootz HD, Chembiochem 2019, 20, 72–77; [DOI] [PubMed] [Google Scholar]; van Rosmalen M, Janssen BM, Hendrikse NM, van der Linden AJ, Pieters PA, Wanders D, de Greef TF, Merkx M, J Biol Chem 2017, 292, 1477–1489; [DOI] [PMC free article] [PubMed] [Google Scholar]; Huang Y, Wiedmann MM, Suga H, Chem Rev 2018; [DOI] [PubMed] [Google Scholar]; Tavassoli A, Curr Opin Chem Biol 2017, 38, 30–35. [DOI] [PubMed] [Google Scholar]
- [13].Qian Z, Upadhyaya P, Pei D, Methods Mol Biol 2015, 1248, 39–53. [DOI] [PubMed] [Google Scholar]
- [14].Derda R, Jafari MR, Protein Pept Lett 2018, 25, 1051–1075; [DOI] [PubMed] [Google Scholar]; Ng S, Derda R, Org Biomol Chem 2016, 14, 5539–5545; [DOI] [PubMed] [Google Scholar]; Jafari MR, Yu H, Wickware JM, Lin YS, Derda R, Org Biomol Chem 2018, 16, 7588–7594. [DOI] [PubMed] [Google Scholar]
- [15].McLafferty MA, Kent RB, Ladner RC, Markland W, Gene 1992, 128, 29–36; [DOI] [PubMed] [Google Scholar]; Koivunen E, Wang B, Ruoslahti E, Biotechnology (N Y) 1995, 13, 265–270; [DOI] [PubMed] [Google Scholar]; O’Neil KT, Hoess RH, Jackson SA, Ramachandran NS, Mousa SA, DeGrado WF, Proteins 1992, 14, 509–515. [DOI] [PubMed] [Google Scholar]
- [16].Desimmie BA, Humbert M, Lescrinier E, Hendrix J, Vets S, Gijsbers R, Ruprecht RM, Dietrich U, Debyser Z, Christ F, Mol. Ther 2012, 20, 2064–2075; [DOI] [PMC free article] [PubMed] [Google Scholar]; Choi DS, Jin HE, Yoo SY, Lee SW, Bioconjugate Chem. 2014, 25, 216–223; [DOI] [PubMed] [Google Scholar]; Meyer SC, Gaj T, Ghosh I, Chem. Biol. Drug Des. 2006, 68, 3–10; [DOI] [PubMed] [Google Scholar]; Katz BA, Biochemistry 1995, 34, 15421–15429; [DOI] [PubMed] [Google Scholar]; Healy JM, Murayama O, Maeda T, Yoshino K, Sekiguchi K, Kikuchi M, Biochemistry 1995, 34, 3948–3955. [DOI] [PubMed] [Google Scholar]
- [17].Heinis C, Rutherford T, Freund S, Winter G, Nat Chem Biol 2009, 5, 502–507; [DOI] [PubMed] [Google Scholar]; Kale SS, Villequey C, Kong XD, Zorzi A, Deyle K, Heinis C, Nat Chem 2018, 10, 715–723; [DOI] [PubMed] [Google Scholar]; Diderich P, Bertoldo D, Dessen P, Khan MM, Pizzitola I, Held W, Huelsken J, Heinis C, ACS Chem Biol 2016, 11, 1422–1427; [DOI] [PubMed] [Google Scholar]; Baeriswyl V, Rapley H, Pollaro L, Stace C, Teufel D, Walker E, Chen S, Winter G, Tite J, Heinis C, ChemMedChem 2012, 7, 1173–1176; [DOI] [PubMed] [Google Scholar]; Angelini A, Cendron L, Chen S, Touati J, Winter G, Zanotti G, Heinis C, ACS Chem Biol 2012, 7, 817–821. [DOI] [PubMed] [Google Scholar]
- [18].Heinis C, Rutherford T, Freund S, Winter G, Nat. Chem. Biol 2009, 7, 502–507; [DOI] [PubMed] [Google Scholar]; Jafari MR, Deng L, Kitov PI, Ng S, Matochko WL, Tjhung KF, Zeberoff A, Elias A, Klassen JS, Derda R, ACS Chem. Biol 2014, 9, 443–450; [DOI] [PubMed] [Google Scholar]; Bellotto S, Chen S, Rebollo IR, Wegner HA, Heinis C, J. Am. Chem. Soc 2014, 136, 5880–5883. [DOI] [PubMed] [Google Scholar]
- [19].Kather I, Bippes CA, Schmid FX, J Mol Biol 2005, 354, 666–678. [DOI] [PubMed] [Google Scholar]
- [20].Tian F, Tsao ML, Schultz PG, J Am Chem Soc 2004, 126, 15962–15963; [DOI] [PubMed] [Google Scholar]; Liu CC, Mack AV, Brustad EM, Mills JH, Groff D, Smider VV, Schultz PG, J Am Chem Soc 2009, 131, 9616–9617; [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu CC, Choe H, Farzan M, Smider VV, Schultz PG, Biochemistry 2009, 48, 8891–8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Smith JM, Frost JR, Fasan R, J Org Chem 2013, 78, 3525–3531; [DOI] [PubMed] [Google Scholar]; Kawakami T, Ishizawa T, Fujino T, Reid PC, Suga H, Murakami H, ACS Chem Biol 2013, 8, 1205–1214; [DOI] [PubMed] [Google Scholar]; Xiang Z, Ren H, Hu YS, Coin I, Wei J, Cang H, Wang L, Nat Methods 2013, 10, 885–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Xiang Z, Lacey VK, Ren H, Xu J, Burban DJ, Jennings PA, Wang L, Angew Chem Int Ed Engl 2014, 53, 2190–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Srinivasan G, James CM, Krzycki JA, Science 2002, 296, 1459–1462. [DOI] [PubMed] [Google Scholar]
- [24].Lee YJ, Wu B, Raymond JE, Zeng Y, Fang X, Wooley KL, Liu WR, ACS Chem Biol 2013, 8, 1664–1670; [DOI] [PMC free article] [PubMed] [Google Scholar]; Lee YJ, Kurra Y, Liu WR, Chembiochem 2016, 17, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang XS, Lee YJ, Liu WR, Chem Commun (Camb) 2014, 50, 3176–3179. [DOI] [PubMed] [Google Scholar]
- [26].Kaya E, Vrabel M, Deiml C, Prill S, Fluxa VS, Carell T, Angew Chem Int Ed Engl 2012, 51, 4466–4469. [DOI] [PubMed] [Google Scholar]
- [27].Chakrabarti A, Oehme I, Witt O, Oliveira G, Sippl W, Romier C, Pierce RJ, Jung M, Trends Pharmacol Sci 2015, 36, 481–492; [DOI] [PubMed] [Google Scholar]; Lopez JE, Haynes SE, Majmudar JD, Martin BR, Fierke CA, J Am Chem Soc 2017, 139, 16222–16227; [DOI] [PMC free article] [PubMed] [Google Scholar]; Tian Y, Wong VW, Wong GL, Yang W, Sun H, Shen J, Tong JH, Go MY, Cheung YS, Lai PB, Zhou M, Xu G, Huang TH, Yu J, To KF, Cheng AS, Chan HL, Cancer Res 2015, 75, 4803–4816. [DOI] [PubMed] [Google Scholar]
- [28].Ingham OJ, Paranal RM, Smith WB, Escobar RA, Yueh H, Snyder T, Porco JA Jr., Bradner JE, Beeler AB, ACS Med Chem Lett 2016, 7, 929–932; [DOI] [PMC free article] [PubMed] [Google Scholar]; Rettig I, Koeneke E, Trippel F, Mueller WC, Burhenne J, Kopp-Schneider A, Fabian J, Schober A, Fernekorn U, von Deimling A, Deubzer HE, Milde T, Witt O, Oehme I, Cell Death Dis 2015, 6, e1657; [DOI] [PMC free article] [PubMed] [Google Scholar]; Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ, Leukemia 2008, 22, 1026–1034; [DOI] [PubMed] [Google Scholar]; Heimburg T, Chakrabarti A, Lancelot J, Marek M, Melesina J, Hauser AT, Shaik TB, Duclaud S, Robaa D, Erdmann F, Schmidt M, Romier C, Pierce RJ, Jung M, Sippl W, J Med Chem 2016, 59, 2423–2435. [DOI] [PubMed] [Google Scholar]
- [29].Wegener D, Wirsching F, Riester D, Schwienhorst A, Chem Biol 2003, 10, 61–68. [DOI] [PubMed] [Google Scholar]
- [30].Dowling DP, Gattis SG, Fierke CA, Christianson DW, Biochemistry 2010, 49, 5048–5056; [DOI] [PMC free article] [PubMed] [Google Scholar]; Marek M, Kannan S, Hauser AT, Moraes Mourao M, Caby S, Cura V, Stolfa DA, Schmidtkunz K, Lancelot J, Andrade L, Renaud JP, Oliveira G, Sippl W, Jung M, Cavarelli J, Pierce RJ, Romier C, PLoS Pathog 2013, 9, e1003645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfi A, De Francesco R, Steinkuhler C, Di Marco S, EMBO Rep 2007, 8, 879–884; [DOI] [PMC free article] [PubMed] [Google Scholar]; Tabackman AA, Frankson R, Marsan ES, Perry K, Cole KE, J Struct Biol 2016, 195, 373–378; [DOI] [PMC free article] [PubMed] [Google Scholar]; Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW, Structure 2004, 12, 1325–1334; [DOI] [PubMed] [Google Scholar]; Porter NJ, Christianson DW, ACS Chem Biol 2017, 12, 2281–2286; [DOI] [PMC free article] [PubMed] [Google Scholar]; Decroos C, Bowman CM, Moser JA, Christianson KE, Deardorff MA, Christianson DW, ACS Chem Biol 2014, 9, 2157–2164; [DOI] [PMC free article] [PubMed] [Google Scholar]; Banerjee S, Adhikari N, Amin SA, Jha T, Eur J Med Chem 2019, 164, 214–240. [DOI] [PubMed] [Google Scholar]
- [32].Xuan W, Li J, Luo X, Schultz PG, Angew Chem Int Ed Engl 2016, 55, 10065–10068; [DOI] [PubMed] [Google Scholar]; Xuan W, Shao S, Schultz PG, Angew Chem Int Ed Engl 2017, 56, 5096–5100; [DOI] [PMC free article] [PubMed] [Google Scholar]; Furman JL, Kang M, Choi S, Cao Y, Wold ED, Sun SB, Smider VV, Schultz PG, Kim CH, J Am Chem Soc 2014, 136, 8411–8417; [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang N, Yang B, Fu C, Zhu H, Zheng F, Kobayashi T, Liu J, Li S, Ma C, Wang PG, Wang Q, Wang L, J Am Chem Soc 2018, 140, 4995–4999; [DOI] [PMC free article] [PubMed] [Google Scholar]; Chen XH, Xiang Z, Hu YS, Lacey VK, Cang H, Wang L, ACS Chem Biol 2014, 9, 1956–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.