

Abstract
A palladium-catalyzed dearomative syn-1,4-oxyamination protocol using non-activated arenes has been developed. This one-pot procedure utilizes arenophile chemistry, and the corresponding para-cycloadducts are treated with oxygen nucleophiles via formal allylic substitution, providing direct access to syn-1,4-oxyaminated products. The reaction conditions permit a range of arenes, as well as different O-nucleophiles, such as oximes and benzyl alcohols. Moreover, this process was established in an asymmetric fashion, delivering products with high enantioselectivity. The dearomatized products are amenable to a multitude of further derivatizations ranging from olefin chemistry to C–H activation, giving rise to a diverse set of new functionalities. Overall, this dearomative functionalization offers rapid and controlled formation of molecular complexity, enabling straightforward access to functionalized small molecules from simple and readily available arenes.
Keywords: arene, arenophile, dearomatization, oxyamination, photochemistry
Graphical Abstract
Escape from flatland:
A dearomative strategy is reported based on combination of arenophile chemistry and Pd catalysis. Simple, non-activated arenes were readily converted into the corresponding syn-1,4-oxyaminated products using oximes or benzyl alcohols as O-nucleophiles. This process delivers products with an exclusive diastereoselectivity, amenable to myriad of further elaborations, and highly enantioselective variant was established with naphthalene.
New synthetic methodologies that introduce heteroatom functionality onto readily available hydrocarbon sources have brought about a paradigm shift in organic synthesis. Recent advances in the areas of C–H activation and functionalization of unsaturated systems have drastically simplified the preparation of small functionalized molecules.[1] The field of π–functionalization chemistry has seen exponential growth, specifically involving alkenes,[2] alkynes,[3] and allenes;[4] however, the formal π–functionalizations of non-activated aromatic systems are underdeveloped.[5] Such processes are particularly challenging as the loss of aromatic stabilization during the functionalization event yields unsaturated products that are more reactive that their parent arenes (Figure 1A). Consequently, overreaction or decomposition are commonly observed. An elegant strategy to control the reactivity of intermediates during the dearomative functionalization is the stoichiometric use of transition metals.[6] For example, certain metal-bound arene complexes can undergo elaborate olefin-like functionalizations that stop at the metal-diene stage, revealing the desired products after oxidative decomplexation (Figure 1B).[7] While powerful, this methodology often requires metal complexes that are toxic or expensive.
Figure 1.

(A) Overreaction and decomposition are the major challenges associated with olefin-like dearomative functionalization. (B) Transition-metal-mediated dearomative functionalizations. (C) Palladium-catalyzed dearomative syn-1,4-oxyamination (this work). (D) Examples of biologically active compounds containing the syn-1-amino-4-hydroxycyclohexane scaffold. TM = transition metal complex; E = electrophile; Nu = nucleophile; DNA = deoxyribonucleic acid.
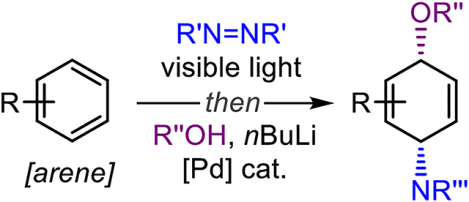
We have introduced a conceptually distinct two-stage dearomative functionalization strategy based on small organic molecules – arenophiles.[8] An arene undergoes visible-light-induced cycloaddition with an arenophile, N-methyl-1,2,4-triazoline-3,5-dione (MTAD, 1, Figure 1C), to induce dearomatization and provide cycloadduct intermediate I. This cycloadduct can be manipulated in situ to furnish a range of different dearomatized products.[9] Salient features of this approach include the mild conditions for dearomatization, the direct application of non-activated arenes, and the diverse elaboration opportunities for the dearomatized products. Since such dearomatizations deliver products that are not readily accessible by any other chemical or biological means,[10] we have been exploring the scope of these transformations further. Herein, we showcase that the use of arenophile chemistry in combination with Pd-catalysis and oxygen nucleophiles provides the means for dearomative syn-1,4-oxyamination (Figure 1C).
We have recently reported arenophile-mediated, Pd-catalyzed dearomatizations that are characterized by exclusive syn-1,4-selectivity.[11] Mechanistically, these transformations proceed through Pd-mediated allylic substitution with a double inversion or net retention stereochemistry.[12] In addition to carbon- and nitrogen-based nucleophiles, we were keen to explore oxygen nucleophiles, as the resulting syn-1,4-hydroxyaminocyclohexane motifs are ubiquitous in natural products and biologically-relevant compounds (Figure 1D). For example, such aminocyclitol subunits are found in glycoside inhibitor conduramine F-1 (2),[13] antifungal natural product salbostatin (3), [14] and metabolite 4,[15] derived from a highly carcinogenic polyaromatic environmental pollutant. Considering the importance of such compounds, as well as the lack of methods to prepare them,[16] we report herein the realization of a Pd-catalyzed dearomative aminohydroxylation strategy. Thus, a range of non-activated aromatic compounds was selectively transformed to the corresponding syn-1,4-aminohydroxylated unsaturated products using oximes or benzylic alcohols as O-nucleophiles. Moreover, in the case of naphthalene, this dearomatization could be conducted in an enantioselective fashion. Finally, the products of this method offer numerous possibilities for further elaboration, and rapidly provide functionalized small molecules that would be difficult to access by other methods.
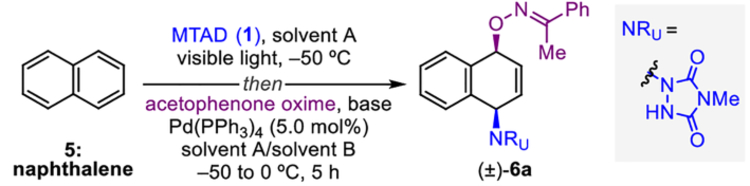
In our initial foray into this chemistry, naphthalene (5) and MTAD (1) underwent cycloaddition under visible light in dichloromethane at −50 °C, followed by the addition of a palladium catalyst and acetophenone oxime, and the reaction was slowly warmed to 0 °C over 5 h (Table 1). While [Pd(PPh3)4] was readily identified as the optimal catalyst, the choice of base played a significant role on the reaction outcome (for full optimization data, see Supporting Information). For example, no reaction was observed in the absence of base, and the use of KOtBu in THF gave only 5% yield of dearomatized product (±)-6a (entry 1). Gratifyingly, stronger bases such as LHMDS and nBuLi gave noticeably higher yields (entries 2 and 3), indicating the necessity of deprotonated oxime. Further optimization studies revealed that solvent effects were crucial for the reaction efficiency. Specifically, propionitrile was found to be the optimal solvent for the cycloaddition step (entries 3–6), and toluene proved ideal for the palladium catalyzed nucleophilic attack (entries 6–8). Thus, an overall 70% isolated yield of the syn-1,4-oxyaminated product was obtained by using EtCN/PhMe system (entry 8). Importantly, the structure of syn-1,4-oxyaminated product (±)-6a was confirmed by X-ray crystallographic analysis (see Table 2), and careful analysis of crude reaction mixture did not reveal the presence of any other constitutional and diastereoisomers.
Table 1.
Optimization of reaction conditions.[a]
 | ||||
|---|---|---|---|---|
| entry | solvent A | solvent B | base | yield (%)[b] |
| 1 | CH2Cl2 | THF | KOtBu | 5 |
| 2 | CH2Cl2 | THF | LHMDS | 35 |
| 3 | CH2Cl2 | THF | nBuLi | 42 |
| 4 | Acetone | THF | nBuLi | 21 |
| 5 | EtOAc | THF | nBuLi | 10 |
| 6 | EtCN | THF | nBuLi | 52 |
| 7 | EtCN | Et2O | nBuLi | 43 |
| 8 | EtCN | PhMe | nBuLi | 71 (70) |
Reaction conditions: naphthalene (5, 1.0 mmol, 2.0 eq.), MTAD (1, 0.5 mmol, 1.0 eq.), solvent A (0.1 M), visible light, −50 °C; then addition of solution of acetophenone oxime (1.0 mmol, 2.0 eq.) and base (0.95 mmol, 1.9 eq.) in solvent B; and Pd(PPh3)4 (5.0 mol%), −50 to 0 °C, 5 h.
Determined by 1H NMR analysis relative to the internal standard. Isolated yield shown in parentheses. THF=tetrahydrofuran, LHMDS=lithium bis(trimethylsilyl)amide
Table 2.
Nucleophile scope for Pd-catalyzed dearomative syn-1,4-oxyamination.[a]
|
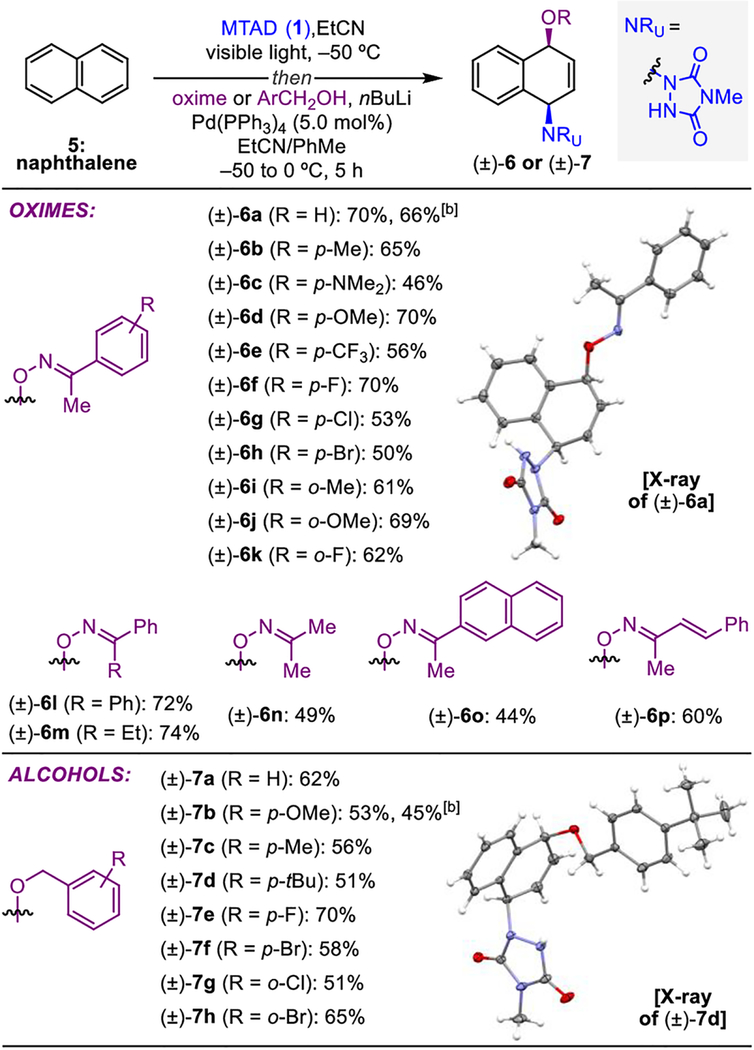
Reaction conditions: naphthalene (5, 1.0 mmol, 2.0 eq.), MTAD (1, 0.5 mmol, 1.0 eq.), EtCN (0.1 M), visible light, −50 °C; then addition of solution of acetophenone oxime or ArCH2OH (1.0 mmol, 2.0 eq.) and nBuLi (0.95 mmol, 1.9 eq.) in PhMe; and Pd(PPh3)4 (5.0 mol%), −50 to 0 °C, 5 h. Yields of isolated product after column chromatography. [b] Yield of reaction conducted on a gram scale. The pictured X-ray structure is shown with thermal ellipsoids at 30% probability.
With optimal conditions established, the scope of oximes was explored in detail using naphthalene (5) as the standard substrate (Table 2, top). In addition to acetophenone-derived oxime (±)-6a, para-substituted derivatives containing electron neutral [(±)-6b], donating [(±)-6c and (±)-6d], and withdrawing groups [(±)-6e] were compatible with this transformation, as well as halogens [(±)-6f–h]. Likewise, dearomatizations with sterically more demanding ortho-substituted analogs [(±)-6i–k] proceeded smoothly. Moreover, oximes derived from other ketones such as benzophenone [(±)-6l], propiophenone [(±)-6m], acetone [(±)-6n], acetonaphthone [(±)-6o], and (E)-4-phenyl-3-buten-2-one [(±)-6p], proved viable to generate the corresponding syn-1,4-oxyaminated products. Finally, the scalability of this process was tested using acetophenone oxime, and a gram-scale reaction provided a comparable yield of product (±)-6a. Importantly, in all cases, syn-1,4-oxyaminated products were obtained as single constitutional and diastereoisomers.
Furthermore, other oxygen-based reagents were tested under the described conditions, leading to the discovery that benzyl alcohols were also viable nucleophiles for this functionalization process (Table 2, bottom).[17] Thus, benzyl alcohols incorporating a range of diverse para-substituents delivered syn-1,4-oxyaminated products [(±)-7a–f]. As above, all products were obtained as single diastereoisomers; in the case of (±)-7d the relative stereochemistry was further unambiguously confirmed by X-ray analysis. Likewise, the ortho-substituted benzyl alcohols afforded the desired products [(±)-7g and (±)-7h], and a gram-scale reaction with 4-methoxybenzyl alcohol provided product (±)-7b without significant erosion in yield.
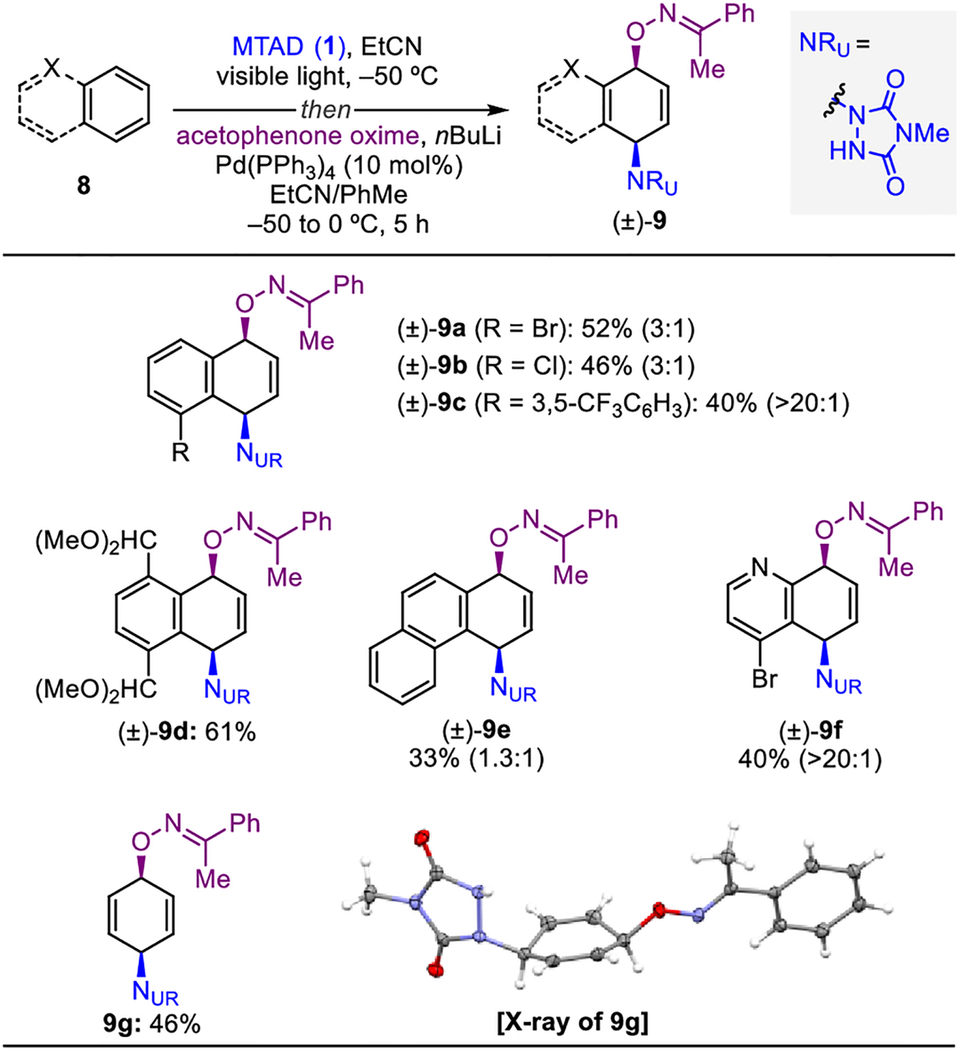
Next, the scope of arenes was examined using acetophenone oxime as the O-nucleophile (Table 3). Exploration of different naphthalene derivatives [(±)-9a–d] revealed lower efficiency compared to the parent naphthalene [(±)-6a, see Table 2]. Though several conditions and Pd-based catalysts were additionally examined with these substrates, none of them resulted in increased yields.[18] Nevertheless, a range of substituted naphthalenes encompassing halides [(±)-9a and (±)-9b], aryl [(±)-9c], and acetal [(±)-9d] functionality gave desired products, with ratio of constitutional isomers ranging from 3:1 to >20:1. The higher selectivity for product (±)-9c, resulting from ring opening at different sites of arene−arenophile cycloadduct, is attributed to a stronger steric bias from the adjacent peri-substituent. Similarly, dearomative syn-1,4-oxyamination involving higher order arenes, such as phenanthrene [(±)-9e], worked under the described conditions, albeit with diminished yield and regioselectivity. Importantly, a heteroaromatic quinoline derivative and benzene were competent substrates for this protocol, with the former delivering highly functionalized product [(±)-9f]. The latter furnished skipped cyclohexadiene derivative 9g, for which the relative syn-1,4-stereochemistry was confirmed by X-ray crystallographic analysis. Unfortunately, although substituted benzene substrates successfully underwent photocycloaddition with MTAD, their corresponding arene-MTAD cycloadducts showed no reactivity towards the Pd-mediated ring-opening step.
Table 3.
Arene scope for Pd-catalyzed dearomative syn-1,4-oxyamination.[a]
|
Reaction conditions: arene (1.0 mmol, 2.0 eq.) or benzene (5.0 mmol, 10 eq.), MTAD (1, 0.5 mmol, 1.0 eq.), EtCN (0.1 M), visible light, −50 °C; then addition of solution of acetophenone oxime (1.0 mmol, 2.0 eq.) and nBuLi (0.95 mmol, 1.9 eq.) in PhMe; and Pd(PPh3)4 (10 mol%), −50 to 0 °C, 5 h. Yields of isolated product after column chromatography. Ratio of the constitutional isomers were determined by 1H NMR analysis of the crude reaction mixture (shown within parentheses). The pictured X-ray structure is shown with thermal ellipsoids at 30% probability.
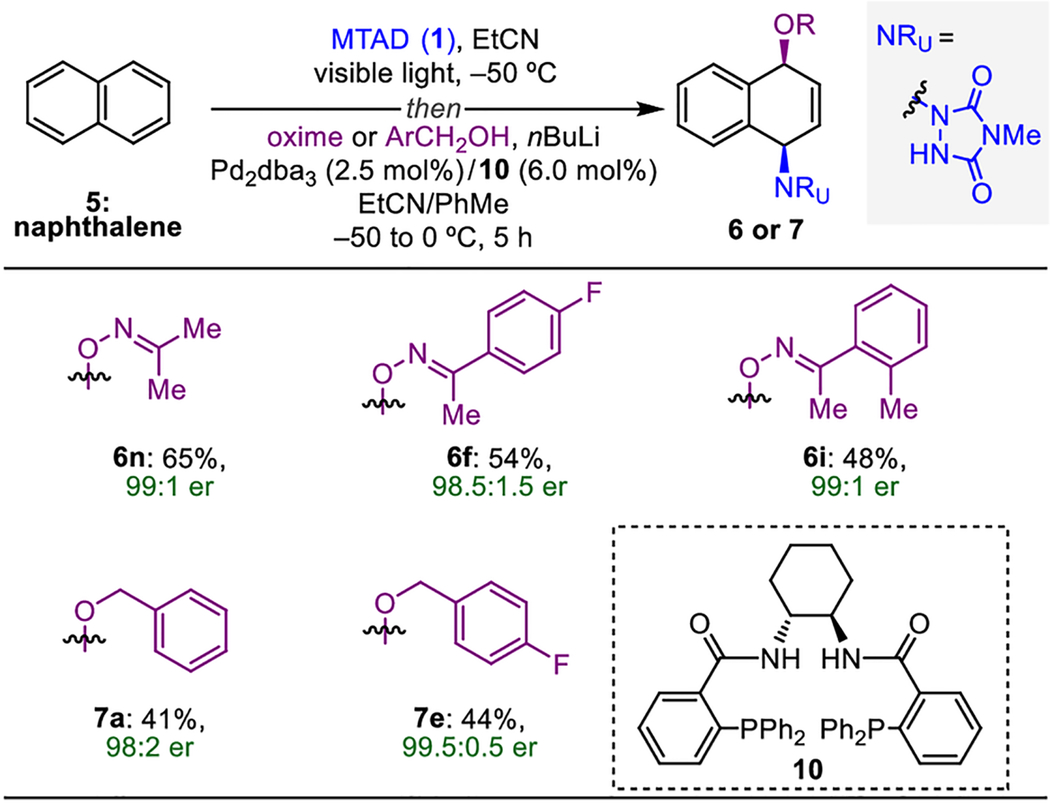
Considering that asymmetric dearomatizations of non-activated arenes are rare and underdeveloped,[19] this catalytic dearomative functionalization was explored via desymmetrization of naphthalene with different O-nucleophiles (Table 4). Thus, upon examination of various chiral complexes, a combination of [Pd2dba3] and (S,S)-DACH-Phenyl Trost ligand (10)[20] was found to be highly selective in this asymmetric transformation. For example, by using the acetone-based oxime, product 6n was obtained with 65% yield and 99:1 er. Moreover, p-fluoro- or o-methyl-substituted acetophenone oximes gave desired products 6f and 6i with >98:2 er. Benzyl alcohol could also be used, as demonstrated with products 7a and 7e (≥98:2 er).
Table 4.
Enantioselective Pd-catalyzed dearomative syn-1,4-oxyamination.[a]
|
Reaction conditions: naphthalene (5, 1.0 mmol, 2.0 eq.), MTAD (1, 0.5 mmol, 1.0 eq.), EtCN (0.1 M), visible light, −50 °C; then addition of solution of oxime or ArCH2OH (1.0 mmol, 2.0 eq.) and nBuLi (0.95 mmol, 1.9 eq.) in PhMe; and Pd2dba3 (2,5 mol%), ligand 10 (6.0 mol%), −50 to 0 °C, 5 h. Yields of isolated product after column chromatography. Enantiomeric excess determined by HPLC analysis on a chiral stationary phase. dba=dibenzylideneacetone.
The dearomative syn-1,4-oxyamination generates products susceptible to rapid elaboration to a diverse set of functionalized small molecules (Figure 2). Overall, alicyclic compounds with various degrees of functionalization were prepared from simple arenes in just a few steps. For example, the oxyaminated product (±)-7b, derived from naphthalene and para-methoxybenzyl alcohol, was readily converted to syn-1,4-oxyaminated tetrahydronaphthalene 12 through a sequence involving chemoselective reduction of the olefin moiety [(±)-7b→11], alkylation of the urazole, and subsequent base-induced urazole fragmentation.[21] On the other hand, osmium-catalyzed dihydroxylation [(±)-7b→13],[22] followed by urazole oxidation with household bleach[23] afforded trihydroxyketone derivative 14.
Figure 2.

Derivatization of dearomatized products (±)-7b, (±)-6a, and (±)-6n. Reagents and conditions: a) Rh/Al2O3 (cat.), H2, 98%; b) 1. α-bromoacetophenone, K2CO3, 85 %; 2. KOH, 95%; c) OsO4, NMO, 72%; d) NaOCl, 50%; e) 1. Rh/Al2O3 (cat.), H2, 85%; f) tBuOCl, H2O 52%; g) tBuOCl, 64%, 4:1 dr; h) 1. Zn, AcOH, 80%; 2. tBuOCl, H2O 60%; i) 1. Rh/Al2O3 (cat.), H2, 82%; 2. MeI, K2CO3, 80%; j) Pd(OAc)2, PIDA, 74%; k) Pd(OAc)2, tBuOOH, PhCHO, 87%. PMB=para-methoxybenzyl; NMO=N-methylmorpholine-N-oxide, PIDA=phenyliodine diacetate.
Likewise, dearomatized products with oxime functionality could be further elaborated, as exemplified by the conversion of compound (±)-6a to different ketones 16–18. Thus, the oxidation of reduced product 15 with aqueous bleach solution gave oxime-protected γ-hydroxytetralone 16, while using tert-butyl hypochlorite under anhydrous conditions provided α–chloro-γ–hydroxytetralone 17. The oxime protecting group could be cleaved using zinc and acetic acid, and subsequent oxidation of urazole under aqueous conditions provided γ-hydroxytetralone 18, a high-value added bioactive compound ($380 g−1, Aldrich).[24] Notably, based on recent advances in C–H activation, the installed oxime could also serve as a powerful directing group for the introduction of functional handles on the neighbouring aromatic ring.[25] This type of oxime exo-directing group is highly versatile;[26] however, it previously required installation through manipulation of the pre-existing alcohol. On the other hand, dearomative oxyamination directly introduces this handle during a single functionalization event. Thus, acetone oxime-derived compound 19, prepared by chemoselective hydrogenation and urazole methylation of (±)-6n, was used for the introduction of O- and C-based functional groups. Oxime directed, Pd-catalyzed C–H bond activation in the presence of PIDA successfully incorporated an acetate group to provide 20.[27] Similarly, Pd-mediated oxidation with tert-butyl hydrogen peroxide in the presence of benzaldehyde enabled direct ortho-aroylation, affording ketone 21.[28]
In summary, a palladium-catalyzed dearomative syn-1,4-oxyamination of simple and nonactivated arenes was developed based on the arenophile-mediated cycloaddition strategy. A range of arenes underwent this process with oximes or benzyl alcohols as O-nucleophiles, providing products with an exclusive syn-1,4-selectivity. Moreover, a highly enantioselective process (≥98:2 er) has been established using naphthalene. This dearomative difunctionalization protocol forms products that are amenable to an array of further derivatizations as demonstrated with facile preparation of highly oxidized alicyclic motifs from simple non-activated aromatic precursors. As numerous bioactive compounds encompass aminocyclitols or similarly functionalized motifs, this method is expected to find application in the synthesis of natural products and medicinally-relevant compounds.
Supplementary Material
Acknowledgements
Financial support for this work was provided by the University of Illinois, the National Science Foundation (CAREER Award No. CHE-1654110), the NIH/National Institute of General Medical Sciences (R01 GM122891), and the donors of the American Chemical Society Petroleum Research Fund (PRF#57175-DNI1). D.S. is an Alfred P. Sloan Fellow. M.O. thanks the Honjo International Scholarship Foundation. Solvias AG is acknowledged for a generous gift of chiral ligands. We also thank Dr. D. Olson and Dr. L. Zhu for NMR spectroscopic assistance, Dr. D. L. Gray and Dr. T. Woods for X-ray crystallographic analysis assistance, and F. Sun for mass spectrometric assistance.
References
- [1].a) Godula AK, Sames D, Science 2006, 312, 67–72; [DOI] [PubMed] [Google Scholar]; b) Kakiuchi F, Chatani N, Adv. Synth. Catal 2003, 345, 1077–1101; [Google Scholar]; c) Davies HML, Du Bois J, Yu J-Q, Chem. Soc. Rev 2011, 40, 1855–1856; [DOI] [PubMed] [Google Scholar]
- [2].a) Beller M, Seayad J, Tillack A, Jiao H, Angew. Chem. Int. Ed 2004, 43, 3368–3398; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2004, 116, 3448–3479; [Google Scholar]; b) Coombs JR, Morken JP, Angew. Chem. Int. Ed 2016, 55, 2636–2649; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 2682–2696. [Google Scholar]
- [3].a) Acetylene Chemistry. Chemistry, Biology and Material Science, Diederich F, Stang PJ and Tykwinski RR, Wiley-VCH, Weinheim, 2005; [Google Scholar]; b) Chinchilla R, Nájera C, Chem. Rev 2014, 114, 1783–1826. [DOI] [PubMed] [Google Scholar]
- [4].Yu S, Ma S, Angew. Chem. Int. Ed 2012, 51, 3074–3112; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2012, 124, 3128–3167. [Google Scholar]
- [5].Wertjes WC, Southgate EH, Sarlah D, Chem. Soc. Rev 2018, 47, 7996–8017. [DOI] [PubMed] [Google Scholar]
- [6].Pape AR, Kaliappan KP, Kündig EP, Chem. Rev 2000, 100, 2917–2940. [DOI] [PubMed] [Google Scholar]
- [7].For a recent review on this topic, see: Liebov BK, Harman WD, Chem. Rev 2017, 117, 13721–13755. [DOI] [PubMed] [Google Scholar]
- [8].a) Hamrock SV, Sheridan RS, J. Am. Chem. Soc 1989, 111, 9247–9249; [Google Scholar]; b) Southgate EH, Pospech J, Fu J, Holycross DR, Sarlah D, Nat. Chem 2016, 8, 922–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Okumura M, Sarlah D, Synlett, 2018, 29, 845–855. [Google Scholar]
- [10].For an overview of dearomative chemistry in synthesis, see: Roche SP, Porco JA Jr, Angew. Chem. Int. Ed 2011, 50, 4068–4093; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2011, 123, 4154–4179. [Google Scholar]
- [11].a) Okumura M, Shved AS, Sarlah D, J. Am. Chem. Soc 2017, 139, 17787–17790; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wertjes WC, Okumura M, Sarlah D, J. Am. Chem. Soc 2019, 141, 163–167; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tang C, Okumura M, Zhu Y, Hooper AR, Zhou Y, Lee Y-H, Sarlah D, Angew. Chem. Int. Ed 2019, 58, 10245–10249; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 10351–10355. [Google Scholar]
- [12].a) Trost BM, Van Vranken DL, Chem. Rev 1996, 96, 395–422; [DOI] [PubMed] [Google Scholar]; b) Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis in Topics in Organometallic Chemistry, Vol. 38, Ed. Kazmaier U, Springer, Heidelberg, 2012, pp. 1–63. [Google Scholar]
- [13].Lysek R, Schutz C, Favre S, O’Sullivan A C, Pillonel C, Krulle T, Jung P M J, Clotet-Codina I, Este J A, Vogel P, Bioorg. Med. Chem 2006, 14, 6255–6282. [DOI] [PubMed] [Google Scholar]
- [14].Vertesy L, Fehlhaber H-W, Schulz A, Angew. Chem. Int. Ed 1994, 33, 1844–1846; [Google Scholar]; Angew. Chem 1994, 106, 1936–1937. [Google Scholar]
- [15].Jeffrey M, Weinstein IB, Jennette K, Grzeskowiak K, Nakanishi K, Harvey RG, Autrup H, Harris C, Nature 1977, 269, 348–350. [DOI] [PubMed] [Google Scholar]
- [16].For selected reviews on preparation of aminocyclitols, see:a) Yang Y, Yu B, Chem. Rev 2017, 117, 12281–12356; [DOI] [PubMed] [Google Scholar]; b) Duchek J, Adams DR, Hudlicky T, Chem. Rev 2011, 111, 4223–4258; [DOI] [PubMed] [Google Scholar]; c) Arjona O, Gómez AM, López JC, Plumet J, Chem. Rev 2007, 107, 1919–2036. [DOI] [PubMed] [Google Scholar]
- [17]. Other type of alcohols, as well as phenols and carboxylic acids did not react under the described conditions. See Supporting Information for full details.
- [18]. Low yields of these substrates were attributed to poor conversion. Only unreacted arenes were recovered, resulting from retrocycloaddition of MTAD-arene adducts. Different reaction times and temperature profile manipulations did not improve conversion.
- [19].Mainly phenols and heteroarenes are subject to such processes. For recent reviews on this topic, see: a) Zheng C, You S-L, Chem, 2016, 1, 830–857; [Google Scholar]; b) Zhuo C-X, Zhang W, You S-L, Angew. Chem. Int. Ed 2012, 51, 12662–12686; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2012, 124, 12834–12858. [Google Scholar]
- [20].Trost BM, VanVranken D, Angew. Chem. Int. Ed 1992, 31, 228–230; [Google Scholar]; Angew. Chem 1992, 104,194–196. [Google Scholar]
- [21].Adam W, Pastor A, Wirth T, Org. Lett 2000, 2, 1295–1298. [DOI] [PubMed] [Google Scholar]
- [22].VanRheenen V, Kelly RC, Cha DY, Tetrahedron Lett 1976, 17, 1973–1976. [Google Scholar]
- [23].Wilson RM, Hengge ACJ, Org. Chem 1990, 55, 197–202. [Google Scholar]
- [24].Fournet A, Barrios A, Muñoz V, Hocquemiller R, Roblot F, Cavé A, Planta Med 1994, 60, 8–12. [DOI] [PubMed] [Google Scholar]
- [25].Lyons TW, Sanford MS, Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].For the use of oximes as exo-directing group, see: a) Ren Z, Mo F, Dong G, J. Am. Chem. Soc 2012, 134, 16991–16994; [DOI] [PubMed] [Google Scholar]; for selected recent applications, see: b) Mao Y-J, Lou S-J, Hao H-Y, Xu D-Q, Angew. Chem. Int. Ed 2018, 57, 14085–14089; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 14281–14285; [Google Scholar]; c) Guo K, Chen X, Guan M, Zhao Y, Org. Lett 2015, 17, 1802–1805; [DOI] [PubMed] [Google Scholar]; d) Guo K, Chen X, Zhang J, Zhao Y, Chem. Eur. J 2015, 21, 17474–17478; [DOI] [PubMed] [Google Scholar]; e) see also Ref. 28.
- [27].Desai LV, Stowers KJ, Sanford MS, J. Am. Chem. Soc 2008, 130, 13285–13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chan C-W, Zhou Z, Chan ASC, Yu W-Y, Org. Lett 2010, 12, 3926–3929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.