

Abstract
Old World (Africa) and New World (South America) arenaviruses are associated with human hemorrhagic fevers. Efforts to develop small molecule therapeutics have yielded several chemical series including the 4-acyl-1,6-dialkylpiperazin-2-ones. Herein, we describe an extensive exploration of this chemotype. In initial Phase I studies, R1 and R4 scanning libraries were assayed to identify potent substituents against Old World (Lassa) virus. In subsequent Phase II studies, R6 substituents and iterative R1, R4 and R6 substituent combinations were evaluated to obtain compounds with improved Lassa and New World (Machupo, Junin, and Tacaribe) arenavirus inhibitory activity, in vitro human liver microsome metabolic stability and aqueous solubility.
Keywords: Arenavirus, Lassa, Junin, Machupo, Piperazinone, Entry inhibitor
The Arenaviridae family of viruses is comprised of a large number of species, some of which are associated with acute hemorrhagic fevers (HF) in humans.1–3 HF arenaviruses, including Lassa (LASV) and the New World species Machupo (MACV) or Junin (JUNV), represent a serious public health risk, especially in endemic regions of Africa and South America.4–6 Due to the high mortality rates and limited therapeutic options these HF viruses are classified as Category A pathogens by the Centers for Disease Control (CDC).7 LASV, in particular, may be responsible for up to 300,000 human disease cases per year.8–10 Mortality rates for hospitalized LASV HF patients are 15–20% and survivors often suffer permanent sequelae including bilateral hearing damage.11–14 The nonspecific antiviral Ribavirin currently offers the sole therapeutic option, however, its use is restricted to high-risk patients due to its variable efficacy and potentially serious adverse effects.15–17 The development of potent and specific agents to treat Lassa and other arenavirus hemorrhagic fevers is therefore, urgently needed.
To mitigate the significant safety and logistical challenges associated with utilizing Biosafety Level 4 (BSL4) arenaviruses in drug discovery, a number of surrogate BSL2 assays have been developed. For example, a screening campaign using the New World arenavirus Tacaribe virus (TCRV), which is a BSL2 Clade B New World arenavirus that is highly-related to Category BSL4 arenaviruses including JUNV and MACV, was used to identify a novel chemical series (including ST-294) of arenavirus inhibitors.18 Cell-based assays specifically targeting the arenavirus glycoprotein (GP) and cell entry, including pseudotyped virus and cell–cell membrane fusion assays, have also been used to identify or characterize the benzimidazole and 4-acyl-1,6-dialkylpiperazin-2-one chemical series, which share a common GP binding site and mechanism of action.19–23
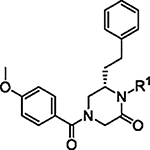
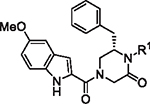
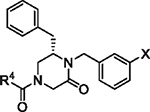
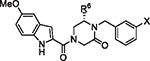
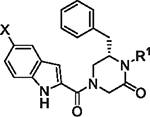
The initial report of the 4-acyl-1,6-dialkylpiperazin-2-one inhibitors identified a set of substituents at the R1, R4 and R6 positions of the 4-acyl-piperazin-2-one core (Fig. 1) that exhibited low micromolar to submicromolar potency against LASV glycoprotein expressed in vesicular stomatitis virus (VSV) pseudotyped viruses (pLASV).20,24,25 Additional investigation of the structure–activity relationships (SAR) of this chemical series identified the carbonyl groups as significant activity determinants and that the enantiomer possessing (S) configuration at C6 (R6 group attachment point) was approximately 15-fold more potent than the (R)-enantiomer.26 The (S)-enantiomers of our initial 4-acyl-1,6-dialkylpiperazin-2-one compounds 1 (16G8) and 2 (17D1) represent eutomers with pLASV EC50 values: rac-16G8 600 nM vs (S)-16G8 300 nM and rac-17D1 500 nM vs (S)-17D1 250 nM.26,27 We therefore utilized the (S)-enantiomers of R6 benzyl, phenethyl and other substituents as starting points for further SAR exploration of this chemical series.
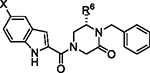
Fig. 1.

Initial 4-acyl-1,6-dialkylpiperazin-2-one starting points and optimization flow scheme.
In Phase I exploration R1 and R4 scanning libraries were synthesized and screened for inhibition of LASV cell entry as determined in a membrane cell–cell fusion assay at 50 nM (Fig. 1). In Phase II studies, iterative SAR modifications at substituent positions R1, R4 and R6 were evaluated in assays with pseudotyped pLASV, pMACV or pJUNV virions expressing LASV, MACV or JUNV glycoproteins, respectively, in order to identify broad-spectrum arenavirus cell entry inhibitors. Selected compounds were subsequently characterized in human liver microsome (HLM) metabolism and solubility assays (Fig. 1).
For R1 scanning library compounds we devised a synthetic route enabling final step diversification at the R1 position starting with an intermolecular Mitsunobu alkylation28 of t-butyl nosylglycine with Boc protected aminoalcohol A to provide B (Scheme 1). The nosyl group was cleaved (thiophenol, K2CO3, DMF) to yield the secondary amine C, which was then coupled with the appropriate carboxylic acid R4COOH (EDCI, HOAt, 2,6-lutidine, DMF) to give the tertiary amide D. Hydrolysis of the t-butyl ester (LiOH, t-BuOH/H2O/THF) followed by Bocdeprotection with TFA (CH2Cl2) provided the amino acid E as the TFA salt. Final step diversification at R1 was completed through a one-pot reductive amination29/EDCI-mediated ring closure starting with intermediate E.
Scheme 1.

Reagents and conditions: (i) PPh3, DIAD, THF, under N2, rt, 2 h; (ii) PhSH, K2CO3, DMF, under N2, rt, 3 h; (iii) R4COOH, HOAt, 2,6-lutidine, EDCI, DMF, rt, 16 h; (iv) LiOH, t-BuOH/H2O/THF, rt, 1 h, then 1 N aq. HCl; (v) TFA, CH2Cl2, rt; (vi) RCHO, NaBH(OAc)3, Et3N, ClCH2CH2Cl, rt, 18 h, then EDCI, NaHCO3, CH2Cl2, rt, 8 h.
A synthetic route enabling final step diversification at the R4 position (Scheme 2) started with commercially available aminoalcohol G. The R1 group was added using a stepwise reductive amination procedure with appropriate aldehyde entailing pre-formation of imine followed by NaBH4 reduction (MeOH) to give intermediate H. Subsequent N-acylation by treatment of H with the acid chloride of nosylglycine (NaHCO3, CH2Cl2) provided I. The 6-membered ring was closed using an intramolecular Mitsunobu alkylation of the sulfonamide28 (PPh3, DIAD, THF) to give J. Nosyl deprotection using thiophenol and K2CO3 (DMF) and subsequent acylation of the free amine with the appropriate carboxylic acid R4COOH (EDCI, HOAt, 2,6-lutidine, DMF) furnished compounds of type K. All compounds were purified by PTLC or flash chromatography and the purity and identity of all compounds was assessed by LC/MS.
Scheme 2.

Reagents and conditions: (i) RCHO, MeOH, rt, 1 h, then NaBH4, 0 °C to rt, 2 h; (ii) 2-(2-nitrophenylsulfonamido)acetyl chloride, NaHCO3, CH2Cl2, rt, 3 h; (iii) PPh3, DIAD, THF, under N2, rt, 2 h; (iv) PhSH, K2CO3, DMF, under N2, rt, 3 h; (v) R4COOH, HOAt, 2,6-lutidine, EDCI, DMF, rt, 14 h.
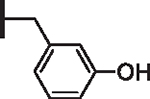
At R1 benzyl was favored over phenethyl or phenylpropyl substituents (Table 1, compare 6, 14 and 16), indicating a defined preference for the linker length at R1. A single aryl ring demonstrated greater activity than naphthalene and biaryl groups (compare 6 with 10, 12, 13 and 15) and the identity of aryl substituents significantly impacted activity while their positional effects were relatively weaker. The incorporation of single methoxy or hydroxy groups revealed a preference for meta-substitution but were well tolerated at any ring position (4 vs. 8 and 9, 5 vs. 7). In contrast, halogen, phenoxy, cyano, nitro, or basic nitrogen substitutions in the ring significantly reduced activity at any position (see Supplementary data, Fig. S1). Overall, the results indicated significant flexibility for incorporation of small lipophilic substituents or polar alcohols on the R1 benzyl ring.
Table 1.
SAR data of selected R1 (left panel) and R4 (right panel) substituent Phase I scanning libraries indicating their % inhibition at 50nM in the LASV GPC cell–cell fusion assay.
|
|
||||
|---|---|---|---|---|---|
| Compd. | R1 | % inh. 50 nM | Compd. | R4 | % inh. 50 nM |
| 3 |  |
86 | 17 | 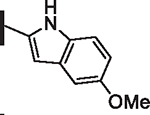 |
66 |
| 4 | 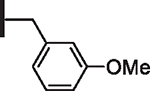 |
82 | 18 | 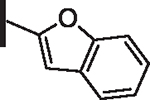 |
61 |
| 5 | 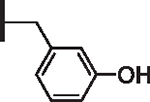 |
81 | 19 | 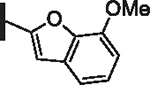 |
59 |
| 6 | 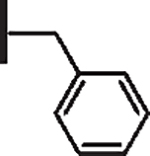 |
71 | 20 | 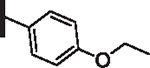 |
58 |
| 7 | 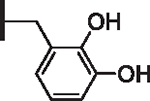 |
64 | 21 | 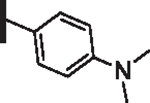 |
56 |
| 8 | 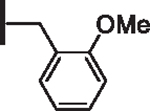 |
61 | 22 | 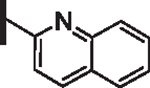 |
54 |
| 9 | 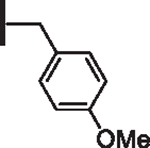 |
60 | 23 | 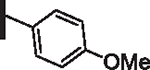 |
50 |
| 10 | 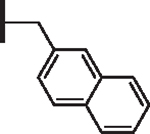 |
58 | 24 | 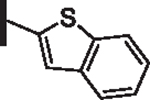 |
45 |
| 11 | 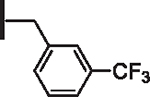 |
53 | 25 | 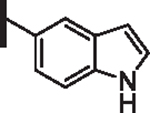 |
40 |
| 12 | 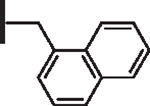 |
51 | 26 | 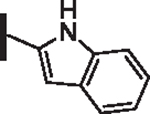 |
26 |
| 13 | 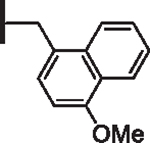 |
36 | 27 | 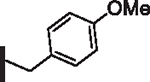 |
18 |
| 14 | 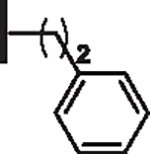 |
34 | 28 | 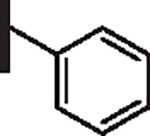 |
15 |
| 15 | 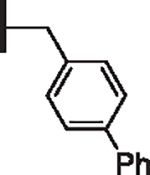 |
33 | 29 | 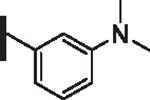 |
14 |
| 16 | 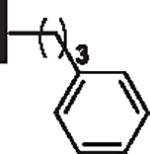 |
31 | 30 | 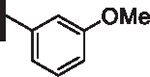 |
0 |
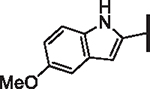
As opposed to R1 substituents, the R4 acyl substituents demonstrated SAR with strong preferences in both the identity and position of aryl functionality (Table 1, Supplementary data, Fig. S2). The two most active classes of aryl groups observed in the original piperazinone study (the 2-heteroaryl (17–19, 22) and the 4-substituted phenyl (20, 21, 23) groups) proved optimal in the R4 scanning library.20 Of note, incorporation of an alkoxy or dimethylamino group at the 4-position of the phenyl ring resulted in a notable increase in potency for compounds 20, 21, and 23 while the absence of a 4-position substituent (28) and/ or the placement of a dimethylamino (29) or alkoxy (30) group at the 3-position resulted in dramatic losses in activity. The addition of a 5-methoxy group to the 2-indole substituent (compounds 17 vs. 26) also provided a notable increase in potency. In contrast, the addition of a single carbon linker (27) to the phenyl ring significantly decreased activity.
Following the individual R1 and R4 replacements, a small combinatorial library was synthesized containing three of the most active R1 substituents (compounds 3–5, Table 1) and R4 acyl groups (compounds 17, 18, and 20, Table 1). These compounds showed low-nanomolar inhibition of LASV GP fusion with no loss of activity when the more polar optimal substituents were combined in the same structure (compounds 31–33, Table 2) suggesting that optimization for lipophilic efficiency (LipE) may lead to potent compounds with improved drug-like properties. These initial scanning libraries allowed the identification of promising R1 and R4 substituents as starting points to initiate optimization of broad-spectrum arenavirus inhibitory activity and drug-like properties.
Table 2.
Analogs incorporating combinations of the most active substituents from the R1and R4 scanning libraries and their respective EC50 values in the LASV GPC cell–cell fusion assay.
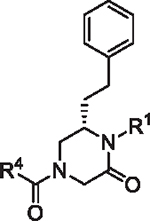
| ||||||
|---|---|---|---|---|---|---|
| R4 | R1 |
|||||
|
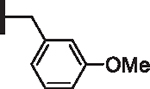
|
|
||||
| Compd. | EC50 (nM) | Compd. | EC50 (nM) | Compd. | EC50 (nM) | |
 |
31 | 5 | 34 | 4 | 37 | 5 |
 |
32 | 8 | 35 | 7 | 38 | 7 |
 |
33 | 7 | 36 | 6 | 39 | 5 |
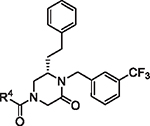
Phase II SAR exploration studies, including characterization of broad-spectrum arenavirus (pseudotyped pLASV, pMACV and pJUNV) activity, human liver microsome (HLM) metabolic stability and solubility, were initiated with the synthesis of analogs containing benzyl or phenethyl at R6, benzyl or m-methoxy benzyl at R1 and 5-methoxyindole at R4. R6 phenethyl was found to exhibit approximately 2–4 fold greater potency over benzyl (Table 3, compare 35, 40, 41 and 42). However, R6 benzyl compounds exhibited greater solubility and metabolic stability in HLM assays. Thus, while 42 was 2–4 fold less potent in pLASV, pMACV and pJUNV assays than 41 it exhibited a substantially increased HLM half-life of 59 vs 10 min and solubility of 36 vs 4 μM.
Table 3.
Comparison of activity of R6 linker length and R1 substituents. Activity is given as EC50 (nM) in the indicated pseudotyped virus assays. Cytotoxicity is given as CC50 in Vero cells (7 days). HLM data is the observed half-life (T1/2 (min)) in a human liver microsome assay. Aqueous solubility (μM) was measured at pH 7.4.
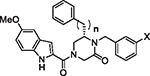
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | n | X | p-LASV | p-MACV | p-JUNV | CC50 (nM) | HLM | Solubility |
| 35 | 2 | OMe | 1.74 | 2.39 | 1.77 | 4,500 | 7.7 | 4.9 |
| 40 | 1 | OMe | 6.9 | 3.6 | 2.52 | 3,300 | 9.8 | 7.4 |
| 41 | 2 | H | 3.3 | 2.13 | 5.8 | > 30,000 | 10 | 4.4 |
| 42 | 1 | H | 8.64 | 11.52 | 4.87 | > 10,000 | 59 | 36.3 |
For Phase II SAR expansion we synthesized R6 (S) benzyl analogs with a limited number of R4 substituents (5-methoxy, 5-methyl, and 5-cyclopropyl indoles) to further explore R1 SAR (80 analogs) as well as a limited number of R1 substituents (benzyl, m-methoxybenzyl, m-isopropoxybenzyl, or m-difluoromethoxybenzyl) to explore additional R4 SAR (68 analogs), respectively. Novel R6 SAR was also explored utilizing a limited number of R1 and R4 substituents (64 analogs). In total 212 additional analogs were synthesized in Phase II modifications at R1, R4 and R6.
All Phase II analogs were first screened against pLASV and pMACV. Those exhibiting EC50 values < 25 nM in both assays were subsequently tested against pJUNV. Of the 132 of compounds displaying < 25 nM potency in the pLASV assay, 118 were also < 25 nM against pMACV and of those 108 were also < 25 nM against pJUNV. Comparative EC50 values and the range of activity spectrums for these Phase II analogs are illustrated in Fig. 2. While a number of interesting pseudotyped-specific SAR trends were observed, our focus was on the identification of compounds exhibiting broad spectrum activity with EC50 values < 10 nM in all three assays (64 compounds).
Fig. 2.

A 3-dimensional plot of EC50 values obtained in the pLASV, pMACV and pJUNV assays for the analogs exhibiting < 25 nM (dark circles) or < 10 nM (yellow circles) EC50 values in each of the pseudtoyped virus assays.
To confirm that the activities observed in pseudotyped virus assays translate to the inhibition of replicative arenaviruses the activities of two compounds (35 and 42, Table 3) were initially tested and confirmed against BSL4 replicative LASV with EC50 values of 33 and 113 nM, respectively. To extend this observation to a broader range of compounds we subsequently tested compounds exhibiting EC50 values < 25 nM against pLASV, pMACV and pJUNV in assays with both pseudotyped (p-TCRV) and replicative Tacaribe (TCRV) virus, a BLS2 virus that is non-pathogenic to humans but highly related to JUNV.18 As shown in Fig. 3 compounds exhibiting < 25 nM EC50 values against the BSL4 pseudotyped viruses also exhibited comparable activities against pTCRV. These data confirm comparable inhibition between the glycoprotein-mediated cell entry of both the BSL4 and BSL2 viruses. Furthermore, the data confirm the translation between pseudotyped and replicative virus inhibition (R2 = 0.61, Fig. 3). Thus, the data further validate the selection of cell entry inhibitors based on their activities in pseudotyped virus assays.
Fig. 3.

Logarithmic plot of pTCRV vs replicative TCRV (immunostaining assay) EC50 values (R2 = 0.61) for analogs selected for EC50 values < 25 nM against each of the pseudotyped viruses (pLASV pMACV and pJUNV) and HLM half-life data.
At R1, SAR trends were observed in both antiviral activity and HLM stability (Table 4). Benzyl groups with less polar meta- or ortho- substituents are consistently potent and broad spectrum (compounds 40, 43, 44, 45, 48, and 49) with a particular preference for lipophilic ethers. Some polar R1 meta-benzyl substituents groups including alcohols (compounds 52, 56, and 59), retain broad spectrum activity despite their increased polarity while others such as sulfonamides (54), and moderately polar groups like 1H-pyrazolyl (53) and alkyl esters (55) retain pMACV and pJUNV activity but lose activity against pLASV. Both carboxylic acids and amides (compounds 60 and 61) are not tolerated for any viral species tested (see data for more analogs in Supplementary data, Table S1).
Table 4.
Phase II SAR of R1 substituents. Activities provided are EC50 values (nM) in the indicated pseudotyped virus assays. Cytotoxicity is given as CC50 in Vero cells (7 days). HLM data is the observed half-life (minutes) in a human liver microsome assay. Aqueous solubility (μM) was measured at pH 7.4.
| |||||||
|---|---|---|---|---|---|---|---|
| Compd. | R1 | p-LASV | p-MACV | p-JUNV | CC50 (nM) | HLM | Solubility |
| 40 | 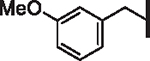 |
6.9 | 3.6 | 2.52 | 3,300 | 9.8 | 7.4 |
| 42 | 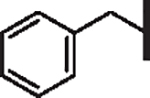 |
8.64 | 11.52 | 4.87 | > 10,000 | 59 | 36.3 |
| 43 | 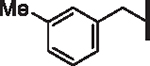 |
2.92 | 2.08 | 3.04 | > 10,000 | 37 | 5.8 |
| 44 | 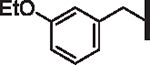 |
1.33 | 1.54 | 1.91 | 41,000 | 12 | 6.9 |
| 45 | 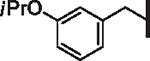 |
2.37 | 2.88 | 3.78 | 49,000 | 8 | 5.7 |
| 46 | 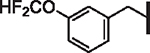 |
0.94 | 1.85 | 0.75 | > 30,000 | 59 | 7.2 |
| 47 | 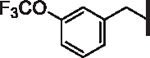 |
5.8 | > 25 | 12.23 | > 30,000 | 72 | 5.6 |
| 48 | 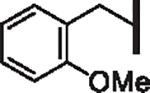 |
5.45 | 2.85 | 1.56 | > 30,000 | 25 | 11.3 |
| 49 | 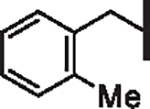 |
5.56 | 6.26 | 11.16 | > 100,000 | 27 | 24.2 |
| 50 | 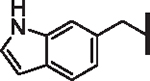 |
0.733 | 1.73 | 2.68 | 5,000 | 36 | 10.21 |
| 51 | 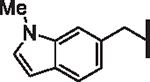 |
4.44 | 1.32 | 3.14 | > 30,000 | 13 | 9.2 |
| 52 | 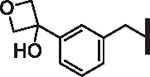 |
2.81 | 3.17 | 6.91 | 79,500 | 11 | 48.8 |
| 53 | 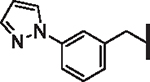 |
22.1 | 1.38 | 1.65 | > 30,000 | N/A | 6.8 |
| 54 | 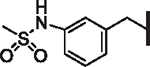 |
> 25 | 5.71 | 3.27 | N/A | N/A | 12 |
| 55 | 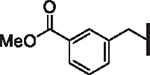 |
23.6 | 5.4 | 1.8 | > 30,000 | N/A | 6.9 |
| 56 | 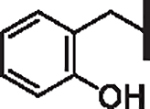 |
2.42 | 2.50 | 5.38 | 10,200 | 18 | 17.2 |
| 57 | 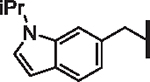 |
3.7 | 2.88 | N/A | > 30,000 | 5.3 | 3.9 |
| 58 | 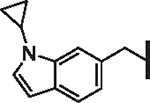 |
2.7 | 2.03 | 2.6 | > 100,000 | 13 | 7.5 |
| 59 | 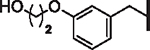 |
4.09 | 1.01 | 1.85 | 39,000 | 29 | 30.2 |
| 60 | 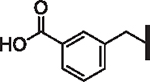 |
> 25 | > 25 | N/A | N/A | N/A | N/A |
| 61 | 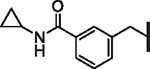 |
> 25 | > 25 | N/A | N/A | N/A | N/A |
Unfortunately, alkyl ethers at R1 pose a significant metabolic liability as demonstrated by the relative HLM stability of compounds 42 and 43 vs. 40, 44 and 45. Stabilized ethers with oxidation-resistant alkyl groups (compounds 46 and 47) have more favorable microsome stability but only the m-difluoromethoxy (46) group maintains broadspectrum activity. The broad-spectrum nature of m-difluoromethoxy group is interesting in light of the poor pJUNV and pMACV activity of more lipophilic m-trifluoromethoxy (47) analog and the general preference for alcohols as R1 substituents, as the difluoromethoxy group has been reported to act as a hydrogen-bond donor and potential isostere for alcohols.30
Both 6-indolyl (50) and N-alkyl 6-indolyl groups (51, 57, 58) also demonstrate good broad-spectrum activity, but the highest activity is derived from the unalkylated analog 50. This is likely due to the hydrogen bonding of indole NeH, similar to the phenols and –OCF2H groups. The unalkylated indoles also possess above average metabolic stability with 50 having an HLM half-life (T1/2) of 36 min, while introduction of alkyl groups causes significant metabolic liability.
Novel R4 analogs were synthesized using the metabolically more stable R1 groups benzyl and m-difluoromethoxy benzyl to further evaluate potential metabolic liabilities stemming from the R4 substituent (Table 5). The majority of Phase II R4 analogs were designed to explore substituents on the most potent aryl and heteroaryl R4 groups (indoles, phenyls, and quinolines) identified in the Phase I exploration, with the intention of further improving metabolic stability while retaining broad-spectrum potency. Although some sterically bulky 5-alkoxy indoles showed dramatic improvements in metabolic stability (compounds 62 and 63) broad-spectrum potency was lost. 5-methylindole and 5-cyclopropylindole substituents (compounds 68 and 69) demonstrated sufficiently increased metabolic stability over 5-methoxyindole analog (42), when paired with a benzyl R1. Of note, compound 66 having 5-cyclopropylindole at R4 and m-difluoromethoxy benzyl at R1 showed some improvement in metabolic stability but at the same time was slightly less potent than the 5-methoxyindole analog 46 (see Table 4).
Table 5.
Phase II SAR of R4 substituents. Activities provided are EC50 values (nM) in the indicated pseudotyped virus assays. Cytotoxicity is given as CC50 in Vero cells (7 days). HLM data is the observed half-life (T1/2 (min)) in a human liver microsome assay. Aqueous solubility (μM) was measured at pH 7.4.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | R4 | X | p-LASV | p-MACV | p-JUNV | CC50 (nM) | HLM | Solubility |
| 42 | 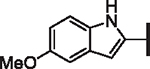 |
H | 8.64 | 11.52 | 4.87 | > 10,000 | 59 | 36.3 |
| 62 | 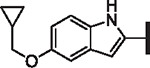 |
H | 9.38 | > 25 | > 25 | > 30,000 | 137 | 8.6 |
| 63 | 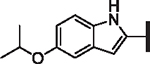 |
H | > 25 | > 25 | 4.235 | N/A | 246 | 14.6 |
| 64 | 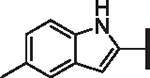 |
OCF2H | 2.34 | 19.52 | 3.8 | > 100,000 | 43 | 5.3 |
| 65 | 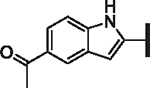 |
H | 25.4 | > 25 | > 25 | > 10,000 | 120 | 15.5 |
| 66 | 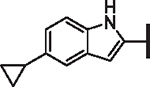 |
OCF2H | 2.12 | 6.81 | 4.05 | 10,400 | 77 | 6.2 |
| 67 | 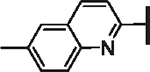 |
OCF2H | 2.29 | 2.99 | 0.52 | 29,700 | 13 | 10 |
| 68 | 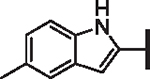 |
H | 1.95 | 21.52 | 2.68 | 11,200 | 119 | 7.7 |
| 69 | 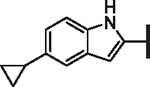 |
H | 14.6 | > 25 | > 25 | > 100,000 | 125 | 8.3 |
| 70 | 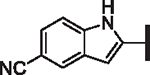 |
H | > 25 | > 25 | > 25 | 16,000 | N/A | 10.8 |
| 71 | 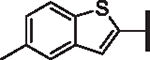 |
H | 5.96 | 12.53 | 4.46 | 9,040 | 42 | 5.7 |
| 72 | 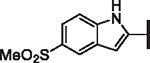 |
H | > 25 | > 25 | > 25 | N/A | N/A | 67.7 |
| 73 |  |
OCF2H | 2.31 | 3.59 | 4.78 | > 100,000 | 16 | 9.5 |
| 74 | OMe | > 25 | > 25 | > 25 | N/A | N/A | > 100 | |
| 75 | OCF2H | 4.68 | 3.98 | 8.82 | 46,700 | 5 | 8.8 | |
| 76 | 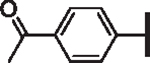 |
H | > 25 | > 25 | > 25 | > 100,000 | 31 | > 100 |
The 7-fold difference in potency against pLASV between 66 and 69 strongly suggested that it should be possible to pair 5-cyclopropylindole and a suitable R1 group to obtain a broadly potent compound that maintained the cyclopropylindole’s metabolic stability gains. While other heteroaryl groups showed acceptable-to-good broad-spectrum potency, such as quinolines 67 and 73 and benzothiophene 71, none of them were found to endow stability or solubility advantages over the indole. Further, the wide range of 4-substituted phenyls synthesized showed significantly reduced broad-spectrum activity with few exceptions, e.g., the 4-piperidinylphenyl group (75), which while potent, exhibited an HLM half-life of only 5 min. Polar substituents such as sulfones (72, 74), nitriles (70), acetyl (65, 76) and a variety of other groups were not tolerated on either phenyl or heteroaryl rings at any position and lead to dramatically reduced potency (see Supplementary data, Table S2). 5-methyl and 5-cyclopropyl indoles were thus selected as the preferred R4 groups to generate R1 and R6 combinatorial compounds optimized for stability and potency.
In parallel with expansion of R1 and R4 SAR we explored additional R6 substituents whereby R4 (5-methoxy indole) and R1 (benzyl, m-methoxybenzyl and m-difluoromethoxybenzyl) substituents were restricted and novel R6 substituent analogs synthesized. The R6 position is the most tolerant of different substitutions and a wide variety of either lipophilic or polar charged groups were synthesized using either Scheme 1, starting with the appropriate commercially available chiral aminoalcohol, or Scheme 3 with the intention of increasing solubility and metabolic stability concurrently. Thus, methyl (O-t-Bu)serinate (L) was first acylated with R1COOH and EDCI (Scheme 3). Subsequent reduction of M using lithium aluminum hydride afforded aminoalcohol N. Acylation of the amine with nosylglycine acyl chloride and magnesium oxide gave intermediate O, which was cyclized under Mitsunobu conditions to afford piperazinone P. Treatment of the t-butyl ether with 4 N HCl in 1,4-dioxane gave free alcohol Q, which was first triflated using triflic anhydride and finally reacted with a desired amine NHR’R” to afford intermediate R. Finally, nosyl deprotection followed by the amide coupling with the appropriate carboxylic acid R4COOH furnished compounds of type S.
Scheme 3.

Reagents and conditions: (i) R1COOH, EDCI, Et3N, DCM, rt, 16 h; (ii) LAH, THF, reflux, 16 h; (iii) 2-(2-nitrophenylsulfonamido)acetyl chloride, MgO, 4:1 THF/H2O, rt, 4 h; (iv) PPh3, DIAD, THF, under N2, rt, 16 h; (v) 4 N HCl/1,4-dioxane, 70 °C, 4 h; (vi) Tf2O, Et3N, DCM, under N2, 0 °C, 1 h, then rt, 3 h; (vii) NHR’R”, Et3N, THF, rt, 16 h; (viii) PhSH, K2CO3, DMF, under N2, rt, 16 h; (ix) R4COOH, 2,6-lutidine, EDCI, DMF, rt, 4 h.
For non-polar R6 substituents (Table 6) potency tracks well with lipophilicity and the physical length of the group is unimportant (compounds 77, 81, 82, 83, and 84). For R6 analogs bearing a basic amine, a linker length of one carbon between the piperazinone and amine (e.g., 79 vs 94) is preferred for activity. Overall lipophilicity of the R6 amine is important for activity, as tertiary amines (90) with fewer carbons than piperidine, and thus lower lipophilicity, resulted in complete loss of potency. Larger amine groups themselves bearing polar substituents such as sulfones and sulfonamides (88, 91), nitriles (92), and even methoxy (87) groups all show significant losses in activity. Additionally, the basicity of the amine plays an important role in determining activity, as difluorinated piperidine analogs, which are more lipophilic yet less basic, exhibit decreased activity compared to the unfluorinated parent piperidine (compound 78 vs. 86 and 93). If the site of fluorination is sufficiently removed from the basic nitrogen, as in the 4-methylpiperidine (85) and its 4-trifluoromethyl analog (89), the inductive withdrawal effect on basicity is minimized and the increased lipophilicity makes 89 more potent.
Table 6.
Phase II SAR of R6 substituents. R6 groups bearing amines were synthesized according to Scheme 3. Activities provided are EC50 values (nM) in the indicated pseudotyped virus assays. Cytotoxicity is given as CC50 in Vero cells (7 days). HLM data is the observed half-life (T1/2 (min)) in a human liver microsome assay. Aqueous solubility (μM) was measured at pH 7.4.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | R6 | X | p-LASV | p-MACV | p-JUNV | CC50 (nM) | HLM | Solubility |
| 77 | 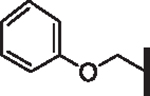 |
H | 5.53 | 20.75 | 5.66 | > 10,000 | 39 | 12.2 |
| 78 | 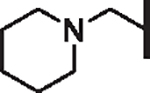 |
H | 4.76 | 4.75 | 5.35 | 76,900 | 17 | > 100 |
| 79 | 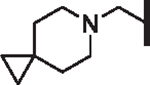 |
H | 2.99 | 4.53 | 1.25 | 17,100 | 52 | 28.2 |
| 80 | 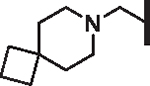 |
H | 12.56 | 11.24 | 12.6 | 4,900 | N/A | 11.8 |
| 81 | 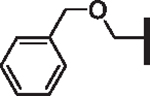 |
OMe | 2.7 | 3.38 | 4.65 | > 10,000 | N/A | 11.7 |
| 82 | 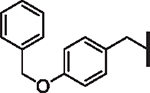 |
OMe | 1.34 | 2.66 | 1.14 | > 10,000 | N/A | 3.8 |
| 83 | 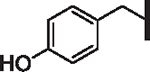 |
OMe | 5.56 | 5.08 | 8.05 | > 10,000 | N/A | 11.8 |
| 84 | 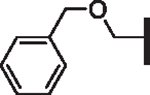 |
H | 5.53 | 4.285 | 4.3 | > 30,000 | N/A | 8.8 |
| 85 | 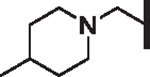 |
H | 11.2 | > 25 | 3.15 | > 30,000 | 16 | 93.1 |
| 86 | 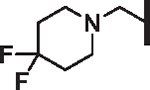 |
H | 11.34 | 20.5 | 23.6 | 95,300 | 23 | 72.4 |
| 87 | 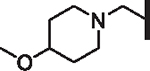 |
H | > 25 | > 25 | N/A | N/A | N/A | > 100 |
| 88 | 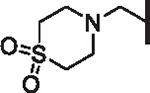 |
H | > 25 | > 25 | N/A | N/A | N/A | > 100 |
| 89 | 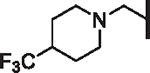 |
H | 3.47 | 8.925 | 3.92 | 17,300 | 32.5 | 16.5 |
| 90 | 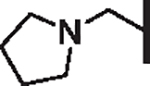 |
H | > 25 | > 25 | N/A | N/A | N/A | > 100 |
| 91 | 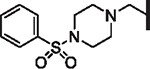 |
H | 21.1 | > 25 | 16.2 | N/A | N/A | 16.9 |
| 92 | 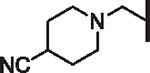 |
OCF2H | 20.69 | 23.24 | > 25 | N/A | N/A | 27.9 |
| 93 | 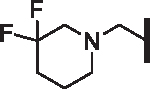 |
OCF2H | 20.25 | 25.52 | 32.7 | > 100,000 | N/A | 53.1 |
| 94 | 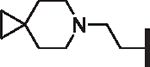 |
H | > 25 | > 25 | > 25 | N/A | N/A | > 100 |
The HLM half-life of all compounds in this R6 exploration series was < 50 min, except 79, which exhibited a half-life of 52 min. Pairing the cyclopropylpiperidine of 79 with an R4 group that is less metabolically labile than 5-methoxy indole could provide further improvement in HLM stability. A number of additional R6 analogs did not exhibit broad-spectrum potency and/or improved HLM stability (see Supplementary data, Table S3).
Initial exploration at positions R3 and R5, as well as attempts to conformationally lock the position of the R4 or R1 substituents (see Supplementary data, Table S4 and Fig. S3) were unable to identify suitably active chemical subseries. Thus, final analog series were synthesized to identify additional R1 (Table 7, Supplementary data, Table S5) and R6 (Table 8) modifications in combination with the most stable, broadly potent complementary R4, R6 and R1 groups. Combining preferred R1 substituents with the 5-methylindole and 5-cyclopropylindole moieties gave a series of compounds with attractive broad-spectrum potency and improved stability.
Table 7.
Secondary Phase II synthesis and SAR of R1 analogs combined with selected R4 and benzyl R6 groups. Activities provided are EC50 values (nM) in the indicated pseudotyped virus assays. Cytotoxicity is given as CC50 in Vero cells (7 days). HLM data is the observed half-life (T1/2 (min)) in a human liver microsome assay. Aqueous solubility (μM) was measured at pH 7.4.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | R1 | X | p-LASV | p-MACV | p-JUNV | CC50 (nM) | HLM | Solubility |
| 95 | 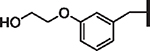 |
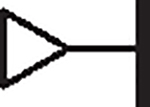 |
7.72 | 1.08 | 2.16 | 4,300 | 50 | 9.4 |
| 96 | 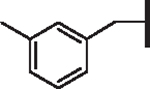 |
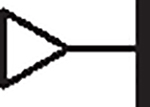 |
0.94 | 1.38 | 1.24 | 9,770 | 46 | 3.2 |
| 97 | 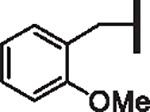 |
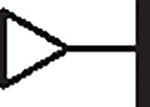 |
2.16 | 1.62 | 0.98 | > 100,000 | 35 | 3.6 |
| 98 | 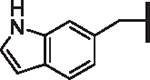 |
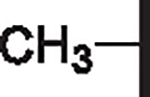 |
6.40 | 3.97 | 3.51 | 8,170 | 102 | 11.7 |
| 99 | 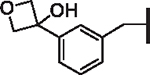 |
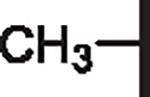 |
7.05 | 4.54 | 3.75 | 23,950 | 20 | 28.5 |
| 100 | 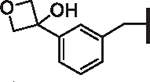 |
 |
3.57 | 3.05 | 1.53 | 5,560 | 22 | 11.75 |
| 101 | 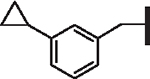 |
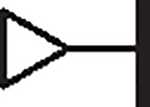 |
3.15 | 1.14 | 0.71 | > 100,000 | 54 | 3.25 |
| 102 | 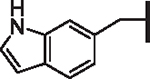 |
 |
3.91 | 3.35 | 3.50 | 9,210 | 187 | 12 |
| 103 | 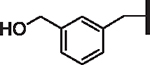 |
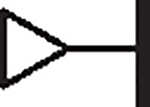 |
1.00 | 2.79 | 1.03 | 27,830 | 19 | 11.34 |
| 104 | 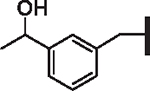 |
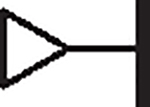 |
0.89 | 2.04 | 0.65 | 4,960 | 6.8 | 7.39 |
Table 8.
Secondary Phase II synthesis and SAR of R6 analogs combined with best R4 and benzyl R1 groups. Activities provided are EC50 values (nM) in the indicated pseudotyped virus assays. Cytotoxicity is given as CC50 in Vero cells (7 days). HLM data is the observed half-life (T1/2 (min)) in a human liver microsome assay. Aqueous solubility (μM) was measured at pH 7.4.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | R6 | X | p-LASV | p-MACV | p-JUNV | CC50 (nM) | HLM | Solubility |
| 105 | 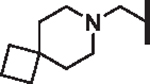 |
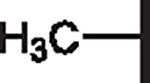 |
11.87 | 17.18 | > 25 | 7,800 | N/A | 12.30 |
| 106 | 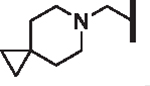 |
 |
10.50 | 15.58 | 2.33 | 3,400 | 156 | 7.70 |
| 107 | 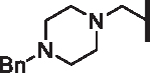 |
 |
15.10 | 5.09 | 5.55 | 5,500 | N/A | 12.86 |
| 108 | 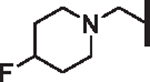 |
 |
4.13 | 11.32 | 3.64 | 24,450 | 23 | 12.35 |
The most active polar compounds in this set (Table 7, compounds 99, 100, 103, and 104) contain R1 groups with benzyl alcohol substituents, as previously suggested by the Phase I and II R1 explorations. While the benzyl alcohols proved to be metabolically labile m-alkyl substituted groups (96, 101) provided moderate HLM stability. Compound 102, containing the 6-indolyl group, demonstrated broad-spectrum potency and an excellent HLM half-life of 187 min. Unfortunately, additional combinations of selected metabolically stable R1 (benzyl) and R4 groups (5-methylindole and 5-cyclopropylindole) with R6 amines intended to increase aqueous solubility did not prove advantageous for either solubility or broad-spectrum antiviral activity (Table 8, compounds 105–108).
In a final functional assay the prioritized compounds (Table 9) were also tested against pseudotyped virus expressing the GP of an attenuated JUNV strain (Candid-1). Each of these analogs exhibited decreased potencies against pJUNV (Candid-1) vs pJUNV, ranging from > 10- (98) to nearly 1000-fold (46). Interestingly, the attenuating mutation (F427I) in the GP of Junin (Candid-1) virus also exhibits resistance to benzimidazole cell entry inhibitors.31,32 Consistent with earlier reports that the 4-acyl-1,6-dialkylpiperazin-2-one and benzimidazole chemical series share a common GP binding site and mechanism of action our results indicate that these distinct chemical series also share at least some resistance variants associated with attenuation.22,23,31
Table 9.
Collection of prioritized compounds expressing potent, broad spectrum activity (< 10 nM EC50 activity against pLASV, pMACV and pJUNV) and HLM metabolic half-life (T1/2) > 45 min with indicated aqueous solubility (μM) at pH 7.4 and pJUNV (Candid-1) EC50 activity.
| Compd. | p-LASV | p-MACV | p-JUNV | p-JUNV (Candid-1) | HLM | Solubility | |
|---|---|---|---|---|---|---|---|
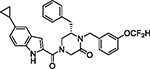 |
66 | 2.12 | 6.81 | 4.05 | N/A | 77 | 6.2 |
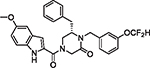 |
46 | 0.94 | 1.85 | 0.75 | 710.3 | 59 | 7.2 |
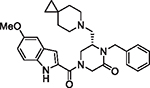 |
79 | 2.99 | 4.53 | 1.25 | 56.2 | 52 | 28.2 |
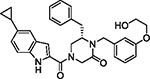 |
95 | 7.72 | 1.08 | 2.16 | 272.9 | 50 | 9.4 |
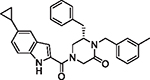 |
96 | 0.94 | 1.38 | 1.24 | 356 | 46 | 3.2 |
 |
98 | 6.40 | 3.97 | 3.51 | 48.4 | 102 | 11.7 |
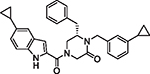 |
101 | 3.15 | 1.14 | 0.71 | 173.2 | 54 | 3.25 |
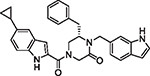 |
102 | 3.91 | 3.35 | 3.5 | 181.5 | 187 | 12 |
In summary, the SAR exploration and optimization of 4-acyl-1,6-dialkylpiperazin-2-one chemical series was initiated with the (S)-enantiomers of compounds 1 and 2 exhibiting pLASV EC50 values in the 250–300 nM range.26 In the current study we generated analogs with up to ~100-fold greater potency including low to sub-nanomolar broad spectrum activity against Old (pLASV) and New World (pMACV, pJUNV and pTCRV) pseudotyped viruses that translate to activity against replicative Lassa and Tacaribe viruses. Attempts to substantially increase solubility while maintaining broad-spectrum potency and other drug like properties met with limited success. However, novel R1, R4 and R6 substituent combinations that improved the HLM metabolic half-lives from < 10 min in our initial broad-spectrum compounds to > 45 min were identified for a subset of analogs. Those compounds exhibiting broad-spectrum arenavirus activity with EC50 values ≤10 nM against each of the pseudotyped viruses and HLM half-lives of > 45 min (Table 9) may be prioritized for further evaluation and advancement.
Supplementary Material
Acknowledgements
We gratefully acknowledge the financial support of the National Institutes of Health (R43AI112097 to K.M and M.B.P.; R44AI112097 to G.H.; CA042056 to D.L.B.; AI074818 and AI065357 to J.H.N) and the generous gifts of arenavirus glycoprotein plasmid constructs and pseudotyped vectors from Juan Carlos de la Torre and Brian Hjelle. The replicative Tacaribe virus TRVL-11573 and monoclonal anti-JUNV, clone MA03-BE06 were obtained from BEI Resources, NIAID, NIH.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2019.08.024.
References
- 1.Isaacson M. Clin Infect Dis. 2001;33:1707. [DOI] [PubMed] [Google Scholar]
- 2.Jahrling PB, Geisbert TW. Nat Med. 2004;10:S110. [DOI] [PubMed] [Google Scholar]
- 3.Emonet SF, de la Torre JC, Domingo E, Sevilla N. Infect Genet Evol. 2009;9:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cajimat MNB, Milazzo ML, Rollin PE, et al. Virus Res. 2009;140:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pontremoli C, Forni D, Sironi M. Curr Opin VIrol. 2018;34:18. [DOI] [PubMed] [Google Scholar]
- 6.Fichet-Calvet E, Rogers DJ. PLoS Negl Trop Dis. 2009;3:e388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borio L, Inglesby T, Peters CJ, et al. JAMA. 2002;287:2391. [DOI] [PubMed] [Google Scholar]
- 8.McCormick JB, Fisher-Hoch SP. Curr Top Microbiol Immunol. 2002;262:75. [DOI] [PubMed] [Google Scholar]
- 9.Ogbu O, Ajuluchukwu E, Uneke CJ. J Vector Dis. 2007;44:1. [PubMed] [Google Scholar]
- 10.Khan SH, Goba A, Chu M, et al. Antiviral Res. 2008;78:103. [DOI] [PubMed] [Google Scholar]
- 11.Cummins D, McCormick JB, Bennett D, et al. JAMA. 1990;264:2093. [PubMed] [Google Scholar]
- 12.Macher AM, Wolfe MS. Emerg Infect Dis. 2006;12:835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Idemyor VJ. Natl Med Assoc. 2010;102:1243. [DOI] [PubMed] [Google Scholar]
- 14.Ibekwe TS, Okokhere PO, Asogun D, et al. Eur Arch Otorhinolaryngol. 2011;268:197. [DOI] [PubMed] [Google Scholar]
- 15.McCormick JB, King IJ, Webb PA, et al. N Engl J Med. 1986;314:20. [DOI] [PubMed] [Google Scholar]
- 16.Nomura H, Tanimoto H, Kajiwara E, et al. J Gastroenterol Hepatol. 2004;19:312. [DOI] [PubMed] [Google Scholar]
- 17.Fuster D, Huertas JA, Gomez G, et al. Antivir Ther. 2005;10:841. [PubMed] [Google Scholar]
- 18.Bolken TC, Laquerre S, Zhang Y, et al. Antiviral Res. 2006;69:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larson RA, Dai D, Hosack VT, et al. J Virol. 2008;82:10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee AM, Rojek JM, Spiropoulou CF, et al. J Biol Chem. 2008;283:18734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cashman KA, Smith MA, Twenhafel, et al. Antiviral Res. 2011;90:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas CJ, Casquilho-Gray HE, York J, et al. J Biol Chem. 2011;286:6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nunberg JH, York J. Viruses. 2012;4:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rojek JM, Spiropoulou CF, Kunz S. Virology. 2006;349:476. [DOI] [PubMed] [Google Scholar]
- 25.Pasquato A, Kunz S. Expert Opin Drug Discov. 2016;11:383. [DOI] [PubMed] [Google Scholar]
- 26.Whitby LR, Lee AM, Kunz S, Oldstone MB, Boger DL. Bioorg Med Chem Lett. 2009;19:3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee AM, Pasquato A, Kunz A. Virology. 2011;411:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kan T, Fukuyama T. Chem Commun. 2004:353. [DOI] [PubMed] [Google Scholar]
- 29.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. J Org Chem. 1996;61:3849. [DOI] [PubMed] [Google Scholar]
- 30.Zafrani Y, Yeffet D, Sod-Moriah G, et al. J Med Chem. 2017;60:797. [DOI] [PubMed] [Google Scholar]
- 31.Madu IG, Files M, Garaibeh DM, et al. PLoS Pathog. 2018;14(12):e1007439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Albarino CG, Bird BH, Chakrabarti AK, et al. J Virol. 2011;19:10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.