

Feline coronavirus (FCoV) is one of the most significant coronaviruses, because this virus induces feline infectious peritonitis (FIP), which is a lethal disease in cats. Tissue culture-adapted type I FCoV often loses pathogenicity, which complicates research on type I FCoV-induced feline infectious peritonitis (FIP). Since we previously found that type I FCoV strain C3663 efficiently induces FIP in specific-pathogen-free cats, we established a reverse genetics system for the C3663 strain to obtain recombinant viruses in the present study. By using a reporter C3663 virus, we were able to examine the inhibitory effect of 68 compounds on C3663 replication in Fcwf-4 cells and infectivity in a canine-derived cell line. Interestingly, one canine cell line, A72, permitted FCoV replication but with low efficiency and aberrant viral gene expression.
KEYWORDS: coronavirus, viral replication
ABSTRACT
Feline infectious peritonitis (FIP) is one of the most important infectious diseases in cats and is caused by feline coronavirus (FCoV). Tissue culture-adapted type I FCoV shows reduced FIP induction in experimental infections, which complicates the understanding of FIP pathogenesis caused by type I FCoV. We previously found that the type I FCoV strain C3663 efficiently induces FIP in specific-pathogen-free cats through the naturally infectious route. In this study, we employed a bacterial artificial chromosome-based reverse genetics system to gain more insights into FIP caused by the C3633 strain. We successfully generated recombinant virus (rC3663) from Fcwf-4 cells transfected with infectious cDNA that showed growth kinetics similar to those shown by the parental virus. Next, we constructed a reporter C3663 virus carrying the nanoluciferase (Nluc) gene to measure viral replication with high sensitivity. The inhibitory effects of different compounds against rC3663-Nluc could be measured within 24 h postinfection. Furthermore, we found that A72 cells derived from canine fibroblasts permitted FCoV replication without apparent cytopathic effects. Thus, our reporter virus is useful for uncovering the infectivity of type I FCoV in different cell lines, including canine-derived cells. Surprisingly, we uncovered aberrant viral RNA transcription of rC3663 in A72 cells. Overall, we succeeded in obtaining infectious cDNA clones derived from type I FCoV that retained its virulence. Our recombinant FCoVs are powerful tools for increasing our understanding of the viral life cycle and pathogenesis of FIP-inducing type I FCoV.
IMPORTANCE Feline coronavirus (FCoV) is one of the most significant coronaviruses, because this virus induces feline infectious peritonitis (FIP), which is a lethal disease in cats. Tissue culture-adapted type I FCoV often loses pathogenicity, which complicates research on type I FCoV-induced feline infectious peritonitis (FIP). Since we previously found that type I FCoV strain C3663 efficiently induces FIP in specific-pathogen-free cats, we established a reverse genetics system for the C3663 strain to obtain recombinant viruses in the present study. By using a reporter C3663 virus, we were able to examine the inhibitory effect of 68 compounds on C3663 replication in Fcwf-4 cells and infectivity in a canine-derived cell line. Interestingly, one canine cell line, A72, permitted FCoV replication but with low efficiency and aberrant viral gene expression.
INTRODUCTION
Coronaviruses (CoVs) are pathogens that infect a wide variety of animals, including humans, and cause respiratory and enteric diseases (1). CoVs are enveloped viruses possessing a large (∼32-kb), single-stranded, positive-sense RNA (2) and are classified as order Nidovirales, family Coronaviridae, and subfamily Coronavirinae. CoVs are further classified into four genera, namely, Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus (3). Feline coronavirus belongs to Alphacoronavirus, together with Canine coronavirus, Porcine transmissible gastroenteritis virus, Porcine epidemic diarrhea virus, and Human coronavirus 229E and NL63 (3).
Feline CoV (FCoV) infections are distributed worldwide in domestic cats and wild Felidae, such as lions (4, 5) and cheetahs (6). On the basis of their pathogenicity, FCoVs can be classified into two biotypes—feline enteric CoV (FECV) and feline infectious peritonitis virus (FIPV). FECV infections are asymptomatic or occasionally induce mild intestinal inflammation in kittens (7). On the other hand, FIPV infections induce the more severe and immune-mediated lethal disease feline infectious peritonitis (FIP) (8, 9).
FCoVs can also be further classified into two types, types I and II, on the basis of their antigenicity (10, 11). Unlike type II FCoV infections, type I FCoV infections occur predominantly in felids worldwide (12–14). Furthermore, their virological features differ, including growth characteristics in cell culture and receptor usage (7, 15). Type II FCoV shows better growth kinetics than type I FCoV and can more easily induce FIP in specific-pathogen-free (SPF) cats. Despite the fact that type II FCoV infections occur with low frequency, many researchers employ type II FCoVs to analyze FIP pathogenesis. Therefore, a type I FCoV strain that can induce FIP is needed to fully understand FIP pathogenesis.
It has been proposed that type I FECV replicates and acquires mutations in its viral genome in kittens and that the mutated FECV then becomes a FIP-associated virus. This hypothesis is known as the “internal mutation theory” (16–18) and is supported by the proposal of the presence of virulent FIP markers. On the basis of epidemiological studies, spike (S) and/or open reading frame (ORF) 3c genes of type I FCoV are thought to be virulence markers (18–20). However, none of the proposed markers have been proven virulent owing to the lack of feasible FIP cat models with type I FCoV. It is difficult for researchers using most type I FCoVs isolated from FIP cats to induce FIP in experimental settings using SPF cats. It is thought that adaptation of type I FCoV in tissue culture results in a loss of pathogenicity (21, 22).
Recently, we discovered C3663, a strain of type I FCoV isolated from FIP cats (23) that retained virulence despite adaptation in Fcwf-4 cells (9). Surprisingly, three (75%) of four SPF cats developed FIP after infection with the C3663 strain (9). These findings suggest that our C3663 strain is a candidate for analysis of FIP pathogenesis induced by type I FCoV in experimental settings.
In this study, we constructed an infectious cDNA clone derived from the type I FCoV C3663 strain by utilizing the bacterial artificial chromosome (BAC) system. Recombinant C3663 (rC3663) virus was easily rescued from Fcwf-4 cells transfected with BAC plasmids carrying the C3663 full-length genome. rC3663 showed growth kinetics similar to those shown by the parental virus. Furthermore, we generated a recombinant virus bearing the nanoluciferase (Nluc) gene in the ORF 3abc region. This rC3663-Nluc reporter virus was useful in investigating the inhibitory effects of compounds and revealed the infectivity of type I FCoV in canine cells. Interestingly, the expression ratio of subgenomic (sg) mRNA was different in canine-derived A72 cells infected with rC3663 virus, suggesting that aberrant viral RNA transcription of the rC3663 virus had occurred in the A72 cells.
RESULTS
Construction of BAC carrying the full-length C3663 genome.
The full-genome sequence of type I FCoV strain C3663 was assembled into the pBeloBAC11 vector to generate infectious cDNA clones under the control of a cytomegalovirus (CMV) immediate early promoter (Fig. 1A). To this end, we separated the genomic sequence of C3663 into 11 fragments and sequentially assembled them into the BAC plasmid (Fig. 1A). The vector backbone bore the CMV promoter followed by the hepatitis delta virus (HDV) ribozyme and bovine growth hormone (BGH) termination sequences (Fig. 1A); the C3663 genomic sequence was cloned into the pBeloBAC11 vector between the CMV promoter and the 25-nucleotide (nt) poly(A), HDV ribozyme, and BGH termination sequences (Fig. 1A). The full-length infectious cDNA clone was designated pBAC-FCoV-C3663, and sequence analysis showed that it possessed 25 nucleotide mutations compared with the C3663 reference sequence (Table 1). Of the 25 mutations, 11 were synonymous and 14 were nonsynonymous (Table 1). Two synonymous mutations at nt 9831 and nt 9834 were introduced as the genetic marker, which disrupts the EcoRI site (ΔEcoRI), confirming virus recovery from the cDNA clone (Fig. 1B; see also Table 1).
FIG 1.
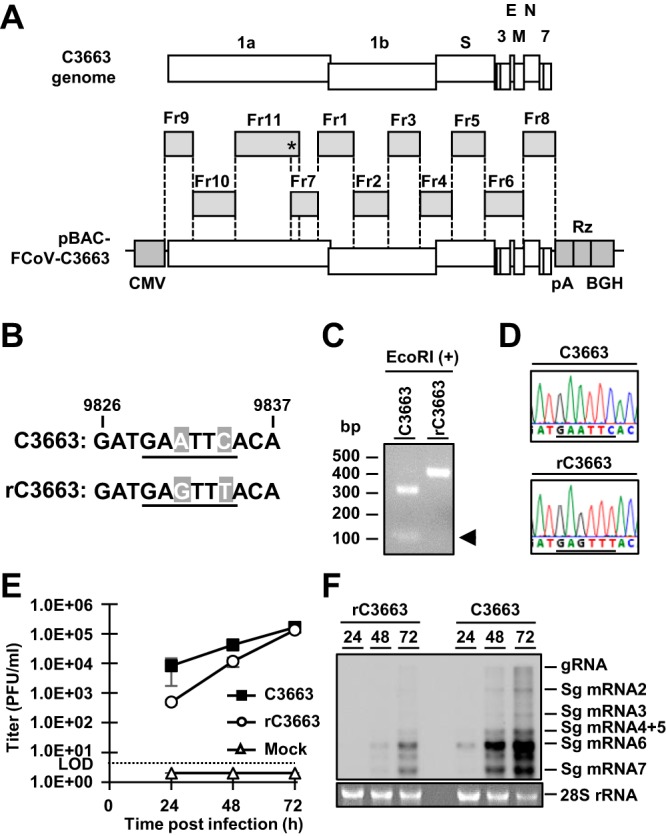
Constructing type I FCoV strain C3663 cDNA clones. (A) Schematic diagram illustrating the strategy for constructing infectious cDNA clones bearing the full-length genome of type I FCoV strain C3663. The full-length C3663 sequence was divided into 11 fragments (Fr1 to Fr11), and each fragment was sequentially assembled into the plasmid backbone. The asterisk (*) indicates the site of the genetic marker. (B) Nucleotide sequence of the C3663 genome between nt 9826 and nt 9837. The EcoRI restriction site is underlined. rFCoV was mutated from GAATTC to GAGTTT for disruption of the EcoRI site (ΔEcoRI) to use as a genetic marker. Mutated nucleotides are shown in gray boxes with white letters. (C) Confirmation of the genetic marker in the rC3663 genome by EcoRI treatment. RT-PCR products of parental virus C3663 and rC3663 that amplified a region that included the genetic marker were treated with EcoRI. The treated samples were subjected to electrophoresis to confirm disruption of the EcoRI site in the rC3663 genome. (D) Sequence analysis of C3663 and rC3663 at the genetic marker site. The EcoRI restriction site and ΔEcoRI genetic marker are underlined. (E) Growth kinetics of parental virus C3663 and rC3663 in Fcwf-4 cells. Each virus was inoculated onto Fcwf-4 cells at an MOI of 0.01 and incubated for 24, 48, and 72 h. Viral titers of culture supernatants were measured by plaque assays using Fcwf-4 cells. LOD, limit of detection. The data represent means ± standard deviations (SD) of results from three independent experiments. (F) Northern blot analysis for detecting viral RNA in C3663-infected or rC3663-infected Fcwf-4 cells. Total RNAs from Fcwf-4 cells infected with parental C3663 or rC3663 were extracted and subjected to electrophoresis. The transferred viral RNAs were hybridized with DIG-labeled RNA targeting ORF 7b and 3ʹ-UTR. gRNA, genomic RNA; sgRNA, subgenomic RNA.
TABLE 1.
Mutations in pBAC-FCoV-C3663
| Mutation no. | Nucleotide position | Gene | Nucleotide mutation | Amino acid mutation |
|---|---|---|---|---|
| 1 | 2889 | 1a | C → T | Synonymous |
| 2 | 6381 | 1a | C → T | Synonymous |
| 3 | 6645 | 1a | G → T | K → N |
| 4 | 9831 | 1a | A → G | Synonymous |
| 5 | 9834 | 1a | C → T | Synonymous |
| 6 | 12841 | 1b | T → C | V → A |
| 7 | 17870 | 1b | C → T | Synonymous |
| 8 | 18065 | 1b | C → T | Synonymous |
| 9 | 18169 | 1b | C → T | A → V |
| 10 | 21563 | S | G → A | D → N |
| 11 | 21976 | S | T → C | Synonymous |
| 12 | 22319 | S | A → G | M → V |
| 13 | 22619 | S | G → A | E → K |
| 14 | 23049 | S | G → A | G → E |
| 15 | 23473 | S | C → T | Synonymous |
| 16 | 23757 | S | G → A | G → E |
| 17 | 24500 | 3c | G → A | R → H |
| 18 | 24608 | 3c | C → T | S → F |
| 19 | 25220 | E | C → T | Synonymous |
| 20 | 25353 | E | C → T | T → M |
| 21 | 25928 | M | A → G | Synonymous |
| 22 | 26279 | N | C → T | Synonymous |
| 23 | 26442 | N | G → A | E → K |
| 24 | 27315 | N | G → A | V → I |
| 25 | 27634 | 7a | T → G | S → A |
Virus recovery by pBAC-FCoV-C3663 transfection.
We produced rC3663 virus from Fcwf-4 cells, which are highly susceptible to FCoV infection, by transfecting the cells with pBAC-FCoV-C3663. Low levels of cytopathic effects (CPE) were observed at 2 days posttransfection (dpt), and the levels became higher by 3 dpt. To determine the rates of rC3663 virus recovery, we employed reverse transcription-PCR (RT-PCR) on isolated RNA from rC3663 and the parental strain. We further confirmed the ΔEcoRI genetic maker by analyzing EcoRI digestion and Sanger sequencing (Fig. 1C and D). Next, we analyzed the virological features of the rC3663 virus by comparing the growth kinetics of rC3663 and parental C3663 in Fcwf-4 cells after inoculating them with viruses at a multiplicity of infection (MOI) of 0.01. The results showed that the growth kinetics of rC3663 was similar to that of the parental C3663 (Fig. 1E). Furthermore, we compared levels of viral RNA replication in parental C3663-infected and rC3663-infected Fcwf-4 cells by Northern blotting analysis (Fig. 1F) and found that the amounts of genomic RNA (gRNA) and of sg mRNAs in rC3663-infected Fcwf-4 cells were similar to those seen with the parental C3663-infected cells (Fig. 1F). Taking the results together, we were able to successfully generate infectious cDNA clones derived from type I FCoV strain C3663 using the BAC system. Our results indicate that the recovered rC3663 virus possesses virological features identical to those possessed by the parental C3663 virus (Fig. 1E and F).
Establishment of reporter rC3663 bearing the Nluc gene.
In virology, recombinant viruses carrying reporter genes (green fluorescent protein [GFP], red fluorescent protein [RFP], or luciferase) present many advantages for analyzing viral characteristics and screening for therapeutic compounds (24, 25). Thus, we attempted to construct an infectious cDNA clone of type I FCoV strain C3663 carrying an Nluc gene. Following the protocols of Tekes et al. (26), we inserted the Nluc gene into pBAC-FCoV-C3663 in place of the ORF 3abc gene to obtain pBAC-FCoV-C3663-Nluc (Fig. 2A). The Nluc gene replaced a region beginning at the start codon of ORF 3a and extending to 71 nt upstream of the ORF 3c stop codon to retain the transcription regulatory sequence (TRS) of the M gene (Fig. 2A).
FIG 2.

Construction and characteristics of the reporter virus carrying the Nluc gene. (A) Nluc gene replacement occurred at ORF 3abc, resulting in pBAC-FCoV-C3663-Nluc. The Nluc gene replaced the sequence from the start codon of ORF 3a to 71 nt upstream of the ORF 3c stop codon to retain the TRS of the E gene. Light gray, gray, and white boxes indicate nonstructural proteins, structural proteins, and accessory proteins, respectively. TRS, transcription regulatory sequence. (B) Luciferase activity of rC3663-Nluc-infected cells. rC3663-Nluc and rC3663 were inoculated onto Fcwf-4 cells at an MOI of 0.01 and incubated for 24, 48, and 72 h. Infected cells were lysed, and luciferase activity was measured. Experiments were carried out in triplicate. (C) Growth kinetics of rC3663-Nluc. rC3663-Nluc and rC3663 were inoculated into Fcwf-4 cells at an MOI of 0.01 and incubated for 24, 48, and 72 h. Viral titers of culture supernatants were measured by plaque assays using Fcwf-4 cells. LOD, limit of detection. (D and E) Evaluation of the inhibitory effects of (D) CsA and (E) lopinavir on rC3663-Nluc. Fcwf-4 cells were inoculated with rC3663-Nluc at an MOI of 0.01. After adsorption, the viruses were removed and replaced by culture medium with or without different concentrations of (D) CsA or (E) lopinavir. After incubation for 24 h, levels of luciferase activities (black circle) or viral RNA (white triangle) were measured. The experiments were carried out in triplicate. (F) Compound screening using rC3663-Nluc and evaluation of the cytotoxicity of protease inhibitors by MTT assays. A total of 68 protease inhibitors were used in this screening. Virus was added at an MOI of 0.01 onto cultured Fcwf-4 cells with a 10 μM concentration of each protease inhibitor or DMSO and further cultured for 24 h. CsA (10 μM) was used as a positive control. Infected cells were lysed, and levels of Nluc activities were measured (bar graphs). For MTT assays, seeded Fcwf-4 cells were cultured with DMEM containing 10% FBS and a 10 μM concentration of each compound for 24 h. Then, cultured cells underwent MTT assays and the absorbance was measured at 570 nm (line graph). The data represent means ± SD of results from three independent experiments.
To examine Nluc expression in Fcwf-4 cells infected with rC3663-Nluc virus, we inoculated Fcwf-4 cells with rC3663-Nluc at an MOI of 0.01. Infection with rC3663 was used as a control. After 24, 48, and 72 h postinfection (hpi), we found that Nluc activity in rC3663-Nluc-infected Fcwf-4 cells—but not in rC3663-infected or mock-infected cells—increased in a time-dependent manner (Fig. 2B).
We further investigated the viral growth of rC3663-Nluc in Fcwf-4 cells by harvesting the supernatants of rC3663-Nluc-infected or rC3663-infected Fcwf-4 cells at 24, 48, and 72 hpi and then determining infectious titers of the supernatants by plaque assays. The level of production of infectious virus particles from rC3663-Nluc-infected cells was comparable to that from rC3663-infected cells (Fig. 2C). As shown in Fig. 2B and C, the increase in Nluc activity was significantly correlated with viral replication in rC3663-Nluc-infected cells. Our data indicate that rC3663 carrying the Nluc reporter gene represents a powerful tool for investigating type I FCoV replication and production.
Application of the rC3663 reporter virus in compound screening.
Before application of the rC3663 reporter virus to compound screening, we determined the sensitivity of the rC3663-Nluc virus to treatment with known inhibitors of CoV replication, namely, cyclosporine (CsA) (27, 28) and lopinavir (29). After adsorption of rC3663-Nluc onto Fcwf-4 cells at an MOI of 0.01, the infected cells were treated with various concentrations of CsA or lopinavir. As shown in Fig. 2D and E, both compounds inhibited luciferase activity in a dose-dependent manner. Furthermore, viral RNA levels in CsA-treated or lopinavir-treated cells were measured by real-time RT-PCR (Fig. 2D and E). Dose-dependent reductions of intracellular viral RNA levels were found for both compounds and were correlated with luciferase activity, suggesting that the sensitivity of detection of luciferase expression levels in rC3663-Nluc-infected cells is comparable to that of viral RNA expression levels.
Next, to determine the usefulness of the rC3663 reporter virus for screening antiviral compounds, we utilized a commercially available library of 68 protease inhibitors. Fcwf-4 cells were inoculated with rC3663-Nluc (MOI = 0.01), and 10 μM concentrations of each protease inhibitor with CsA and with dimethyl sulfoxide (DMSO) were used as positive and negative controls, respectively. The side-effects of protease inhibitors were determined by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assays (Fig. 2F). Together with the MTT assay results, 15 inhibitors were found to exhibit a greater than 75% reduction in Nluc activity compared with that of the DMSO control and without any accompanying cytotoxicity (Fig. 2F; compounds 2, 25, 29, 31, 34, 35, 48, 50, 56, 58, 64 to 67, and 69). Indeed, compound 31 (lopinavir) inhibited luciferase activity, which is consistent with the results presented in Fig. 2E. Overall, our results support the idea of the suitability of rC3663-Nluc for compound screening.
Identification of permissive cell lines for type I FCoV.
In vitro propagation of type I FCoV is limited to a few cell lines, including Fcwf-4 cells, AKD cells, and CRFK cells, because type I FCoV shows a CPE only in such cell lines (23, 30, 31). Thus, it is difficult to investigate the infectivity of type I FCoV in cell lines derived from other animals, such as dogs. Nevertheless, we explored novel cell lines for propagation of type I FCoV by inoculating three canine-derived cell lines, A72 (canine fibroblasts), MDCK (canine kidney epithelial cells), and DH82 (canine macrophages), with the rC3663-Nluc virus (MOI = 0.1) and investigated infectivity by measuring Nluc activity. Although no CPE was observed for rC3663-Nluc-infected A72 cells, Nluc activity was significantly high at 24 hpi and increased in a time-dependent manner (Fig. 3A and C). On the other hand, rC3663-Nluc-infected MDCK and DH82 cells did not exhibit detectable Nluc activity (Fig. 3B).
FIG 3.
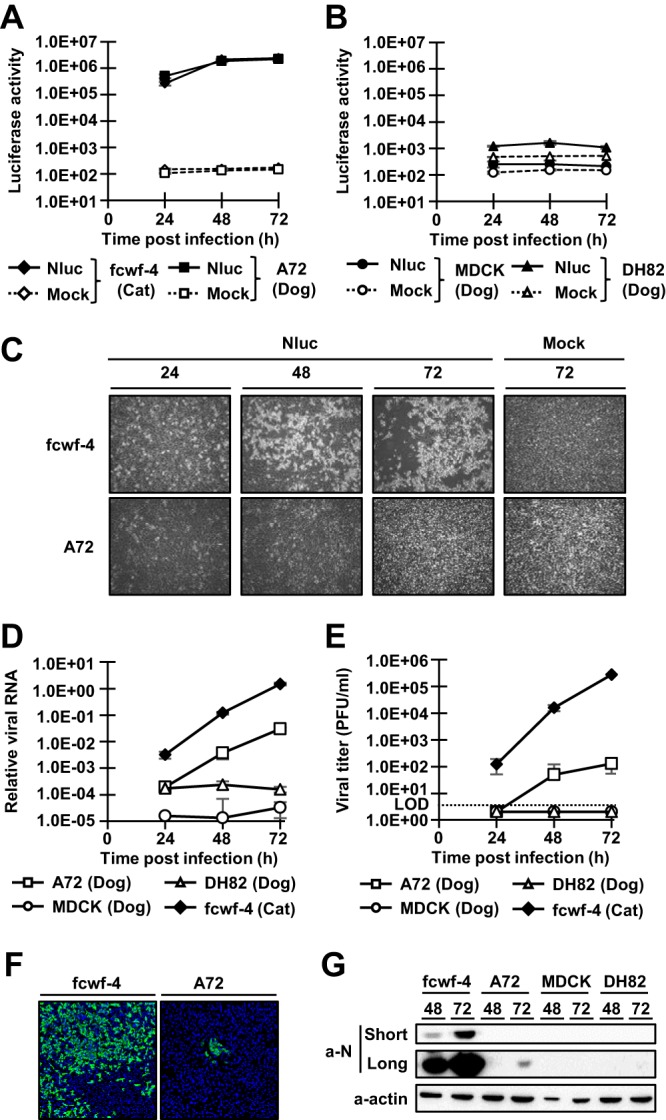
Investigation of the infectivity of type I FCoV in canine-derived cell lines. (A and B) Infectivity of rC3663-Nluc in canine-derived cell lines. Fcwf-4 cells (A) and canine-derived A72 as well as MDCK and DH82 cells (B) were subjected to mock inoculation or were inoculated with rC3663-Nluc (Nluc) at an MOI of 0.1 and incubated for 24, 48, and 72 h. After incubation, infected cells were lysed and Nluc activities were measured. The experiments were carried out in triplicate. (C) Cytopathic effects in Fcwf-4 and A72 cells infected with rC3663-Nluc. Fcwf-4 and A72 cells were subjected to mock inoculation or were inoculated with rC3663-Nluc (Nluc) at an MOI of 0.1 and incubated for 24, 48, and 72 h. (D) Real-time RT-PCR for the evaluation of viral RNA replication. Fcwf-4, A72, MDCK, and DH82 cells were inoculated with rC3663 at an MOI of 0.01. Total RNA was extracted from the infected cells, and real-time RT-PCR targeting the 3ʹ-UTR was carried out. (E) Growth kinetics of rC3663 in Fcwf-4, A72, MDCK, and DH82 cells. rC3663 was used to inoculate the cells at an MOI of 0.01, and the cells were incubated for 24, 48, and 72 h. The culture supernatants were collected at each time point, and viral titers were measured by plaque assays using Fcwf-4 cells. LOD, limit of detection. (F) Detection of rC3663 N protein in Fcwf-4 and A72 cells by IFA. rC3663 was used to inoculate Fcwf-4 and A72 cells at an MOI of 0.1. Infected cells were incubated for 48 h. Then, infected cells were fixed with 4% paraformaldehyde. Fixed cells were treated with mouse anti-FCoV N monoclonal antibody (primary antibody) and CF488-conjugated anti-mouse IgG (secondary antibody). (G) Western blot analysis for the detection of rC3663 N protein. Cell lysates of Fcwf-4, A72, MDCK, and DH82 cells infected with rC3663 were subjected to Western blot analysis using anti-FCoV N monoclonal antibody (a-N) and anti-actin antibody (a-actin). “Short” and “Long” indicate short and long exposure times, respectively. The data represent means ± SD of results from three independent experiments.
To determine viral RNA replication in A72 cells, A72, MDCK, DH82, and Fcwf-4 cells were infected with rC3663-Nluc virus at an MOI of 0.01, followed by real-time RT-PCR analysis of RNA extracted at 24, 48, and 72 hpi. Although the amount of viral RNA in rC3663-Nluc-infected A72 cells was lower than in Fcwf-4 cells, the amount of viral RNA in A72 cells (but not in DH82 and MDCK cells) was still significantly high at 48 and 72 hpi (Fig. 3D). These results indicate that A72 cells permit replication of type I FCoV C3663 virus.
Next, we determined the levels of production of infectious virus particles from rC3663-Nluc-infected A72 cells by collecting the culture supernatants at 24, 48, and 72 hpi and measuring viral titers by plaque assays with Fcwf-4 cells (Fig. 3E). The supernatant infectious titers of Fcwf-4 cells reached 1.67 × 105 PFU/ml at 72 hpi, and the amount of viral RNA determined by real-time RT-PCR increased in a time-dependent manner in Fcwf-4 cells (Fig. 3D and E). As shown in Fig. 3A and D, A72 cells supported rC3663 virus replication, but the production of infectious viruses was lower than that seen with Fcwf-4 cells. Meanwhile, infectious viruses were not produced by infected MDCK and DH82 cells (Fig. 3E). These results indicate that A72 cells produce progeny viruses—albeit with low efficiency—while MDCK and DH82 cells are not permissive cell lines for type I FCoV.
To further determine the low level of production of progeny virus by rC3663-Nluc-infected A72 cells, we examined the propagation of rC3663 virus (MOI = 0.1) in A72 and Fcwf-4 cells by indirect immunofluorescence assays (IFA) using confocal microscopy analysis. Using an anti-FCoV N monoclonal antibody (MAb), N protein expression in A72 cells was observed by IFA, and the A72 cells exhibited small foci at 48 hpi compared with those exhibited by Fcwf-4 cells (Fig. 3F). Therefore, infectious particles were found to have been generated in A72 cells at a low efficiency of infection expansion by progeny particles to neighboring cells. We also examined N protein expression levels using immunoblotting. As expected, N protein expression levels in rC3663 virus-infected A72 cells were significantly low (Fig. 3G). Although the levels of production of progeny virus and N protein were low, our results suggest that canine-derived A72 cells represent a permissive cell line for type I FCoV infections without cytotoxic effects.
MDCK cells do not permit viral replication during type I FCoV infection.
As shown in Fig. 3, and unlike the results seen with A72 cells, neither viral RNA replication nor progeny virus production was observed in MDCK and DH82 cells infected with rC3663 virus. These results led us to speculate that the entry receptor for type I FCoV is perhaps not expressed in MDCK and DH82 cells or that viral RNA replication of type I FCoV is not permitted in these cell lines. Unfortunately, the type I FCoV receptor remains unknown. Thus, we examined viral replication levels in cells transfected with pBAC-FCoV-C3663-Nluc. As a negative control, we generated pBAC-FCoV-C3663-Nluc-PolDead by mutating the active site of viral RNA-dependent RNA polymerase (RdRp; nsp12) from SDD to SAA (Fig. 4A) and confirmed that virus rescue did not occur in Fcwf-4 cells transfected with pBAC-FCoV-C3663-Nluc-PolDead because of disrupted RdRp activity (data not shown). After transfecting MDCK cells with pBAC-FCoV-C3663-Nluc or pBAC-FCoV-C3663-Nluc-PolDead together with pcDNA3.1-fluc, we determined the levels of luciferase activity in cell lysates at 24, 48, and 72 h posttransfection (Fig. 4B, with firefly luciferase reporter plasmid pcDNA3.1-fluc (32) used as an internal control. As shown in Fig. 4B, Nluc expression levels in cells transfected with pBAC-FCoV-C3663-Nluc were comparable to those in cells transfected with pBAC-FCoV-C3663-Nluc-PolDead. Consistent with the luciferase assay results (Fig. 4B), no progeny virus was produced in MDCK cells transfected with pBAC-FCoV-C3663-Nluc (Fig. 4C). In addition, no increases in the levels of viral RNA (Fig. 4D) or N protein expression (Fig. 4E) were observed in MDCK cells transfected with pBAC-FCoV-C3663-Nluc. Thus, our results indicate that MDCK cells do not permit replication of C3663 virus RNA.
FIG 4.
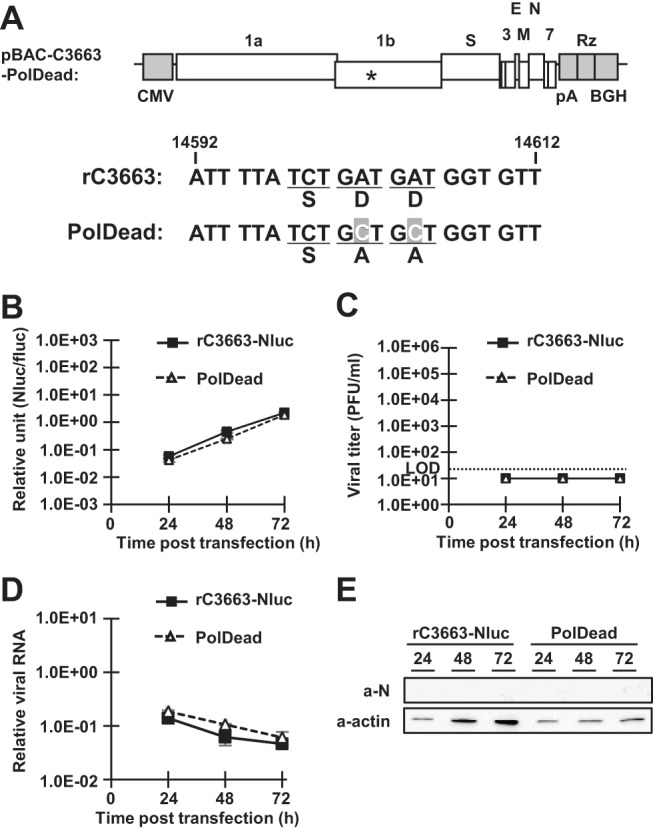
Investigation of resistance to type I FCoV infection. (A) Strategy for construction of polymerase dead mutant cDNA clones (pBAC-FCoV-C3663-Nluc-PolDead [PolDead]). The asterisk (*) indicates the active site of viral RNA-dependent RNA polymerase (RdRp; nsp12). The diagram represents the nucleotide sequence of the C3663 genome between nt 14592 and nt 14612; mutated nucleotides are shown in gray boxes with white letters. pBAC-FCoV-C3663-Nluc (rC3663-Nluc) or pBAC-FCoV-C3663-Nluc-PolDead (PolDead) was transfected into seeded MDCK cells. Transfected cells were incubated for 24, 48, and 72 h. (B) At each time point, the transfected cells were lysed and levels of Nluc activities measured. As an internal control, a firefly luciferase reporter plasmid (pcDNA3.1-fluc) was cotransfected with BAC plasmids. Nluc activity was normalized to the activity of firefly luciferase. (C) The culture supernatants were collected at each time point, and viral titers were measured by plaque assays using Fcwf-4 cells. LOD, limit of detection. The experiments were carried out in triplicate. (D) Total RNA was extracted from transfected MDCK cells, and the levels of viral RNA were determined by real-time RT-PCR. (E) Cell lysates of transfected MDCK cells were subjected to Western blot analysis using anti-FCoV N monoclonal antibody (a-N) and anti-actin antibody (a-actin). The data represent means ± SD of results from three independent experiments.
Aberrant expression of type I FCoV RNA in A72 cells.
As shown in Fig. 3G, expression levels of N protein in infected A72 cells were significantly low. Thus, to determine expression levels of sg N mRNA (sg mRNA6), total RNAs extracted from rC3663 virus-infected or mock-infected Fcwf-4 and A72 cells at 48 and 72 hpi were subjected to Northern blot analysis with a specific type I FCoV 3ʹ untranslated region (3ʹUTR) probe (Fig. 5A). We found that all viral RNAs, including gRNA and sg mRNA2 to mRNA7, were detected in Fcwf-4 cells infected with rC3663 virus (Fig. 5A). However, the expression levels of gRNA and sg mRNA2 to mRNA5 were significantly lower in infected A72 cells than in infected Fcwf-4 cells (Fig. 5A).
FIG 5.
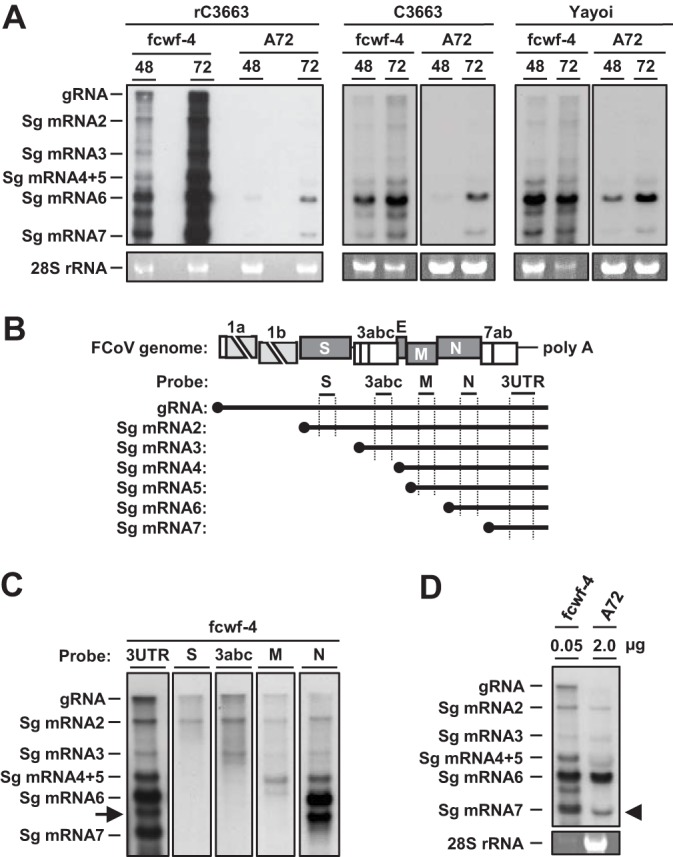
Aberrant expression of viral RNA of type I FCoV in A72 cells. (A) Northern blot analysis for the detection of viral RNAs in rC3663-infected, parental C3663-infected, or Yayoi-infected A72 and Fcwf-4 cells. Total RNA was extracted from infected cells and subjected to electrophoresis. Viral RNAs were then hybridized with DIG-labeled RNA targeting ORF 7b and 3ʹ-UTR. gRNA, genomic RNA; sgRNA, subgenomic RNA. (B) Diagram illustrating the DIG-labeled RNA probes used in Northern blot analysis. (C) Northern blot analysis using S, 3abc, M, and N probes for detecting viral RNA in rC3663-infected Fcwf4-cells. The arrow indicates an unknown RNA signal. (D) Northern blot analysis for detecting viral RNA in rC3663-infected Fcwf-4 and A72 cells. First lane, 0.05 μg of total RNA extracted from infected Fcwf-4 cells; second lane, 2 μg of total RNA extracted from infected A72 cells.
Next, to determine the specific RNA signal in infected A72 cells, we generated a set of specific digoxigenin (DIG)-labeled RNA probes against S, 3abc, M, and N genes (Fig. 5B). As shown in Fig. 5C, we identified seven specific viral mRNAs (gRNA and sg mRNA2 to mRNA7) using a combination of S, 3abc, M, and N-specific RNA probes in infected Fcwf-4 cells. An unexpected RNA signal was observed between sg mRNA6 and sg mRNA7 (Fig. 5C). These results indicate that the two RNAs detected in A72 cells infected with rC3663 were sg mRNA6 and sg mRNA7.
We further examined the expression levels of viral sg mRNAs in cells infected with the parental C3663 strain or type I FCoV strain Yayoi using Northern blot analysis with specific RNA probes against the 3ʹ-UTR. Although gRNA and sg mRNA2 to mRNA7 were detected in Fcwf-4 cells infected with the C3663 strain or the Yayoi strain, two specific mRNAs—sg mRNA6 and sg mRNA7—were observed in A72 cells infected with C3663 or Yayoi (Fig. 5A). These results suggest that the decreased synthesis of viral mRNAs was not specific to the infectious clone of C3663.
Although the expression levels of sg mRNA6 and sg mRNA7 in A72 cells infected with rC3663 were low (Fig. 5A), the ratio of sg mRNA6 expression to sg mRNA7 expression in infected A72 cells was different from that in infected Fcwf-4 cells. The level of sg mRNA7 in infected A72 cells determined on the basis of the level of sg mRNA6 was lower than that in infected Fcwf-4 cells (Fig. 5A). To compare these ratios, we used a smaller amount of total RNA extracted from infected Fcwf-4 cells for Northern blot analysis to adjust the expression levels of sg mRNA6 between infected Fcwf-4 and A72 cells. As a result, the expression levels of sg mRNA6 in A72 cells were identical to those in Fcwf-4 cells, whereas the expression levels of sg mRNA7 in A72 cells were lower than those in Fcwf-4 cells (Fig. 5D). These findings indicate that aberrant expression of viral RNA occurred in infected A72 cells.
DISCUSSION
Epidemiological research conducted on type I FCoV has led to the proposal of the presence of a virulent factor(s) in its viral genome (19, 20). However, experimental confirmation of the presence of a virulent factor(s) is difficult in vivo due to the lack of a feasible FIP cat model of type I FCoV. Recently, we found that type I FCoV strain C3663 has the ability to induce FIP in SPF cats (9). Thus, in the present study, we constructed an infectious cDNA clone derived from the virulence-retaining type I FCoV strain C3663 by utilizing the BAC system. As a result, we successfully rescued recombinant type I FCoV by transfection of the BAC cDNA clone into Fcwf-4 cells, where the recovered rC3663 showed growth kinetics similar to that of the parental virus (Fig. 1). In addition, we generated recombinant type I FCoV carrying Nluc as a reporter gene and applied our reporter rC3663 virus to drug screening (Fig. 2).
Several host proteases, such as furin, TMPRSS2, and cathepsins, are required for the entry step of CoVs as mediated by cleaving spike proteins at the cell surface or in endosomes (33). Yamamoto et al. showed that nafamostat inhibited Middle East respiratory syndrome coronavirus (MERS-CoV) infection by inhibiting TMPRSS2 (34). Consistent with these findings, we identified nafamostat mesylate (compound 38) as an FCoV inhibitor that shows no cytotoxic effects, suggesting that FCoV utilizes the host protease TMPRSS2 for entry. Another compound, lopinavir, is an inhibitor of the human immunodeficiency virus type 1 (HIV-1) protease and is used for AIDS treatment (35). In coronavirus infections, lopinavir inhibits MERS-CoV, severe acute respiratory syndrome coronavirus (SARS-CoV), and human CoV (HCoV) 229E infection by inhibiting the CoV 3C-like protease (29). HCoV-229E belongs to the same genus as FCoV—Alphacoronavirus (3). Unsurprisingly, we identified lopinavir (compound 31) as an FCoV inhibitor that does not exhibit cytotoxicity. Our results suggest nafamostat and lopinavir as potential therapeutic agents against FCoV infection.
Specific chemical compounds that are active against type I FCoV can be applied in two different strategies against FIP, namely, cure and prevention. Several studies have attempted to develop curative therapeutic agents against FIP (36–38), and although these compounds suppressed FCoV replication in vitro, the compounds failed to suppress FIP in vivo (39, 40). While the differences between the in vitro and in vivo activities of these compounds remain controversial, the highly complicated pathological mechanism of FIP, including such components as antibody-dependent enhancement of infection (ADE) (41, 42) and type III hypersensitivity (43, 44), makes developing efficient and curative therapeutics against FIP difficult.
On the other hand, the emergence mechanism of FIPV is best explained by an internal mutation theory (16–18) where type I FCoV has the potential to become type I FIPV by acquiring mutations in its viral genome during replication in kittens. Therefore, controlling the viral load of type I FCoV in kittens is important in preventing the onset of FIP disease. If chemical compounds can suppress the viral load of type I FCoV in kittens, then the probability of disease onset should be reduced. Thus, the use of therapeutics against type I FCoV would be an efficient method of controlling FIP. In this regard, our reporter virus represents a powerful tool for finding compounds for use in the cure and/or prevention of FIP, as the virus can be utilized in high-throughput screening.
Several reports have addressed the possibility that some viruses among the members of alphacoronavirus-1, such as type II FCoV (45, 46), type I canine CoV (CCoV) (47), and type IIb CCoV (48), emerged as chimeric viruses via recombination events. Feline aminopeptidase N (APN) works as a viral receptor for type II CCoV and porcine transmissible gastroenteritis virus (TGEV) (15, 49), suggesting that cats, and especially kittens, play an important role in producing new chimeric viruses. However, the possibility of recombination in other animals, such as dogs, has not been well-studied. Furthermore, to the best of our knowledge, no reports have shown type I FCoV induction of apparent CPE in cell culture except for Fcwf-4 cells. Indeed, the C3663 strain of type I FCoV did not exhibit CPE in canine-derived cells (Fig. 3C). However, the level of luciferase activity in A72 cells infected with the rC3663-Nluc virus was significantly increased without concomitant CPE (Fig. 3A and C), and although the progeny virus production levels were low, A72 cells permitted type I FCoV replication (Fig. 3D). We reasoned that investigations of type I FCoV infectivity in other cell lines should not rely solely on CPE-based assays. Furthermore, our findings indicate that events of recombination between type I FCoV and CCoV may occur in dogs. Specifically, type I FCoV may be transmitted from cats to dogs and a new CoV may emerge in dogs through recombination events. In fact, CCoV replicates in A72 cells (50).
Like other positive-strand viruses, CoVs require host factors for replication (51–53) in infected cells. Unlike A72 cells, MDCK and DH82 cells did not permit viral replication of type I FCoV from pBAC-FCoV-C3663-Nluc (Fig. 3). Thus, we hypothesized that essential host factors exist in canine-derived A72 cells but not in MDCK cells. Future research should elucidate the host factors that support replication of type I FCoV in A72 cells, as these factors would be attractive targets for the development of therapeutics against type I FCoV.
Recently, many researchers have focused on bats as a major reservoir of novel viruses (54). Several novel viruses have been identified and/or isolated from bats, and some are related to human-pathogenic viruses, such as SARS-CoV (55) and MERS-CoV (56). However, other animal species are also potential novel virus reservoirs; for example, Lau et al. reported that the Deltacoronavirus porcine CoV HKU15 (PorCoV HKU15) can be transmitted from birds to pigs (57). In addition, HCoV OC43 is known to originate from bovine CoV (58). Therefore, not only bats but also other animal species should be investigated for their potential as reservoirs or sources of emerging viruses. Indeed, we found that the canine-derived A72 cell line permitted replication of type I FCoV (Fig. 3). Although the host receptors of type I FCoV remain unclear, use of our reporter virus for FCoV infection may provide insights on viral host jumping and emergence of novel viruses.
Generally, expression levels of sg mRNA6 are the highest among all viral mRNAs in CoV-infected cells (59). While sg mRNA6 expression levels in A72 cells were lower than in Fcwf-4 cells, the expression ratio of sg mRNA6 to sg mRNA7 in infected A72 cells was different from that in infected Fcwf-4 cells (Fig. 5A). In fact, the expression levels of sg mRNA6 in 2 μg of total RNA extracted from infected A72 cells were similar to its expression levels in 0.05 μg of total RNA extracted from Fcwf-4 cells (Fig. 5D). In contrast, the expression levels of sg mRNA7 in A72 cells were lower than in Fcwf-4 cells (Fig. 5D), suggesting that synthesis of sg mRNAs is impaired in A72 cells.
Our real-time RT-PCR method amplifies the 3ʹ-UTR of viral RNA, which is present in all viral RNAs. Therefore, the increase in viral RNA in A72 cells (Fig. 3D) was possibly caused by the synthesis of sg mRNA6 (Fig. 5A). To our knowledge, there are no reports on aberrant RNA transcription of CoVs in infected cells, such as in A72 cells. Several host proteins, such as members of the heterogeneous nuclear ribonucleoprotein (hnRNP) family or the DEAD box RNA helicase family, interact with the TRS of CoV RNA and regulate transcription/replication (60). Thus, our data indicate that a host factor(s) present in Fcwf-4 cells, but not in A72 cells, regulates viral RNA transcription. On the basis of the aberrant expression of viral mRNA seen in rC3663 virus-infected A72 cells, further studies are required for uncovering the CoV RNA transcription mechanisms in A72 cells.
In conclusion, we generated recombinant type I FCoV strain C3663 using a BAC-based reverse genetics system. Our recombinant virus can potentially be used to expand research on type I FCoV-induced FIP. Furthermore, infection of the A72 cells with the rC3663 virus revealed unusual viral mRNA expression patterns that may provide novel insights into the mechanisms of CoV transcription. Although further study is required to test whether the recombinant virus can induce FIP in vivo, our established type I FCoV clone has the potential to be a powerful tool for understanding FIP pathogenesis. For example, because the S gene is a candidate for FCoV pathogenesis (19, 20), a chimeric S gene virus corresponding to our recombinant virus may provide novel insights into type I FCoV pathogenesis. Additionally, other groups have previously established type I FCoV reverse genetics using the Black strain (26) and type I field virus (61); thus, exploiting multiple approaches can synergistically further our understanding of FIP.
MATERIALS AND METHODS
Cells and viruses.
Cat-derived Fcwf-4 cells (CRL-2787) (9, 62) and the canine-derived cell lines A72 (CRL-1542) (63, 64), MDCK (CCL-34) (65), and DH82 (CRL-10389) (66) were grown in Dulbecco’s modified Eagle’s medium (DMEM; Nacalai Tesque, Kyoto, Japan) supplemented with 10% (for Fcwf-4, A72, and MDCK cells) or 15% (DH82 cells) heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (Nacalai Tesque). Cells were maintained in a humidified 5% CO2 incubator at 37°C. The type I FCoV strains C3663 and Yayoi were used in this study (9, 23) and propagated in Fcwf-4 cells as described previously (14).
BAC construction.
The BAC DNA of SARS-CoV-Rep (67) was kindly provided by Luis Enjuanes (Spanish National Center for Biotechnology [CNB-CSIC], Madrid, Spain) and was used as a backbone BAC sequence to generate infectious cDNA carrying the full-length type I FCoV strain C3663 sequence. The full-length genomic sequence of C3663 was divided into 11 fragments that were then assembled into pBeloBAC11 plasmids in sequential order (Fig. 1A). Fragments Fr1, Fr2, Fr3, Fr4, Fr5, Fr6, Fr7, Fr8, Fr9, Fr10, and Fr11 correspond to nt 11218 to 13811, nt 13812 to 16360, nt 16361 to 18675, nt 18676 to 20998, nt 20999 to 23400, nt 23401 to 26227, nt 9210 to 11217, nt 26228 to 28545, nt 1 to 2069, nt 2070 to 5152, and nt 5153 to 9834, respectively. Red/ET recombination was employed for fragment integration with a Red/ET recombination system counterselection BAC modification kit (Gene Bridges, Heidelberg, Germany) according to the manufacturer’s instructions (32). The cDNA corresponding to the 3ʹ end of the genomic RNA between nt 28046 and 28545 with 25 nt of adenine (pA) and a partial HDV ribozyme (Rz) sequence was generated by chemical synthesis (Eurofins, Brussels, Belgium); this cDNA was used as a PCR template for synthesizing Fr8 (Fig. 1A). For synthesizing other fragments, RT-PCR was carried out using genomic RNA of the C3663 strain as described under “RNA extraction and RT-PCR” (see below). For Fr11 amplification, reverse primer YT648 (5ʹ-TTTGCCTTATAACTTCCGTAGGTGTAAACTCATCACATAATGAGCCATAAGACA-3ʹ) was designed to disrupt the EcoRI site of C3663 between nt 9829 and nt 9834 (Fig. 1B). The cDNA clone carrying the full-length C3663 sequence was designated pBAC-FCoV-C3663. Sequence analysis of the full-genome sequence of C3663 in pBAC-FCoV-C3663 was carried out by Eurofins. For construction of reporter rC3663, we replaced the ORF 3abc genes of pBAC-FCoV-C3663 with Nluc (Promega, Fitchburg, WI) using the recombination method described above (Fig. 2A) and designated the infectious cDNA clone pBAC-FCoV-C3663-Nluc. The same recombination method was applied for the construction of pBAC-FCoV-C3663-Nluc-PolDead, which possesses amino acid substitutions (SDD to SAA) at the active site of RdRp (nsp12; Fig. 4A).
RNA extraction and RT-PCR.
Viral RNAs from the viral stock of type I FCoV strain C3663 and strain rC3663 were extracted using a PureLink RNA minikit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. RNA was subjected to reverse transcription using a SuperScript III first-strand synthesis system for RT-PCR (Invitrogen, Waltham, MA) and random hexamers according to the manufacturer’s instructions, and PCR was carried out using PrimeSTAR GXL DNA polymerase (TaKaRa, Shiga, Japan). C3663 cDNA was used as a PCR template for synthesizing the recombination template as described above. rC3663 cDNA was used for genetic marker confirmation.
Total RNA from rC3663 virus-infected or mock-infected Fcwf-4 cells and A72 cells was isolated and subjected to reverse transcription as described above.
Plasmid construction.
To obtain DIG-labeled riboprobes for detecting FCoV RNA via Northern blot analysis, we constructed the pSPT18-FCoV plasmid. The nucleotide sequence between nt 27803 and nt 28329 of the C3663 genome was cloned into the pSPT18 plasmid through ligation using HindIII and EcoRI restriction sites (DNA ligation kit Mighty Mix; TaKaRa). Furthermore, we constructed Escherichia coli expression plasmid pGEX6P-1-3663N181-377/GST to produce a glutathione S-transferase (GST)-fused partial N protein. The nucleotide sequence between nt 26745 and nt 27335, encoding amino acid residues 181 to 377 of C3663, was inserted into pGEX6P-1 through ligation using BamHI and XhoI restriction sites (DNA ligation kit Mighty Mix).
Rescue of rC3663 from susceptible Fcwf-4 cells.
Fcwf-4 cells were seeded onto 6-well plates (Violamo; Misumi, Schaumberg, IL) at 4.0 × 105 cells/well. After incubation at 37°C overnight, the cells were transfected using 4 μg cDNA clones and XtremeGene 9 DNA transfection reagent (Roche, Basel, Switzerland) according to the manufacturer’s instructions. After 3 days of culturing at 37°C, CPE were observed and culture supernatants were harvested. The supernatants were stored as passage 0 (P0) viruses. P0 viruses were passaged twice in fresh Fcwf-4 cells in 10-cm-diameter dishes (Violamo), and then P2 viruses were used for conducting experiments.
rC3663 genetic marker confirmation.
To determine the genetic marker of rC3663, rC3663 cDNA was used as a template for PCR using primer pair YT649 (5ʹ-GCATGCAACTGGAGGGTACT-3ʹ) and YT650 (5ʹ-AGAGGATAGCCAAAGCGGTC-3ʹ) and PrimeSTAR GXL DNA polymerase. PCR products were purified using a HighPure PCR purification kit (Roche), and DNA samples were treated with EcoRI at 37°C overnight. Treated samples were then subjected to electrophoresis and cleavage was verified. Purified DNA samples were also used for sequence analysis to confirm the genetic marker. Sequence analyses were carried out by Eurofins using primer YT649.
Measurement of viral growth in Fcwf-4 cells and canine-derived A72, MDCK, and DH82 cells.
Fcwf-4, A72, MDCK, and DH82 cells were seeded onto 6-well plates at 4.0 × 105, 2.0 × 105, 2.0 × 105, and 2.0 × 105 cells/well, respectively, and cultured at 37°C overnight. Viruses were used to inoculate each cell line at an MOI of 0.01. After adsorption at 37°C for 1 h, the cells were washed twice with DMEM and then fresh DMEM containing 10% FBS was added. Infected cells were cultured at 37°C for 24, 48, and 72 h, and culture supernatants were collected and stored at –80°C until further use in titration assays. Next, infected cells were collected and washed once using phosphate-buffered saline (PBS) and the cell pellets divided into two aliquots for real-time RT-PCR and Western blot analysis.
Measurement of viral titers by the plaque assay.
Fcwf-4 cells were seeded onto 6-well plates at 1.0 × 106 cells/well and cultured at 37°C overnight. Samples were diluted with a 10-fold serial dilution from a 10× dilution using DMEM. Diluted viruses (400 μl) were added to the Fcwf-4 cells, and the mixture was incubated at 37°C for 1 h for adsorption. After adsorption, supernatants were removed, and the cells were washed twice using DMEM. Next, a mixture of 0.8% agarose (SeaPlaque GTG agarose; Lonza, Basel, Switzerland) with DMEM containing 10% FBS was overlaid on to the infected cells, and the cells were incubated at 37°C until CPE were observed. Finally, infected cells were fixed with phosphate-buffered formalin (Nacalai Tesque) and stained with crystal violet. The number of plaques was then determined, and viral titers were calculated as numbers of PFU per milliliter.
Northern blot analysis.
RNAs from C3663-infected or rC3663-infected Fcwf-4 cells were used for Northern blot analysis as described elsewhere (32). RNA samples were diluted to 2 μg in 5 μl by the use of UltraPure distilled water (DW) (Invitrogen) and then mixed with 5 μl 2× loading dye (New England Biolabs, Ipswich, MA). After heating was performed at 65°C for 5 min, 10-μl RNA samples were subjected to electrophoresis with 1.2% denaturing agarose gel and transferred onto a positively charged nylon membrane (Roche). Northern blot analysis was performed using a DIG wash and block buffer set and a DIG luminescence detection kit (Roche). DIG-labeled riboprobes for detecting viral RNAs were generated using pSPT18-FCoV and a DIG RNA labeling kit (SP6/T7; Roche) as described previously (68, 69).
Measurement of Nluc activity.
Fcwf-4 cells (1.0 × 105 cells/well) and A72, MDCK, and DH82 cells (all 1.5 × 105 cells/well) were seeded onto 24-well plates (Violamo) and cultured at 37°C overnight. After washing, rC3663-Nluc or rC3663 was inoculated at an MOI of 0.01 or 0.1. After adsorption, the cells were washed twice using DMEM and then fresh DMEM containing 10% FBS was added. The cells were then incubated at 37°C for 24, 48, and 72 h, after which culture supernatants were removed and cells lysed using passive buffer (Promega). Luciferase activity levels were measured using a Nano-Glo luciferase assay system (Promega) on a PowerScan HT luminometer (DS Pharma Biomedical, Osaka, Japan). The experiments were carried out in triplicate.
Measurement of inhibitory effects on type I FCoV replication.
Fcwf-4 cells were seeded onto 24-well plates at 1.0 × 105 cells/well and cultured at 37°C overnight. After washing of the cells was performed, 100 μl rC3663-Nluc was used for inoculation at an MOI of 0.01. After adsorption at 37°C for 1 h, the infected cells were washed twice with DMEM and then fresh DMEM supplemented with 10% FBS and different concentrations of CsA (Sigma-Aldrich, St. Louis, MO) or lopinavir (Sigma-Aldrich) was added. Using a 2-fold serial dilution performed with DMSO, CsA was diluted from 50 μM to 3.125 μM and lopinavir from 30 μM to 3.75 μM. DMSO was also used as a control. After incubation at 37°C for 24 h, culture supernatants were removed, and the cells lysed with passive buffer. Nluc activity was then measured as described above.
Compound screening using a protease inhibitor library.
We used a chemical library of 68 compounds (L1100) (Protease Inhibitor Library; TargetMol) (70) for compound screening (Table 2). Fcwf-4 cells were seeded onto 96-well plates (Violamo) at 2.0 × 104 cells/well and cultured at 37°C overnight. Mixtures containing rC3663-Nluc at an MOI of 0.01 and a 10 μM concentration of each compound from the library were prepared in DMEM and added to the cultured Fcwf-4 cells. CsA (10 μM) and DMSO were used as the positive and negative controls, respectively. After incubation for 24 h, the culture supernatants were removed and the cells lysed in passive buffer. Nluc activity was measured as described above.
TABLE 2.
Compound library of protease inhibitorsa
| Compound | ID | Name | CAS no. | Formula |
|---|---|---|---|---|
| 1 | T2316 | MK3102 | 1226781-44-7 | C17H20F2N4O3S |
| 2 | T1772 | Apoptosis activator 2 | 79183-19-0 | C15H9Cl2NO2 |
| 3 | T1581 | Picolamine | 3731-52-0 | C6H8N2 |
| 4 | T2893 | Muscone | 541-91-3 | C16H30O |
| 5 | T0429 | Glucosamine | 3416-24-8 | C6H13NO5 |
| 6 | T0372 | Gabexate mesylate | 56974-61-9 | C17H27N3O7 |
| 7 | T0087L | Sulfacetamide sodium | 127-56-0 | C8H9N2NaO3S |
| 8 | T0127 | Glimepiride | 93479-97-1 | C24H34N4O5S |
| 9 | T0178 | Saxagliptin hydrate | 945667-22-1 | C18H25N3O2.H2O |
| 10 | T0191 | Linagliptin | 668270-12-0 | C25H28N8O2 |
| 11 | T0242 | Sitagliptin | 486460-32-6 | C16H15F6N5O |
| 12 | T0984 | Fluorouracil (5-fluoracil [5-FU]) | 51-21-8 | C4H3FN2O2 |
| 13 | T1140 | Doxycycline HCl | 10592-13-9 | C22H24N2O8.HCl |
| 14 | T1149 | Fenofibrate | 49562-28-9 | C20H21ClO4 |
| 15 | T1366 | 3-Pyridylacetic acid hydrochloride | 6419-36-9 | C7H8ClNO2 |
| 16 | T2731 | Usnic acid | 125-46-2 | C18H16O7 |
| 17 | T2728 | Limonin | 1180-71-8 | C26H30O8 |
| 18 | T2830 | Betulinic acid | 472-15-1 | C29H46O3 |
| 19 | T2754 | Oxymatrine | 16837-52-8 | C15H24N2O2 |
| 20 | T2888 | Pterostilbene | 537-42-8 | C16H16O3 |
| 21 | T0789 | PMSF | 329-98-6 | C7H7FO2S |
| 22 | T0951 | Hydroxychloroquine sulfate | 747-36-4 | C18H26ClN3O.H2SO4 |
| 23 | T1402 | Fenofibric acid | 42017-89-0 | C17H15ClO4 |
| 24 | T1462 | Captopril | 62571-86-2 | C9H15NO3S |
| 25 | T1525 | Ritonavir | 155213-67-5 | C37H48N6O5S2 |
| 26 | T1564 | Cisplatin | 15663-27-1 | H6Cl2N2Pt |
| 27 | T2843 | Aloe-emodin | 481-72-1 | C15H10O5 |
| 28 | T2401 | Alogliptin benzoate | 850649-62-6 | C25H27N5O4 |
| 29 | T2399 | Bortezomib (PS-341) | 179324-69-7 | C19H25BN4O4 |
| 30 | T1592 | Acetohydroxamic acid | 546-88-3 | C2H5NO2 |
| 31 | T1623 | Lopinavir | 192725-17-0 | C37H48N4O5 |
| 32 | T2296 | SYR472 | 1029877-94-8 | C22H26FN5O6 |
| 33 | T2262 | GHF-5074 | 749269-83-8 | C16H11Cl2FO2 |
| 34 | T2016 | MLN9708 | 1201902-80-8 | C20H23BCl2N2O9 |
| 35 | T2122 | MLN2238 (ixazomib) | 1072833-77-2 | C14H19BCl2N2O4 |
| 36 | T2239 | Raltegravir potassium | 871038-72-1 | C20H20FN6O5.K |
| 37 | T2117 | PSI6206 | 863329-66-2 | C10H13FN2O5 |
| 38 | T2392 | Nafamostat mesylate | 82956-11-4 | C19H17N5O2.2CH4O3S |
| 39 | T1786 | Daclatasvir (BMS790052) | 1009119-65-6 | C40H52Cl2N8O6 |
| 40 | T2324 (T3335) | Darunavir ethanolate | 635728-49-3 | C27H37N3O7S.C2H5OH |
| 41 | T2743 | Ilomastat (GM6001 [Galardin]) | 142880-36-2 | C20H28N4O4 |
| 42 | T2332 | Elvitegravir (GS-9137 [JTK-303]) | 697761-98-1 | C23H23ClFNO5 |
| 43 | T2329 | Dolutegravir (GSK1349572) | 1051375-19-9 | C20H18F2N3NaO5 |
| 44 | T2834 | Nobiletin | 478-01-3 | C21H22O8 |
| 45 | T3028 | Celastrol | 34157-83-0 | C29H38O4 |
| 46 | T2792 | Glucosamine sulfate | 29031-19-4 | C6H13NO5.H2SO4 |
| 47 | T0100 | Atazanavir sulfate | 229975-97-7 | C38H52N6O7.H2SO4 |
| 48 | T1853 | NMS 873 | 1418013-75-8 | C27H28N4O3S2 |
| 49 | T1822 | Clemizole | 442-52-4 | C19H20ClN3 |
| 50 | T1795 | Carfilzomib (PR-171) | 868540-17-4 | C40H57N5O7 |
| 51 | T0100L | Atazanavir | 198904-31-3 | C38H52N6O7 |
| 52 | T1502 | Vildagliptin (LAF-237) | 274901-16-5 | C17H25N3O2 |
| 54 | T2009 | SB-3CT | 292605-14-2 | C15H14O3S2 |
| 55 | T1757 | ML323 | 1572414-83-5 | C23H24N6 |
| 56 | T2424 | P22077 | 1247819-59-5 | C12H7F2NO3S2 |
| 57 | T2493 | PD 151746 | 179461-52-0 | C11H8FNO2S |
| 58 | T2503 | PAC1 | 315183-21-2 | C23H28N4O2 |
| 59 | T2393 | Efavirenz | 154598-52-4 | C14H9ClF3NO2 |
| 60 | T1883 | Des(benzylpyridyl) Atazanavi | 1192224-24-0 | C26H43N5O7 |
| 61 | T1862 | PR-619 | 2645-32-1 | C7H5N5S2 |
| 62 | T2625 | MK0752 | 471905-41-6 | C21H21ClF2O4S |
| 63 | T2639 | LY2811376 | 1194044-20-6 | C15H14F2N4S |
| 64 | T3075 | FLI-06 | 313967-18-9 | C25H30N2O5 |
| 65 | T1969 | DBEQ | 177355-84-9 | C22H20N4 |
| 66 | T1932 | B-AP15 | 1009817-63-3 | C22H17N3O6 |
| 67 | T1924 | LDN-57444 | 668467-91-2 | C17H11Cl3N2O3 |
| 68 | T1891 | NSC 405020 | 7497-07-6 | C12H15Cl2NO |
| 69 | T2154 | MG-132 | 133407-82-6 | C26H41N3O5 |
ID, identifier; CAS, Chemical Abstracts Service; PMSF, phenylmethylsulfonyl fluoride.
MTT assay.
Fcwf-4 cells were seeded onto 96-well plates at 1.0 × 104 cells/well and cultured at 37°C overnight. Each compound (10 μM in DMEM) from the protease inhibitor library was added to the cultured Fcwf-4 cells. After incubation for 24 h, MTT assays were carried out using an MTT cell count kit (Nacalai Tesque) according to the manufacturer’s instructions. Absorbance at 570 nm was then measured with the PowerScan HT luminometer.
Real-time RT-PCR.
Cell pellets of infected Fcwf-4, A72, MDCK, and DH82 cells were lysed, and then total RNA was extracted using a PureLink RNA minikit. RNA was then used for real-time RT-PCR with Thunderbird Probe one-step quantitative PCR (qPCR) mix (Toyobo, Osaka, Japan) and run on a StepOne real-time PCR system (Applied Biosystems, Waltham, MA). For quantification of FCoV RNA, we used 3ʹ-UTR targeting primers FcoV1128f (5ʹ-GATTTGATTTGGCAATGCTAGATTT-3ʹ; nt 28398 to 28422 of C3663) and FcoV1229r (5ʹ-AACAATCACTAGATCCAGACGTTAGCT-3ʹ; nt 28473 to 28499 of C3663) as well as a specific probe (5ʹ-TCCATTGTTGGCTCGTCATAGCGGA-3ʹ; nt 28446 to 28470 of C3663) labeled with 6-carboxyfluorescein (FAM) (71). For quantification of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA, wk1288 (5′-GAAGGTGAAGGTCGGAGT-3′) and wk1289 (5′-GAAGATGGTGATGGGATTTC-3′) were used as primers and FAM-labeled wk1290 (5′-CAAGCTTCCCGTTCTCAGCC-3′) was used as a probe (32). Cycling conditions were 95°C for 1 min followed by 40 cycles of 95°C for 15 s and 58°C for 45 s.
Construction of monoclonal antibody against N protein of type I FCoV.
A GST-fused partial N protein, C3663N181-377/GST, was expressed in Escherichia coli carrying pGEX6P-1-C3663N181-377/GST and purified using glutathione Sepharose 4B beads (GE Healthcare, Chicago, IL). The purified protein was used as an antigen for immunization of BALB/c mice. The method of monoclonal antibody (MAb) establishment was described previously (72). Screening of hybridomas was performed by enzyme-linked immunosorbent assays (ELISA) using purified C3663N181-377/GST or GST proteins. ELISA was carried out as described previously (73). The experiments yielded a clone (designated 4D10) which produced MAbs against type I FCoV N protein.
Western blot analysis.
Infected Fcwf-4, A72, MDCK, and DH82 cells were lysed in lysis buffer (100 mM Tris-HCl [pH 8.0], 150 mM NaCl, and 1% Triton X-100) and centrifuged at 16,000 × g for 10 min at 4°C. Supernatants were then collected and mixed with 2× sample buffer (0.1 M Tris-HCl [pH 6.8], 4% sodium dodecyl sulfate [SDS], 20% glycerol, 0.004% bromophenol blue, 10% 2-mercaptoethanol). Next, the boiled samples were subjected to electrophoresis by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Merck Millipore, Billerica, MA). The membranes were blocked in 3% skim milk–PBS containing 0.05% Tween 20 (PBS-T; Nacalai Tesque). The primary antibodies were mouse anti-FCoV-N monoclonal antibody (4D10) and mouse anti-β-actin antibody (Sigma-Aldrich), while goat anti-mouse IgG horseradish peroxidase (HRP; Sigma-Aldrich) was used as a secondary antibody. ChemiLumi One Ultra (Nacalai Tesque) was used for visualization (32).
IFA.
Fcwf-4 and A72 cells were seeded onto 35-mm-diameter glass-bottom dishes (Matsunami Glass, Osaka, Japan) at 2.0 × 105 and 1.0 × 105 cells/well, respectively. After incubation at 37°C overnight, the cells were inoculated with rC3663 at an MOI of 0.1. Following adsorption at 37°C for 1 h, the supernatants were removed and fresh DMEM containing 10% FBS was added. The infected cells were cultured at 37°C for 48 h and then fixed with 4% paraformaldehyde–PBS. After washing was performed once with PBS, the cells were permeabilized for 15 min at room temperature with PBS containing 0.5% Triton X-100. The cells were then incubated with mouse anti-FCoV-N 4D10 monoclonal antibody at 4°C overnight, after which they were washed three times with PBS and incubated with CF488-conjugated anti-mouse IgG (Sigma-Aldrich) (1:500) for 1 h at room temperature. Finally, the cells were washed three times with PBS and then Fluoroshield with DAPI (4′,6-diamidino-2-phenylindole; ImmunoBioScience, Mukilteo, WA) was used as a mounting medium and for counterstaining of nuclei with DAPI. The cells were observed with a FluoView FV1000 laser scanning confocal microscope (Olympus, Tokyo, Japan).
Transfection of MDCK cells with pBAC-FCoV-Nluc.
MDCK cells were seeded onto 24-well plates at 1.5 × 105 cells/well and cultured at 37°C overnight. The cells were then transfected with 1 μg pBAC-FCoV-Nluc and 0.1 μg pcDNA3.1-fluc (32) using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. pBAC-FCoV-Nluc-PolDead was used as a control. Transfected cells were incubated at 37°C for 6 h, after which the supernatants were removed and fresh DMEM containing 10% FBS was added. After incubation was performed for an additional 18, 42, and 66 h (i.e., 24, 48, and 72 h after transfection), culture supernatants were removed and the cells lysed in passive buffer. Levels of Nluc and firefly luciferase activity were measured as described above and with a luciferase assay system (Promega), respectively. Nluc activity was normalized to firefly luciferase activity. For other virological tests (Fig. 4B to D), the experiment was scaled up from 24-well plates to 6-well plates.
ACKNOWLEDGMENTS
We thank Luis Enjuanes (CNB-CSIC, Madrid, Spain) for providing the SARS-CoV-Rep BAC DNA. We also thank Kaede Yukawa for secretarial assistance and Kanako Yoshizawa and Nozomi Shimada for their technical assistance.
This work was supported in part by grants-in-aid for the Research Program on Emerging and Re-Emerging Infectious Diseases from the Japan Agency for Medical Research and Development and Japanese Society for the Promotion of Science (JSPS) (KAKENHI grant JP16K08811, JP18fk0108058, and JP15J07066). Y.T. was supported by a JSPS Research Fellowship for young scientists. We have no potential conflicts of interest in relation to this study.
REFERENCES
- 1.Weiss SR, Navas-Martin S. 2005. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 69:635–664. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woo PC, Huang Y, Lau SK, Yuen KY. 2010. Coronavirus genomics and bioinformatics analysis. Viruses 2:1804–1820. doi: 10.3390/v2081803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed). 2012. Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses. Academic Press, London, United Kingdom. [Google Scholar]
- 4.Biek R, Ruth TK, Murphy KM, Anderson CR Jr, Johnson M, DeSimone R, Gray R, Hornocker MG, Gillin CM, Poss M. 2006. Factors associated with pathogen seroprevalence and infection in Rocky Mountain cougars. J Wildl Dis 42:606–615. doi: 10.7589/0090-3558-42.3.606. [DOI] [PubMed] [Google Scholar]
- 5.Hofmann-Lehmann R, Fehr D, Grob M, Elgizoli M, Packer C, Martenson JS, O’Brien SJ, Lutz H. 1996. Prevalence of antibodies to feline parvovirus, calicivirus, herpesvirus, coronavirus, and immunodeficiency virus and of feline leukemia virus antigen and the interrelationship of these viral infections in free-ranging lions in east Africa. Clin Diagn Lab Immunol 3:554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heeney JL, Evermann JF, McKeirnan AJ, Marker-Kraus L, Roelke ME, Bush M, Wildt DE, Meltzer DG, Colly L, Lukas J. 1990. Prevalence and implications of feline coronavirus infections of captive and free-ranging cheetahs (Acinonyx jubatus). J Virol 64:1964–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedersen NC, Evermann JF, McKeirnan AJ, Ott RL. 1984. Pathogenicity studies of feline coronavirus isolates 79-1146 and 79-1683. Am J Vet Res 45:2580–2585. [PubMed] [Google Scholar]
- 8.Holzworth J. 1963. Some important disorders of cats. Cornell Vet 53:157–160. [PubMed] [Google Scholar]
- 9.Terada Y, Shiozaki Y, Shimoda H, Mahmoud HY, Noguchi K, Nagao Y, Shimojima M, Iwata H, Mizuno T, Okuda M, Morimoto M, Hayashi T, Tanaka Y, Mochizuki M, Maeda K. 2012. Feline infectious peritonitis virus with a large deletion in the 5’-terminal region of the spike gene retains its virulence for cats. J Gen Virol 93:1930–1934. doi: 10.1099/vir.0.043992-0. [DOI] [PubMed] [Google Scholar]
- 10.Fiscus SA, Teramoto YA. 1987. Antigenic comparison of feline coronavirus isolates: evidence for markedly different peplomer glycoproteins. J Virol 61:2607–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hohdatsu T, Sasamoto T, Okada S, Koyama H. 1991. Antigenic analysis of feline coronaviruses with monoclonal antibodies (MAbs): preparation of MAbs which discriminate between FIPV strain 79-1146 and FECV strain 79-1683. Vet Microbiol 28:13–24. doi: 10.1016/0378-1135(91)90096-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benetka V, Kübber-Heiss A, Kolodziejek J, Nowotny N, Hofmann-Parisot M, Möstl K. 2004. Prevalence of feline coronavirus types I and II in cats with histopathologically verified feline infectious peritonitis. Vet Microbiol 99:31–42. doi: 10.1016/j.vetmic.2003.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kummrow M, Meli ML, Haessig M, Goenczi E, Poland A, Pedersen NC, Hofmann-Lehmann R, Lutz H. 2005. Feline coronavirus serotypes 1 and 2: seroprevalence and association with disease in Switzerland. Clin Diagn Lab Immunol 12:1209–1215. doi: 10.1128/CDLI.12.10.1209-1215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shiba N, Maeda K, Kato H, Mochizuki M, Iwata H. 2007. Differentiation of feline coronavirus type I and II infections by virus neutralization test. Vet Microbiol 124:348–352. doi: 10.1016/j.vetmic.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hohdatsu T, Izumiya Y, Yokoyama Y, Kida K, Koyama H. 1998. Differences in virus receptor for type I and type II feline infectious peritonitis virus. Arch Virol 143:839–850. doi: 10.1007/s007050050336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pedersen NC, Liu H, Dodd KA, Pesavento PA. 2009. Significance of coronavirus mutants in feces and diseased tissues of cats suffering from feline infectious peritonitis. Viruses 1:166–184. doi: 10.3390/v1020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poland AM, Vennema H, Foley JE, Pedersen NC. 1996. Two related strains of feline infectious peritonitis virus isolated from immunocompromised cats infected with a feline enteric coronavirus. J Clin Microbiol 34:3180–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vennema H, Poland A, Foley J, Pedersen NC. 1998. Feline infectious peritonitis viruses arise by mutation from endemic feline enteric coronaviruses. Virology 243:150–157. doi: 10.1006/viro.1998.9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang HW, Egberink HF, Halpin R, Spiro DJ, Rottier PJ. 2012. Spike protein fusion peptide and feline coronavirus virulence. Emerg Infect Dis 18:1089–1095. doi: 10.3201/eid1807.120143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Licitra BN, Millet JK, Regan AD, Hamilton BS, Rinaldi VD, Duhamel GE, Whittaker GR. 2013. Mutation in spike protein cleavage site and pathogenesis of feline coronavirus. Emerg Infect Dis 19:1066–1073. doi: 10.3201/eid1907.121094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiss I, Poland AM, Pedersen NC. 2004. Disease outcome and cytokine responses in cats immunized with an avirulent feline infectious peritonitis virus (FIPV)-UCD1 and challenge-exposed with virulent FIPV-UCD8. J Feline Med Surg 6:89–97. doi: 10.1016/j.jfms.2003.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pedersen NC, Black JW. 1983. Attempted immunization of cats against feline infectious peritonitis, using avirulent live virus or sublethal amounts of virulent virus. Am J Vet Res 44:229–234. [PubMed] [Google Scholar]
- 23.Mochizuki M, Mitsutake Y, Miyanohara Y, Higashihara T, Shimizu T, Hohdatsu T. 1997. Antigenic and plaque variations of serotype II feline infectious peritonitis coronaviruses. J Vet Med Sci 59:253–258. doi: 10.1292/jvms.59.253. [DOI] [PubMed] [Google Scholar]
- 24.Eckert N, Wrensch F, Gartner S, Palanisamy N, Goedecke U, Jager N, Pohlmann S, Winkler M. 2014. Influenza A virus encoding secreted Gaussia luciferase as useful tool to analyze viral replication and its inhibition by antiviral compounds and cellular proteins. PLoS One 9:e97695. doi: 10.1371/journal.pone.0097695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran V, Moser LA, Poole DS, Mehle A. 2013. Highly sensitive real-time in vivo imaging of an influenza reporter virus reveals dynamics of replication and spread. J Virol 87:13321–13329. doi: 10.1128/JVI.02381-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tekes G, Hofmann-Lehmann R, Stallkamp I, Thiel V, Thiel HJ. 2008. Genome organization and reverse genetic analysis of a type I feline coronavirus. J Virol 82:1851–1859. doi: 10.1128/JVI.02339-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Wilde AH, Zevenhoven-Dobbe JC, van der Meer Y, Thiel V, Narayanan K, Makino S, Snijder EJ, van Hemert MJ. 2011. Cyclosporin A inhibits the replication of diverse coronaviruses. J Gen Virol 92:2542–2548. doi: 10.1099/vir.0.034983-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pfefferle S, Schöpf J, Kögl M, Friedel CC, Müller MA, Carbajo-Lozoya J, Stellberger T, von Dall’Armi E, Herzog P, Kallies S, Niemeyer D, Ditt V, Kuri T, Züst R, Pumpor K, Hilgenfeld R, Schwarz F, Zimmer R, Steffen I, Weber F, Thiel V, Herrler G, Thiel H-J, Schwegmann-Weßels C, Pöhlmann S, Haas J, Drosten C, von Brunn A. 2011. The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog 7:e1002331. doi: 10.1371/journal.ppat.1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Wilde AH, Jochmans D, Posthuma CC, Zevenhoven-Dobbe JC, van Nieuwkoop S, Bestebroer TM, van den Hoogen BG, Neyts J, Snijder EJ. 2014. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob Agents Chemother 58:4875–4884. doi: 10.1128/AAC.03011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyle JF, Pedersen NC, Evermann JF, McKeirnan AJ, Ott RL, Black JW. 1984. Plaque assay, polypeptide composition and immunochemistry of feline infectious peritonitis virus and feline enteric coronavirus isolates. Adv Exp Med Biol 173:133–147. doi: 10.1007/978-1-4615-9373-7_12. [DOI] [PubMed] [Google Scholar]
- 31.McKeirnan AJ, Evermann JF, Davis EV, Ott RL. 1987. Comparative properties of feline coronaviruses in vitro. Can J Vet Res 51:212–216. [PMC free article] [PubMed] [Google Scholar]
- 32.Terada Y, Kawachi K, Matsuura Y, Kamitani W. 2017. MERS coronavirus nsp1 participates in an efficient propagation through a specific interaction with viral RNA. Virology 511:95–105. doi: 10.1016/j.virol.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Millet JK, Whittaker GR. 2015. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res 202:120–134. doi: 10.1016/j.virusres.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue JI, Matsuda Z. 2016. Identification of nafamostat as a potent inhibitor of Middle East respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother 60:6532–6539. doi: 10.1128/AAC.01043-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walmsley S, Bernstein B, King M, Arribas J, Beall G, Ruane P, Johnson M, Johnson D, Lalonde R, Japour A, Brun S, Sun E, Team MS. 2002. Lopinavir-ritonavir versus nelfinavir for the initial treatment of HIV infection. N Engl J Med 346:2039–2046. doi: 10.1056/NEJMoa012354. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh LE, Lin CN, Su BL, Jan TR, Chen CM, Wang CH, Lin DS, Lin CT, Chueh LL. 2010. Synergistic antiviral effect of Galanthus nivalis agglutinin and nelfinavir against feline coronavirus. Antiviral Res 88:25–30. doi: 10.1016/j.antiviral.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim Y, Mandadapu SR, Groutas WC, Chang KO. 2013. Potent inhibition of feline coronaviruses with peptidyl compounds targeting coronavirus 3C-like protease. Antiviral Res 97:161–168. doi: 10.1016/j.antiviral.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takano T, Katoh Y, Doki T, Hohdatsu T. 2013. Effect of chloroquine on feline infectious peritonitis virus infection in vitro and in vivo. Antiviral Res 99:100–107. doi: 10.1016/j.antiviral.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fischer Y, Ritz S, Weber K, Sauter-Louis C, Hartmann K. 2011. Randomized, placebo controlled study of the effect of propentofylline on survival time and quality of life of cats with feline infectious peritonitis. J Vet Intern Med 25:1270–1276. doi: 10.1111/j.1939-1676.2011.00806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pedersen NC. 2014. An update on feline infectious peritonitis: diagnostics and therapeutics. Vet J 201:133–141. doi: 10.1016/j.tvjl.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olsen CW, Corapi WV, Ngichabe CK, Baines JD, Scott FW. 1992. Monoclonal antibodies to the spike protein of feline infectious peritonitis virus mediate antibody-dependent enhancement of infection of feline macrophages. J Virol 66:956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takano T, Kawakami C, Yamada S, Satoh R, Hohdatsu T. 2008. Antibody-dependent enhancement occurs upon re-infection with the identical serotype virus in feline infectious peritonitis virus infection. J Vet Med Sci 70:1315–1321. doi: 10.1292/jvms.70.1315. [DOI] [PubMed] [Google Scholar]
- 43.Jacobse-Geels HE, Daha MR, Horzinek MC. 1980. Isolation and characterization of feline C3 and evidence for the immune complex pathogenesis of feline infectious peritonitis. J Immunol 125:1606–1610. [PubMed] [Google Scholar]
- 44.Jacobse-Geels HE, Daha MR, Horzinek MC. 1982. Antibody, immune complexes, and complement activity fluctuations in kittens with experimentally induced feline infectious peritonitis. Am J Vet Res 43:666–670. [PubMed] [Google Scholar]
- 45.Herrewegh AA, Smeenk I, Horzinek MC, Rottier PJ, de Groot RJ. 1998. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J Virol 72:4508–4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terada Y, Matsui N, Noguchi K, Kuwata R, Shimoda H, Soma T, Mochizuki M, Maeda K. 2014. Emergence of pathogenic coronaviruses in cats by homologous recombination between feline and canine coronaviruses. PLoS One 9:e106534. doi: 10.1371/journal.pone.0106534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pratelli A, Martella V, Decaro N, Tinelli A, Camero M, Cirone F, Elia G, Cavalli A, Corrente M, Greco G, Buonavoglia D, Gentile M, Tempesta M, Buonavoglia C. 2003. Genetic diversity of a canine coronavirus detected in pups with diarrhoea in Italy. J Virol Methods 110:9–17. doi: 10.1016/S0166-0934(03)00081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Decaro N, Mari V, Campolo M, Lorusso A, Camero M, Elia G, Martella V, Cordioli P, Enjuanes L, Buonavoglia C. 2009. Recombinant canine coronaviruses related to transmissible gastroenteritis virus of swine are circulating in dogs. J Virol 83:1532–1537. doi: 10.1128/JVI.01937-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tresnan DB, Levis R, Holmes KV. 1996. Feline aminopeptidase N serves as a receptor for feline, canine, porcine, and human coronaviruses in serogroup I. J Virol 70:8669–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pratelli A, Decaro N, Tinelli A, Martella V, Elia G, Tempesta M, Cirone F, Buonavoglia C. 2004. Two genotypes of canine coronavirus simultaneously detected in the fecal samples of dogs with diarrhea. J Clin Microbiol 42:1797–1799. doi: 10.1128/jcm.42.4.1797-1799.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. 2010. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol 84:12658–12664. doi: 10.1128/JVI.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verheije MH, Raaben M, Mari M, Te Lintelo EG, Reggiori F, van Kuppeveld FJ, Rottier PJ, de Haan CA. 2008. Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated ARF1 activation. PLoS Pathog 4:e1000088. doi: 10.1371/journal.ppat.1000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu CH, Chen PJ, Yeh SH. 2014. Nucleocapsid phosphorylation and RNA helicase DDX1 recruitment enables coronavirus transition from discontinuous to continuous transcription. Cell Host Microbe 16:462–472. doi: 10.1016/j.chom.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han HJ, Wen HL, Zhou CM, Chen FF, Luo LM, Liu JW, Yu XJ. 2015. Bats as reservoirs of severe emerging infectious diseases. Virus Res 205:1–6. doi: 10.1016/j.virusres.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 56.Annan A, Baldwin HJ, Corman VM, Klose SM, Owusu M, Nkrumah EE, Badu EK, Anti P, Agbenyega O, Meyer B, Oppong S, Sarkodie YA, Kalko EK, Lina PH, Godlevska EV, Reusken C, Seebens A, Gloza-Rausch F, Vallo P, Tschapka M, Drosten C, Drexler JF. 2013. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg Infect Dis 19:456–459. doi: 10.3201/eid1903.121503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lau SKP, Wong EYM, Tsang CC, Ahmed SS, Au-Yeung RKH, Yuen KY, Wernery U, Woo P. 2018. Discovery and sequence analysis of four deltacoronaviruses from birds in the Middle East reveal interspecies jumping with recombination as a potential mechanism for avian-to-avian and avian-to-mammalian transmission. J Virol 92. doi: 10.1128/JVI.00265-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vijgen L, Keyaerts E, Moes E, Thoelen I, Wollants E, Lemey P, Vandamme AM, Van Ranst M. 2005. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J Virol 79:1595–1604. doi: 10.1128/JVI.79.3.1595-1604.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Hemert MJ, van den Worm SH, Knoops K, Mommaas AM, Gorbalenya AE, Snijder EJ. 2008. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog 4:e1000054. doi: 10.1371/journal.ppat.1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sola I, Mateos-Gomez PA, Almazan F, Zuñiga S, Enjuanes L. 2011. RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA Biol 8:237–248. doi: 10.4161/rna.8.2.14991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ehmann R, Kristen-Burmann C, Bank-Wolf B, Konig M, Herden C, Hain T, Thiel HJ, Ziebuhr J, Tekes G. 2018. Reverse genetics for type I feline coronavirus field isolate to study the molecular pathogenesis of feline infectious peritonitis. mBio 9:e01422-18. doi: 10.1128/mBio.01422-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacobse-Geels HE, Horzinek MC. 1983. Expression of feline infectious peritonitis coronavirus antigens on the surface of feline macrophage-like cells. J Gen Virol 64:1859–1866. doi: 10.1099/0022-1317-64-9-1859. [DOI] [PubMed] [Google Scholar]
- 63.Binn LN, Marchwicki RH, Stephenson EH. 1980. Establishment of a canine cell line: derivation, characterization, and viral spectrum. Am J Vet Res 41:855–860. [PubMed] [Google Scholar]
- 64.Nakano H, Kameo Y, Andoh K, Ohno Y, Mochizuki M, Maeda K. 2009. Establishment of canine and feline cells expressing canine signaling lymphocyte activation molecule for canine distemper virus study. Vet Microbiol 133:179–183. doi: 10.1016/j.vetmic.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gaush CR, Hard WL, Smith TF. 1966. Characterization of an established line of canine kidney cells (MDCK). Proc Soc Exp Biol Med 122:931–935. doi: 10.3181/00379727-122-31293. [DOI] [PubMed] [Google Scholar]
- 66.Wellman ML, Krakowka S, Jacobs RM, Kociba GJ. 1988. A macrophage-monocyte cell line from a dog with malignant histiocytosis. In Vitro Cell Dev Biol 24:223–229. doi: 10.1007/BF02623551. [DOI] [PubMed] [Google Scholar]
- 67.Almazán F, Dediego ML, Galán C, Escors D, Álvarez E, Ortego J, Sola I, Zuñiga S, Alonso S, Moreno JL, Nogales A, Capiscol C, Enjuanes L. 2006. Construction of a severe acute respiratory syndrome coronavirus infectious cDNA clone and a replicon to study coronavirus RNA synthesis. J Virol 80:10900–10906. doi: 10.1128/JVI.00385-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kamitani W, Narayanan K, Huang C, Lokugamage K, Ikegami T, Ito N, Kubo H, Makino S. 2006. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc Natl Acad Sci U S A 103:12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tanaka T, Kamitani W, DeDiego ML, Enjuanes L, Matsuura Y. 2012. Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J Virol 86:11128–11137. doi: 10.1128/JVI.01700-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tamura T, Fukuhara T, Uchida T, Ono C, Mori H, Sato A, Fauzyah Y, Okamoto T, Kurosu T, Setoh YX, Imamura M, Tautz N, Sakoda Y, Khromykh AA, Chayama K, Matsuura Y. 2018. Characterization of recombinant Flaviviridae viruses possessing a small reporter tag. J Virol 92:e01582-17. doi: 10.1128/JVI.01582-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gut M, Leutenegger CM, Huder JB, Pedersen NC, Lutz H. 1999. One-tube fluorogenic reverse transcription-polymerase chain reaction for the quantitation of feline coronaviruses. J Virol Methods 77:37–46. doi: 10.1016/S0166-0934(98)00129-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shimoda H, Mahmoud HY, Noguchi K, Terada Y, Takasaki T, Shimojima M, Maeda K. 2013. Production and characterization of monoclonal antibodies to Japanese encephalitis virus. J Vet Med Sci 75:1077–1080. doi: 10.1292/jvms.12-0558. [DOI] [PubMed] [Google Scholar]
- 73.Shimoda H, Inthong N, Noguchi K, Terada Y, Nagao Y, Shimojima M, Takasaki T, Rerkamnuaychoke W, Maeda K. 2013. Development and application of an indirect enzyme-linked immunosorbent assay for serological survey of Japanese encephalitis virus infection in dogs. J Virol Methods 187:85–89. doi: 10.1016/j.jviromet.2012.09.022. [DOI] [PubMed] [Google Scholar]