

Viruses establish infection in hosts by targeting highly permissive cell types. The retrovirus Friend murine leukemia virus (FrMLV) infects a subtype of B cells called B-1 cells that permit robust virus replication. The reason for their susceptibility had remained unknown. We found that innate sensing of incoming virus and the ensuing type I interferon response within B-1 cells are responsible for their observed susceptibility. Our data provide insights into how retroviruses coevolved with the host to co-opt innate immune sensing pathways designed to fight virus infections for establishing infection. Understanding early events in viral spread can inform antiviral intervention strategies that prevent the colonization of a host.
KEYWORDS: retrovirus, FrMLV, B-1 cells, B-2 cells, popliteal lymph node, TLR7, bumble mice
ABSTRACT
Lymph-borne Friend murine leukemia virus (FrMLV) exploits the sentinel macrophages in the draining popliteal lymph node (pLN) to infect highly permissive innate-like B-1 cells and establish infection in mice. The reason for FrMLV sensitivity of B-1 cells and their impact on viral spread is unknown. Here we demonstrate that Toll-like receptor 7 (TLR7) sensing and type I interferon (IFN-I) signaling in B-1 cells contribute to FrMLV susceptibility. FrMLV infection in B-1 cell-deficient mice (bumble; IκBNS dysfunctional) was significantly lower than that in the wild-type mice and was rescued by adoptive transfer of wild-type B-1 cells. This rescue of FrMLV infection in bumble mice was dependent on intact TLR7 sensing and IFN-I signaling within B-1 cells. Analyses of infected cell types revealed that the reduced infection in bumble mice was due predominantly to compromised virus spread to the B-2 cell population. Our data reveal how FrMLV exploits innate immune sensing and activation in the B-1 cell population for infection and subsequent spread to other lymphocytes.
IMPORTANCE Viruses establish infection in hosts by targeting highly permissive cell types. The retrovirus Friend murine leukemia virus (FrMLV) infects a subtype of B cells called B-1 cells that permit robust virus replication. The reason for their susceptibility had remained unknown. We found that innate sensing of incoming virus and the ensuing type I interferon response within B-1 cells are responsible for their observed susceptibility. Our data provide insights into how retroviruses coevolved with the host to co-opt innate immune sensing pathways designed to fight virus infections for establishing infection. Understanding early events in viral spread can inform antiviral intervention strategies that prevent the colonization of a host.
INTRODUCTION
Murine leukemia virus (MLV) is a mouse gammaretrovirus that can cause leukemia and lymphoma in mice (1–3). The virus is transmitted vertically from mother to offspring and horizontally between fighting mice (4, 5). We have studied early events during subcutaneous (s.c.) infection of mice as a model for horizontal transmission (6). We observed that Friend murine leukemia virus (FrMLV) exploits the ability of CD169+ subcapsular sinus (SCS) macrophages (Mϕ) in lymph nodes to be captured and then trans-infect a specific B cell subpopulation called B-1 cells (6). Unlike the major naive B-2 cell population in popliteal lymph nodes (pLNs), B-1 cells are highly susceptible to FrMLV (6) and represent the first B cell subpopulation to be infected during FrMLV infection. However, the reason for B-1 cell susceptibility and its contribution to virus spread are unknown.
Simple retroviruses such as FrMLV require the breakdown of the nuclear envelope that occurs during mitosis to integrate proviral DNA into the host genome (7). Therefore, cell division plays a critical role in FrMLV susceptibility of lymphocytes (7). However, the low homeostatic proliferation rate of B-1 cells is similar to that of other naive B cell populations in peripheral lymphoid tissues and does not explain their heightened susceptibility to FrMLV (8). Due to the low proliferation rate, naive follicular B cells display reduced susceptibility to FrMLV infection ex vivo. However, their permissivity can be prominently enhanced upon stimulation with Toll-like receptor (TLR) agonists, such as lipopolysaccharide (LPS) and imiquimod, suggesting a potential function for TLR sensing in enhancing B cell activation and the susceptibility of B cells to FrMLV infection (9).
Viruses can repurpose antiviral TLR innate immune signaling to promote their infection of the host (10). Mouse mammary tumor virus (MMTV) can sequester bacterial LPS in the gut during mother-to-offspring transmission (11, 12). Virus-bound LPS triggers TLR4 signaling in dendritic cells (DCs) and macrophages that stimulated secretion of the immunosuppressive cytokine interleukin-10 (IL-10) by B cells (11–13). IL-10 can suppress host immune responses in general and facilitate MMTV infection. The innate-like B-1 cells are known to secrete IL-10 homeostatically, as well as upon TLR stimulation, as a means to self-regulate their autoreactivity (14, 15). Whether IL-10 plays a role in increasing or decreasing permissivity of B-1 cells to FrMLV infection is unknown.
Innate and adaptive immune signaling can activate lymphocytes and contribute to the susceptibility of B-1 cells to FrMLV. TLR7 senses incoming murine retroviral single-stranded RNA (ssRNA) upon viral entry into target cells (16, 17). B-1 cells naturally express TLR7 and can be activated with TLR7 agonist in vitro (18). Upon ligand recognition, TLR7 signaling induces type I interferon (IFN-I) production, which normally initiates an antiviral program (19). Whether innate immune sensing of virus and signaling in resident cells of pLN or within B-1 cells contribute to B-1 cell susceptibility to FrMLV infection remains to be addressed.
To understand the role of B-1 cells in murine retrovirus infection in vivo, we investigated FrMLV infection in the bumble mouse model that has a point mutation in the atypical IκB gene (IκBNS), resulting in a dysfunctional protein (20). Bumble mice are deficient in B-1 cells, as transitional B cells require IκBNS to develop into B-1 cells (20). Here we show that B-1 cells are highly susceptible to FrMLV infection by comparing infection in wild-type and bumble mice. Through a series of B-1 cell adoptive transfer experiments in bumble mice, we demonstrate that B-1 cell-intrinsic TLR7 sensing and type I IFN signaling contribute to their FrMLV susceptibility. Infected B-1 cells then facilitate FrMLV spread to the B-2 cell population, thus fomenting infection within the pLN. Our study highlights how a murine retrovirus exploits innate immune sensing in an intrinsically susceptible lymphocyte subpopulation to facilitate the establishment of viral infection in an animal.
RESULTS
B-1 cells are highly susceptible to FrMLV infection and are required for robust infection in the popliteal lymph nodes.
We determined the cellular tropism of FrMLV after s.c. delivery of a green fluorescent protein (GFP)-encoding reporter virus. Wild-type C57BL/6J (B6) mice were challenged through intrafootpad (i.f.) injection, and the infected cells (GFP+) in the draining popliteal lymph node (pLN) were identified 3 days postinfection (dpi) by surface marker staining (B-1 cells, CD19+ IgDlo CD43+; B-2 cells, CD19+ IgDhi; and CD4+ T cells, CD3+ CD4+). B-1 cells, B-2 cells, and CD4+ T cells became infected at 3 dpi (Fig. 1A). However, when we compared percentages of infected cells within each population, it became apparent that B-1 cells were highly susceptible to FrMLV (up to 2 orders of magnitude more susceptible than B-2 and CD4+ T cells) (Fig. 1B).
FIG 1.

B-1 cells are highly susceptible to FrMLV infection and are required for robust infection in the popliteal lymph nodes (pLNs). (A) Total numbers of GFP+ FrMLV-infected cells (LTR-GFP) in the B-1 (CD19+ IgDlo CD43+), B-2 (CD19+ IgDhi), and CD4+ T (CD3+ CD4+) cell populations within pLNs (n = 4) of wild-type C57BL/6J (B6) mice, 3 dpi after subcutaneous (s.c.) challenge. (B) Analyses of data presented in panel A showing the percentage of FrMLV-infected cells in pLNs (n = 4) at 3 dpi (s.c.) within each indicated cell population. (C) Gating strategy for characterizing the B-2, B-1a, and B-1b cell populations in the pLNs of B6 and bumble (bmb) mice. N.D., not detected in 5 out of 8 lymph nodes analyzed. (D) Number of B-1a, B-1b, B-2, CD4+ T cells, CD11b+ cells, and subcapsular sinus macrophages (SCS Mϕ; CD169+ CD11b+) in pLNs (n = 4 to 8) of uninfected B6 and bmb mice. (E) Mean fluorescence intensity of CD169 staining in SCS Mϕ (CD169+ CD11b+) in pLNs (n = 4 or 6) of uninfected B6 and bmb mice. ns, P > 0.05. (F) Percentages of cells capturing Gag-GFP-labeled FrMLV particles 1 h p.i. (hpi) (s.c.) in pLNs (n = 6) of B6 and bmb mice. ns, P > 0.05. (G) FrMLV-infected cells (glycoGag+) at indicated time points in pLNs (n = 4 to 10) of B6 and bmb mice after s.c. infection. *, P = 0.0159; ****, P < 0.0001. Statistical comparisons were performed using nonparametric Mann-Whitney tests (two-tailed).
To study the role of B-1 cells in FrMLV infection, we took advantage of bumble mice, a mouse strain that is deficient in mature B-1 cell populations (20). Our analyses confirmed that these mice lacked B-1 cells in the pLNs but retained numbers of B-2 cells, CD4+ T cells, and CD11b+ cells similar to those of wild-type B6 mice (Fig. 1C and D). Importantly, the numbers of virus-capturing SCS macrophages (CD169+ CD11b+) and CD169 expression levels on these cells were similar between the two groups of mice (Fig. 1D and E). Accordingly, the levels of virus capture (% Gag-GFP-positive cells) were comparable in bumble and B6 mice (Fig. 1F). Despite similar levels of virus capture, an in vivo time course of FrMLV infection revealed that bumble mice exhibited a significantly reduced number of early infected cells at 3 dpi in the pLNs (Fig. 1G) compared to B6 controls. Notably, the pronounced expansion in infected cells from 3 to 5 dpi in B6 mice was not observed in bumble mice, indicating that B-1 cells contributed to the local spread of FrMLV in the pLNs (Fig. 1G).
IL-10 does not significantly contribute to the observed reduction of FrMLV infection in bumble mice after subcutaneous challenge.
Innate-like B-1 cells are potent spontaneous producers of the immunosuppressive cytokine interleukin-10 (IL-10) (14). Therefore, reduced FrMLV infection in bumble mice can be due to an exuberant immune response resulting from diminished IL-10 levels (14, 15). Indeed, the number of IL-10-secreting cells (Fig. 2A) and the amount of IL-10 secreted by lymphocytes (Fig. 2B) were reduced in the pLNs of bumble mice. However, FrMLV infection at the pLN after s.c. challenge was enhanced in IL-10−/− mice (Fig. 2C), similarly to a previously observed detrimental effect of IL-10 on infection levels with the closely related pathogenic retrovirus, Friend virus complex (FVC) (21). Thus, our data suggest that IL-10-mediated immune suppression within pLNs adversely affects FrMLV infection, likely by dampening the activation of target lymphocytes.
FIG 2.

Interleukin-10 (IL-10) does not significantly contribute to the observed reduction of FrMLV infection in bumble (bmb) mice after subcutaneous challenge. (A) Percentage of IL-10+ cells (n = 6) from pLNs of FrMLV-infected B6 and bmb mice (s.c., 5 dpi) after ex vivo stimulation with FrMLV-specific peptide for 44 h and phorbol myristate acetate (PMA)/ionomycin for the last 4 h. (B) IL-10 concentration (pg/ml) was determined by ELISA in the supernatants of ex vivo-cultured cells isolated from pLNs (n = 5 or 6) of FrMLV-infected B6 and bmb mice (s.c., 5 dpi) after stimulation with FrMLV-specific peptide for 24 h. (C) Total numbers of FrMLV-infected (GFP+, LTR-GFP) cells in the pLNs (n = 6) of B6 and IL-10−/− mice (s.c., 5 dpi). ns, P > 0.05. Statistical comparisons were performed using nonparametric Mann-Whitney tests (two-tailed).
CD8+ T cell activity and neutralizing antibody responses do not contribute to reduced FrMLV infection in bumble mice.
IκBNS belongs to the nuclear IκB-like family of proteins and is classified as an inhibitor of NF-κB (22). The dysfunctionality of IκBNS in bumble mice could potentially result in elevated cell-mediated and humoral immune responses against FrMLV (20, 23–26). We tested a possible role for CD8+ T cells by depleting them in B6 and bumble mice using antibodies against CD8α. CD8+ T cell depletion resulted in similar fold increases in FrMLV infection (s.c.) in the pLNs of B6 and bumble mice (Fig. 3A). CD8+ T cell degranulation activities monitored by surface exposure of lysosomal marker CD107a after in vitro stimulation with FrMLV Gag peptide were also similar between the two mouse strains (Fig. 3B) (27). In addition, we did not observe any significant differences in the number of granzyme A- and B-expressing CD8+ T cells (Fig. 3C and D) (28). These data indicate that CD8+ T cell responses are not altered in bumble mice due to IkBNS-related dysfunction.
FIG 3.

CD8+ T cell activity and neutralizing antibody responses do not contribute to reduced FrMLV infection in bumble mice. (A) FrMLV-infected cells in the pLNs (n = 4) of B6 and bumble (bmb) mice treated with isotype or CD8+ T cell-depleting antibodies (s.c., 5 dpi). (B to D) Percentage of CD8+ T cells that are CD107a+ (surface staining) after stimulation with FrMLV-specific peptide for 17 h (B) or granzyme A+ (C) or granzyme B+ (D) in pLNs (n = 5) of FrMLV-infected B6 and (bmb) mice (s.c., 6 dpi). Cells from pLNs from uninfected (UI) B6 and bmb mice are shown as controls. ns, P > 0.05. (E) FrMLV-neutralizing activity in serially diluted sera from B6 and bmb mice (n = 7 to 9) at indicated time points after FrMLV infection. (F) Neutralizing titers (IC50) in sera from the experiment shown in panel E. ns, P > 0.05. Statistical comparisons were performed using nonparametric Mann-Whitney tests (two-tailed).
We tested the possibility of an altered antibody response in bumble mice by measuring FrMLV-specific neutralizing antibody titers at 3 and 7 dpi. We were not able to detect any neutralizing antibody responses in either B6 or bumble mice at these early time points postinfection (data not shown). Although there were differences at 21 dpi, likely due to reduced antigen loads (Fig. 1G, 3E, and 3F), we concluded that humoral immune responses could not explain the reduced infection levels observed in bumble mice at the 3- and 5-day time points postinfection used in our study. Rather, our data suggest that a specific deficiency in B-1 cells is responsible for reduced FrMLV infection in bumble mice.
Adoptively transferred wild-type B-1 cells restore FrMLV infection in bumble mice.
We reconstituted bumble mice with wild-type B-1 cells by adoptively transferring them through s.c. (hock) injection to directly test their role in establishing FrMLV infection. The numbers of B-1 cells that homed to the pLN within a day of transfer increased with the ascending numbers of B-1 cells used for adoptive transfer (Fig. 4A). We observed a concomitant increase in the number of FrMLV-infected cells in the pLN (s.c., 3 dpi) (Fig. 4B). We observed ∼400 B-1 cells in the pLN (a number that naturally exist in the pLN of a B6 mouse) when 4 × 105 B-1 cells were used for transfer (Fig. 4A and 1D). Under this condition, we partially rescued FrMLV infection levels in recipient bumble mice at 3 dpi and completely rescued them at 5 dpi (Fig. 4B and C). Interestingly, when B-1 cell repopulation reached supraphysiological levels, infection levels were higher than that seen in B6 control mice (3 dpi) (Fig. 4B). The direct correlation between B-1 cell numbers and virus infection attested to their importance in supporting FrMLV infection. The rescue of infection was B-1 cell specific, as adoptively transferred B-2 cells had no effect on FrMLV infection levels in bumble mice (Fig. 4C). Also, as expected, B-1 cells from IL-10−/− mice rescued FrMLV infection in the pLNs of bumble mice, indicating that B-1 cell-derived IL-10 did not play a role in virus permissivity (Fig. 4C). Together, our data demonstrate that B-1 cell reconstitution in bumble mice is a suitable system to interrogate host factors that potentially contribute to FrMLV infection of B-1 cells.
FIG 4.

Adoptively transferred wild-type B-1 cells restore FrMLV infection in bumble (bmb) mice. (A) Experimental design for determining the effect of adoptively transferred wild-type B-1 cells on FrMLV infection in bmb mice. Representative flow cytometry plots of cells from a pLN 1 day after adoptive transfer (s.c. hock). The numbers of B-1 cells that homed to the pLN (shown below the DsRed+ gate) when increasing numbers of adoptively transferred B-1 cells were used (top). (B and C) FrMLV-infected cells (GFP+ [B] or glycoGag+ [C]) in the pLNs (n = 4 to 9) of bmb mice adoptively transferred with the indicated numbers of B-1 cells or B-2 cells isolated from B6 or IL-10−/− mice at 3 dpi (B) and 5 dpi (C). Infection levels in the pLNs of B6 and bmb mice determined in parallel are shown as controls. ns, P > 0.05. Statistical comparisons were performed using nonparametric Mann-Whitney tests (two-tailed).
Cell-intrinsic TLR7-mediated sensing of FrMLV and autocrine type I interferon signaling contribute to susceptibility of B-1 cells.
TLR7 senses murine retroviral RNA during viral entry (16, 17). To test the possibility that TLR7 sensing is important for FrMLV infection in target cells, we compared FrMLV infection in Tlr7−/− mice and B6 mice. FrMLV infection was significantly diminished in Tlr7−/− mice and to a greater extent in the B-1 cell population at both 3 and 5 dpi (Fig. 5A and B). The reduction in the number of infected cells was more prominent at 5 dpi than at 3 dpi, probably due to a combination of a lack of initial infection in the B-1 cell population and a compromised secondary viral spread (Fig. 5A). We adoptively transferred B-1 cells isolated from Tlr7−/− mice into bumble mice to directly test whether B-1 cell-intrinsic TLR7-mediated sensing was critical to FrMLV infection. In contrast to wild-type B-1 cells, Tlr7−/− B-1 cells were not able to restore FrMLV infection in bumble mice even when an excessive number of B-1 cells were transferred (Fig. 5C). Our data indicate that the sensing of incoming viral RNA by TLR7 contributes to high FrMLV permissivity of B-1 cells and boosts the local spread of FrMLV.
FIG 5.

Cell-intrinsic TLR7-mediated sensing of FrMLV and autocrine type I interferon signaling contributes to susceptibility of B-1 cells. (A and B) Total numbers of FrMLV-infected cells (glycoGag+) (A) or FrMLV-infected B-1 cells (glycoGag+ CD19+ IgDlo CD43+) (B) at 3 and 5 dpi (s.c.) in pLNs (n = 4 to 9) of B6 and Tlr7−/− mice. (C) FrMLV-infected cells in the pLNs (n = 2 to 6) of bumble (bmb) mice adoptively transferred with the indicated numbers of B-1 cells from B6 or Tlr7−/− mice at 5 dpi (s.c.). Infection levels in the pLNs (n = 10) of B6 and bmb mice determined in parallel are shown for statistical comparison. ns, P > 0.05. (D and E) Total numbers of FrMLV-infected cells (glycoGag+) (D) or FrMLV-infected B-1 cells (glycoGag+ CD19+ IgDlo CD43+) (E) at 3 and 5 dpi (s.c.) in pLNs (n = 4 or 10) of B6 and Ifnar1−/− mice. (F) FrMLV-infected cells in the pLNs (n = 7 to 8) of specified mouse strains after reconstitution with (4 × 105) B-1 cells from B6 or Ifnar1−/− mice at 5 dpi (s.c.). Infection levels in the pLNs (n = 10) of B6, bmb, and Ifnar1−/− mice determined in parallel are shown as controls. ns, P > 0.05. Statistical comparisons were performed using nonparametric Mann-Whitney tests (two-tailed).
TLR7 signaling triggers the expression of type I interferons (IFN-I), which can subsequently activate cells to proliferate and potentially become susceptible to MLV (19, 29). We used mice that lack functional IFN-I receptors (Ifnar1−/−) to test the contribution of IFN-I-mediated signaling in FrMLV infection. Surprisingly, the total number of FrMLV-infected cells as well as infected B-1 cells in Ifnar1−/− mice was significantly reduced compared to that in B6 mice (Fig. 5D and E), suggesting a proviral role for IFN-I. We next reconstituted Ifnar1−/− B-1 cells in bumble mice. This experiment allowed us to determine if IFN-I signaling was required in other cell populations or within B-1 cells. B-1 cells from Ifnar1−/− mice were unable to restore FrMLV infection in bumble mice (Fig. 5F). In line with these data, adoptive transfer of wild-type B-1 cells into Ifnar1−/− mice completely restored the FrMLV infection (Fig. 5F). Together, these data strongly suggest that autocrine IFN-I signaling in B-1 cells is required for FrMLV permissivity.
TLR7–IFN-I axis promotes B-1 cell activation and FrMLV infection.
TLR7 sensing and intrinsic type I IFN signaling may contribute to the activation of B-1 cells, which renders them susceptible to cell division-dependent FrMLV infection. We therefore analyzed B-1 cells for the upregulation of an early activation marker, CD69, a type II C-type lectin receptor (17, 30). Forty percent of the FrMLV-infected (glycoGag+) B-1 cells were CD69+ in pLNs at 5 dpi (Fig. 6A and B). In comparison, only 20% of the uninfected B-1 cells in these pLNs were CD69+, indicating that infected B-1 cells were preferentially activated (Fig. 6A and B). We next tested if B-1 cell activation (CD69+) was dependent on TLR7 and IFN-I signaling. Our analyses revealed that the numbers of CD69+-activated B-1 cells were significantly enhanced in B6 mice compared to Tlr7−/− and Ifnar1−/− mice at 5 dpi (Fig. 6C). These data suggest that the TLR7–IFN-I axis promotes the activation of B-1 cells to render them susceptible to FrMLV infection.
FIG 6.

TLR7–IFN-I axis promotes B-1 cell activation and FrMLV infection. (A and B) Gating strategy (A) and numbers (B) of CD69+ cells in infected (glycoGag+) and uninfected (glycoGag–) B-1 cell populations (CD19+ IgDlo CD43+) in the pLNs (n = 6) of FrMLV-infected B6 mice (s.c., 5 dpi). (C) Numbers of CD69+ B-1 cells in pLNs (n = 6) of FrMLV-infected B6, Tlr7−/−, and Ifnar1−/− mice (s.c., 5 dpi). Cells from pLNs (n = 2) of uninfected (UI) B6, Tlr7−/−, and Ifnar1−/− mice are shown as controls. Statistical comparisons were performed using nonparametric Mann-Whitney tests (two-tailed).
B-1 cells promote FrMLV infection of B-2 cells.
B-1 cells comprise less than 0.1% of the B cell population in mouse peripheral lymph nodes (31). However, a lack of B-1 cells led to a profound decrease in FrMLV infection levels. We therefore evaluated how the presence or absence of B-1 cells influenced FrMLV infection in other lymphocyte subpopulations, such as B-2 cells and CD4+ T cells. We infected B6 and bumble mice and compared the increases in number of infected cells in the mentioned lymphocyte populations as a function of time (3 dpi versus 5 dpi). We observed a 67.8-fold increase, compared to a 17.7-fold increase, in the number of infected B-2 cells in B6 and bumble mice, respectively (Fig. 7A). In contrast, the expansion in the number of infected CD4+ T cells was only slightly compromised in bumble mice (6.8-fold compared to 8.8-fold in B6 mice) (Fig. 7A). Moreover, adoptive transfer of wild-type B-1 cells into bumble mice rescued FrMLV infection in B-2 cells (Fig. 7B and C). Collectively, these data suggest that B-1 cells promote FrMLV infection of B-2 cells without significantly affecting the initial CD4+ T cell infection.
FIG 7.
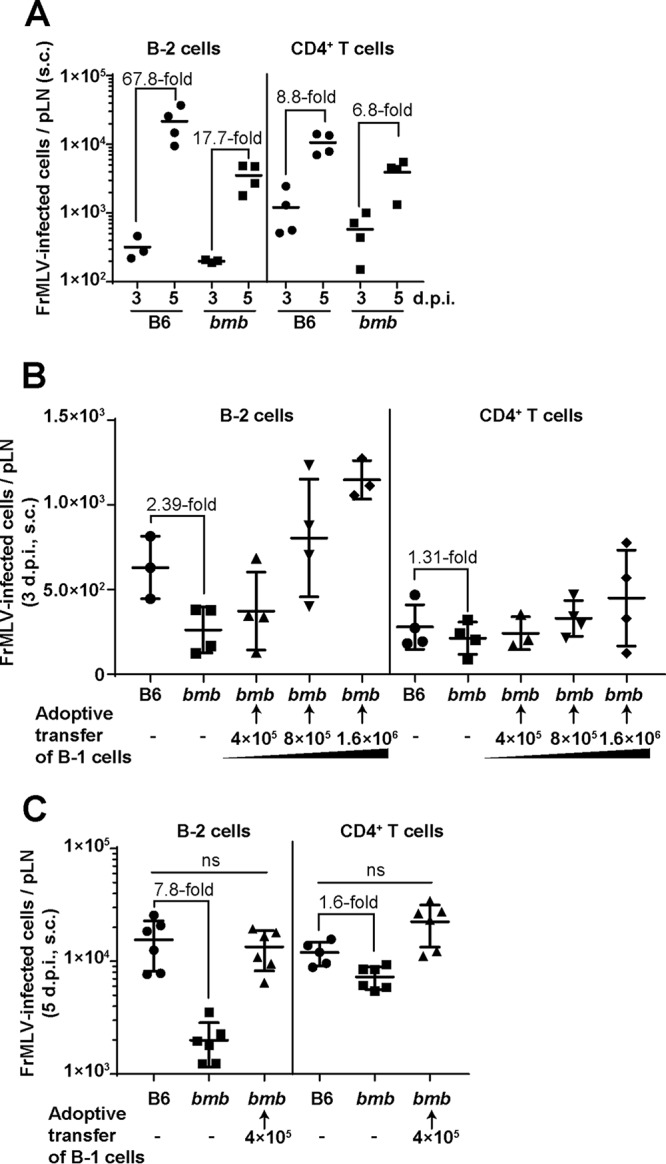
B-1 cells promote FrMLV infection of B-2 cells. (A) Temporal analyses of the indicated FrMLV-infected cell types in pLNs (n = 3 or 4) of B6 and bumble (bmb) mice (s.c., 3 and 5 dpi). (B and C) Analyses of indicated FrMLV-infected (GFP+, LTR-GFP for 3 dpi, glycoGag+ for 5 dpi) cell types in the pLNs (n = 3 to 6) of bmb mice with the indicated numbers of adoptively transferred B-1 cells (s.c., 3 dpi). Infection levels in the pLNs of B6 and bmb mice determined in parallel are shown for statistical comparison. ns, P > 0.05. Statistical comparisons were performed using nonparametric Mann-Whitney tests (two-tailed).
FrMLV-infected cells localize to the medullary region of pLNs.
We explored the role of infected B-1 cells in promoting virus spread in B cell populations by adoptively transferring GFP+ B-1 cells into the pLNs of B6 mice and examined their location by immunostaining pLN cryosections. In our previous study, B-1 cells had localized mainly to the SCS and interfollicular regions within the pLN at 2 dpi (6). As infection progressed, we observed that B-1 cells had migrated to the medullary region at 5 dpi (Fig. 8A), Interestingly, the majority of FrMLV-infected cells (glycoGag+) were also found in the medullary region (Fig. 8A and B). We estimated the numbers of infected B cells (B220+) and CD4+ T cells (CD4+) that resided within a 15-μm distance from each infected B-1 cell (GFP+ glycoGag+). This analysis allowed us to evaluate whether infected B-1 cells preferentially spread FrMLV to specific lymphocyte populations (Fig. 8C). The relative frequency of distribution for one to three infected B220+ cells around an infected B-1 cell was ∼60%. In comparison, the infected CD4+ T cell distribution was only ∼40% (Fig. 8C). Importantly, 5% of infected B-1 cells resided within a 15-μm radius from four to six infected B220+ cells (Fig. 8C). In contrast, this number was zero for CD4+ T cells. These results suggested that B-1 cells preferentially spread FrMLV to B cell populations in the medullary region of the pLN. Notably, our current data indicate that B-1 cells may not be the primary source of CD4+ T cell infection in vivo. The path of virus flow and spread within the CD4+ T cell population after its capture at the SCS needs further investigation.
FIG 8.

FrMLV-infected cells localize to the medullary region of popliteal lymph nodes. (A) Merged images of immunostained pLN tissue cryosections from a B6 mouse adoptively transferred with GFP+ B-1 cells (green) and infected with FrMLV (5 dpi, s.c.) (glycoGag+, white). B cells, CD4+ T cells, and CD169+ macrophages were identified using antibodies for surface markers B220 (blue), CD4 (blue), and CD169 (red), respectively. Scale bars, 100.00 μm. (B) Merged images of medullary regions containing infected cells (glycoGag+, red, 5 dpi, s.c.) in immunostained pLN tissue cryosections from B6 mice adoptively transferred with GFP+ B-1 cells (green). B cells and CD4+ T cells were identified using antibodies for surface markers B220 (blue) and CD4 (blue), respectively. Scale bars, 50.00 μm. (C) Histograms compiled from 36 or 15 FrMLV-infected (GFP+ glycoGag+) B-1 cells in medullary regions of two pLNs (as shown in panel B) (5 dpi, s.c.), indicating a distribution of the number of infected B or CD4+ T cells located proximally to infected B-1 cells. The proximity was defined as located within a 15-μm distance.
DISCUSSION
In this work, we have demonstrated that the ability of FrMLV to establish infection in mice via the s.c. route depends on B-1 cell-intrinsic innate TLR7 sensing and IFN-I signaling (Fig. 9). Innate immune sensing that culminates in IFN synthesis is designed to protect host cells from viral infection through the induction of interferon-stimulated genes (ISGs). Here we demonstrated that a retrovirus, FrMLV, can exploit the innate immune activation to its advantage and render one of the early target cells like B-1 cells permissive for infection. Efficient infection of B-1 cells was required for subsequent amplification of infection in B-2 cells. In this scenario, B-1 cells functioned like cellular superspreaders of FrMLV infection (32). Our data highlight the importance of characterizing early target cells and factors that govern their permissivity to develop effective antiviral strategies for curtailing virus colonization of the host.
FIG 9.

Scheme showing a hypothetical model of FrMLV infection in B-1 cells and spread to B-2 cells. Lymph-borne FrMLV is captured by CD169+ subcapsular sinus macrophages (SCS Mϕ) in the pLN. Panel 1, B-1 cells acquire viral particles when interacting with FrMLV-laden SCS macrophages (6). Panel 2, TLR7 expressed by B-1 cells senses viral RNA upon viral entry and induces downstream signaling through adaptor protein MyD88, which culminates in the synthesis of IFN-I. IFN-I signaling activates B-1 cells and enhances their susceptibility to FrMLV. Panel 3, Productively infected B-1 cells function as cellular superspreaders for the subsequent expansion of FrMLV in B-2 cell populations.
Innate sensing of retroviruses is mediated by multiple nucleic acid sensors, including TLR7, cyclic GMP-AMP synthase (cGAS), DEAD/H-box helicase 41 (DDX41), and the interferon γ-inducible protein 203 (IFI203) (33–35). These sensors recognize retroviral components, including genomic ssRNA, RNA/DNA hybrids, and double-stranded DNA (dsDNA), that are exposed at different steps before integration (12, 34, 35). It has been reported that TLR7 sensing of incoming RNA within CD4+ T cells increases the intracellular calcium flux and facilitates human immunodeficiency virus type 1 (HIV-1) infection in vitro (36). Our data revealed that TLR7 sensing and autocrine IFN-I signaling within B-1 cells are required to render them permissive to robust FrMLV infection in mouse pLNs. We hypothesize that these innate immune responses activate B-1 cells, which leads to cell proliferation and facilitates cell division-dependent FrMLV transduction (Fig. 6) (7). Similarly, the subsequent spread to B-2 cells must depend on cellular activation, since naive B-2 cells are not permissive to FrMLV infection (6). It is possible that FrMLV-specific B-2 cells that interact with B-1 cells are particularly prone to infection due to B cell receptor-induced signaling. Future work should define the cytokines and signaling events instigated by B-1 cells that promote activation and infection of B-2 cells.
The role of TLR7 in sensing incoming retroviral RNA and promoting antiviral humoral immune response is well known (16, 17, 21). However, our data with FrMLV reveal a proviral facet of TLR7 sensing and type I IFN signaling. Unlike simple retroviruses like FrMLV and Moloney murine leukemia virus (MoMLV), pathogenic retroviral complexes like Friend virus complex (FVC) and LP-BM5 induce robust and systemic type I IFN responses in the host that are antiviral and result in induction of interferon-induced genes like the tetherin gene (37, 38). Accordingly, when innate immune responses were compromised, infection of FVC and LP-BM5 was enhanced (21, 38, 39). In contrast, FrMLV exploited localized and autocrine innate immune activation within the B-1 cell compartment to promote its infection (Fig. 5). Exploitation of TLR7-induced inflammatory responses for replicative advantage was also previously documented for influenza A viruses (40). Thus, our data highlight how fine-tuning of host innate immune signaling can tip the balance of host responses from antiviral to proviral outcomes.
Mouse mammary tumor virus (MMTV), another simple retrovirus, encodes a superantigen (SAg) that activates target cells (41). MMTV-infected B cells present SAg to the appropriate T cell subset, leading to a strong “cognate” T cell-B cell interaction and preferential clonal expansion of infected B cells (42, 43). Some infected B cells differentiate into long-lived memory cells, which results in stable infection of the host (43). Thus, MMTV exploits both immunosuppressive cytokine IL-10 and strong SAg-induced immune activation to establish infection in mice. However, for viruses like FrMLV that do not encode SAgs and rely on immune activation for transduction, the presence of immunosuppressive cytokine IL-10 can detrimentally affect overall infection (Fig. 2). In contrast to MLV and MMTV, lentiviruses such as HIV and simian immunodeficiency virus (SIV) do not depend on cell division for the import of proviral DNA into cell nuclei (44). Nevertheless, they depend on target cell activation for productive infection (45). Furthermore, at the primary site of infection in vaginal epithelium, SIV-infected plasmacytoid dendritic cells (pDCs) secrete chemokines that recruit CD4+ T cells (46). The infiltration of activated CD4+ T cells facilitates the rapid expansion of SIV, which is also critical for the subsequent systemic spread (46). Thus, while mechanistically distinct, immunotropic viruses such as HIV, MMTV, and murine leukemia virus (MLV), which cause chronic infection, have learned to utilize immune activation to support their replication and spread.
From an evolutionary standpoint, the apparent paradox that FrMLV uses innate immune signaling to support its replication is not surprising. A recent investigation showed that mammals have co-opted retroelements to function as IFN-inducible enhancers for regulation of ISG transcription (47). These data show an evolutionary relationship between retroviral long terminal repeats (LTRs) and the modern interferon-based innate immune system, a prime example being the HIV LTR, which has an NF-κB binding site (48). Possibly, this coevolution and dependence on innate immune signaling is still visible in present-day MLVs that have been in the mouse for at least one million years (49).
MATERIALS AND METHODS
Mice.
C57BL/6 (B6), NagyDsRed.T3, and UBI-GFP mice were obtained from The Jackson Laboratory. Bumble mice (B6 background) were obtained from Mutant Mouse Resources & Research Centers supported by NIH (MMRRC). Tlr7−/− mice (B6 background) and Ifnar1−/− mice (B6 background) were from Akiko Iwasaki and Priti Kumar, Yale University (50). IL-10−/− mice (B6 background) were from Jorge Galán and Ruslan Medzhitov, Yale University. All animals were housed under specific-pathogen-free conditions in Yale Animal Resources Center (YARC) facilities. All experiments were approved by the Institutional Animal Care and Use Committees (IACUC) and Institutional Biosafety Committee of Yale University. Six- to 12-week-old male and female mice were used for all experiments. B6, NagyDsRed.T3, and UBI-GFP mice of the same sex as the recipient mice were used for B-1 and B-2 cell adoptive transfer.
Virus preparation.
Replication-competent FrMLV was generated by cotransfecting HEK293 cells with pLRB303-FrMLV that encodes full-length replication competent Friend 57 MLV and pLZRS-FrMLV Env to enhance the infectivity of the virus (51). Infected cells were detected by staining for cell surface glycoGag protein. Reporter MLVs to indicate infection were prepared by cotransfecting HEK293 cells with pLRB303-FrMLV and pMMP-LTR-GFP, which express cytoplasmic GFP at a ratio of 10:1 (9). Compared to staining with antibodies to glycoGag, cytoplasmic GFP expression offered better reliability, especially when the level of infection was relatively low at an early time point (3 dpi). Gag-GFP-labeled MLV particles were prepared as previously described (51) by cotransfecting pLRB303-GagGFP (with a large fragment of Pol deleted in pLRB303-FrMLV and enhanced GFP [EGFP] fused to the C terminus of Gag), pLZRS-GagPol (to compensate for the deletion of Pol in pLRB303-GagGFP), and pLZRS-FrMLV Env. Culture supernatants from HEK293 cells were harvested 48 h after cotransfection, filtered with 0.45-μm nylon membrane filters, aliquoted, and stored at –80°C. Viruses in supernatant were concentrated by sedimentation with a 15% sucrose cushion at 4°C, 32,000 × g, for 2 h. Titers of concentrated viruses were determined by infecting S49.1 cells for 24 h, and infected cells were quantified by flow cytometry analysis for glycoGag+ cells (for full-length replication-competent FrMLV) or GFP+ cells (for LTR-GFP reporter FrMLV) (9). A total of 4 × 105 infectious units (IU) of concentrated viral particles were resuspended in sterile phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin (BSA) and injected into animals through the intrafootpad (i.f.) subcutaneous (s.c.) route with a 31-gauge insulin syringe. For Gag-GFP MLV that was s.c. injected for capture experiments, a virus amount equivalent to 2 × 106 IU was ascertained by comparison of gag signals in sedimented virus using antibodies to MLV gag p30 (clone R187, ATCC CRL-1912) by Western blotting (6).
CD8+ T cell depletion.
CD8+ T cells were depleted by administering 150 μg of in vivo-grade antibody (anti-CD8α, clone YTS 169.4; Bio X Cell) intraperitoneally (i.p.) starting 1 day before viral challenge and every 3 days during the course of infection, as described earlier (39). More than 99.5% of the CD8+ T cells were confirmed to be depleted in the pLNs by flow cytometric analysis.
Single-cell preparation from mouse tissue.
pLNs were isolated, briefly disrupted with forceps, and treated with 0.2 mg/ml of Liberase and 20 μg/ml of DNase I in serum-free RPMI 1640 medium at 37°C for 20 min (9). The reaction was terminated by adding excess serum-containing RPMI 1640 medium. The tissue was thoroughly minced with a plunger and passed through a 70-μm cell strainer. Suspended single cells were used for ex vivo stimulation or fixed with 4% paraformaldehyde (PFA) and stained before flow cytometric analyses.
B cell isolation and adoptive transfer.
B-1 cells were isolated from the indicated mouse strains as previously described (6). Briefly, cells were collected from mouse peritoneum with 1× PBS sterile solution containing 1% BSA and 2 mM EDTA. B-1 cells were purified by negative selection using a cocktail of biotinylated antibodies against mouse F4/80, CD4, CD8, Gr-1, and CD23, followed by separation with streptavidin RapidSphere magnetic beads (Stemcell Technologies). B-2 cells were isolated from mouse splenocytes with the EasySep negative-selection mouse B cell enrichment kit (Stemcell Technologies). Indicated numbers of B-1 and B-2 cells (as mentioned in the figure legends) were adoptively transferred into recipient mice through s.c. hock injection with a 27-gauge insulin syringe. We were able to detect more reproducible numbers of adoptively transferred cells in the pLN through hock injections than through footpad deliveries. Recipient mice were challenged with virus 24 h after the adoptive transfer, as indicated.
Flow cytometric analyses.
PFA-fixed cells from lymph nodes were blocked and stained with 1× PBS containing 2% BSA, 5% fetal bovine serum, and 0.2% gelatin. FrMLV infection was determined either by detecting cytoplasmic GFP or by staining the cells with CF647-labeled anti-MLV glycoGag antibody (clone mab34), as indicated. Analyses of infected cell populations were carried out by costaining for B-1 cells (CD19+ IgDlo CD43+), B-2 cells (CD19+ IgDhi), and CD4+ T cells (CD3+ CD4+) with anti-CD19-BV605 (clone 6D5), anti-IgD-BV711 (clone 11-26c.2a), anti-CD43-phycoerythrin (PE), anti-CD43-allophycocyanin (APC), or anti-CD43-BV421 (clone S7) or with anti-CD3-APC/Cy7 (clone 17A2) or anti-CD4-PE/Cy7 (clone GK1.5). We distinguished two B-1 cell subpopulations, B-1a cells (CD5+) and B-1b cells (CD5−), by costaining with anti-CD5-BUV737 (clone 53-7.3). Analyses of subcapsular sinus macrophages (SCS Mϕ, CD169+ CD11b+) was carried out by costaining with anti-CD169-PE (clone 3D6.112) and anti-CD11b-APC (clone M1/70). We determined the proportion of granzyme A- and granzyme B-expressing CD8+ T cells by staining with anti-CD3-APC/Cy7, anti-CD8-AF488 (clone 53-6.7), and anti-granzyme A-PE (clone 3G8.5) or anti-granzyme B-PE (clone QA16A02). We determined early activation in B-1 cells by staining for CD69 with anti-CD69-PE (clone H1.2F3). Data were acquired on an Accuri C6 (BD Biosciences) and LSRII (BD Biosciences) and analyzed with Accuri CSampler or FlowJo software (Treestar). A total of 200,000 to 300,000 viable, singlet, and nonautofluorescent cells were acquired from each pLN sample. Each data point represents a single lymph node.
Immunostaining and imaging of pLN cryosections.
FrMLV-infected pLNs were dissected at 3 or 5 dpi and fixed overnight with 1× PBS containing 1% PFA (Electron Microscopy Sciences), 100 mM l-lysine, and 10 mM sodium M-peroxidate at 4°C (6). The fixed lymph nodes were washed three times with 1× PBS and dehydrated with a series of 10%, 20%, and 30% sucrose in 1× PBS. Lymph node samples were embedded in Tissue-Tek O.C.T. compound (Sakura), snap-frozen at –80°C, and sectioned at a 10- to 15-μm thickness using a cryostat (CM1950; Leica). The sections were washed with 1× PBS, blocked with 1× PBS with 2% BSA for 1 h, and stained for CD169+ macrophages and infected B and CD4+ T cells with anti-CD169-AF594 (clone 3D6.112), CF647-conjugated anti-MLV glycoGag, anti-B220-eFluor450 (clone RA3-6B2), and anti-CD4-eFluor450 (clone GK1.5). B220 staining detected predominantly B-2 cells, since B-1 cells express low levels of this surface marker (20). Stained samples were treated with ProLong Gold antifade reagent and observed with a Leica TCS SP8 microscope. Images were processed and analyzed with Volocity software (PerkinElmer, Waltham, MA, USA).
Image analyses.
The center of an infected B-1 cell (GFP+ glycoGag+) was designated the middle point of the longest axis that crosses the cytoplasm. A circle with a radius of 15 μm was drawn from the center of the B-1 cell. Infected B (glycoGag+ B220+) or CD4+ T (glycoGag+ CD4+) cells were enumerated within each circle, and their distribution was plotted as a histogram.
Ex vivo stimulation of lymphocytes.
We evaluated the proportion of CD8+ T cells that actively secreted cytotoxic enzymes by stimulating 1 × 106 cells from pLNs of uninfected or FrMLV-infected mice (4 × 105 IU, s.c., 6 dpi) with 6 μM FrMLV Gag-specific peptide (KKCCLCLTVFL) in complete RPMI 1640 medium at 37°C. Surface exposure of CD107A was monitored by adding a 1:500 dilution of AF647-conjugated anti-mouse CD107A. GolgiStop (BD Biosciences) was added 1 h later, and the cells were cultured for another 16 h. The cells were harvested, fixed with 4% PFA, and stained as mentioned above for CD8+ T cells. We measured the percentage of IL-10-secreting cells in FrMLV-infected pLNs (4 × 105 IU, s.c., 5 dpi). In addition, 1 × 106 cells from each pLN were treated with 6 μM FrMLV Gag-specific peptide (KKCCLCLTVFL) for 44 h. The cells were further stimulated for the last 4 h with 50 ng/ml phorbol myristate acetate, 1 μM ionomycin, and GolgiStop. The cells were harvested, fixed with 4% PFA, permeabilized with 0.2% TX-100, and stained with anti-IL-10-AF647 (clone JES5-16E3) before flow cytometric analyses.
Measuring IL-10 concentration via ELISA.
A total of 1 × 106 cells from FrMLV-infected (4 × 105 IU, i.f., 5 dpi) pLNs were stimulated with 6 μM FrMLV Gag-specific peptide (KKCCLCLTVFL) in complete RPMI 1640 medium in a U-bottom 96-well plate at 37°C for 24 h. IL-10 concentrations in culture supernatants were measured quantitatively with an ELISA kit (catalog no. 88-7105-22; Invitrogen). A standard curve was interpolated, and IL-10 concentrations were calculated using GraphPad Prism v7.0a (La Jolla, CA, USA).
Neutralizing antibody titers.
FrMLV (s.c., 4 × 105 IU)-infected mice were bled retro-orbitally (r.o.) at 14 and 21 dpi with FrMLV and allowed to clot at room temperature for 1 h. Blood samples were sedimented at 32,000 × g for 30 min at 25°C to collect sera and stored at –80°C. Serial 2-fold dilutions of heat-inactivated (56°C for 30 min) serum samples were incubated for 1 h at 37°C with MLV-expressing Antares luciferase (8.3 × 106 luciferase units) and 1 μl of guinea pig complement (MP Biomedical;1:64 hemolytic titer) in a total volume of 50 μl (serum-free medium) in a 96-well plate. Equal volumes of medium with 2× serum containing S49.1 T cells (2 × 105) were added to each well and incubated further at 37°C for 24 h. Sedimented cells were lysed using 25 μl of 1× passive lysis buffer (Promega Corp) and mixed with 25 μl of Nano-Glo luciferase assay buffer containing NanoLuc substrate (diluted 1:50) (Nano-Glo luciferase assay system; Promega Corp). Luciferase activity was measured in 96-well white microplates (Costar catalog no. 3917) with a luminometer (Berthold Technologies). Luciferase activity in samples with pooled sera from uninfected mice was set at 100%. The log(dose-dependent inhibition/serum dilution) was plotted against the log serum dilution to fit a linear regression. The slope was used to calculate 50% inhibitory concentration (IC50) values, defined as the amount of serum that reduces MLV infection by 50%. The IC50 values were analyzed for statistical significance by applying correction for multiple comparisons using nonparametric Mann-Whitney tests (two-tailed) available in GraphPad Prism v7.0a.
Quantification and statistical analysis.
Statistical comparisons were performed using nonparametric Mann-Whitney tests (two-tailed) available in GraphPad Prism software v7.0a (La Jolla, CA, USA). Exact P values and numbers of independent replicates (n) are mentioned in the figures or figure legends. A difference was considered significant if the P value was <0.05.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 CA098727 and P50-AI150464 to W.M. and Flow Cytometry Shared Resource of the Yale Cancer Center grant P30 CA016359, Yale Center for Cellular and Molecular Imaging grant S10 OD020142, and a fellowship from the China Scholarship Council-Yale World Scholars to R.P. A.I. is a Howard Hughes Medical Institute Investigator.
We thank Jorge Galan, Ruslan Medzhitov and Priti Kumar for providing IL-10−/− and Ifnar1−/− mice. We thank Kelsey Haugh for critical readings of the manuscript.
R.P., A.I., X.S., W.M., and P.D.U. designed the experiments. R.P., X.S., and P.D.U. performed the experiments and analyzed the data. R.P., W.M., and P.D.U. wrote the manuscript, and R.P., A.I., X.S., W.M., and P.D.U. contributed to the interpretation and discussion of the work.
REFERENCES
- 1.Troxler DH, Scolnick EM. 1978. Rapid leukemia induced by cloned Friend strain of replicating murine type-C virus. Association with induction of xenotropic-related RNA sequences contained in spleen focus-forming virus. Virology 85:17–27. doi: 10.1016/0042-6822(78)90408-7. [DOI] [PubMed] [Google Scholar]
- 2.Ru M, Shustik C, Rassart E. 1993. Graffi murine leukemia virus: molecular cloning and characterization of the myeloid leukemia-inducing agent. J Virol 67:4722–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pepersack L, Lee JC, McEwan R, Ihle JN. 1980. Phenotypic heterogeneity of Moloney leukemia virus-induced T cell lymphomas. J Immunol 124:279–285. [PubMed] [Google Scholar]
- 4.Portis JL, McAtee FJ, Hayes SF. 1987. Horizontal transmission of murine retroviruses. J Virol 61:1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buffett RF, Grace JT Jr, DiBerardino LA, Mirand EA. 1969. Vertical transmission of murine leukemia virus. Cancer Res 29:588–595. [PubMed] [Google Scholar]
- 6.Sewald X, Ladinsky MS, Uchil PD, Beloor J, Pi R, Herrmann C, Motamedi N, Murooka TT, Brehm MA, Greiner DL, Shultz LD, Mempel TR, Bjorkman PJ, Kumar P, Mothes W. 2015. Retroviruses use CD169-mediated trans-infection of permissive lymphocytes to establish infection. Science 350:563–567. doi: 10.1126/science.aab2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roe T, Reynolds TC, Yu G, Brown PO. 1993. Integration of murine leukemia virus DNA depends on mitosis. EMBO J 12:2099–2108. doi: 10.1002/j.1460-2075.1993.tb05858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deenen GJ, Kroese FG. 1993. Kinetics of peritoneal B-1a cells (CD5 B cells) in young adult mice. Eur J Immunol 23:12–16. doi: 10.1002/eji.1830230104. [DOI] [PubMed] [Google Scholar]
- 9.Sewald X, Gonzalez DG, Haberman AM, Mothes W. 2012. In vivo imaging of virological synapses. Nat Commun 3:1320. doi: 10.1038/ncomms2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson JM, Iwasaki A. 2008. Toll-like receptors regulation of viral infection and disease. Adv Drug Deliv Rev 60:786–794. doi: 10.1016/j.addr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilks J, Lien E, Jacobson AN, Fischbach MA, Qureshi N, Chervonsky AV, Golovkina TV. 2015. Mammalian lipopolysaccharide receptors incorporated into the retroviral envelope augment virus transmission. Cell Host Microbe 18:456–462. doi: 10.1016/j.chom.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kane M, Case LK, Kopaskie K, Kozlova A, MacDearmid C, Chervonsky AV, Golovkina TV. 2011. Successful transmission of a retrovirus depends on the commensal microbiota. Science 334:245–249. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jude BA, Pobezinskaya Y, Bishop J, Parke S, Medzhitov RM, Chervonsky AV, Golovkina TV. 2003. Subversion of the innate immune system by a retrovirus. Nat Immunol 4:573–578. doi: 10.1038/ni926. [DOI] [PubMed] [Google Scholar]
- 14.O’Garra A, Chang R, Go N, Hastings R, Haughton G, Howard M. 1992. Ly-1 B (B-1) cells are the main source of B cell-derived interleukin 10. Eur J Immunol 22:711–717. doi: 10.1002/eji.1830220314. [DOI] [PubMed] [Google Scholar]
- 15.Sindhava V, Woodman ME, Stevenson B, Bondada S. 2010. Interleukin-10 mediated autoregulation of murine B-1 B-cells and its role in Borrelia hermsii infection. PLoS One 5:e11445. doi: 10.1371/journal.pone.0011445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kane M, Case LK, Wang C, Yurkovetskiy L, Dikiy S, Golovkina TV. 2011. Innate immune sensing of retroviral infection via Toll-like receptor 7 occurs upon viral entry. Immunity 35:135–145. doi: 10.1016/j.immuni.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Browne EP. 2011. Toll-like receptor 7 controls the anti-retroviral germinal center response. PLoS Pathog 7:e1002293. doi: 10.1371/journal.ppat.1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gururajan M, Jacob J, Pulendran B. 2007. Toll-like receptor expression and responsiveness of distinct murine splenic and mucosal B-cell subsets. PLoS One 2:e863. doi: 10.1371/journal.pone.0000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwasaki A, Medzhitov R. 2004. Toll-like receptor control of the adaptive immune responses. Nat Immunol 5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 20.Pedersen GK, Adori M, Khoenkhoen S, Dosenovic P, Beutler B, Karlsson Hedestam GB. 2014. B-1a transitional cells are phenotypically distinct and are lacking in mice deficient in IkappaBNS. Proc Natl Acad Sci U S A 111:E4119–4126. doi: 10.1073/pnas.1415866111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Browne EP. 2013. Toll-like receptor 7 inhibits early acute retroviral infection through rapid lymphocyte responses. J Virol 87:7357–7366. doi: 10.1128/JVI.00788-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto M, Takeda K. 2008. Role of nuclear IkappaB proteins in the regulation of host immune responses. J Infect Chemother 14:265–269. doi: 10.1007/s10156-008-0619-Y. [DOI] [PubMed] [Google Scholar]
- 23.Arnold CN, Pirie E, Dosenovic P, McInerney GM, Xia Y, Wang N, Li X, Siggs OM, Karlsson Hedestam GB, Beutler B. 2012. A forward genetic screen reveals roles for Nfkbid, Zeb1, and Ruvbl2 in humoral immunity. Proc Natl Acad Sci U S A 109:12286–12293. doi: 10.1073/pnas.1209134109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Touma M, Keskin DB, Shiroki F, Saito I, Koyasu S, Reinherz EL, Clayton LK. 2011. Impaired B cell development and function in the absence of IkappaBNS. J Immunol 187:3942–3952. doi: 10.4049/jimmunol.1002109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pedersen GK, Adori M, Stark JM, Khoenkhoen S, Arnold C, Beutler B, Karlsson Hedestam GB. 2016. Heterozygous mutation in IkappaBNS leads to reduced levels of natural IgM antibodies and impaired responses to T-independent type 2 antigens. Front Immunol 7:65. doi: 10.3389/fimmu.2016.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Presa M, Racine JJ, Dwyer JR, Lamont DJ, Ratiu JJ, Sarsani VK, Chen YG, Geurts A, Schmitz I, Stearns T, Allocco J, Chapman HD, Serreze DV. 2018. A hypermorphic Nfkbid allele contributes to impaired thymic deletion of autoreactive diabetogenic CD8(+) T cells in NOD mice. J Immunol 201:1907–1917. doi: 10.4049/jimmunol.1800465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. 2003. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 28.Zelinskyy G, Balkow S, Schimmer S, Werner T, Simon MM, Dittmer U. 2007. The level of Friend retrovirus replication determines the cytolytic pathway of CD8+ T-cell-mediated pathogen control. J Virol 81:11881–11890. doi: 10.1128/JVI.01554-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat Rev Immunol 14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purtha WE, Chachu KA, Virgin HW IV, Diamond MS. 2008. Early B-cell activation after West Nile virus infection requires alpha/beta interferon but not antigen receptor signaling. J Virol 82:10964–10974. doi: 10.1128/JVI.01646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumgarth N. 2011. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol 11:34–46. doi: 10.1038/nri2901. [DOI] [PubMed] [Google Scholar]
- 32.Talbert-Slagle K, Atkins KE, Yan KK, Khurana E, Gerstein M, Bradley EH, Berg D, Galvani AP, Townsend JP. 2014. Cellular superspreaders: an epidemiological perspective on HIV infection inside the body. PLoS Pathog 10:e1004092. doi: 10.1371/journal.ppat.1004092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. 2013. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stavrou S, Aguilera AN, Blouch K, Ross SR. 2018. DDX41 recognizes RNA/DNA retroviral reverse transcripts and is critical for in vivo control of murine leukemia virus infection. mBio 9:e00923-18. doi: 10.1128/mBio.00923-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stavrou S, Blouch K, Kotla S, Bass A, Ross SR. 2015. Nucleic acid recognition orchestrates the anti-viral response to retroviruses. Cell Host Microbe 17:478–488. doi: 10.1016/j.chom.2015.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dominguez-Villar M, Gautron AS, de Marcken M, Keller MJ, Hafler DA. 2015. TLR7 induces anergy in human CD4(+) T cells. Nat Immunol 16:118–128. doi: 10.1038/ni.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrett BS, Smith DS, Li SX, Guo K, Hasenkrug KJ, Santiago ML. 2012. A single nucleotide polymorphism in tetherin promotes retrovirus restriction in vivo. PLoS Pathog 8:e1002596. doi: 10.1371/journal.ppat.1002596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liberatore RA, Bieniasz PD. 2011. Tetherin is a key effector of the antiretroviral activity of type I interferon in vitro and in vivo. Proc Natl Acad Sci U S A 108:18097–18101. doi: 10.1073/pnas.1113694108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uchil PD, Pi R, Haugh KA, Ladinsky MS, Ventura JD, Barrett BS, Santiago ML, Bjorkman PJ, Kassiotis G, Sewald X, Mothes W. 2019. A protective role for the lectin CD169/Siglec-1 against a pathogenic murine retrovirus. Cell Host Microbe 25:87–100.e110. doi: 10.1016/j.chom.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pang IK, Pillai PS, Iwasaki A. 2013. Efficient influenza A virus replication in the respiratory tract requires signals from TLR7 and RIG-I. Proc Natl Acad Sci U S A 110:13910–13915. doi: 10.1073/pnas.1303275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi Y, Kappler JW, Marrack P. 1991. A superantigen encoded in the open reading frame of the 3′ long terminal repeat of mouse mammary tumour virus. Nature 350:203–207. doi: 10.1038/350203a0. [DOI] [PubMed] [Google Scholar]
- 42.Held W, Shakhov AN, Izui S, Waanders GA, Scarpellino L, MacDonald HR, Acha-Orbea H. 1993. Superantigen-reactive CD4+ T cells are required to stimulate B cells after infection with mouse mammary tumor virus. J Exp Med 177:359–366. doi: 10.1084/jem.177.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Held W, Waanders GA, Acha-Orbea H, MacDonald HR. 1994. Reverse transcriptase-dependent and -independent phases of infection with mouse mammary tumor virus: implications for superantigen function. J Exp Med 180:2347–2351. doi: 10.1084/jem.180.6.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lewis P, Hensel M, Emerman M. 1992. Human immunodeficiency virus infection of cells arrested in the cell cycle. EMBO J 11:3053–3058. doi: 10.1002/j.1460-2075.1992.tb05376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA. 1990. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J 9:1551–1560. doi: 10.1002/j.1460-2075.1990.tb08274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, Reilly CS, Peterson ML, Schultz-Darken N, Brunner KG, Nephew KR, Pambuccian S, Lifson JD, Carlis JV, Haase AT. 2009. Glycerol monolaurate prevents mucosal SIV transmission. Nature 458:1034–1038. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chuong EB, Elde NC, Feschotte C. 2016. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 351:1083–1087. doi: 10.1126/science.aad5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawakami K, Scheidereit C, Roeder RG. 1988. Identification and purification of a human immunoglobulin-enhancer-binding protein (NF-kappa B) that activates transcription from a human immunodeficiency virus type 1 promoter in vitro. Proc Natl Acad Sci U S A 85:4700–4704. doi: 10.1073/pnas.85.13.4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Young GR, Yap MW, Michaux JR, Steppan SJ, Stoye JP. 2018. Evolutionary journey of the retroviral restriction gene Fv1. Proc Natl Acad Sci U S A 115:10130–10135. doi: 10.1073/pnas.1808516115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Treger RS, Tokuyama M, Dong H, Salas-Briceno K, Ross SR, Kong Y, Iwasaki A. 24 July 2019. Human APOBEC3G prevents emergence of infectious endogenous retrovirus in mice. J Virol doi: 10.1128/JVI.00728-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin J, Li F, Mothes W. 2011. Viral determinants of polarized assembly for the murine leukemia virus. J Virol 85:7672–7682. doi: 10.1128/JVI.00409-11. [DOI] [PMC free article] [PubMed] [Google Scholar]