

Influenza A viruses (IAV) and influenza B viruses (IBV) cause significant morbidity and mortality during seasonal outbreaks. Cleavage of the viral surface glycoprotein hemagglutinin (HA) by host proteases is a prerequisite for membrane fusion and essential for virus infectivity. Inhibition of relevant proteases provides a promising therapeutic approach that may avoid the development of drug resistance. HA of most influenza viruses is cleaved at a monobasic cleavage site, and a number of proteases have been shown to cleave HA in vitro. This study demonstrates that the transmembrane protease TMPRSS2 is the major HA-activating protease of IAV in primary human bronchial cells and of both IAV and IBV in primary human type II pneumocytes. It further reveals that human and murine airway cells can differ in their HA-cleaving protease repertoires. Our data will help drive the development of potent and selective protease inhibitors as novel drugs for influenza treatment.
KEYWORDS: TMPRSS2, hemagglutinin cleavage, host cell proteases, influenza virus, murine airway cells, primary human airway cells, protease inhibitors
ABSTRACT
Cleavage of influenza virus hemagglutinin (HA) by host cell proteases is essential for virus infectivity and spread. We previously demonstrated in vitro that the transmembrane protease TMPRSS2 cleaves influenza A virus (IAV) and influenza B virus (IBV) HA possessing a monobasic cleavage site. Subsequent studies revealed that TMPRSS2 is crucial for the activation and pathogenesis of H1N1pdm and H7N9 IAV in mice. In contrast, activation of H3N2 IAV and IBV was found to be independent of TMPRSS2 expression and supported by an as-yet-undetermined protease(s). Here, we investigated the role of TMPRSS2 in proteolytic activation of IAV and IBV in three human airway cell culture systems: primary human bronchial epithelial cells (HBEC), primary type II alveolar epithelial cells (AECII), and Calu-3 cells. Knockdown of TMPRSS2 expression was performed using a previously described antisense peptide-conjugated phosphorodiamidate morpholino oligomer, T-ex5, that interferes with splicing of TMPRSS2 pre-mRNA, resulting in the expression of enzymatically inactive TMPRSS2. T-ex5 treatment produced efficient knockdown of active TMPRSS2 in all three airway cell culture models and prevented proteolytic activation and multiplication of H7N9 IAV in Calu-3 cells and H1N1pdm, H7N9, and H3N2 IAV in HBEC and AECII. T-ex5 treatment also inhibited the activation and spread of IBV in AECII but did not affect IBV activation in HBEC and Calu-3 cells. This study identifies TMPRSS2 as the major HA-activating protease of IAV in human airway cells and IBV in type II pneumocytes and as a potential target for the development of novel drugs to treat influenza infections.
IMPORTANCE Influenza A viruses (IAV) and influenza B viruses (IBV) cause significant morbidity and mortality during seasonal outbreaks. Cleavage of the viral surface glycoprotein hemagglutinin (HA) by host proteases is a prerequisite for membrane fusion and essential for virus infectivity. Inhibition of relevant proteases provides a promising therapeutic approach that may avoid the development of drug resistance. HA of most influenza viruses is cleaved at a monobasic cleavage site, and a number of proteases have been shown to cleave HA in vitro. This study demonstrates that the transmembrane protease TMPRSS2 is the major HA-activating protease of IAV in primary human bronchial cells and of both IAV and IBV in primary human type II pneumocytes. It further reveals that human and murine airway cells can differ in their HA-cleaving protease repertoires. Our data will help drive the development of potent and selective protease inhibitors as novel drugs for influenza treatment.
INTRODUCTION
Influenza A viruses (IAV) and influenza B viruses (IBV) cause acute respiratory disease (flu) that results in significant human morbidity and mortality in annual epidemics. IAV and IBV are enveloped viruses with a segmented, negative-sense, single-stranded RNA genome and belong to the family Orthomyxoviridae. IAV circulate in a broad range of avian and mammalian hosts, including poultry, pigs, and humans (1). Aquatic birds provide the natural reservoir of IAV, and based on antigenic criteria, 16 hemagglutinin (HA) subtypes (H1 to H16) and 9 neuraminidase (NA) subtypes (N1 to N9) have been identified. Two more HA subtypes (H17 and H18) and NA subtypes (N10 and N11) have been found in bats. In contrast, IBV infections are restricted primarily to humans. IBV are divided into two antigenic lineages, Victoria and Yamagata, and currently cocirculate with IAV of subtypes H1N1 and H3N2 in the human population (2, 3). Annual seasonal flu vaccines are designed to protect against these four influenza viruses. In addition to seasonal outbreaks, the recurrent transmission of IAV between various species provides the basis for the emergence of novel influenza viruses which pose unpredictable challenges for public health. In 2009, the emergence of a new H1N1 virus (H1N1pdm) caused the first influenza pandemic of the 21st century, with an estimated 151,000 to 575,000 deaths (4). Since February 2013, human infections with an avian IAV of subtype H7N9 have caused over 1,500 confirmed cases of severe influenza disease in China, with a mortality rate of 39% (WHO, September 2018).
Influenza virus infection is initiated by the major surface glycoprotein HA through binding to sialic acid-containing cell surface receptors. Upon endocytosis from the cell surface, fusion of the viral lipid envelope and endosomal membrane delivers the virus genome into the cytoplasm of the host cell. HA is synthesized as the fusion-incompetent precursor protein HA0, which requires cleavage by a host cell protease into the subunits HA1 and HA2 in order to gain fusion capacity. This posttranslational cleavage has been recognized for more than 40 years as a requirement for IAV and IBV infectivity and as a major determinant of avian IAV pathogenicity (reviewed in references 5 and 6). Low-pathogenicity avian IAV possess a single arginine (R) at the cleavage site, designated the “monobasic cleavage site,” which can be activated by trypsin-like proteases present in the respiratory and intestinal tracts. In contrast, HA of highly pathogenic avian IAV is cleaved at a multibasic motif of the consensus sequence R-X-R/K-R by the proprotein convertases (PC) furin and PC5/6. The ubiquitous expression of these enzymes supports virus activation and replication in multiple organs and tissues, causing systemic infection with an often fatal outcome.
Mammalian and human IAV as well as IBV are also activated at a monobasic HA cleavage site. However, until a few years ago, the human proteases involved in activation had not been identified. We identified transmembrane protease serine S1 member 2 (TMPRSS2), a protease expressed in human airway epithelial cells, as being able to cleave IAV and IBV HA having a monobasic cleavage site (7, 8). More recent studies demonstrated that TMPRSS2 (also designated epitheliasin in mice) is essential for activation, multicycle replication, and, consequently, pathogenesis of H1N1pdm and H7N9 IAV in mice (9–11). Intriguingly, mice deficient in expression of TMPRSS2 were protected from pathogenesis and lethal outcomes resulting from infection with H7N9 or H1N1pdm IAV. In contrast, the same mice were not refractory to infection by a number of H3N2 strains, suggesting that H3 activation can be independent of TMPRSS2 (9–11). Similar observations have been made with IBV (12). To date, however, the cellular proteases responsible for HA activation in H3N2 and IBV in mice have not been clearly determined. Together, these previous studies have made clear that activation of HA is essential for virus spread and pathogenesis in mammalian hosts. Furthermore, the data have established that HA having a monobasic cleavage site can differ in its sensitivity to host cell proteases in mice in vivo.
Peptide-conjugated phosphorodiamidate morpholino oligomers (PPMO) are nucleic acid-like antisense agents composed of a morpholino oligomer covalently conjugated to a cell-penetrating peptide. PPMO are water soluble, enter cells readily without assistance, and have been used in clinical trials (reviewed in reference 13). Recently, a morpholino oligomer designed to treat Duchenne muscular dystrophy (eteplirsen) received approval by the U.S. FDA. PPMO targeting influenza virus or host factor RNA have been demonstrated to be able to reduce the expression of their respective targets and significantly affect viral titers in vitro and in vivo (14–20). PPMO have been shown to enter numerous cell types in vitro and in vivo in a benign manner, including airway epithelial and primary alveolar cells (16, 21).
We previously developed a PPMO (T-ex5) that interferes with the splicing of TMPRSS2 pre-mRNA, resulting in the production of mature mRNA lacking exon 5 (17). This truncated form of TMPRSS2 lacks the low-density lipoprotein receptor class A (LDLRA) domain and is consequently enzymatically inactive. Knockdown of active TMPRSS2 expression by T-ex5 prevented HA cleavage of both the H1N1 2009 pandemic virus A/Hamburg/05/09 (Hamburg/H1N1pdm) and the H3N2 1968 pandemic virus A/Aichi/2/68 and strongly suppressed virus replication in Calu-3 human airway epithelial cells (17). The data imply that both H1N1pdm and H3N2 IAV are activated predominantly by TMPRSS2 in Calu-3 cells. However, in experiments intended to elucidate protease expression in Calu-3 cells, reverse transcription-PCR (RT-PCR) analyses revealed that Calu-3 cells lack the expression of human airway trypsin-like protease (HAT) (also referred to as TMPRSS11D), an enzyme which, in vitro, is capable of cleaving IAV and IBV HA having a monobasic cleavage site. Calu-3 cells likely also lack the expression of other potential HA-cleaving proteases present in various cell types of the human respiratory tract (8, 17). Hence, the role of TMPRSS2 in influenza virus activation in the human airways overall still remained to be examined in greater detail.
This study was carried out using three types of experimental human airway cell cultures. Calu-3 is an immortalized human airway epithelial cell line which supports proteolytic activation of HA with a monobasic cleavage site by endogenous proteases. Well-differentiated human bronchial epithelial cells (HBEC) grown under air-liquid interface conditions provide a cell culture system considered to have high relevance to the human respiratory epithelium. Alveolar type II epithelial cells (AECII) are the major target cells of influenza viruses in the human lung (22, 23), and hence, primary AECII provide another relevant in vitro airway model. This study was designed to use PPMO-mediated knockdown of TMPRSS2 to investigate its role in proteolytic activation of IAV and IBV in Calu-3 cells, HBEC, and AECII.
We show that T-ex5 PPMO treatment produced efficient knockdown of the expression of active TMPRSS2 in all three types of cell cultures and prevented the activation and spread of H1N1pdm, H7N9, as well as H3N2 IAV. Furthermore, knockdown of active TMPRSS2 by T-ex5 inhibited proteolytic activation of IBV in AECII, while activation and spread of IBV in Calu-3 cells and HBEC were not affected. Our data provide strong evidence that TMPRSS2 is the major HA-activating protease of IAV in the human lower respiratory tract and of IBV in the human lung and that it constitutes a potential target for the development of drugs to address influenza infections.
RESULTS
Knockdown of enzymatically active TMPRSS2 by T-ex5 treatment inhibits replication of H7N9 IAV in Calu-3 airway epithelial cells.
In a previous study, we demonstrated that knockdown of expression of enzymatically active TMPRSS2 by T-ex5 prevented HA cleavage of H1N1pdm 2009 virus and H3N2 1968 pandemic virus and strongly suppressed virus replication in Calu-3 cells (17). Here, we analyzed the role of TMPRSS2 in the activation of zoonotic H7N9, as well as IBV, in Calu-3 cells and various IAV and IBV in primary HBEC and AECII culture systems.
Calu-3 cells were incubated with T-ex5 PPMO for 24 h prior to infection with A/Anhui/1/2013 (H7N9) (Anhui/H7N9), in order to reduce the production of normal TMPRSS2 mRNA and deplete the endogenous enzymatically active TMPRSS2 protein present in the cells. The cells were then inoculated at a low multiplicity of infection (MOI) and further incubated without PPMO for 72 h. At different time points postinfection (p.i.), virus titers were determined by a plaque assay. As shown in Fig. 1A, multicycle replication of Anhui/H7N9 was almost completely blocked by T-ex5 treatment, whereas the virus replicated efficiently in untreated cells. To confirm that the inhibition of virus replication was specifically caused by a block of HA cleavage, Calu-3 cells were treated with T-ex5 as described above and then infected with Anhui/H7N9 at a high MOI of 1 for 24 h, followed by SDS-PAGE and Western blot analysis. As expected, cleavage of HA0 was detected in the lysates of untreated cells (Fig. 1B). In contrast, only the uncleaved HA0 precursor was detected in T-ex5-treated cells.
FIG 1.

Knockdown of TMPRSS2 expression by PPMO T-ex5 inhibits activation and multicycle replication of IAV but not of IBV in Calu-3 cells. (A) Multicycle replication of Anhui/H7N9 in T-ex5-treated Calu-3 cells. Confluent Calu-3 cells were treated with 25 μM T-ex5 PPMO for 24 h or remained untreated (without [w/o]), inoculated with Anhui/H7N9 at an MOI of 0.001, and further incubated in the absence of T-ex5 for 72 h. Virus titers were determined as PFU per milliliter by a plaque assay of supernatants taken at the indicated time points. Results are mean values ± standard deviations (SD) from two independent experiments. (B) Analysis of HA cleavage in T-ex5-treated Calu-3 cells. Calu-3 cells treated with or without T-ex5 for 24 h were inoculated with Anhui/H7N9 at an MOI of 1 and incubated for 24 h in the absence of T-ex5. Cell lysates were subjected to SDS-PAGE and Western blotting using H7-specific antibodies. HA1 is not detected by the antibody. Beta-actin was used as a loading control. (C) Analysis of TMPRSS2-specific mRNA in Calu-3 cells. Cells were treated with 25 μM T-ex5 for 24 h and then incubated without T-ex5 for 72 h. Total RNA was isolated and analyzed by RT-PCR using primers designed to amplify 1,228 nucleotides of full-length TMPRSS2 mRNA. Full-length and truncated Δex5 PCR products are indicated. (D) Multicycle replication of IBV in Calu-3 cells with or without T-ex5 treatment. Confluent Calu-3 monolayers were treated with 25 μM PPMO T-ex5 or remained untreated. Cells were then inoculated with Malaysia/B or Massachusetts/B at a low MOI of 0.01 and incubated for 72 h in the absence of PPMO T-ex5. At the indicated time points, virus titers were analyzed by a plaque assay. Data are mean values ± SD from three independent experiments. (E) Analysis of HA cleavage in T-ex5-treated Calu-3 cells. Cells incubated with or without T-ex5 as described above were infected with Malaysia/B or Massachusetts/B at an MOI of 1 and incubated without further PPMO treatment for 48 h. Cell lysates were subjected to SDS-PAGE and Western blotting using IBV HA-specific antibodies. Beta-actin was used as a loading control.
Finally, the effect of T-ex5 on TMPRSS2 expression was studied by analysis of TMPRSS2-specific mRNA (17). Confluent Calu-3 monolayers were treated with 25 μM T-ex5 PPMO for 24 h, infected, and further incubated for 72 h in the absence of PPMO. Total RNA was isolated and analyzed with primers designed to amplify nucleotides 108 to 1336 of TMPRSS2 mRNA. As shown in Fig. 1C, a full-length PCR product of 1,228 bp was amplified from untreated Calu-3 cells, whereas a shorter PCR fragment of about 1,100 bp was amplified from T-ex5 PPMO-treated cells. Sequencing revealed that the truncated TMPRSS2 mRNA lacked the entire exon 5 (data not shown). The data demonstrate that T-ex5 was highly effective at producing exon skipping in TMPRSS2 pre-mRNA in Calu-3 cells and, thus, at inhibiting the expression of the functional protease.
Together, the data indicate that in Calu-3 cells, TMPRSS2 is the major HA-activating protease for H7N9 having a monobasic cleavage site.
TMPRSS2 is not critical for proteolytic activation and multicycle replication of IBV in Calu-3 cells.
We next investigated the role of TMPRSS2 in the proteolytic activation of IBV in Calu-3 cells. IBV HA is cleaved at a monobasic cleavage site and has been shown to be activated by TMPRSS2 in vitro (8). However, Sakai and coworkers demonstrated that IBV activation and replication are independent of TMPRSS2 expression in a mouse model (12). Rather, IBV activation was associated with as-yet-unspecified proteases.
To examine the role of TMPRSS2 in proteolytic activation of IBV in Calu-3 cells, confluent cell monolayers were treated with T-ex5 PPMO prior to infection, inoculated with B/Malaysia/2506/2004 (Malaysia/B) or B/Massachusetts/71 (Massachusetts/B) at a low MOI, and incubated in the absence of further T-ex5 treatment for 72 h. At 16, 24, 48, and 72 h p.i., virus propagation was analyzed by plaque titration. Interestingly, IBV replicated efficiently in T-ex5-treated Calu-3 cells. Virus titers of Massachusetts/B were similar in T-ex5-treated and untreated control cells. Cells infected with Malaysia/B and treated with T-ex5 had a 10-fold reduction of the virus titer compared to control cells (Fig. 1D). To analyze cleavage of IBV HA, Calu-3 cells were treated with T-ex5 for 24 h, infected with Malaysia/B or Massachusetts/B at an MOI of 1, and incubated in the absence of T-ex5 for 48 h. Untreated Calu-3 cells were used as controls. Cell lysates were subjected to SDS-PAGE and immunoblotting with IBV HA-specific antibodies. Cleavage of HA0 was observed in both untreated and T-ex5-treated cells (Fig. 1E). Similar amounts of viral proteins were present in T-ex5-treated and control cells at 48 h p.i., indicating replication of both viruses. These data indicate that the proteolytic activation of IBV in Calu-3 cells is independent of TMPRSS2 expression and thus differs from the activation of IAV, at least in this cell type. TMPRSS2-independent activation of IBV in Calu-3 cells is consistent with results previously observed in mice (12).
Expression of HA-activating proteases in Calu-3 cell, HBEC, and AECII systems.
In various in vitro settings, the type II transmembrane serine proteases TMPRSS2, TMPRSS4, HAT (TMPRSS11D), matriptase (ST14), and TMPRSS13 and the soluble proteases kallikrein 5 (KLK5) and KLK12 have all been shown to be competent to cleave IAV HA having a monobasic cleavage site (reviewed in reference 24). Therefore, we specifically determined the expression of these proteases in Calu-3 cells using RT-quantitative PCR (qPCR) analysis. Results for individual protease-specific mRNAs were normalized to the value for tubulin mRNA. As shown in Fig. 2, mRNAs specific for TMPRSS2, TMPRSS4, and matriptase/ST14 were expressed in Calu-3 cells. Interestingly, although the expression level of TMPRSS2 mRNA was low compared to that of tubulin mRNA, it was sufficient to support efficient activation of IAV in the cells. The TMPRSS2 mRNA level was about 9-fold lower than the TMPRSS4 mRNA level and 2.5-fold lower than the matriptase-specific mRNA level. We did not detect expression of HAT/TMPRSS11D, TMPRSS13, KLK5, and KLK12 mRNA in Calu-3 cells. Taken together, these data suggest that Calu-3 cells express the HA-cleaving proteases TMPRSS4 and matriptase at higher levels than TMPRSS2. The lack of Anhui/H7N9 multiplication in T-ex5 PPMO-treated cells indicates that, at least in Calu-3 cells, TMPRSS4 and matriptase do not support the activation of H7N9 IAV.
FIG 2.

RT-qPCR analysis of HA-cleaving protease transcripts in Calu-3 cells, HBEC, and AECII. Total RNA was isolated from Calu-3 cells, HBEC cultures, or AECII. Relative mRNA expression levels (percent) of selected proteases were measured by RT-qPCR and normalized by using tubulin as an internal control. Data are mean values ± SD (n = 3).
Next, we examined the expression of proteases capable of cleaving HA having a monobasic cleavage site in primary HBEC and AECII cultures. By virtue of their morphological and architectural complexity, these two types of in vitro model systems provide a higher degree of physiological relevance to the human respiratory tract than do Calu-3 cells. In HBEC, we found that mRNAs specific for TMPRSS2, TMPRSS4, HAT/TMPRSS11D, TMPRSS13, matriptase/ST14, and KLK5 were present, yet only low levels of TMPRSS13 and KLK5 mRNAs were detected (Fig. 2). The expression level of TMPRSS2 mRNA was about 8-fold higher than that of HAT/TMPRSS11D mRNA but 2.6-fold lower than that of TMPRSS4-specific mRNA. Expression levels of TMPRSS2 and matriptase/ST14 mRNAs were similar in HBEC. In AECII, mRNAs of all tested proteases were expressed, although TMPRSS13, KLK5, and KLK12 mRNAs were present at only very low levels. Interestingly, TMPRSS2 mRNA showed the highest expression level among the proteases tested in AECII, followed by TMPRSS4, matriptase/ST14, and HAT/TMPRSS11D. The data indicate that HBEC and AECII express more potential HA-cleaving proteases than do Calu-3 cells. We therefore wished to examine the role of TMPRSS2 in the activation of IAV in HBEC and AECII.
Knockdown of active TMPRSS2 inhibits multicycle replication of IAV, but not IBV, in primary human bronchial epithelial cells.
Primary HBEC cultivated under air-liquid interface conditions for 3 to 4 weeks form a pseudostratified epithelium comprised of ciliated, goblet, and basal cells and provide a cell culture system that closely approximates the human airway epithelium. First, we examined the expression of TMPRSS2 in HBEC cultures by immunofluorescence analysis. Well-differentiated HBEC cultures were immunostained for mucin 5AC (MUC5AC) to indicate goblet cells and acetylated tubulin to indicate ciliated cells (Fig. 3A). Using nonpermeabilized HBEC, we found that TMPRSS2 is expressed on the surface of ciliated cells (Fig. 3A). By using cross sections of permeabilized HBEC cultures, TMPRSS2 was seen to be expressed in basal cells in addition to ciliated cells. We note that the high level of signal colocalization for TMPRSS2 and cilia on the plasma membrane (Fig. 3A, cross section) is most likely due to the folding over of cilia during the preparation of HBEC cross sections, rather than expression of TMPRSS2 along the entire length of the cilia (Fig. 3A, top view, nonpermeabilized cells).
FIG 3.

PPMO T-ex5 inhibits multicycle replication of IAV but not of IBV in primary human bronchial epithelial cells (HBEC). (A) Immunofluorescence analysis of the expression of TMPRSS2 in HBEC cultures. Permeabilized HBEC were immunostained using antibodies against mucin 5AC (MUC5AC) (goblet cells) and acetylated tubulin (cilia), and nonpermeabilized HBEC were immunostained against cilia and TMPRSS2. Sections of permeabilized HBEC cultures were immunostained against TMPRSS2 and cilia. Coexpression is presented as merged false color (yellow). The nuclei were stained using DAPI (blue) as a counterstain. ci, ciliated cell; go, goblet cell; ba, basal cell. (B) RT-PCR analysis of TMPRSS2 mRNA in T-ex5-treated HBEC cultures. Cells were treated with the indicated concentrations of PPMO T-ex5 for 24 h, the medium was then replaced, and the cells were incubated in the absence of T-ex5 for 24 h. Total RNA was isolated and amplified with TMPRSS2-specific primers to amplify a full-length mature mRNA fragment of 1,228 bp and the truncated Δex5 fragment. (C) Evaluation of the effect of PPMO treatment on cell viability. HBEC cultures were treated with 25 μM T-ex5 or a nonsense-sequence negative-control PPMO (scramble) for 24 h. Cell viability of untreated cells (w/o) was set as 100%. Results are mean values ± SD (n = 3). (D) Multicycle replication of IAV and IBV in PPMO-treated HBEC. HBEC cultures were treated with 25 μM T-ex5 or scramble for 24 h or remained untreated (w/o); infected with Hamburg/H1N1pdm, Anhui/H7N9, Aichi/H3N2/PR8, or Malaysia/B at a low MOI of 0.01 to 0.005; and further incubated in the absence of PPMO for 24 h. Cells were fixed, permeabilized, and immunostained against the viral nucleoprotein (NP) and cilia. Coexpression is presented as merged false color (yellow). The nuclei were stained using DAPI as a counterstain. Representative images from two (H7N9 and IBV) or three (H1N1pdm and H3N2) independent experiments are shown. Bars, 100 μm. (E) Plaque assays of progeny virus released from H7N9/Anhui- and Malaysia/B-infected HBEC at 24 h p.i. Results show virus titers from one experiment (H7N9/Anhui) and mean values ± SD from three independent experiments (Malaysia/B). (F) Analysis of HA cleavage in PPMO-treated HBEC. HBEC cultures were treated with 25 μM T-ex5 or scramble PPMO for 24 h and then infected with Anhui/H7/PR8 or Malaysia/B at an MOI of 1 for 48 h. Cell lysates were subjected to SDS-PAGE and Western blotting with H7- or IBV HA-specific antibodies. Beta-actin served as a loading control.
Next, to determine whether T-ex5 PPMO efficiently enters the cytoplasm and provides efficient knockdown of enzymatically active TMPRSS2 expression in HBEC, the cultures were treated with 25 μM T-ex5 PPMO for 24 h in a manner similar to that carried out in the experiments using Calu-3 cells described above. Subsequently, total RNA was isolated and analyzed by RT-PCR. As shown in Fig. 3B, T-ex5 treatment caused skipping of exon 5 in approximately 80% of TMPRSS2 mRNA. We found that the use of higher concentrations of T-ex5 PPMO (30 μM or 40 μM) did not increase the exon skipping efficiency, as an estimated 20% of the TMPRSS2 mRNA still remained as full-length mRNA in the HBEC cultures. This is in contrast to the dose-dependent knockdown of TMPRSS2 expression previously observed in monolayer Calu-3 cells (17), where concentrations above 20 μM produced higher levels of splice alteration. We suspect that this difference is likely due to the stratified structure of differentiated HBEC cultures and that full-length TMPRSS2 mRNA was derived mostly from basal cells, which reside below the ciliated and goblet cells (Fig. 3A). It is likely that T-ex5 PPMO did not penetrate sufficiently through the layers of other cell types to the extent necessary to efficiently access the basal cells. Thus, based on the observed overall knockdown, the exon skipping efficiency of T-ex5 in the upper cell layers was most likely higher than 80%. Based on these observations, we decided to use a dose of 25 μM T-ex5 PPMO for further experiments in HBEC cultures.
To evaluate whether PPMO treatment affected cell viability, HBEC were treated with 25 μM T-ex5 PPMO for 24 h under the same conditions as the ones used in the virus multicycle replication experiments reported below. As a control, a PPMO of a nonsense sequence designated “scramble” was used. The cell viability of PPMO-treated HBEC was compared to that of untreated HBEC cultures. As shown in Fig. 3C, HBEC cultures treated with T-ex5 or scramble PPMO displayed no loss in viability.
To examine the replication efficiency of IAV in HBEC cultures in the absence or presence of active TMPRSS2, cells were treated with 25 μM T-ex5 or scramble PPMO or remained untreated for 24 h prior to infection. Cells were then inoculated with either Hamburg/H1N1pdm, Aichi/H3N2/PR8 [HA and NA of A/Aichi/2/68 (H3N2) and 6 genes of A/PR8/34 (H1N1) (PR8)], Anhui/H7N9, or Malaysia/B, at a low MOI of 0.01 to 0.001, and incubated for 24 h to allow multiple cycles of viral replication. At 24 h p.i., cells were fixed and immunostained against the viral nucleoprotein (NP). In addition, ciliated cells were immunostained. As shown in Fig. 3D, foci of infection were visible for all four viruses at 24 h postinfection (p.i.) in untreated or scramble PPMO-treated HBEC cultures. In contrast, the spread of Hamburg/H1N1pdm, Anhui/H7N9, and Aichi/H3N2/PR8 was strongly suppressed by T-ex5 treatment, with only a few sporadic single cells apparently infected. We suspect that these individual virus-positive cells were the result of direct infection by the initial inoculation. However, the spread of Malaysia/B was unaffected by T-ex5 treatment in HBEC cultures, in congruence with that observed in Calu-3 cells. We also quantified progeny virus released from Anhui/H7N9- and Malaysia/B-infected HBEC at 24 h p.i. by using apical washes of the cultures for plaque titration. Consistent with our observations of PPMO-treated Calu-3 cells (Fig. 1A), a 100-fold reduction of the virus titer was determined for Anhui/H7N9 in T-ex5-treated HBEC compared to control PPMO-treated cells, whereas virus titers of Malaysia/B were similar in T-ex5- and scramble-treated cells (Fig. 3E). To analyze HA cleavage in HBEC, the cells were treated with PPMO as described above, infected with Anhui/H7/PR8 or Malaysia/B at an MOI of 1, and incubated for 48 h. Infection of HBEC for HA cleavage analysis was performed under biosafety level 2 (BSL2) conditions, and therefore, the recombinant Anhui/H7/PR8 virus, which contains the HA of Anhui/H7N9 along with 7 genes of PR8, was used instead of the Anhui/H7N9 virus. Analysis of cell lysates by SDS-PAGE and immunoblotting demonstrated that cleavage of H7 HA0 was inhibited in T-ex5-treated HBEC, while IBV HA0 cleavage was not significantly inhibited in either T-ex5- or scramble-treated HBEC (Fig. 3F).
Together, the data indicate that in primary HBEC, TMPRSS2 is the primary protease responsible for the activation of IAV HA having a monobasic cleavage site. In contrast, activation of IBV in HBEC is independent of TMPRSS2 expression and instead supported by an as-yet-undetermined protease(s).
Upregulation of HA-activating protease transcripts in HBEC upon IAV and IBV infection.
A recent study demonstrated that KLK5 mRNA is upregulated in air-liquid interface cultures of primary human bronchial epithelial cells infected with A/Scotland/20/74 (H3N2), thus suggesting that KLK5 may be involved in H3N2 activation in human bronchi (25). Here, we aimed to examine whether mRNA expression levels of the different HA-activating protease candidates mentioned above are altered in HBEC cultures upon influenza virus infection and whether differences in protease mRNA expression are observed between the different viruses used in the present study. HBEC cultures were inoculated with Aichi/H3N2/PR8, Hamburg/H1N1pdm, or Malaysia/B at an MOI of 1 and incubated for 24 h. Subsequently, mRNA expression of TMPRSS2, TMPRSS4, HAT/TMPRSS11D, TMPRSS13, matriptase/ST14, and KLK5 was analyzed by RT-qPCR. Upon infection of HBEC with H3N2 IAV, an approximately 1.6-fold upregulation of KLK5 mRNA was detected (Fig. 4). Furthermore, significant increases in mRNA levels of matriptase (2.4-fold), TMPRSS4 (1.7-fold), and HAT/TMPRSS11D (1.8-fold) were observed in H3N2-infected HBEC compared to uninfected cells. H1N1pdm infection caused a slight increase in KLK5 and TMPRSS4 mRNA levels and a significant increase in matriptase/ST14 and HAT/TMPRSS11D mRNA levels. IBV infection resulted in a significant increase (2-fold) in KLK5 mRNA expression and an ∼1.4-fold increase in mRNA levels of matriptase/ST14 and HAT/TMPRSS11D. Interestingly, the TMPRSS2 mRNA level was not altered upon infection with any of the three viruses. The same was observed for TMPRSS13 mRNA. The data suggest that expression levels of TMPRSS4, HAT, KLK5, and matriptase are slightly increased in virus-infected HBEC. However, the lack of IAV multicycle replication in T-ex5-treated HBEC (Fig. 3) indicates that these proteases are not able to support IAV activation in HBEC.
FIG 4.
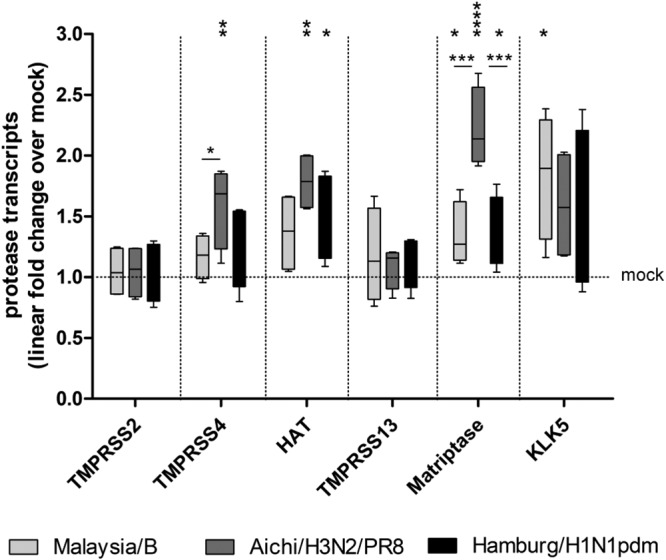
RT-qPCR analysis of HA-activating protease transcripts in virus-infected HBEC. Well-differentiated HBEC cultures were infected with Malaysia/B, Aichi/H3N2/PR8, or Hamburg/H1N1pdm at an MOI of 1 for 24 h, and total RNA was subsequently isolated. Relative mRNA expression levels of selected proteases were measured by RT-qPCR and normalized using tubulin as an internal control. Protease transcript expression levels are expressed as fold change values obtained with the uninfected mock control (set as 1). Data are shown as 2−ΔΔCT values in box plots (minimum to maximum) (n = 4). Data (ΔCT values) were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison test according to the number of parameters and groups being compared. A P value of <0.05 was considered significant (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.00001).
TMPRSS2 is essential for proteolytic activation of both IAV and IBV in primary human type II alveolar epithelial cells.
AECII account for approximately 15% of total cells in the human lung and have been shown to be the major target cells for IAV infection in human lung (22, 23, 26). Therefore, we also examined the role of TMPRSS2 in proteolytic activation and multiplication of Hamburg/H1N1pdm, Aichi/H3N2/PR8, and Anhui/H7/PR8 IAV and IBV in primary human AECII.
In contrast to the experiments performed in Calu-3 cells and HBEC, efficient knockdown of TMPRSS2 expression in AECII required treatment of cells with 30 μM PPMO both pre- and postinfection. The reasons are not entirely clear but may be due to the more elaborate surface composition in AECII than in other cell types, which could represent a more challenging physical barrier to cellular entry by PPMO.
AECII cultures were grown on filter devices, treated with 30 μM T-ex5 or scramble PPMO for 24 h, inoculated with recombinant Anhui/H7/PR8 at a low MOI, and further incubated in the presence of 30 μM PPMO for 72 h. Untreated cells were used as a further control. At different time points p.i., multicycle replication was analyzed by a plaque assay. Efficient replication of up to 107 PFU/ml of Anhui/H7/PR8 was observed in untreated and scramble PPMO-treated AECII, whereas virus growth was strongly inhibited in T-ex5-treated cells (Fig. 5A). To demonstrate inhibition of HA0 cleavage by T-ex5 treatment in AECII, cells treated with or without T-ex5 for 24 h were infected with Anhui/H7/PR8 at an MOI of 0.1 and further incubated with PPMO for 72 h. As shown in Fig. 5B, only the precursor HA0 was detected in T-ex5-treated cells, while cleavage of HA0 into HA2 was observed in control cells. RT-PCR analysis of TMPRSS2 mRNA revealed efficient exon skipping in T-ex5-treated AECII, with almost 100% of TMPRSS2 mRNA being expressed as a truncated form lacking exon 5 (Fig. 5C). We next analyzed multicycle replication of Hamburg/H1N1pdm and Aichi/H3N2/PR8 in AECII T-ex5-treated or untreated cells. The spread of both IAVs was inhibited in T-ex5-treated cells, with only a few infected cells detected at 24 h p.i. (Fig. 5D). The virus titer of Aichi/H3N2/PR8 was reduced 100-fold in T-ex5-treated AECII compared to control cells (Fig. 5E), similar to the titer reduction observed with Anhui/H7/PR8 (Fig. 5A).
FIG 5.

Knockdown of TMPRSS2 activity inhibits activation of both IAV and IBV with a monobasic HA cleavage site in primary human type II alveolar epithelial cells (AECII). (A) Multicycle replication of Anhui/H7/PR8 in T-ex5-treated AECII. Cells were treated with 30 μM T-ex5 or scramble PPMO or remained untreated (w/o) for 24 h. Cells were then infected with Anhui/H7/PR8 at an MOI of 0.005 and incubated in the presence of 30 μM PPMO for 72 h. At the indicated time points, virus titers were determined by a plaque assay. Data are mean values from two independent experiments ± SD. (B) Analysis of HA cleavage in PPMO-treated AECII. AECII treated with T-ex5 for 24 h were infected with Anhui/H7/PR8 at an MOI of 0.1 and incubated for 72 h in the presence of T-ex5. Untreated cells were used as controls. Cell lysates were analyzed for HA cleavage by SDS-PAGE and Western blotting. HA1 is not detected by the antibody. (C) RT-PCR analysis of TMPRSS2 mRNA in Anhui/H7/PR8-infected AECII with or without T-ex5 treatment for 72 h. Total RNA was isolated and amplified with TMPRSS2-specific primers to amplify a full-length mature mRNA fragment and the truncated Δex5 fragment. (D) Spread of IAV and IBV in T-ex5-treated cells. AECII cultures treated with T-ex5 for 24 h were inoculated with the indicated virus at an MOI of 0.01 to 0.001 and incubated in the presence of T-ex5 for 24 h. Cells were fixed and immunostained against viral NP, prosurfactant protein C (pro-SP-C) (AECII marker), and DAPI. Untreated cells were used as controls. Representative images from two independent experiments are shown. Bars, 100 μm. (E) Progeny virus released from Aichi/H3N2/PR8- and Malaysia/B-infected AECII was quantified using a plaque assay at 24 h postinfection. Virus titers from a representative experiment are shown. (F) Analysis of HA cleavage in T-ex5-treated AECII. AECII were treated with T-ex5 for 24 h, infected at an MOI of 0.5 (Aichi/H3N2/PR8) or 0.01 (Malaysia/B), and further incubated in the presence of T-ex5 for 24 and 72 h, respectively. Untreated cells were used as controls. Cell lysates were analyzed for HA cleavage by immunoblotting using H7- and IBV HA-specific antibodies, respectively. Beta-actin was used as a loading control. Lanes are spliced together from one immunoblot from one experiment. (G) Spread of FPV/H7N1 in T-ex5-treated AECII. Cells with or without T-ex5 treatment for 24 h were infected with FPV/H7N1 and incubated in the presence or absence of T-ex5 for 24 h. Cells were immunostained against viral NP, pro-SP-C, and DAPI. (H) Effect of PPMO treatment on AECII viability. AECII were treated with PPMO for 24 h. Cell viability of untreated (w/o) cells was set as 100%. Results are mean values ± SD (n = 3).
We also examined whether the TMPRSS2-independent activation of IBV observed in Calu-3 cells and HBEC cultures also occurs in AECII. Surprisingly, the spread of Malaysia/B and Massachusetts/B was inhibited by knockdown of TMPRSS2 expression in T-ex5-treated AECII, with only a few single infected cells visible (Fig. 5D). This result is in contrast to what has been observed for HBEC and Calu-3 cell cultures and is also in contrast to what has been described in mice (12). The strong suppression of Malaysia/B replication in T-ex5-treated cells was further confirmed by plaque titration of progeny virus at 24 h p.i. where we observed a 1,000-fold reduction of virus titers in T-ex5 versus untreated cells (Fig. 5E). In addition, large amounts of HA were detected in untreated AECII infected with Aichi/H3N2/PR8 or Malaysia/B, where cleavage of HA0 was observed (Fig. 5F). In contrast, only a low level of, if any, HA was detected in T-ex5-treated AECII infected with either virus. Finally, to further confirm that the suppression of virus propagation by T-ex5 PPMO was due to inhibition of cleavage of HA with a monobasic cleavage site by TMPRSS2, we investigated the multicycle replication of A/FPV/Rostock/34 (H7N1) (FPV/H7N1) in T-ex5-treated AECII. FPV/H7N1 contains a multibasic HA cleavage site, which can be activated by ubiquitously expressed furin and PC5/6 and is therefore unlikely to be affected by knockdown of TMPRSS2 activity. As expected, FPV/H7N1 spread was efficient and similar in both T-ex5-treated and untreated cells (Fig. 5G). In addition, evaluation of cell viability in PPMO-treated AECII revealed no significant cytotoxicity in the cells, with 3% and 5% losses in viability in scramble- and T-ex5-incubated cells, respectively (Fig. 5H).
Taken together, our data demonstrate that TMPRSS2 is the major HA-activating protease of H1N1pdm, H7N9, and H3N2 IAV in human bronchial epithelial cells and type II pneumocytes and is essential for virus multiplication. Furthermore, our data show that TMPRSS2 is essential for the activation and spread of IBV in human type II pneumocytes, whereas IBV activation in HBEC and Calu-3 cells is carried out, at least in part, by a protease(s) other than TMPRSS2. Thus, the TMPRSS2-independent activation of IBV previously observed in mice was also observed here in human bronchial cells but not in human type II pneumocytes.
Murine AECII support TMPRSS2-independent activation of H3N2 IAV and IBV.
As described above, TMPRSS2-dependent activation of H3N2 IAV and IBV in primary human AECII differs from TMPRSS2-independent activation of these viruses in mice. The protease(s) that activates H3N2 and IBV, but not H1N1pdm and H7N9 IAV, in murine lung is still not determined, and it remains unknown whether it is expressed in murine AECII or produced and made available by other cell types in the murine lung. We therefore investigated multicycle replication of Hamburg/H1N1pdm, Aichi/H3N2/PR8, Anhui/H7/PR8, and Malaysia/B in primary AECII from TMPRSS2-deficient mice (Tmprss2−/−) and their wild-type littermates (Tmprss2+/+) (27). As shown in Fig. 6A and B, Hamburg/H1N1pdm and Anhui/H7/PR8 were able to undergo multicycle replication and grow to high titers in AECII from wild-type mice, whereas virus spread and efficient multiplication in AECII from TMPRSS2-deficient mice were inhibited. Western blot analysis of cell lysates confirmed virus multiplication and cleavage of H1 and H7 in AECII from Tmprss2+/+ mice, whereas a very little, if any, H1 or H7 was detected in AECII from TMPRSS2-deficient mice (Fig. 6C). In contrast, multicycle replication of Aichi/H3N2/PR8 and Malaysia/B and cleavage of H3 and IBV HA were similar in AECII of TMPRSS2-deficient mice and wild-type littermates (Fig. 6). These data suggest that the protease(s) that supports TMPRSS2-independent activation of H3N2 IAV and IBV in murine lung is expressed in murine AECII. Moreover, the data indicate that mouse and human AECII differ in their protease repertoires capable of activating H3 and IBV HA.
FIG 6.

TMPRSS2-independent activation of H3N2 IAV and IBV in murine AECII. (A) Primary AECII isolated from lungs of TMPRSS2-deficient mice (Tmprss2−/−) or wild-type littermates (Tmprss2+/+) were infected with the indicated virus at an MOI of 0.01 to 0.001. At 24 h p.i., cells were fixed, permeabilized, and stained for viral NP and pro-SP-C (AECII marker). DAPI was used as a counterstain. Representative images from three independent experiments are shown. Bars, 100 μm. (B) Multicycle replication of IAV and IBV in AECII of Tmprss2−/− and Tmprss2+/+ mice. Primary murine AECII were infected with the indicated virus at a low MOI. At the indicated time points, virus multiplication was analyzed by a plaque assay. Data are mean values ± SD from three independent experiments. (C) HA cleavage in AECII of Tmprss2+/+ and Tmprss2−/− mice. At 72 h p.i., cell lysates were subjected to SDS-PAGE and immunoblotting using HA-specific antibodies. Beta-actin served as a loading control.
In conclusion, our data indicate that TMPRSS2 is the major HA-activating protease of IAV and IBV in human type II pneumocytes. In contrast, HA of H3N2 IAV and IBV is activated in murine type II pneumocytes by a different protease that has not yet been determined.
DISCUSSION
Previous studies by us and others identified TMPRSS2 as a host cell factor essential for activation and spread of H1N1pdm and H7N9 IAV in murine airways and demonstrated that knockout of TMPRSS2 prevents pathogenesis from these influenza viruses in mice. In contrast, activation and pathogenesis of a number of H3N2 IAV and IBV strains in mice were largely independent of TMPRSS2 expression and were instead performed by as-yet-unspecified proteases. The present study demonstrates that TMPRSS2 is crucial for proteolytic activation and spread of H1N1pdm, H7N9, as well as H3N2 IAV in primary human bronchial epithelial cells and for both IAV and IBV activation in primary human type II pneumocytes. Thus, our data indicate that TMPRSS2 is the major HA-activating protease of IAV in the lower respiratory tract and of IBV in human lung overall and appears to be a promising host factor target for influenza drug development.
We found that the TMPRSS2 dependence of H3N2 IAV and IBV activation is different in primary human airway cells and mice. The identities of the specific proteases involved in the activation of H3 and IBV HA in murine airways remain to be determined. Furthermore, it is unknown whether H3N2 IAV and IBV are activated by the same protease(s) in mice. Here, we show that AECII from human and mouse differ in their repertoires of H3 and IBV HA-cleaving proteases. H3N2 IAV and IBV were able to undergo multicycle replication in AECII isolated from both TMPRSS2-deficient mice and their wild-type littermates. In contrast, knockdown of TMPRSS2 activity in primary human AECII using the T-ex5 PPMO prevented the spread of both viruses. The differences in the HA-activating protease repertoires between mice and humans may be related to a larger number or higher expression level of appropriate proteases in murine airways and/or due to the tight regulation of protease activities by endogenous inhibitors in human cells. Furthermore, differences in the substrate specificity of murine and human proteases may contribute to the differences in the TMPRSS2 dependence of H3 cleavage in human and murine airways. Human KLK5 has been shown to cleave IAV H3 in vitro (25, 28). In contrast, murine KLK5 was not able to cleave H3 in vitro, and replication of H3N2 IAV was similar in wild-type and KLK5-deficient mice, demonstrating that KLK5 does not contribute to H3N2 IAV activation in mice (29). Vice versa, it may be that an undetermined murine protease cleaves H3 or IBV HA, whereas its human orthologue does not. Notably, the human genome encodes 588 proteases, whereas 672 protease genes have been annotated in the murine genome (30) (Mammalian Degradome Database). Hence, TMPRSS2-independent activation of H3N2 IAV or IBV in murine airways may be due to a mouse-specific protease. A recent study showed that TMPRSS4 contributes to H3N2 IAV activation in mice (31). Nonetheless, H3N2 virus was still proteolytically activated in TMPRSS2-TMPRSS4 knockout mice, and 30% of mice succumbed to infection, suggesting that at least one other undetermined H3-cleaving protease is present in murine airways. Our RT-qPCR analysis indicated that TMPRSS4 is also expressed in human AECII, HBEC, and Calu-3 cells. However, TMPRSS4 was not able to compensate for the lack of TMPRSS2 in the activation of H3N2, as shown by the lack of multicycle replication of H3N2 virus in T-ex5-treated human airway cells. It remains to be investigated why TMPRSS4 supports IAV activation in murine airways yet fails to do so in primary human airway cells. Overall, it remains to be investigated which protease(s), in addition to TMPRSS2, supports the activation of H3N2 IAV and IBV in murine AECII. Notably, it appears that the unidentified protease either is not present, as an active enzyme, in human AECII or is incapable of cleaving influenza virus HA in these cells. The differences in the TMPRSS2 dependence of H3N2 IAV and IBV activation between primary human airway cells and mice exemplify limitations in using the mouse as a model to identify and characterize HA-cleaving proteases. Such species-specific differences should be considered when testing the efficacy of protease inhibitors as influenza drugs in an animal model. However, we also note that different mouse strains could differ in their protease repertoires or that genetic differences of the human donors could contribute to the species-specific discrepancies in the expression of HA-cleaving proteases that we observed in this study.
Orthologues of TMPRSS2 have been cloned from chicken, swine, and nonhuman primates and have been shown to be capable of activating IAV with a monobasic HA cleavage site, indicating that TMPRSS2 can support HA activation in various species (32–34). The characteristic role of TMPRSS2 in IAV activation in these hosts, however, remains to be investigated. Comparative genomic analysis of zebra finch, chicken, human, and mouse indicates that avian genomes encode a considerably lower number of proteases (including tryptases and kallikreins) than do human and mouse genomes (35). However, it cannot be concluded from that study how important TMPRSS2 is for the activation of IAV in chicken. Because of the importance of avian hosts for the global ecology and epidemiology of IAV, it would be of particular interest to examine the role of TMPRSS2 in IAV activation in the respiratory and intestinal tracts of birds.
Here, we found that activation of IBV in primary HBEC and Calu-3 cells was independent of TMPRSS2 and due to a so-far-unspecified protease(s), a result similar to that reported previously regarding IBV activation in mice (12). Interestingly, it was also previously observed that IBV, but not IAV, is proteolytically activated by undetermined endogenous proteases in specific MDCK cell lines (36, 37). Thus, it appears that, compared to IAV, IBV HA can be cleaved by a broader range of host proteases. IBV HA cleavage in the airways has not been well characterized, but proteases capable of cleaving IAV HA are probably involved. Our data indicate that Calu-3 cells and HBEC express an IBV-activating protease that is not present (at least in an enzymatically active form) in human AECII. Analysis of the expression of different HA-activating proteases in AECII by RT-qPCR, however, revealed no obvious candidate. High levels of TMPRSS4 mRNA were detected in Calu-3 cells and HBEC compared to other protease mRNAs, and TMPRSS4 may therefore represent a protease candidate responsible for the activation of IBV in HBEC and Calu-3 cells. On the other hand, TMPRSS4 mRNA has also been detected in AECII, and it seems unlikely that it supports IBV activation in HBEC and Calu-3 cells but not in AECII. The same is true for matriptase, HAT, TMPRSS13, KLK5, and KLK12. Expression levels of TMPRSS13, KLK5, and KLK12 were very low in primary human airway cells and not detected in Calu-3 cells. Notably, low levels of TMPRSS2 mRNA were also detected in Calu-3 cells and are likely sufficient to support efficient proteolytic activation of H1N1pdm, H7N9, and H3N2 IAV in these cells (Fig. 1) (17). Thus, influenza virus activation apparently does not require high expression levels of relevant proteases. In sum, our data show that activation of IBV can be carried out by various proteases at various regions along the respiratory tract, with TMPRSS2 being crucial for IBV activation in human AECII but not in human bronchi. It remains to be investigated whether one of the above-mentioned protease candidates supports the activation of IBV in HBEC and Calu-3 cells.
A recent study showed upregulation of KLK5 mRNA in air-liquid interface cultures of HBEC upon infection with H3N2 IAV and elevated levels of KLK5 in tracheal aspirates of influenza patients (25). Enhanced shedding of HAT and TMPRSS2 has been observed for differentiated human nasal epithelial cells upon exposure to ozone in vitro (38). Moreover, ozone exposure enhanced the replication of H3N2 IAV in these cells. HAT was originally isolated as an enzymatically active protease from sputa of patients with chronic airway diseases (39). Thus, stress or disease conditions in human airway cells may enhance the release of HA-cleaving proteases and increase susceptibility to influenza virus infection. Here, we observed a slight increase in mRNA levels of KLK5, matriptase/ST14, HAT/TMPRSS11D, and TMPRSS4 in HBEC cultures inoculated with H1N1pdm, H3N2, or IBV. Interestingly, TMPRSS2 mRNA levels were not altered upon IAV or IBV infection in HBEC. It remains to be investigated whether upregulation of protease mRNAs resulted in overall elevated protease activity in HBEC cultures. Protease activities can be tightly regulated by different mechanisms, including protease inhibitors, zymogen activation, activation cascades, subcellular localization, and cofactors, and it is not possible to directly correlate protease mRNA levels with protease expression and activity. Nevertheless, the lack of H3N2 activation in T-ex5-treated HBEC suggests that KLK5, matriptase, HAT, and TMPRSS4 play a negligible role in the activation of H3N2 IAV in HBEC. Still, these proteases may be involved in the activation of IBV in human bronchial cells.
In 1998, the crystal structure of H3 HA0 was solved and revealed that the cleavage site is located in a prominent surface loop (40). Structural analysis of the H1 HA0 of the 1918 pandemic virus showed that the cleavage site loop is less exposed than in H3 HA0 (41). The third HA0 structure solved to date, H16 of a low-pathogenicity avian IAV, contains an α-helix structure in the cleavage site loop, with the arginine residue being hidden behind the helix (42). Interestingly, H16 was shown to be resistant to cleavage by trypsin but is cleaved by TMPRSS2 in vitro (42, 43). Thus, it appears that the conformation and exposure of the HA cleavage site loop may differ more than generally assumed among IAV subtypes and that the cleavage site of H3 may be more accessible to various proteases. A recent study showed that replacement of amino acids 320 to 328 (H3 numbering) of the C terminus of HA1 of pandemic H1 (L-R-N-I-P-S-I-Q-S-R↓) by the H3 sequence (M-R-N-V-P-E-K-Q-T-R↓) did not support proteolytic activation of the mutated H1 in TMPRSS2 knockout mice (44). However, proteolytic activation of H1 in TMPRSS2-deficient mice was facilitated by replacement of amino acid E31 of H1 with D31 of H3 together with substitution of amino acids 320 to 328 of the HA1 C terminus. E31 of H1 is predicted to form a salt bridge with R321, which is in close proximity to the cleavage loop. Sakai and coworkers found that the H3N2 virus strain A/Guizhou/54/89 required loss of an oligosaccharide at Asn8 of HA1 by N8K mutation to undergo TMPRSS2-independent activation in mice (45). Enhanced HA cleavage and, thus, virulence due to the loss of a steric blocking oligosaccharide side chain have also been described for highly pathogenic avian IAV (46). Taken together, these studies suggest that TMPRSS2-independent cleavage of H3, but not H1, in mice is determined by exposure and accessibility of the cleavage site loop rather than specific HA amino acid motifs. The crystal structure of cleaved IBV HA was solved in 2008 (47), but to date, no structural information is available for the cleavage site of IBV HA0. However, activation of IBV HA by proteases in addition to TMPRSS2 suggests that the cleavage site loop is readily accessible to different proteases. Notably, it seems that the as-yet-undetermined H3/IBV HA-activating protease(s) present in mice and/or HBEC possesses a narrower substrate-binding pocket than TMPRSS2. Endogenous TMPRSS2 activates H1, H3, H7, and IBV HA in airway cells. Furthermore, transiently expressed TMPRSS2 has been shown to cleave IAV HA of almost all subtypes, including H16, which is resistant to cleavage by trypsin, and bat-associated H17 and H18 (43, 48, 49). The role of TMPRSS2 in the activation of low-pathogenicity avian IAV of different HA subtypes as well as bat-associated H17 and H18 in human airway cells remains to be examined.
Previous results showing that TMPRSS2-deficient mice do not develop symptoms of disease after H1N1pdm and H7N9 IAV infection by virtue of inhibition of virus activation and spread along the respiratory tract clearly demonstrate that HA cleavage is a process which could potentially be targeted for drug development to treat influenza infections (9–11). The concept of targeting HA cleavage for influenza treatment was first addressed by Zhirnov and coworkers using the broad-range serine protease inhibitor aprotinin isolated from bovine lungs (50, 51). Aprotinin efficiently inhibited influenza virus activation and multiplication in embryonated chicken eggs, human airway epithelial cells, and lungs of infected mice. In a clinical trial, inhalation of aerosolized aprotinin in patients with influenza and parainfluenza markedly reduced the duration of symptoms without causing side effects (51). To date, the development of protease inhibitors as a preventative and/or therapeutic strategy for influenza treatment has been minimal. This lack of development has been largely due to limited knowledge about the specific physiological functions and characteristics of relevant host cell proteases and concerns about potential side effects resulting from their downregulation. In this study, we provide strong evidence that TMPRSS2 is the major HA-activating protease of IAV in human lower airways and IBV in human lung and that it appears to be a suitable target for the development of inhibitors able to block HA cleavage. This concept is further supported by the observation that a single nucleotide polymorphism in the TMPRSS2 gene resulting in higher protease expression levels correlated with increased susceptibility to H1N1pdm and H7N9 IAV infection and a higher likelihood of severe disease (52). TMPRSS2 has been shown to be sensitive to a number of broad-spectrum serine protease inhibitors, including aprotinin, ovomucoid trypsin inhibitor, and camostat (53–55). Furthermore, a number of potent peptide or peptide-mimetic inhibitors of TMPRSS2 have been described during the last decade and were shown to prevent IAV and IBV activation and multiplication in cell cultures (reviewed in reference 56). However, all of these compounds inhibit numerous trypsin-like proteases and thus may cause various adverse effects. In contrast, specific inhibition of TMPRSS2 during acute influenza infection is expected to be well tolerated. TMPRSS2-deficient mice show no discernible phenotype, indicating functional redundancy or compensation of physiological functions by another protease(s) in the host (27). The development of highly selective peptide or peptide-mimetic protease inhibitors of TMPRSS2 is hampered by the lack of structural information on its catalytic domain. PPMO are highly selective inhibitors of target gene expression. They bind to a complementary sequence in target mRNA and can affect gene expression by steric blockage of translation initiation or pre-mRNA splicing. Based on our findings here, further toxicity and antiviral evaluations of TMPRSS2-specific PPMO in animal models are warranted.
Current measures to control and treat influenza are annual vaccination and the use of inhibitors of the viral neuraminidase (NA) (oseltamivir, zanamivir, and peramivir). Recently, an inhibitor of the cap-dependent endonuclease of the viral polymerase acidic protein (PA) (baloxavir) was approved for treatment of IAV and IBV infections in Japan and the United States. The first-generation antiviral influenza drugs amantadine and rimantadine, both of which block the ion channel protein M2, are no longer recommended due to the widespread antiviral resistance in circulating IAVs. The development of drug resistance to current antiviral treatment options is a major concern in influenza control. Inhibition of HA cleavage by specific protease inhibitors will likely avert the emergence of drug resistance. The C-terminal amino acid sequence of HA1 upstream of the cleavage site differs among different IAV subtypes but is highly conserved within each subtype. The development of escape mutants arising from selective pressure imposed by inhibition of TMPRSS2-mediated HA cleavage is unlikely.
There is accumulating evidence that TMPRSS2 may play a role in other respiratory virus infections. TMPRSS2 has been shown to activate the spike protein (S) of different human coronaviruses (CoV), including severe acute respiratory syndrome CoV (SARS-CoV) and Middle East respiratory syndrome CoV (MERS-CoV), and the fusion protein (F) of human parainfluenza viruses and human metapneumovirus upon coexpression in vitro (reviewed in reference 24). Remarkably, a recent study demonstrated that mice deficient in the expression of TMPRSS2 are protected from severe pathogenesis upon SARS-CoV and MERS-CoV infection, similar to what has been observed for H1N1pdm and H7N9 IAV in TMPRSS2 knockout mice (57). The contribution of TMPRSS2 to the activation of SARS-CoV and MERS-CoV in human respiratory cells, however, remains to be demonstrated.
The present study provides strong evidence that TMPRSS2 is a host cell factor essential for activation and multiplication of IAV in the human lower respiratory tract and IBV in human lung. The development of potent TMPRSS2 inhibitors appears to be a promising approach for therapeutic intervention against influenza virus infections. Further advancement of T-ex5 could lead to a highly selective inhibitor of TMPRSS2 expression with therapeutic potential.
MATERIALS AND METHODS
Cells.
All cell growth and incubations were carried out at 37°C with 5% CO2. Calu-3 human airway epithelial cells (ATCC HTB55) were cultured in Dulbecco’s modified Eagle’s medium (DMEM)–Ham F-12 medium (1:1) (Gibco) supplemented with 10% fetal calf serum (FCS), penicillin, streptomycin, and glutamine, with fresh culture medium replenished every 2 to 3 days. 293F human embryonic kidney cells and Madin-Darby canine kidney II [MDCK(II)] cells were maintained in DMEM supplemented with 10% FCS, antibiotics, and glutamine.
Primary human bronchial epithelial cells (HBEC) (obtained with necessary approvals; originating from patients [n = 3] undergoing thoracic surgery at the University Hospital of Giessen and Marburg) were isolated and cultivated under air-liquid interface conditions to form well-differentiated, pseudostratified cultures as described previously (58). Briefly, isolated HBEC were maintained and expanded (1 passage) in T75 flasks in hormone- and growth factor-supplemented airway epithelial cell growth medium (AEGM) (ready to use; PromoCell). At 80% confluence, cells were detached with 0.05% trypsin-EDTA (Gibco) and seeded onto membrane supports (12-mm Transwell culture inserts with a 0.4-μm pore size; Costar) coated with 0.05 mg collagen from calf skin (Sigma-Aldrich) in ready-to-use AEGM supplemented with 1% penicillin-streptomycin. HBEC were cultured for 2 days until they reached complete confluence. Apical medium was then removed, and basal medium was replaced by a 1:1 mixture of DMEM (Sigma) and ready-to-use AEGM supplemented with 60 ng/ml retinoic acid (Sigma). Cultures were maintained under air-liquid interface conditions by changing the medium in the basal filter chamber three times a week. Fully differentiated 4-week-old cultures were used for the experiments. Mucociliary differentiation was assessed by the presence of beating cilia, and mucus production was observed as a visible mucus layer on the apical surface of the cultures.
Primary human type II alveolar epithelial cells (AECII), also obtained from patients (n = 6) undergoing lung surgery at the University Hospital of Giessen and Marburg, were isolated as described previously (59). A total of 400,000 isolated cells were seeded onto collagen-coated membrane supports (described above) in small airway epithelial cell growth medium (PromoCell) supplemented with hormones, growth factors, 1% penicillin-streptomycin, and 2% FCS. The AECII were cultured for 7 days, with fresh medium in the apical and basal chambers replenished every 2 to 3 days.
Viruses and plasmids.
The influenza viruses used in this study were A/Hamburg/5/09 (H1N1pdm) (Hamburg/H1N1pdm) (kindly provided by Mikhail Matrosovich, Institute of Virology, Marburg, Germany), A/Anhui/1/2013 (H7N9) (Anhui/H7N9) (kindly provided by John McCauley, Division of Virology, MRC National Institute for Medical Research, London, UK), A/FPV/Rostock/34 (H7N1) (FPV/H7N1), B/Malaysia/2506/2004 (Malaysia/B), and B/Massachusetts/71 (Massachusetts/B). Recombinant influenza viruses used were Anhui/H7/PR8 [containing HA of A/Anhui/1/13 (H7N9) along with 7 genes of A/PR8/34 (H1N1) (PR8)] and Aichi/H3N2/PR8 [HA and NA of A/Aichi/2/68 (H3N2) and 6 genes of PR8]. Recombinant IAV were generated by reverse genetics using the pHW2000-based eight-plasmid system described previously by Hoffmann et al. (60). Briefly, 293F cells were cotransfected with eight pHW2000 plasmids containing all eight influenza virus gene segments. Forty-eight hours after transfection, the cell supernatant was treated with 1 μg/ml tosyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Sigma) for 1 h at 37°C and then incubated on MDCK(II) cells for 1 h. Afterwards, cells were washed and maintained in infection medium containing 1 μg/ml TPCK-trypsin for 2 to 3 days. Virus-containing supernatants were analyzed by a plaque assay and sequencing, and stock virus was propagated on MDCK(II) cells as described below.
All IAVs were propagated in MDCK(II) cells in infection medium containing 1 μg/ml TPCK-treated trypsin. IBVs were grown in the allantoic cavity of 11-day-old embryonated chicken eggs. Cell supernatants and allantoic fluid were cleared by low-speed centrifugation and stored at −80°C. Virus stocks grown in MDCK(II) cells with exogenous trypsin as well as in embryonated chicken eggs possess cleaved HA and are infectious. All work with A/Anhui/1/13 (H7N9) was performed under biosafety level 3 (BSL3) conditions. For logistical reasons, infection experiments using AECII were performed under BSL2 conditions, and therefore, the recombinant Anhui/H7/PR8 virus was used instead of A/Anhui/1/13 (H7N9).
pHW2000 plasmids encoding the gene segments of PR8 were kindly provided by Erich Hoffman and Robert Webster (St. Jude Children’s Research Hospital, Memphis, TN, USA) and were described previously (60).
Antibodies.
Polyclonal rabbit sera against HA of A/Anhui/1/13 (H7N9) and HA of IBV were purchased from Sino Biological Inc., and a polyclonal rabbit serum against HA of H3N2 was purchased from GeneTex (catalogue number GTX127363). A monoclonal mouse anti-beta-actin antibody was purchased from Abcam (catalogue number ab6276). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Dako.
Immunofluorescence analyses were performed using the following antibodies: monoclonal mouse anti-IAV nucleoprotein (NP) (Abcam), monoclonal anti-IBV NP (Thermo Fisher Scientific), monoclonal mouse Cy3-labeled anti-beta-tubulin (catalogue number C4585; Sigma), monoclonal mouse anti-acetylated tubulin (catalogue number T7451; Sigma), polyclonal anti-prosurfactant protein C (pro-SP-C) (catalogue number ab40879; Abcam), and polyclonal anti-MUC5AC (catalogue number sc-20118; Santa Cruz). Polyclonal rat serum against TMPRSS2 was generated by DNA vaccination (Genovac; Aldevron). Species-specific fluorescein isothiocyanate (FITC)-conjugated and Alexa Fluor dye-conjugated secondary antibodies were purchased from Dako and Life Technologies, respectively.
PPMO.
Phosphorodiamidate morpholino oligomers (PMO) were synthesized at Gene Tools LLC (Corvallis, OR, USA). PMO sequences (5′ to 3′) were CAGAGTTGGAGCACTTGCTGCCCA for T-ex5 and CCTCTTACCTCAGTTACAATTTATA for scramble. The cell-penetrating peptide (RXR)4 (where R is arginine and X is 6-aminohexanoic acid) was covalently conjugated to the 3′ end of each PMO through a noncleavable linker, to produce peptide-PMO (PPMO), by methods described previously (61).
RNA isolation, RT-PCR analysis of exon skipping, and RT-qPCR analysis of protease transcripts.
For analysis of TMPRSS2 mRNA in experiments where PPMO were used to skip exon 5 in Calu-3 cells, HBEC, and AECII, the cells were incubated with the indicated concentrations of T-ex5 or scramble PPMO or no PPMO in infection medium for 24 h. Total RNA was isolated at the indicated time points using the RNeasy minikit (Qiagen) according to the manufacturer’s protocol. Reverse transcription-PCR (RT-PCR) was carried out with total RNA using the one-step RT-PCR kit (Qiagen) according to the supplier’s protocol. For detection of TMPRSS2 mRNAs, to analyze exon skipping, primers TMPRSS2-108fwd (5′-CTA CGA GGT GCA TCC-3′) and TMPRSS2-1336rev (5′-CCA GAG GCC CTC CAG CGT CAC CCT GGC AA-3′), designed to amplify a full-length PCR product of 1,228 bp from control cells and a shorter PCR fragment of about 1,100 bp (Δex5) from T-ex5-treated cells, were used (17). RT-PCR products were resolved on a 0.8% agarose gel stained with ethidium bromide.
To quantify protease transcripts in the different airway cell systems, RT-quantitative PCR (qPCR) was performed. Total RNA was isolated from Calu-3 cells, HBEC, or AECII using the RNeasy minikit according to the manufacturer’s protocol. Extracted RNA (7.5 ng and 2.5 ng of RNA isolated from Calu-3 cells/HBEC and AECII, respectively) was reverse transcribed and amplified using the Luna universal one-step RT-qPCR kit (New England Biolabs). Primer sequences are available upon request. Transcripts of the housekeeping gene α-tubulin were used for normalizations. The maximum reliable ΔCT value was set to 30. Higher ΔCT levels were defined as protease transcript not being present. Data expressed as a percentage compared to α-tubulin are means ± standard deviations (SD) from three replicates.
To quantify protease transcripts in HBEC upon virus infection, HBEC cultures were inoculated with the indicated virus at a multiplicity of infection (MOI) of 1 for 1 h as described below, the inoculum was then removed, and cells were washed with phosphate-buffered saline (PBS) and further incubated for 24 h. Total RNA was isolated, and 7.5 ng of the RNA was used for RT-qPCR with protease-specific primers as described above. Uninfected HBEC cultures were used as a control. Transcripts of the housekeeping gene α-tubulin served as controls for normalizations. Protease transcript levels of uninfected HBEC were set to a value of 1. Data expressed as n-fold changes over uninfected cells, 2−ΔΔCT, are means ± SD from four replicates.
Infection of cells and multicycle viral replication.
Infection experiments and PPMO treatment of Calu-3 cells were performed using infection medium (DMEM supplemented with 0.1% bovine serum albumin [BSA], glutamine, and antibiotics). Infection and PPMO treatment of HBEC and AECII were performed under serum-free conditions using the airway cell media defined above.
For analysis of multicycle replication kinetics in Calu-3 cells with or without T-ex5 treatment, cells were seeded in 24-well plates and grown to confluence. Cells were incubated with 25 μM T-ex5 PPMO or scramble PPMO in infection medium for 24 h or remained untreated. After removal of preinfection medium, the cells were then inoculated with virus at an MOI of 0.01 to 0.001 in infection medium for 1 h, washed with PBS, and incubated in infection medium without PPMO for 72 h. At 16, 24, 48, and 72 h postinfection (p.i.), supernatants were collected, and viral titers were determined by a plaque assay on MDCK(II) cells with an Avicel overlay as described previously (17).
For analysis of multicycle replication and spread of IAV and IBV in HBEC and AECII grown on membrane supports, the cells were treated apically with 25/30 μM (HBEC/AECII) PPMO in medium for 24 h. Cells were washed with PBS, inoculated apically with virus at an MOI of 0.01 to 0.001 for 1 h, washed with PBS, and incubated in fresh infection medium (apical chamber) containing 30 μM PPMO (AECII) or without medium in the apical chamber and in the absence of PPMO (HBEC) for 24 h. At 24 h postinfection, cells were fixed with methanol-acetone (1:1) on ice and immunostained against viral NP, cilia, or pro-SP-C as described below. To quantify progeny virus released from HBEC grown under air-liquid interface conditions at 24 h p.i., the cells were washed apically with 150 μl medium for 10 min at 37°C with 5% CO2 to harvest released virus particles. To quantify progeny virus released from Aichi/H3N2/PR8- or Malaysia/B-infected AECII at 24 h p.i., cell supernatants were collected prior to fixation of cells. Virus titers in apical washes (HBEC) and cell supernatants (AECII) were determined in PFU per milliliter by a plaque assay.
For analysis of virus growth kinetics in AECII grown on membrane supports, the cells were treated apically with 30 μM T-ex5 PPMO in medium for 24 h. Cells were washed with PBS, inoculated apically with virus at an MOI of 0.005 for 1 h, washed with PBS, and incubated in fresh medium apically with 30 μM T-ex5 or scramble PPMO for 72 h. At 24, 48, and 72 h p.i., supernatants were collected, and viral titers were determined by plaque titration as described above.
To analyze HA cleavage in PPMO-treated Calu-3 cells, HBEC, and AECII, cells were treated with T-ex5 or scramble PPMO for 24 h as described above. Untreated cells were used as controls. Cells were then inoculated with virus at the indicated MOI for 1 h, washed with PBS, and incubated for 24 to 72 in fresh infection medium without PPMO (Calu-3 cells), without medium apically and without PPMO treatment (HBEC), or in fresh medium apically containing 30 μM PPMO (AECII) for 24 to 72 h. Cells were subjected to SDS-PAGE and Western blot analysis as described below.
SDS-PAGE and Western blot analysis.
Cells were washed with PBS, lysed in reducing SDS-PAGE sample buffer, and heated at 95°C for 10 min. Proteins were subjected to SDS-PAGE (12% gel), transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare), and detected by incubation with primary antibodies and species-specific peroxidase-conjugated secondary antibodies. Proteins were visualized using the ChemiDoc XRS+ system with Image Lab software (Bio-Rad).
Animals and murine alveolar epithelial type II cell isolation.
TMPRSS2-deficient mice (strain Tmprss2tm1Psn; mixed C57BL/6J-129 background) were kindly provided by Peter Nelson (Fred Hutchinson Cancer Research Center, Seattle, WA, USA) and have been described previously (27). Homozygous TMPRSS2 knockout mice (Tmprss2−/−) and wild-type littermates (Tmprss2+/+) were bred and housed in the animal facility of the Philipps University Marburg. All protocols involving mice have been approved by the Commission on Animal Protection and Experimentation at the Philipps University Marburg. Preparation of primary AECII was conducted according to previously described procedures (62, 63). Briefly, Tmprss2−/− mice and wild-type littermates were sacrificed using isoflurane and subsequent severing of the vena cava. The chest was opened, and sterile PBS was injected via the right ventricle into the heart until the lungs appeared bloodless. The trachea was exposed and incised with a small opening between cartilage braces to insert a venous catheter of 1.10 by 33 mm without a needle (B. Braun Melsungen), which was fixed with a sterile string. A total of 1 to 1.5 ml of a dispase solution (Corning) was gently injected until lungs were inflated and closed by the addition of prewarmed 1% low-melting-point agarose in sterile distilled water (dH2O). After hardening of the agarose, lungs were dissected and incubated (completely covered) in dispase solution for 45 min. Lungs were mechanically crushed with tweezers in culture dishes filled with 7 ml DMEM supplemented with sterile 25 mM HEPES buffer (Sigma-Aldrich), 0.01% DNase (Serva), 5% glutamine, and 5% penicillin-streptomycin (Gibco), followed by incubation for 10 min at room temperature with shaking. The lung suspension was further homogenized by pipetting (up and down 15 times) and filtering via 100-μm and 40-μm cell strainers (Greiner Bio One). Filtered cells were harvested by centrifugation (140 × g for 10 min at 4°C), the supernatant was carefully discarded, and cells were resuspended in 5 ml growth medium (DMEM supplemented with 10% FCS, antibiotics, and glutamine) and incubated with biotinylated monoclonal rat anti-mouse antibodies (9.5 ng CD16/32, 7.1 ng CD31, and 4.2 ng CD45 [BD Biosciences]) for 30 min at 37°C. Afterwards, 5 ml DMEM, supplemented with 5% glutamine and 5% penicillin-streptomycin, was added to the cells, followed by centrifugation (140 × g for 15 min at 4°C), discarding of the supernatant, and resuspension in 4.5 ml DMEM. Streptavidin-coated magnetic beads (230 μl per lung) washed and equilibrated with PBS to 0.5 ml (Thermo Fisher Scientific) were added and incubated for 30 min at room temperature with shaking. Depletion of leukocytes was performed with a magnetic separator (Thermo Fisher Scientific) for 15 min at room temperature and careful recovery of the cell suspension freed from the magnetic beads. Finally, medium of the cell suspension was changed by centrifugation (140 × g for 15 min at 4°C) to 2 ml growth medium. Isolated cells originating from one lung were equally split, seeded into 6 membrane supports (12-mm Transwell culture inserts with a 0.4-μm pore size; Costar), coated with 0.05 mg collagen type I from calf skin (Sigma-Aldrich) per well, and cultivated under liquid-liquid culture conditions for 24 h in growth medium at 37°C with 5% CO2. Cells were then washed and incubated under air-liquid interface conditions for 24 h. All experiments were performed with cells 48 h after isolation.
Multicycle replication in primary murine alveolar epithelial type II cells.
For analysis of multicycle replication and spread of IAV and IBV in murine AECII, cells were washed apically and basally with PBS and infected apically with virus at an MOI of 0.01 to 0.001 in infection medium for 1 h. The inoculum was removed, and cells were washed and incubated further with infection medium (under liquid-liquid conditions) for 24 h. Cells were fixed, and virus spread was visualized by immunofluorescence staining of IAV or IBV NP as described below.
For quantification of virus multiplication, cells were washed with PBS and infected apically with virus at a low MOI in infection medium for 1 h. The inoculum was removed, and cells were washed and incubated further in infection medium under liquid-liquid conditions for 72 h. At the indicated time points p.i., virus titers in the apical supernatant were determined by a plaque assay on MDCK(II) cells as described above. At 72 h postinfection, cell lysates were subjected to SDS-PAGE and immunoblotting using HA-specific antibodies as described above.
Immunofluorescence staining and microscopy.
Immunofluorescence staining of goblet cells and ciliated cells was performed using HBEC cultures fixed and permeabilized with methanol-acetone (1:1) for 20 min on ice. Cells were then blocked with 2% BSA and incubated with primary antibodies against MUC5AC (mucus) and acetylated tubulin (cilia) for 1 h at room temperature. Cells were washed and incubated with species-specific Alexa Fluor dye-conjugated secondary antibodies for 1 h at room temperature. The nuclei were stained using DAPI (4′,6-diamidino-2-phenylindole). Cell surface immunofluorescence staining of TMPRSS2 was performed using nonpermeabilized and nonfixed HBEC cultures, since the TMPRSS2-specific antibody used in this study was not able to detect TMPRSS2 after paraformaldehyde fixation. Therefore, HBEC cultures were washed with PBS and then incubated with a TMPRSS2-specific antibody for 1 h at 4°C. Subsequently, cells were washed, fixed with 4% paraformaldehyde for 30 min, and then incubated with species-specific FITC-labeled secondary antibody and anti-beta-tubulin Cy3-labeled antibody (cilia) for 1 h at room temperature. For HBEC sections, HBEC cultures were embedded in TissueTek (Sakura), sectioned (4 μm) using a cryostat, and frozen at −80°C. For immunofluorescence staining, sections were rinsed in water to remove the TissueTek, fixed, and permeabilized using methanol-acetone (1:1) for 20 min on ice. Sections were incubated with a TMPRSS2-specific antibody for 1 h, washed, and incubated with a species-specific FITC-labeled antibody and anti-beta-tubulin Cy3-labeled antibody for 1 h. The nuclei were stained using DAPI. All cells were mounted in Fluoroshield (Sigma) and analyzed on a Zeiss Axiophot fluorescence microscope.
To analyze virus spread in HBEC and AECII, the cells were fixed and permeabilized using methanol-acetone (1:1); blocked with a solution containing 2% BSA, 5% glycerol, and 0.2% Tween 20; and incubated with primary antibodies against NP, beta-tubulin (cilia; Cy3-labeled antibody), or pro-SP-C (AECII marker). Subsequently, cells were washed and incubated with species-specific Alexa Fluor dye-conjugated secondary antibodies. DAPI was used as a counterstain. Cells were mounted in Fluoroshield, and images were captured using a fluorescence microscope (Zeiss Axiophot or Zeiss Axiovert 200M) and camera.
Cell viability assay.
Cell viability was assessed by measuring the cellular ATP content using the Cell TiterGlo luminescent cell viability assay (Promega). HBEC and confluent AECII monolayers grown on membrane supports were incubated with the indicated concentrations of PPMO for 24 h. Subsequently, cells were incubated (apical chamber) with the substrate according to the manufacturer’s protocol. Luminescence was measured using a black 96-well plate (Nunc) with a luminometer (Centro LB 960; Berthold Technologies). The absorbance values of PPMO-treated cells were converted to percentages by comparison to untreated control cells, which were set at 100% cell viability.
Statistical analysis.
Statistical analysis was performed by using the freeware R-Studio version 3.4.4. by applying the R package “companion to applied regression” (car) (version 2.1-6). Statistical significance between the different values (ΔCT values) was analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison test according to the number of parameters and groups being compared. A P value of 0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Thomas Damm, Uta Eule, and Guido Schemken for technical assistance and support in generating this work.
This work was funded by Deutsche Forschungsgemeinschaft (DFG) (German Research Foundation), project number 197785619, SFB 1021 to E.B.-F., SFB/TR-84 TP C01 to B.S., and SFB/TR-84 TP C10 to L.S.; Bundesministerium für Bildung und Forschung (BMBF) (German Ministry for Education and Research) e:Med CAPSYS (FKZ 01ZX1604E); JPI-AMR Restrict Pneumo AMR (FKZ 01Kl1702), and ERACoSysMed2-SysMed-COPD (FKZ 031L0140) to B.S.; and the Von-Behring Röntgen Stiftung to E.B.-F. and B.S.
REFERENCES
- 1.Yoon SW, Webby RJ, Webster RG. 2014. Evolution and ecology of influenza A viruses. Curr Top Microbiol Immunol 385:359–375. doi: 10.1007/82_2014_396. [DOI] [PubMed] [Google Scholar]
- 2.Rota PA, Wallis TR, Harmon MW, Rota JS, Kendal AP, Nerome K. 1990. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 175:59–68. doi: 10.1016/0042-6822(90)90186-u. [DOI] [PubMed] [Google Scholar]
- 3.Koutsakos M, Nguyen TH, Barclay WS, Kedzierska K. 2016. Knowns and unknowns of influenza B viruses. Future Microbiol 11:119–135. doi: 10.2217/fmb.15.120. [DOI] [PubMed] [Google Scholar]
- 4.Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng PY, Bandaranayake D, Breiman RF, Brooks WA, Buchy P, Feikin DR, Fowler KB, Gordon A, Hien NT, Horby P, Huang QS, Katz MA, Krishnan A, Lal R, Montgomery JM, Mølbak K, Pebody R, Presanis AM, Razuri H, Steens A, Tinoco YO, Wallinga J, Yu H, Vong S, Bresee J, Widdowson MA. 2012. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis 12:687–695. doi: 10.1016/S1473-3099(12)70121-4. [DOI] [PubMed] [Google Scholar]
- 5.Steinhauer DA. 1999. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 258:1–20. doi: 10.1006/viro.1999.9716. [DOI] [PubMed] [Google Scholar]
- 6.Böttcher-Friebertshäuser E, Garten W, Matrosovich M, Klenk HD. 2014. The hemagglutinin—a determinant of pathogenicity. Curr Top Microbiol Immunol 385:3–34. doi: 10.1007/82_2014_384. [DOI] [PubMed] [Google Scholar]
- 7.Böttcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. 2006. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol 80:9896–9898. doi: 10.1128/JVI.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Böttcher-Friebertshäuser E, Lu Y, Meyer D, Sielaff F, Steinmetzer T, Klenk HD, Garten W. 2012. Hemagglutinin activating host cell proteases provide promising drug targets for the treatment of influenza A and B virus infections. Vaccine 30:7374–7380. doi: 10.1016/j.vaccine.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Hatesuer B, Bertram S, Mehnert N, Bahgat MM, Nelson PS, Pöhlman S, Schughart K. 2013. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog 9:e1003774. doi: 10.1371/journal.ppat.1003774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarnow C, Engels G, Arendt A, Schwalm F, Sediri H, Preuss A, Nelson PS, Garten W, Klenk HD, Gabriel G, Böttcher-Friebertshäuser E. 2014. TMPRSS2 is a host factor that is essential for pneumotropism and pathogenicity of H7N9 influenza A virus in mice. J Virol 88:4744–4751. doi: 10.1128/JVI.03799-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakai K, Ami Y, Tahara M, Kubota T, Anraku M, Abe M, Nakajima N, Sekizuka T, Shirato K, Suzaki Y, Ainai A, Nakatsu Y, Kanou K, Nakamura K, Suzuki T, Komase K, Nobusawa E, Maenaka K, Kuroda M, Hasegawa H, Kawaoka Y, Tashiro M, Takeda M. 2014. The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J Virol 88:5608–5616. doi: 10.1128/JVI.03677-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakai K, Ami Y, Nakajima N, Nakajima K, Kitazawa M, Anraku M, Takayama I, Sangsriratanakul N, Komura M, Sato Y, Asanuma H, Takashita E, Komase K, Takehara K, Tashiro M, Hasegawa H, Odagiri T, Takeda M. 2016. TMPRSS2 independency for haemagglutinin cleavage in vivo differentiates influenza B virus from influenza A virus. Sci Rep 6:29430. doi: 10.1038/srep29430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moulton HM, Moulton JD. 2010. Morpholinos and their peptide conjugates: therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim Biophys Acta 1798:2296–2303. doi: 10.1016/j.bbamem.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 14.Ge Q, Pastey M, Kobasa D, Puthavathana P, Lupfer C, Bestwick RK, Iversen PL, Chen J, Stein DA. 2006. Inhibition of multiple subtypes of influenza A virus in cell cultures with morpholino oligomers. Antimicrob Agents Chemother 50:3724–3733. doi: 10.1128/AAC.00644-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gabriel G, Nordmann A, Stein DA, Iversen PL, Klenk HD. 2008. Morpholino oligomers targeting the PB1 and NP genes enhance the survival of mice infected with highly pathogenic influenza A H7N7 virus. J Gen Virol 89:939–948. doi: 10.1099/vir.0.83449-0. [DOI] [PubMed] [Google Scholar]
- 16.Lupfer C, Stein DA, Mourich DV, Tepper SE, Iversen PL, Pastey M. 2008. Inhibition of influenza A H3N8 virus infections in mice by morpholino oligomers. Arch Virol 153:929–937. doi: 10.1007/s00705-008-0067-0. [DOI] [PubMed] [Google Scholar]
- 17.Böttcher-Friebertshäuser E, Stein DA, Klenk HD, Garten W. 2011. Inhibition of influenza virus infection in human airway cell cultures by an antisense peptide-conjugated morpholino oligomer targeting the hemagglutinin-activating protease TMPRSS2. J Virol 85:1554–1562. doi: 10.1128/JVI.01294-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajsbaum R. 2017. Intranasal delivery of peptide-morpholinos to knockdown influenza host factors in mice. Methods Mol Biol 1565:191–199. doi: 10.1007/978-1-4939-6817-6_16. [DOI] [PubMed] [Google Scholar]
- 19.Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, Yángüez E, Andenmatten D, Pache L, Manicassamy B, Albrecht RA, Gonzalez MG, Nguyen Q, Brass A, Elledge S, White M, Shapira S, Hacohen N, Karlas A, Meyer TF, Shales M, Gatorano A, Johnson JR, Jang G, Johnson T, Verschueren E, Sanders D, Krogan N, Shaw M, König R, Stertz S, García-Sastre A, Chanda SK. 2015. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18:723–735. doi: 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soonthornvacharin S, Rodriguez-Frandsen A, Zhou Y, Galvez F, Huffmaster NJ, Tripathi S, Balasubramaniam VR, Inoue A, de Castro E, Moulton H, Stein DA, Sánchez-Aparicio MT, De Jesus PD, Nguyen Q, König R, Krogan NJ, García-Sastre A, Yoh SM, Chanda SK. 2017. Systems-based analysis of RIG-I-dependent signalling identifies KHSRP as an inhibitor of RIG-I receptor activation. Nat Microbiol 2:17022. doi: 10.1038/nmicrobiol.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai SH, Stein DA, Guerrero-Plata A, Liao SL, Ivanciuc T, Hong C, Iversen PL, Casola A, Garofalo RP. 2008. Inhibition of respiratory syncytial virus infections with morpholino oligomers in cell cultures and in mice. Mol Ther 16:1120–1128. doi: 10.1038/mt.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Riel D, Munster VJ, de Wit E, Rimmelzwaan GF, Fouchier RA, Osterhaus AD, Kuiken T. 2007. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am J Pathol 171:1215–1223. doi: 10.2353/ajpath.2007.070248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinheimer VK, Becher A, Tönnies M, Holland G, Knepper J, Bauer TT, Schneider P, Neudecker J, Rückert JC, Szymanski K, Temmesfeld-Wollbrueck B, Gruber AD, Bannert N, Suttorp N, Hippenstiel S, Wolff T, Hocke AC. 2012. Influenza A viruses target type II pneumocytes in the human lung. J Infect Dis 206:1685–1694. doi: 10.1093/infdis/jis455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Böttcher-Friebertshäuser E. 2018. Membrane-anchored serine proteases: host cell factors in proteolytic activation of viral glycoproteins, p 153–203. In Böttcher-Friebertshäuser E, Garten W, Klenk H (ed), Activation of viruses by host proteases. Springer, Cham, Switzerland. [Google Scholar]
- 25.Magnen M, Gueugnon F, Guillon A, Baranek T, Thibault VC, Petit-Courty A, de Veer SJ, Harris J, Humbles AA, Si-Tahar M, Courty Y. 2017. Kallikrein-related peptidase 5 contributes to H3N2 influenza virus infection in human lungs. J Virol 91:e00421-17. doi: 10.1128/JVI.00421-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knepper J, Schierhorn KL, Becher A, Budt M, Tönnies M, Bauer TT, Schneider P, Neudecker J, Rückert JC, Gruber AD, Suttorp N, Schweiger B, Hippenstiel S, Hocke AC, Wolff T. 2013. The novel human influenza A (H7N9) virus is naturally adapted to efficient growth in human lung tissue. mBio 4:e00601-13. doi: 10.1128/mBio.00601-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim TS, Heinlein C, Hackman RC, Nelson PS. 2006. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol Cell Biol 26:965–975. doi: 10.1128/MCB.26.3.965-975.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamilton BS, Whittaker GR. 2013. Cleavage activation of human-adapted influenza virus subtypes by kallikrein-related peptidases 5 and 12. J Biol Chem 288:17399–17407. doi: 10.1074/jbc.M112.440362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magnen M, Elsässer BM, Zbodakova O, Kasparek P, Gueugnon F, Petit-Courty A, Sedlacek R, Goettig P, Courty Y. 2018. Kallikrein-related peptidase 5 and seasonal influenza viruses, limitations of the experimental models for activating proteases. Biol Chem 399:1053–1064. doi: 10.1515/hsz-2017-0340. [DOI] [PubMed] [Google Scholar]
- 30.Puente XS, Sánchez LM, Overall CM, López-Otín C. 2003. Human and mouse proteases: a comparative genomic approach. Nat Rev Genet 4:544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- 31.Kühn N, Bergmann S, Kösterke N, Lambertz RL, Keppner A, van den Brand JM, Pöhlmann S, Weiß S, Hummler E, Hatesuer B, Schughart K. 2016. The proteolytic activation of (H3N2) influenza A virus hemagglutinin is facilitated by different type II transmembrane serine proteases. J Virol 90:4298–4307. doi: 10.1128/JVI.02693-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertram S, Heurich A, Lavender H, Gierer S, Danisch S, Perin P, Lucas JM, Nelson PS, Pöhlmann S, Soilleux EJ. 2012. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS One 7:e35876. doi: 10.1371/journal.pone.0035876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peitsch C, Klenk HD, Garten W, Böttcher-Friebertshäuser E. 2014. Activation of influenza A viruses by host proteases from swine airway epithelium. J Virol 88:282–291. doi: 10.1128/JVI.01635-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zmora P, Molau-Blazejewska P, Bertram S, Walendy-Gnirß K, Nehlmeier I, Hartleib A, Moldenhauer AS, Konzok S, Dehmel S, Sewald K, Brinkmann C, Curths C, Knauf S, Gruber J, Mätz-Rensing K, Dahlmann F, Braun A, Pöhlmann S. 2017. Non-human primate orthologues of TMPRSS2 cleave and activate the influenza virus hemagglutinin. PLoS One 12:e0176597. doi: 10.1371/journal.pone.0176597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quesada V, Velasco G, Puente XS, Warren WC, López-Otín C. 2010. Comparative genomic analysis of the zebra finch degradome provides new insights into evolution of proteases in birds and mammals. BMC Genomics 11:220. doi: 10.1186/1471-2164-11-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noma K, Kiyotani K, Kouchi H, Fujii Y, Egi Y, Tanaka K, Yoshida T. 1998. Endogenous protease-dependent replication of human influenza viruses in two MDCK cell lines. Arch Virol 143:1893–1909. doi: 10.1007/s007050050428. [DOI] [PubMed] [Google Scholar]
- 37.Lugovtsev VY, Melnyk D, Weir JP. 2013. Heterogeneity of the MDCK cell line and its applicability for influenza virus research. PLoS One 8:e75014. doi: 10.1371/journal.pone.0075014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kesic MJ, Meyer M, Bauer R, Jaspers I. 2012. Exposure to ozone modulates human airway protease/antiprotease balance contributing to increased influenza A infection. PLoS One 7:e35108. doi: 10.1371/journal.pone.0035108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yasuoka S, Ohnishi T, Kawano S, Tsuchihashi S, Ogawara M, Masuda K, Yamaoka K, Takahashi M, Sano T. 1997. Purification, characterization, and localization of a novel trypsin-like protease found in the human airway. Am J Respir Cell Mol Biol 16:300–308. doi: 10.1165/ajrcmb.16.3.9070615. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC. 1998. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 95:409–417. doi: 10.1016/s0092-8674(00)81771-7. [DOI] [PubMed] [Google Scholar]
- 41.Stevens J, Corper AL, Basler CF, Taubenberger JK, Palese P, Wilson IA. 2004. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 303:1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- 42.Lu X, Shi Y, Gao F, Xiao H, Wang M, Qi J, Gao GF. 2012. Insights into avian influenza virus pathogenicity: the hemagglutinin precursor HA0 of subtype H16 has an alpha-helix structure in its cleavage site with inefficient HA1/HA2 cleavage. J Virol 86:12861–12870. doi: 10.1128/JVI.01606-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galloway SE, Reed ML, Russell CJ, Steinhauer DA. 2013. Influenza HA subtypes demonstrate divergent phenotypes for cleavage activation and pH of fusion: implications for host range and adaptation. PLoS Pathog 9:e1003151. doi: 10.1371/journal.ppat.1003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lambertz RLO, Pippel J, Gerhauser I, Kollmus H, Anhlan D, Hrincius ER, Krausze J, Kühn N, Schughart K. 2018. Exchange of amino acids in the H1-haemagglutinin to H3 residues is required for efficient influenza A virus replication and pathology in Tmprss2 knock-out mice. J Gen Virol 99:1187–1198. doi: 10.1099/jgv.0.001128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakai K, Sekizuka T, Ami Y, Nakajima N, Kitazawa M, Sato Y, Nakajima K, Anraku M, Kubota T, Komase K, Takehara K, Hasegawa H, Odagiri T, Tashiro M, Kuroda M, Takeda M. 2015. A mutant H3N2 influenza virus uses an alternative activation mechanism in TMPRSS2 knockout mice by loss of an oligosaccharide in the hemagglutinin stalk region. J Virol 89:5154–5158. doi: 10.1128/JVI.00124-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawaoka Y, Naeve CW, Webster RG. 1984. Is virulence of H5N2 influenza viruses in chickens associated with loss of carbohydrate from the hemagglutinin? Virology 139:303–316. doi: 10.1016/0042-6822(84)90376-3. [DOI] [PubMed] [Google Scholar]
- 47.Wang Q, Cheng F, Lu M, Tian X, Ma J. 2008. Crystal structure of unliganded influenza B virus hemagglutinin. J Virol 82:3011–3020. doi: 10.1128/JVI.02477-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferrara F, Molesti E, Böttcher-Friebertshäuser E, Cattoli G, Corti D, Scott S, Temperton N. 2013. The human transmembrane protease serine 2 is necessary for the production of group 2 influenza A virus pseudotypes. J Mol Genet Med 7:309–314. [PMC free article] [PubMed] [Google Scholar]
- 49.Hoffmann M, Krüger N, Zmora P, Wrensch F, Herrler G, Pöhlmann S. 2016. The hemagglutinin of bat-associated influenza viruses is activated by TMPRSS2 for pH-dependent entry into bat but not human cells. PLoS One 11:e0152134. doi: 10.1371/journal.pone.0152134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ovcharenko AV, Zhirnov OP. 1994. Aprotinin aerosol treatment of influenza and paramyxovirus bronchopneumonia of mice. Antiviral Res 23:107–118. doi: 10.1016/0166-3542(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 51.Zhirnov OP, Klenk HD, Wright PF. 2011. Aprotinin and similar protease inhibitors as drugs against influenza. Antiviral Res 92:27–36. doi: 10.1016/j.antiviral.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 52.Cheng Z, Zhou J, To KK, Chu H, Li C, Wang D, Yang D, Zheng S, Hao K, Bossé Y, Obeidat M, Brandsma CA, Song YQ, Chen Y, Zheng BJ, Li L, Yuen KY. 2015. Identification of TMPRSS2 as a susceptibility gene for severe 2009 pandemic A(H1N1) influenza and A(H7N9) influenza. J Infect Dis 212:1214–1221. doi: 10.1093/infdis/jiv246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Böttcher E, Freuer C, Steinmetzer T, Klenk HD, Garten W. 2009. MDCK cells that express proteases TMPRSS2 and HAT provide a cell system to propagate influenza viruses in the absence of trypsin and to study cleavage of HA and its inhibition. Vaccine 27:6324–6329. doi: 10.1016/j.vaccine.2009.03.029. [DOI] [PubMed] [Google Scholar]
- 54.Shirato K, Matsuyama S, Ujike M, Taguchi F. 2011. Role of proteases in the release of porcine epidemic diarrhea virus from infected cells. J Virol 85:7872–7880. doi: 10.1128/JVI.00464-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shirato K, Kawase M, Matsuyama S. 2013. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol 87:12552–12561. doi: 10.1128/JVI.01890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steinmetzer T, Hardes K. 2018. The antiviral potential of host protease inhibitors, p 279–325. In Böttcher-Friebertshäuser E, Garten W, Klenk H (ed), Activation of viruses by host proteases. Springer, Cham, Switzerland. [Google Scholar]
- 57.Iwata-Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N. 2019. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol 93:e01815-18. doi: 10.1128/JVI.01815-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gray TE, Guzman K, Davis CW, Abdullah LH, Nettesheim P. 1996. Mucociliary differentiation of serially passaged normal human tracheobronchial epithelial cells. Am J Respir Cell Mol Biol 4:104–112. doi: 10.1165/ajrcmb.14.1.8534481. [DOI] [PubMed] [Google Scholar]
- 59.Mao P, Wu S, Li J, Fu W, He W, Liu X, Slutsky AS, Zhang H, Li Y. 2015. Human alveolar epithelial type II cells in primary culture. Physiol Rep 3:e12288. doi: 10.14814/phy2.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A 97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abes S, Moulton HM, Clair P, Prevot P, Youngblood DS, Wu RP, Iversen PL, Lebleu B. 2006. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J Control Release 116:304–313. doi: 10.1016/j.jconrel.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 62.Herold S, von Wulffen W, Steinmueller M, Pleschka S, Kuziel WA, Mack M, Srivastava M, Seeger W, Maus UA, Lohmeyer J. 2006. Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: impact of chemokines and adhesion molecules. J Immunol 177:1817–1824. doi: 10.4049/jimmunol.177.3.1817. [DOI] [PubMed] [Google Scholar]
- 63.Corti M, Brody AR, Harrison JH. 1996. Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol 14:309–315. doi: 10.1165/ajrcmb.14.4.8600933. [DOI] [PubMed] [Google Scholar]