

The HIV-1 genome contains a gene expressed in the opposite, or antisense, direction to all other genes. The protein product of this antisense gene, called ASP, is poorly characterized, and its role in viral replication remains unknown. We provide evidence that the antisense protein, ASP, of HIV-1 is found within the cell nucleus in unstimulated cells. In addition, we show that after PMA treatment, ASP exits the nucleus and localizes on the cell membrane. Moreover, we demonstrate that ASP is present on the surfaces of viral particles. Altogether, our studies identify ASP as a new structural component of HIV-1 and show that ASP is an accessory protein that promotes viral replication. The presence of ASP on the surfaces of both infected cells and viral particles might be exploited therapeutically.
KEYWORDS: HIV-1, antisense protein ASP, cell surface protein, viral envelope protein
ABSTRACT
The negative strand of HIV-1 encodes a highly hydrophobic antisense protein (ASP) with no known homologs. The presence of humoral and cellular immune responses to ASP in HIV-1 patients indicates that ASP is expressed in vivo, but its role in HIV-1 replication remains unknown. We investigated ASP expression in multiple chronically infected myeloid and lymphoid cell lines using an anti-ASP monoclonal antibody (324.6) in combination with flow cytometry and microscopy approaches. At baseline and in the absence of stimuli, ASP shows polarized subnuclear distribution, preferentially in areas with low content of suppressive epigenetic marks. However, following treatment with phorbol 12-myristate 13-acetate (PMA), ASP translocates to the cytoplasm and is detectable on the cell surface, even in the absence of membrane permeabilization, indicating that 324.6 recognizes an ASP epitope that is exposed extracellularly. Further, surface staining with 324.6 and anti-gp120 antibodies showed that ASP and gp120 colocalize, suggesting that ASP might become incorporated in the membranes of budding virions. Indeed, fluorescence correlation spectroscopy studies showed binding of 324.6 to cell-free HIV-1 particles. Moreover, 324.6 was able to capture and retain HIV-1 virions with efficiency similar to that of the anti-gp120 antibody VRC01. Our studies indicate that ASP is an integral protein of the plasma membranes of chronically infected cells stimulated with PMA, and upon viral budding, ASP becomes a structural protein of the HIV-1 envelope. These results may provide leads to investigate the possible role of ASP in the virus replication cycle and suggest that ASP may represent a new therapeutic or vaccine target.
IMPORTANCE The HIV-1 genome contains a gene expressed in the opposite, or antisense, direction to all other genes. The protein product of this antisense gene, called ASP, is poorly characterized, and its role in viral replication remains unknown. We provide evidence that the antisense protein, ASP, of HIV-1 is found within the cell nucleus in unstimulated cells. In addition, we show that after PMA treatment, ASP exits the nucleus and localizes on the cell membrane. Moreover, we demonstrate that ASP is present on the surfaces of viral particles. Altogether, our studies identify ASP as a new structural component of HIV-1 and show that ASP is an accessory protein that promotes viral replication. The presence of ASP on the surfaces of both infected cells and viral particles might be exploited therapeutically.
INTRODUCTION
Retroviral gene expression has been mostly limited to a primary transcript responsible for the expression of numerous genes subsequent to a series of alternative splicing events and frameshift reading (1). However, many studies have described the presence of conserved open reading frames (ORF) in the complementary strand of human and animal retroviruses, suggesting the existence of viral antisense mRNAs expressed from the 3′ long terminal repeat (LTR). The most intensely investigated retroviral antisense protein is the human T-cell leukemia virus type 1 (HTLV-1) bZIP (HBZ) factor (for a comprehensive review, see reference 2). HBZ is a c-Fos-like nuclear factor that attenuates the activation of AP-1 and downregulates viral transcription (2–5). In addition to maintaining HTLV-1 latency, HBZ promotes T cell proliferation and inhibits apoptosis and autophagy (2). Similarly, HTLV-2, -3, and -4 encode antisense proteins (called APH-2, -3, and -4, respectively, for antisense protein of HTLV-2, -3, and -4) (6–9). All three proteins lack the bZIP consensus found in HBZ, but they block 5′ LTR transactivation driven by their respective Tax proteins (7). In addition, while APH-2 and APH-4 display nuclear localization, APH-3 is both nuclear and cytoplasmic (7). Finally, the presence of antisense proteins has also been predicted in the genomes of feline immunodeficiency virus (FIV) and bovine leukemia virus (BLV) (10, 11).
The existence of an ORF in the negative strand of the human immunodeficiency virus type 1 (HIV-1) genome was first predicted 30 years ago (12). The ORF is located at the gp120/gp41 junction of the env gene and encodes a highly hydrophobic protein of ∼189 amino acids (aa), possibly associated with membrane structures (12). Indeed, the product of the HIV-1 antisense ORF (termed antisense protein [ASP]) presents two putative transmembrane domains, two closely spaced cysteine triplets, and an SH3 domain-binding motif (PXXPXXP, where P is proline and X is any amino acid) (13). Several lines of evidence suggest that ASP is expressed in vivo in the course of HIV-1 infection. First, an intact ORF encoding ASP is found only in HIV-1 strains belonging to group M, but not other groups (N, O, or P). This indicates that ASP was created de novo with the emergence of group M, which is responsible for the worldwide pandemic (14). Second, computer simulation and modeling studies showed that preservation of the asp ORF in group M HIV-1 (i.e., maintenance of the start codon and avoidance of early stop codons) did not occur by chance, but rather, under selective pressure, which suggests a role—albeit nonessential—of the protein in viral spread (14). Finally, several reports have documented the presence of humoral and cellular immune responses to ASP in the peripheral blood of HIV-1-infected individuals (15–17).
Defining the role of ASP in HIV-1 replication has remained elusive. Unlike its counterparts encoded by other retroviruses, ASP has no known homologs that might help shed light on its function (14). Several reports, including our own, have shown that antisense transcripts produced by HIV-1 inhibit viral transcription (18–23), but this effect does not require expression of the ASP protein that they encode (18–20, 22). Possible clues about the function of ASP could come from its patterns of expression, subcellular localization, and intracellular dynamics. In keeping with its hydrophobicity, previous reports found that ASP is associated with various cellular membrane structures and possibly with viral particles (13, 24). However, these studies were based on the analysis of a single cell type, utilized a single technique, or relied on transient-transfection approaches.
Here, we used a combination of flow cytometry and microscopy techniques to track the expression and subcellular localization of ASP in a panel of seven lymphoid and two myeloid cell lines chronically infected with full-length, replication-competent HIV-1, both at baseline and after stimulation with phorbol 12-myristate 13-acetate (PMA). Our results show that ASP dwells in the nuclei of unstimulated cells, displaying a polarized, nonhomogeneous distribution. In contrast, we provide evidence that after PMA treatment, ASP translocates to the cytoplasm and is expressed on the plasma membrane. Moreover, after viral budding and release, ASP is incorporated into the viral envelope and becomes a structural protein of the HIV-1 virion. Altogether, our results suggest that ASP may play a role in HIV-1 replication and/or spread and identify ASP as a possible new target for therapeutic and vaccine interventions.
RESULTS
Nuclear expression of ASP in unstimulated, chronically infected lymphoid and myeloid cell lines.
Previous reports investigating the expression of ASP were limited to the use of a single cell line, transient-overexpression techniques, or acute infection (13, 24–26). The ability to rely on a specific monoclonal antibody (MAb) against ASP (clone 324.6) (see Fig. S1 in the supplemental material) allowed us to circumvent these limitations and to systematically investigate ASP expression in multiple cell lines, using multiple techniques, and during several phases of the HIV-1 replication cycle. For our studies, we employed nine different chronically infected cell lines: two of myeloid origin (U1 and OM-10.1) (27–29) and seven of lymphoid origin (J1.1, ACH-2, 8E5, H9-IIIB, H9-CC, H9-MN, and H9-RF) (30–35). It should be noted that U1, OM-10.1, J1.1, ACH-2, 8E5, and H9-IIIB are infected with the same HIV-1 strain (HIV-1LAV/IIIB). The amino acid sequence of the ASP epitope recognized by the 324.6 MAb (aa 97 to 110) is identical to that of the immunogen peptide (see Materials and Methods). The cell lines H9-CC, H9-MN, and H9-RF are infected with HIV-1 strains (CC, MN, and RF) in which the ASP epitope recognized by the 324.6 MAb diverges from the immunogen peptide by 3/14, 2/14, and 4/14 amino acids, respectively. In all three cases, two of the diverging amino acids are the last two C-terminal residues of the 14-mer sequence. The parental uninfected cell lines U937, HL-60, Jurkat, and H9 were utilized as controls. In addition, background staining in flow cytometry and microscopy was minimized by directly conjugating the anti-ASP 324.6 MAb to Alexa Fluor 647 (AF647). After conjugation, we purified the antibody (Ab) from unreacted fluorescent dye, and we assessed the dye-to-antibody ratio (routinely 5:1, except where specified).
We carried out cell surface and intracellular staining (with two different fixation/permeabilization techniques) of all chronically infected cell lines and their parental uninfected lines using AF647-labeled anti-ASP 324.6 MAb and isotype IgG control Abs. We then analyzed the samples by flow cytometry. ASP was detectable in all the chronically infected cell lines—albeit at variable levels—only after permeabilization with buffers that allow the detection of nuclear proteins (Fig. 1). On the other hand, we could not detect ASP on the cell surface or within the cytoplasm of any of the cell lines analyzed (data not shown). No fluorescence signal above the background could be detected in the nuclei of the uninfected parental cell lines (Fig. 1).
FIG 1.

Intranuclear flow cytometry analysis of ASP expression in multiple cell lines. Chronically infected myeloid (U1 and OM-10.1) and lymphoid (J1.1, ACH2, 8E5, H9/IIIB, H9/CC, H9/MN, and H9/RF) cell lines and their uninfected parental myeloid (U937 and HL-60) and lymphoid (Jurkat, A3.01, and H9) cell lines were fixed and permeabilized with a FoxP3 staining kit to allow detection of nuclear proteins. Then, the cells were stained with the anti-ASP 324.6 (light-blue curve and line) or a control mouse IgG (open curve with red line) labeled with Alexa Fluor 647. Finally, the cells were washed, fixed, and analyzed by flow cytometry. All the chronically infected cell lines analyzed showed nuclear expression of ASP to varying degrees. The results shown are representative of several independent experiments. The mean fluorescence intensity (MFI) of samples stained with the 324.6 MAb is reported in each panel.
Since antisense transcription of HIV-1 is independent of Tat and is driven exclusively by cellular transcription factors (21, 36), we utilized U1 cells to investigate whether the rate of cell division influenced the expression levels of ASP. Thus, U1 cells were maintained for several days at either low or high cell density to induce high or low cell division rates, respectively. These experiments were also carried out with a clone of U1 cells isolated by limiting dilution (U1 clone 8 [U1C8]), which was found to express ASP at higher levels than the original U1 cell line. ASP expression was analyzed by intranuclear flow cytometry in U1 and U1C8 cells cultured under the two conditions described above. We found that ASP expression was markedly higher in cultures maintained at low density and undergoing high cell division rates (see Fig. S2 in the supplemental material).
These results show that ASP expression is a common feature of all the chronically infected cell lines we analyzed and that in the absence of external stimuli ASP is exclusively a nuclear protein.
Subnuclear distribution of ASP in unstimulated, chronically infected cell lines.
The highly hydrophobic nature of ASP suggests that it might be tightly associated with cellular membranes. We performed confocal microscopy studies to investigate whether ASP is expressed in the nuclear membranes of unstimulated U1C8 cells. In addition to the 324.6 MAb, cells were also stained with antibodies against lamin B receptor (LBR), an integral protein of the nuclear envelope (37), and with the nucleic acid dye DAPI (4′,6-diamidino-2-phenylindole). We found that ASP does not colocalize with LBR, and thus, it is not associated with the nuclear membrane. Rather, ASP is present underneath the nuclear envelope, within the nucleoplasm, and interspersed with the nucleic acids (Fig. 2). Moreover, ASP displays a nonhomogeneous and polarized distribution (Fig. 2A and D). A clear intranuclear, nonhomogeneous, and polarized distribution of ASP was also observed in OM-10.1 cells (data not shown).
FIG 2.
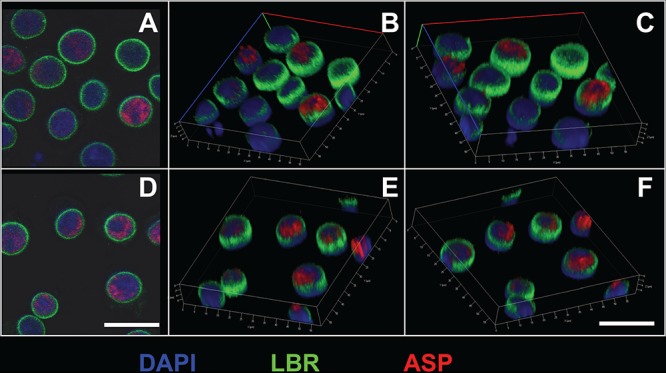
Intranuclear confocal microscopy analysis of ASP expression in unstimulated U1C8 cells. Cells were fixed and permeabilized with a FoxP3 staining kit to allow detection of nuclear proteins. Then, the cells were stained with Alexa Fluor 647-labeled anti-ASP 324.6 (red), FITC-labeled anti-LBR (lamin B receptor) (green), and DAPI (blue). Finally, the cells were washed, fixed, and analyzed by confocal microscopy as described in Materials and Methods. The cells display a nonuniform, polarized nuclear distribution of ASP. Bars, 5 μm. Panels B, C, E, and F are three-dimensional representations reconstructed from z-stack images 0.7 μm thick. The results shown are representative of at least three independent experiments.
The organization and distribution of the genetic material within the nucleus is highly regulated (38, 39). Indeed, heterochromatin localizes in the outer layer of the nucleus, whereas euchromatin is found at the center of the nucleus, as well as in the outer layer in close proximity to the nucleopores. Heterochromatin shows a high content of suppressive epigenetic marks, such as histone H3 trimethylated on lysine 27 (H3K27me3). To investigate whether the distribution of ASP within the nucleus correlated with that of active or inactive chromatin, we performed nuclear staining of U1C8 cells with MAbs recognizing ASP and H3K27me3 and with the nucleic acid dye DAPI. Our results showed that ASP is excluded from areas of the nucleus containing transcriptionally silent chromatin and identified by high levels of H3K27me3 and stronger DAPI staining (indicating more tightly packed chromatin) (Fig. 3; see Movie S1 in the supplemental material).
FIG 3.
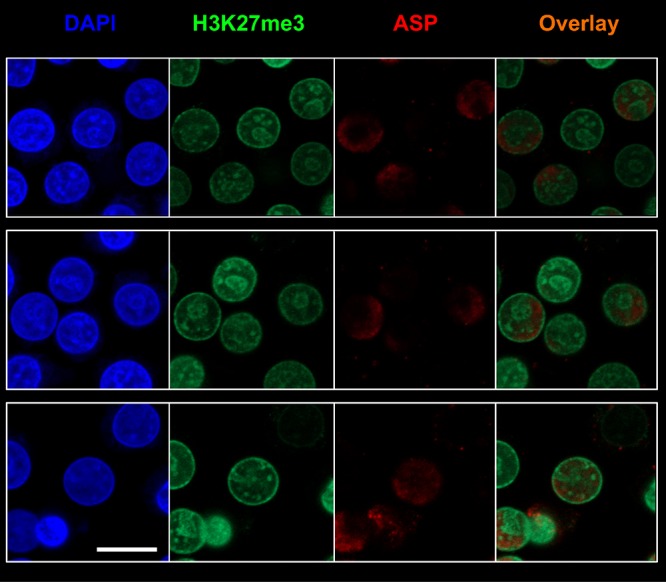
ASP accumulates in nuclear areas lacking suppressive epigenetic marks in unstimulated infected U1C8 cells. Chronically infected U1C8 cells were fixed and permeabilized with a FoxP3 staining kit to allow detection of nuclear proteins. Then, the cells were stained with Alexa Fluor 647-labeled anti-ASP 324.6 (red), Alexa Fluor 488-labeled anti-H3K27me3 (green), and DAPI (blue). Finally, the cells were washed, fixed, and analyzed by confocal microscopy. The overlay of the images taken in the green (H3K27me3) and red (ASP) channels shows that the subnuclear distribution of the two markers is mutually exclusive, indicating that ASP is enriched in areas of the nucleus that contain actively transcribed chromatin. Bar, 5 μm. The results shown are representative of at least three independent experiments.
The confocal microscopy images described above appear to show that ASP is not expressed in every cell, but this could also be due to the polarized distribution of ASP, which in some cells may be visible on a different focal plane than the one shown. To test this, we took serial images of the same cell over multiple focal planes (a z-stack) in order to construct detailed three-dimensional (3D) figures. As shown in Fig. S3 in the supplemental material, the nucleus at the center of the field is clearly positive for ASP from image 1 through image 18, after which ASP is no longer detectable. These images indicate that ASP visibility is dependent on the focal plane being analyzed. Confirmation of this is given by the nucleus visible in the lower-left corner of the field: there, ASP is visible from the appearance of the nucleus (image 6) and throughout the remainder of the z-stack. However, ASP is present exclusively in the lower-left half of the nucleus, not the upper-right half (see Fig. S3).
The confocal images reveal that, despite its hydrophobicity and the predicted transmembrane domains, ASP is not associated with the nuclear envelope, but rather localizes inside the nucleus, displays a polarized distribution, and is present preferentially in areas containing actively transcribed chromatin.
Cytoplasmic translocation and cell surface expression of ASP in PMA-treated, chronically infected cells.
Next, we sought to investigate the subcellular localization of ASP following PMA stimulation. U1C8 cells were cultured in the presence of PMA for 12 to 24 h to reactivate HIV-1 expression; then, we performed intracellular staining with MAbs to ASP and to H3K27me3, and we analyzed the samples by confocal microscopy. We found that upon HIV-1 reactivation, a significant proportion of ASP translocated from the nucleus to the cytoplasm (Fig. 4; see Movies S2 and S3 in the supplemental material). Confocal microscopy analysis of PMA-treated cells stained with MAbs to ASP and HIV-1 p24 showed that the presence of both proteins in the cytoplasm (Fig. 5). This can be further appreciated in the z-stack analyses of these images (see Fig. S4 in the supplemental material).
FIG 4.

ASP exits the nucleus and localizes in the cytoplasm of PMA-treated U1C8 cells. Chronically infected U1C8 cells were treated with PMA to reactivate HIV-1 expression, as described in Materials and Methods. After fixation and permeabilization with a BD Cytofix/Cytoperm kit to allow detection of cytoplasmic proteins, the cells were stained with Alexa Fluor 647-labeled anti-ASP 324.6 (red), Alexa Fluor 488-labeled anti-H3K27me3 (green), and DAPI (blue). Finally, the cells were washed, fixed, and analyzed by confocal microscopy. The overlay of the images taken in the green (H3K27me3) and red (ASP) channels shows that PMA stimulation caused ASP to exit the nucleus and to translocate into the cytoplasm. Bar, 10 μm (panel A, low magnification) and 5 μm (panel B, high magnification). The results shown are representative of at least three independent experiments.
FIG 5.
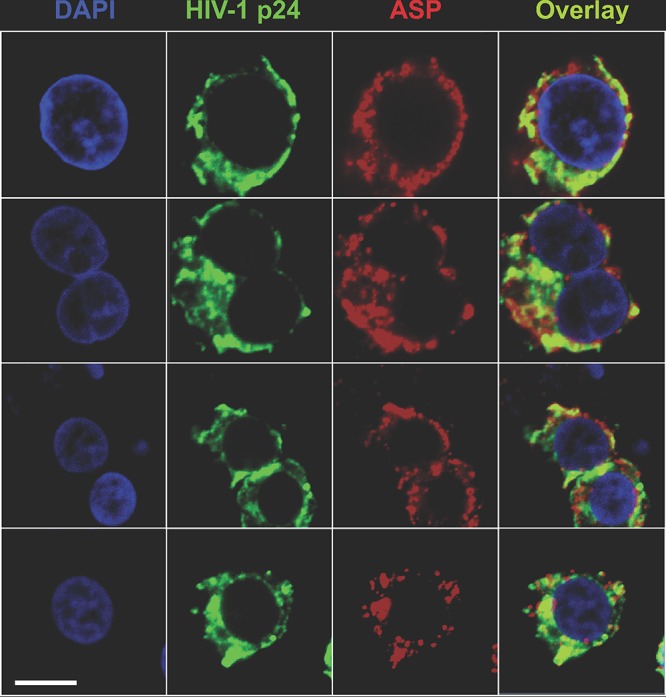
ASP and HIV-1 p24 colocalize in the cytoplasm of PMA-treated U1C8 cells. Chronically infected U1C8 cells were treated with PMA to reactivate HIV-1 expression as described in Materials and Methods. After fixation and permeabilization with a BD Cytofix/Cytoperm kit to allow detection of cytoplasmic proteins, the cells were stained with Alexa Fluor 647-labeled anti-ASP 324.6 (red), Alexa Fluor 488-labeled anti-HIV-1 p24 (green), and DAPI (blue). Finally, the cells were washed, fixed, and analyzed by confocal microscopy. The overlay of the images taken in the green (HIV-1 p24) and red (ASP) channels shows that PMA treatment induced HIV-1 expression, as shown by HIV-1 p24 presence in the cytoplasm. In addition, ASP and HIV-1 p24 present a significant degree of colocalization within the cytoplasm. Bar, 2 μm. The results shown are representative of at least three independent experiments.
We next investigated whether PMA treatment also leads to cell surface expression of ASP. Untreated and PMA-treated U1C8 cells and the parental uninfected cell line, U937, were surface stained with anti-ASP MAb or an isotype control antibody and then analyzed by confocal microscopy. We found that ASP was readily detectable on the surfaces of U1C8 cells following HIV-1 reactivation (Fig. 6, top; see Fig. S5 and Movie S4 in the supplemental material), but not on untreated U1C8 cells (see Fig. S5). Cell surface expression of ASP was also observed on OM-10.1 cells (Fig. 6, bottom). Indeed, flow cytometry experiments showed that ASP was present on the surface in all the PMA-treated cell lines we analyzed (Fig. 7). Moreover, the ability to detect ASP on the surfaces of these cells without the need for membrane permeabilization demonstrates that the epitope recognized by the 324.6 MAb is exposed in the extracellular milieu and supports the notion that ASP is an integral protein of the plasma membrane in PMA-treated cells.
FIG 6.

ASP is an integral protein of the plasma membrane in PMA-treated U1C8 and OM-10.1 cells. Chronically infected U1C8 (top) and OM-10.1 (bottom) cells were treated with PMA to reactivate HIV-1 expression, as described in Materials and Methods. The cells were stained with Alexa Fluor 647-labeled anti-ASP 324.6 (red) and DAPI (blue) without prior fixation and permeabilization. Finally, the cells were washed, fixed, and analyzed by confocal microscopy. The overlay of the images taken in the red (ASP) and blue (DAPI) channels shows that PMA treatment induced ASP translocation to the cell membrane. Detection of ASP on the cell surface without prior permeabilization of the plasma membrane indicates that the 324.6 mAb recognizes an ASP epitope exposed in the extracellular milieu. Bar, 2 μm. The results shown are representative of at least three independent experiments.
FIG 7.

Flow cytometry analysis of ASP expression on the surface in multiple cell lines. Chronically infected myeloid (U1C8 and OM-10.1) and lymphoid (J1.1, ACH2, 8E5, H9/IIIB, H9/CC, H9/MN, and H9/RF) cell lines and their uninfected parental myeloid (U937 and HL-60) and lymphoid (Jurkat, A3.01, and H9) cell lines were treated with PMA as indicated in Materials and Methods to reactivate HIV-1 expression. Then, the cells were surface stained with anti-ASP 324.6 (light-blue curve and line) or a control mouse IgG (open curve with red line) labeled with Alexa Fluor 647. After washing, the cells were fixed and analyzed by flow cytometry. All the chronically infected cell lines analyzed showed cell surface expression of ASP to varying degrees. The results shown are representative of several independent experiments.
Colocalization of ASP and gp120 on the surfaces of PMA-treated cells.
To gain clues about the possible role of ASP in the HIV-1 life cycle, we investigated the relative localizations of ASP and the HIV-1 gp120 protein on the plasma membranes of PMA-treated cells. Thus, U1C8 cells were treated with PMA to induce HIV-1 reactivation and ASP translocation to the cell membrane. After staining with anti-ASP and anti-gp120 MAbs, the cells were analyzed by confocal microscopy. We found that the two proteins presented a high degree of colocalization on the plasma membranes of cells treated with PMA (Fig. 8; see Fig. S6 and Movie S5 in the supplemental material). We determined that the Manders overlap coefficient (MOC) was ∼76%. Analysis of OM-10.1 cells produced results in agreement with those shown above (see Fig. S7 in the supplemental material). Moreover, super-resolution microscopy confirmed the original observations made by confocal microscopy (see Fig. S8 in the supplemental material). Finally, we confirmed that ASP is also expressed on the surfaces of primary human CD4+ T cells and monocyte-derived macrophages (MDM) infected in vitro with HIV-1Ada-M (Fig. 9 and 10). Altogether, these results demonstrate that ASP and gp120 present a high degree of colocalization and are tightly associated on the surfaces of chronically infected cells treated with PMA.
FIG 8.

ASP and HIV-1 gp120 colocalize on the surfaces of PMA-treated cells. U1C8 cells were treated with PMA as described in Materials and Methods to reactivate HIV-1 expression. Then, the cells were surface stained with Alexa Fluor 488-labeled anti-gp120 (green) and Alexa Fluor 647-labeled anti-ASP 324.6 (red). In addition, the cells were stained with DAPI to identify the nuclei. After washing, the cells were fixed and analyzed by confocal microscopy. The overlay of the images taken in the green (HIV-1 gp120), red (ASP), and blue (DAPI) channels shows that PMA treatment induced HIV-1 expression, as shown by the presence of both HIV-1 gp120 and ASP on the cell surface. In addition, ASP and HIV-1 gp120 present a significant degree of colocalization. Bar, 2 μm for all panels. The results shown are representative of several independent experiments.
FIG 9.

ASP and HIV-1 gp120 colocalize on the surfaces of primary human CD4+ T cells infected with HIV-1Ada-M. CD4+ T cells were isolated, activated, and infected as described in Materials and Methods. At day 7 postinfection, the cells were surface stained with Alexa Fluor 488-labeled anti-gp120 (green) and Alexa Fluor 647-labeled anti-ASP 324.6 (red). After washing, the cells were fixed and analyzed by confocal microscopy. The overlay of the images taken in the green (HIV-1 gp120) and red (ASP) channels shows the presence of both HIV-1 gp120 and ASP on the cell surface. In addition, ASP and HIV-1 gp120 present a significant degree of colocalization. Bar, 5 μm for all panels. The results shown are representative of three independent experiments.
FIG 10.

ASP and HIV-1 gp120 colocalize on the surfaces of primary human monocyte-derived macrophages infected with HIV-1Ada-M. Macrophages were generated from freshly isolated PBMC and infected as described in Materials and Methods. At day 15 postinfection, the cells were surface stained with Alexa Fluor 488-labeled anti-gp120 (green) and Alexa Fluor 647-labeled anti-ASP 324.6 (red). In addition, the cells were stained with DAPI to identify the nuclei. After washing, the cells were fixed and analyzed by confocal microscopy. The overlay of the images taken in the green (HIV-1 gp120), red (ASP), and blue (DAPI) channels shows the presence of both HIV-1 gp120 and ASP on the cell surface. In addition, ASP and HIV-1 gp120 present a significant degree of colocalization. Bar, 5 μm for all panels. The results shown are representative of three independent experiments.
ASP is a structural protein of HIV-1.
The close proximity of ASP and gp120 on the surfaces of PMA-treated cells raises the possibility that ASP might become incorporated in the envelopes of HIV-1 viral particles during budding and release. To address this hypothesis, we performed two analyses. First, we employed fluorescence correlation spectroscopy (FCS) to measure the degree of association between the anti-ASP MAb and cell-free HIV-1 particles. Therefore, we recovered HIV-1 particles from culture supernatants of U1C8 cells treated with PMA for 24 to 48 h (HIV-1U1C8). Then, we incubated equal amounts of viral suspension with AF647-labeled anti-ASP (324.6), anti-gp120 (b12), and anti-Rous sarcoma virus (RSV) protein F (palivizumab [Synagis]) MAbs. In these experiments, antibodies were conjugated to AF647 at a dye/Ab ratio of ≤2:1. FCS analyses of these samples showed that the binding efficiency of the 324.6 MAb to cell-free virions in suspension was ∼28%, as assessed by the fraction of rapidly diffusing free versus the more slowly diffusing virion-bound anti-ASP 324.6-AF647 MAb (Fig. 11A). Similar results were obtained with the positive-control anti-gp120 b12-AF647 MAb, while the negative-control anti-RSV protein F palivizumab-AF647 did not show any binding (Fig. 11A). Importantly, the diffusion coefficient of antibody-bound particles derived from the FCS studies allowed us to discriminate between viral particles and extracellular vesicles. Indeed, the diffusion coefficient derived from the fitting of autocorrelation plots of 324.6-bound or b12-bound virion fractions (6 μm2/s) indicates that the particles are about 100 nm in size and rules out the presence of particles larger than 200 nm. This result is consistent with the size of HIV-1 virions (100 to 150 nm) rather than extracellular vesicles (50 to 1,000 nm). Moreover, binding of MAbs to extracellular vesicles would have been nonspecific, and thus, palivizumab (our negative-control MAb) would have also shown binding.
FIG 11.

ASP is an integral protein of the viral envelope. (A) HIV-1 viral particles concentrated from culture supernatants of U1C8 cells (HIV-1U1C8) treated with PMA were incubated with Alexa Fluor 647-labeled anti-gp120 (b12), anti-ASP (324.6), or anti-protein F of RSV (palivizumab). The samples were then analyzed by fluorescence correlation spectroscopy as described in Materials and Methods. (B) HIV-1 viral particles were captured from culture supernatants of PMA-treated U1C8 cells using anti-gp120 (VRC01), anti-ASP (324.6), or control mouse IgG conjugated to protein G Dynabeads. After extensive washes, the samples were analyzed by RT-PCR for the presence of virion-associated HIV-1 RNA. Both assays showed that ASP is expressed on the surfaces of HIV-1 particles as an integral protein of the viral envelope. The results shown are representative of three independent experiments.
To confirm these results, we performed virion capture assays (VCA). Equal amounts of anti-ASP 324.6, anti-gp120 VRC01, and isotype control IgG antibodies were immobilized on protein G-Dynabeads and then incubated with an HIV-1U1C8 suspension prepared as described above. After extensive washes of the virus-antibody-bead complexes, we extracted and quantified HIV-1 vRNA by reverse transcription-quantitative PCR (RT-qPCR). As shown in Fig. 11B, the anti-ASP 324.6 MAb was able to capture viral particles with an efficiency above the background levels of IgG-Dynabead conjugates or Dynabeads alone and similar to that of the anti-gp120 VRC01 antibodies. Although we used two different anti-gp120 antibodies in the FCS (b12) and VCA (VRC01) assays, they are both directed against the CD4 binding site of gp120.
Altogether, the FCS analyses of fluorescently tagged 324.6 MAb bound to an individual virion particle and the VCA analyses of virion particles captured with bead-bound 324.6 MAb clearly suggest that ASP is an integral protein expressed in the envelopes of HIV-1 particles, alongside the envelope glycoproteins gp41 and gp120.
DISCUSSION
While the role of the HTLV-1 antisense protein HBZ in ATL pathogenesis has been intensely investigated and has been defined (for a comprehensive review, see reference 2), the HIV-1 antisense protein ASP has received much less attention, and its existence is still not fully recognized despite multiple lines of evidence that support it. Indeed, the presence of an open reading frame in the HIV-1 genome encoding a new antisense protein was first reported by Roger Miller in 1988 (12). Experimental evidence corroborating that prediction came a few years later, when it was reported that sera of HIV-1 patients contained antibodies against ASP (15). Since then, multiple studies have confirmed the expression of ASP in vivo (16, 17, 40) and have investigated its expression in vitro (13, 24, 25, 41). However, the use of a single cell type, a single technique, or tagged ASP overexpressed ectopically through transient transfection (in part due to the lack of specific antibodies against ASP) did not permit general conclusions about its expression in the context of HIV-1 infection to be reached. Moreover, the role of ASP in the HIV-1 life cycle remains to be elucidated (26, 42).
In the present study, we sought to generate a clear picture of the expression pattern and dynamics of ASP in the context of HIV-1 infection rather than following ectopic overexpression, with the aim of gaining clues about the possible role of ASP in the HIV-1 life cycle. Thus, we investigated ASP expression in several chronically infected myeloid and lymphoid cell lines both at baseline and after PMA stimulation using a specific anti-ASP monoclonal antibody applied by multiple techniques. Our results show that in untreated cells, ASP is contained in the nucleus, where it shows a polarized (i.e., nonuniform) distribution and accumulates in areas with low content of repressive epigenetic marks and less tightly packed chromatin. Sequence analysis has failed to reveal the presence of typical lysine- or arginine-rich nuclear localization signals in ASP. Therefore, the mechanism that leads to ASP nuclear import will require a more thorough investigation. After cell treatment with phorbol esters, ASP exits the nucleus, translocates to the cytoplasm, and becomes associated with the cell membrane, where it is found in close proximity to gp120. A previous study utilizing transient transfection of Jurkat cells with a vector overexpressing FLAG-tagged ASP found that ASP localized to the cell membrane, showing heterogeneous staining patterns (13). Moreover, we demonstrate through in vitro FCS analyses of cell-free single HIV-1 particles that ASP is an integral protein of the viral envelope. This conclusion was consistent with the results we obtained from virion capture assays. Thus, during viral budding and release, ASP becomes incorporated into the HIV-1 envelope and should be regarded as a structural component of the HIV-1 viral particle. Our results are in line with previously reported electron microscopy analyses (24).
Overall, our studies uncovered a complex and dynamic pattern of ASP expression in unstimulated versus PMA-treated cells. The relationship between cell activation, HIV-1 expression, and ASP localization is a complex issue. In the absence of stimuli, we observed nuclear localization of ASP in all the cell lines tested despite variable levels of baseline HIV-1 expression, suggesting that nuclear localization of ASP may not be associated with HIV-1 expression. However, while measurable at baseline in some cell lines, HIV-1 expression could be significantly upregulated by PMA treatment in all the cell lines. Under conditions of maximal HIV-1 expression, ASP presents cytoplasmic and cell surface localization. This suggests that ASP cell surface localization may be associated with maximal HIV-1 expression. One possible interpretation of these apparently contrasting conclusions is that ASP subcellular localization reflects the overall activation state of the cell in unstimulated versus PMA-stimulated cells rather than HIV-1 expression. The ability to induce ASP translocation without upregulating HIV-1 expression would provide evidence in support of this interpretation. Alternatively, while some cell lines express HIV-1 to some degree even at baseline, maximal HIV-1 expression can be induced in response to PMA treatment. Measurable expression of HIV-1 at baseline may be due to several factors: the HIV-1 integration site, the levels of some of the positive and negative transcription factors (e.g., Sp1, YY1, and LSF), leaky expression of Tat, etc. Thus, under conditions of latent or minimal HIV-1 expression, ASP is nuclear; under conditions of fully induced or maximal HIV-1 expression, ASP is membrane associated. Therefore, ASP localization correlates with expression levels of HIV-1. These critical questions are the focus of current investigations.
The subcellular localization of ASP allows us to formulate hypotheses about its possible role(s) in the virus life cycle. Previous studies have shown that HIV-1 integration occurs in the outer shell of the nucleus in close proximity to the nucleopore (43), where actively transcribed chromatin resides (38, 39). It is intriguing to speculate that ASP might associate—either directly or indirectly—with the integrated provirus, contributing to the determination of its subnuclear localization and, consequently, its expression. An alternative hypothesis suggests that ASP may be involved in determining the fate of full-length HIV-1 genomic-RNA molecules between translation and virion packaging through recognition of qualitative and quantitative differences in epigenetic modifications (e.g., N6-methylation, pseudouridylation, or 2′-O-methylation) of the RNA molecule. Moreover, the demonstration that ASP is an integral protein of the cellular and viral envelope also suggests a possible role during viral budding and/or entry. Two recent studies have shown that ASP from all HIV-1 clades has the ability to induce autophagy (26, 42). However, the advantage that triggering this pathway would provide to HIV-1 replication was not investigated. Indeed, autophagy is a process required for antigen presentation by dendritic cells and thus is intimately involved in adaptive immune responses (44), and the HIV-1 Nef protein was shown to inhibit autophagy (45, 46). A possible interpretation is that autophagy may be induced in response to overexpression of ASP under the strong cytomegalovirus (CMV) promoter in transient-transfection assays (26, 42). Therefore, it would be interesting to assess whether expression of ASP under its weaker natural antisense promoter located in the proviral 3′ LTR also promotes autophagy (21, 36). The same studies elegantly demonstrated that ASP displays a propensity to form multimers and aggregates and that the N-terminal cysteine-rich region plays an important role in these events (26, 42). The formation of aggregates may in part explain our observation that a highly hydrophobic protein such as ASP is able to reside in the nucleoplasm and that it is confined to limited areas of the nucleus rather than showing a diffused distribution. More importantly, the demonstration that ASP is an integral protein of the cell membrane and the viral envelope identifies ASP as a potential therapeutic or vaccine target.
Open reading frames created de novo (i.e., not derived by horizontal transmission) and overlapping another ORF most often originated by “overprinting,” a process whereby mutations within a genomic region allow the expression of a new (overprinting) protein from the same DNA sequence of an existing (ancestral, overprinted) protein using one of the other five reading frames (47–49). This is most often the case in viruses, because of the evolutionary constraints that small genomes impose on the virus (50). Compared to their cognate ancestral gene and the genome they belong to, genes created de novo by overprinting display a more restricted phylogenetic distribution, different codon usage, and a greater content of amino acids with high codon degeneracy (47, 51). Viral genes created de novo by overprinting often play roles in virus spreading or pathogenicity (47, 48). Examples of retroviral genes created de novo are rex and p12 and p30 of HTLV-1 (51), as well as rev, the 3′ exon of tat, and vpu of HIV-1 (52).
Based on these criteria, asp also appears to be a gene created de novo by overprinting of env. The truly unique feature of asp is that it is a very “young” ORF, created ∼100 years ago with the emergence of group M HIV-1, which gave rise to the pandemic (14, 53, 54). Indeed, ASP has no known homologs and is absent in all out-of-M HIV-1 and simian immunodeficiency virus (SIV) strains (14). Thus, there is a strong correlation between the presence of a full-length asp ORF and strain prevalence (14). At the same time, ASP is absent in ∼16% of the most prevalent group M HIV-1 strains, a frequency that is similar to that of nef (14, 55). Moreover, the analysis of the presence of the start and stop codons indicates that in group M strains—but not in all other HIV-1 and SIV strains—there has been strong selection pressure to maintain the asp ORF (14). This is particularly striking because ASP is present in a very busy genomic region that also contains env and the Rev response element (RRE).
Altogether, the evidence we have presented here defines a complex and dynamic pattern of ASP expression under baseline and PMA-stimulated conditions, and it identifies ASP as an integral protein of the cell membrane and viral envelope (14). These studies open several avenues of investigation to further define the role of ASP in the virus life cycle.
MATERIALS AND METHODS
Cell culture.
All the cell lines were obtained from the National Institutes of Health (NIH) AIDS Reagent Program, except for HL-60 cells, which were purchased from the ATCC. The cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS) plus penicillin, streptomycin, and l-glutamine. HL-60 cells were cultured in Iscove modified Dulbecco medium (IMDM) supplemented with 25% FBS. All reagents were obtained from Thermo Fisher. Cell lines were maintained at 5 × 105 cells/ml. Where indicated, cells were treated with 10 to 20 ng/ml of PMA (Sigma-Aldrich) for 12 to 24 h. In the experiments comparing ASP expression in cultures maintained at low and high cell densities, U1 and U1C8 cells were split to 2 × 105 (low density) or 1 × 106 (high density) live cells/ml every 3 days for 8 to 10 passages prior to intracellular staining with 324.6 MAb.
Isolation and infection of primary monocyte-derived macrophages and CD4+ T cells.
Mononuclear cells were isolated from peripheral blood of HIV-1-negative donors by Ficoll centrifugation. For generation of MDM, 3.5 × 107 freshly isolated peripheral blood mononuclear cells (PBMC) were seeded in T25 flasks in RPMI 1640 supplemented with 20% heat-inactivated fetal bovine serum and 10% heat-inactivated human serum (Gemcell) plus penicillin, streptomycin, and l-glutamine and incubated at 37°C in 5% CO2. The cultures were examined every day by microscopy. At day 7, the cells were washed 3 times and cultured for at least 1 h in the same medium described above but without human serum. Each T25 flask was then infected with 100 ng p24 of HIV-1ADA-M and incubated at 37°C for 1 h. After infection, the cells were washed three times and cultured in fresh medium without human serum. For isolation of CD4+ T cells, PBMC were plated at 1 × 106 cells/ml in RPMI 1640 supplemented with 10% FBS plus penicillin, streptomycin, and l-glutamine and kept in an incubator overnight. The next day, the CD4+ T cells were isolated by negative selection (Miltenyi Biotec) and activated with CD3/CD28 Dynabeads (1 bead/cell) and 100 ng/ml interleukin 2 (IL-2). After 3 to 4 days, the cells were infected with 20 ng p24 of HIV-1ADA-M per 106 cells. The cells were incubated at 37°C for 30 min and then spun at 1,200 × g for 70 min. Then, the cells were washed 4 times with medium, seeded at 1 × 106 cells/ml in complete medium with 25 ng/ml IL-2, and incubated at 37°C and 5% CO2. HIV-1 replication in both MDM and CD4+ T cell cultures was monitored by p24 enzyme-linked immunosorbent assay (ELISA).
Generation of anti-ASP 324.6 monoclonal antibody.
The hybridoma cell line expressing the anti-ASP 324.6 monoclonal antibody was generated at the Albert Einstein College of Medicine Hybridoma Core Facility on a fee-for-service basis using a synthetic 14-aa peptide (GenScript) with sequence identical to residues 97 to 110 of ASP from HIV-1NL4-3 (NH2-SLISPPPGLKISDP-COOH) and conjugated to keyhole limpet hemocyanin. A detailed description of how the antigen was chosen can be found in the supplemental material. The anti-ASP antibody 324.6 was produced in house by culturing the hybridoma cell line in RPMI 1640 supplemented with 10% FBS, 12.5 mM HEPES, 1 mM sodium pyruvate, 55 μM β-mercaptoethanol, 0.1% gentamicin. IgG was purified from the culture supernatant passed over a HiTrap protein A column (GE Healthcare) equilibrated with phosphate-buffered saline (PBS), pH 7.2. The column was washed with PBS, and the IgG was eluted with 0.1 M glycine, pH 3.0. Eluted fractions were immediately diluted 10:1 with 1 M Tris-HCl, pH 8.5, to raise the pH; concentrated; and then loaded on a Superdex 200 gel filtration column (GE Healthcare) equilibrated in 25 mM Tris-HCl, pH 8.5, and 150 mM sodium chloride. The IgG elution peak corresponding to a molecular weight of approximately 150 kDa was collected and concentrated for use in experiments.
Antibody staining.
Intranuclear staining was carried out with a FoxP3 permeabilization kit (Thermo Fisher). Briefly, cells were washed with Dulbecco's phosphate-buffered saline (DPBS) supplemented with 2% FBS (DPBS-FBS), permeabilized with a FoxP3 permeabilization kit for 45 min, washed with DPBS-FBS, and resuspended in cold DPBS. After addition of fluorescently labeled antibodies (10 ng), the cells were incubated at 2°C for 45 min, washed with cold DPBS containing 30 μg/ml DAPI (Thermo Fisher), and fixed with 1% paraformaldehyde (PFA). For cytoplasmic-antigen detection, we used a BD Cytofix/Cytoperm kit following the instructions of the manufacturer. Cell surface detection of ASP was carried out for 45 min at 2°C with 10 ng of each antibody. The cells were then washed with DPBS and fixed with PFA. The anti-gp120 polyclonal antibody was from Bioss, the anti-H3K27me3 antibody was from Abcam, the anti-gp120 monoclonal antibodies b12 and VRC01 were obtained from the NIH AIDS Reagent Program, and the anti-p24 antibody was from Abcam. When needed, antibodies were fluorescently labeled at dye/Ab ratios of ∼5:1 (except where indicated) using kits from Thermo Fisher. To avoid autofluorescence in confocal and super-resolution microscopy analyses, cells were treated with TrueBlack Lipofuscin autofluorescence quencher (Biotium) and then washed three times with cold DPBS before fixation with PFA.
Sample analysis.
For flow cytometry, samples were analyzed with a Millipore Guava flow cytometer, and the data were analyzed with FlowJo. Confocal images of cell-associated fluorescence were acquired using the Zeiss LSM 800 confocal system (Carl Zeiss Microscopy, Germany). Three laser lines, 405 nm (blue, for DAPI), 488 nm (green, for lamin B receptor, H3K27me3, p24, and gp120), and 647 nm (red, for ASP), were utilized in the imaging experiments. The blue, green, and red signals were originally separated by a quad-DAPI-fluorescein isothiocyanate (FITC)-tetramethyl rhodamine isocyanate (TRITC)-Cy5 dichroic beam splitter. The signals were then further acquired with a Gasp detector (LSM 800; Carl Zeiss, Germany). A C-Apochromat 40×/1.2 Water Korr objective (LSM 800; Carl Zeiss, Germany) was used to visualize the multicolor-labeled cell samples. Zen Blue software (Carl Zeiss Microscopy, Germany) was used to generate and analyze original images. The Manders overlap coefficient of colocalization for double-stained samples was calculated using single-color crosshair values of red and green set in x and y coordinates of the crosshair panel from a given scatterplot. Crosshair values of red and green were averaged from 14 (crosshair average value, 3,933) and 20 (crosshair average value, 15,549) single images, respectively. The average values of single-color images were then computed as the vertical red and horizontal green crosshair x and y coordinates, respectively, and used as a reference on double-color images of red and green as the baseline to generate the Manders colocalization coefficient of green and red pixels together. The Manders overlap coefficient of colocalization was determined by averaging the values from 21 double-color images. Super-resolution images were acquired using Nikon’s direct STORM (dSTORM) with a total internal-reflection fluorescence (TIRF) microscope setup. The system is equipped with electron-multiplying charge-coupled device (EMCCD) technology (Andor iXon Ultra DU987 EMCCD camera) that is capable of single-molecule detection sensitivity. The TIRF angle was adjusted in order to maximize signal-to-noise detection for the 3D STORM images. The TIRF angle was shifted during sample bleaching in order to have angles just below and above the indicated TIRF angle of imaging interest. The sample bleaching is required to ensure that target fluorophores are in a ground state and to eliminate any nonspecific signals from the valid signal of the designated fluorophores. Solid-state lasers (488 nm at 80 mW and 647 nm at 125 mW) were utilized at 100% power during image acquisition to activate the highest possible number of fluorescence molecules and to maximize the signal of interest. Images were acquired using a Nikon 100×/1.49 chrome-free infinity (CFI) Apo TIRF oil immersion objective for TIRF imaging and processed using Nikon’s NIS Elements software.
Fluorescence correlation spectroscopy.
FCS experiments were performed with HIV-1 preparations obtained from U1C8 cells induced with 20 ng/ml PMA for 24 to 36 h. The virus preparations were diluted to 2 μg/ml p24 equivalent. gp120-to-p24 antigen ratios were typically 1:10. The virions were initially incubated with 25 μg/ml of IgG1 (Calbiochem) in PBS to block nonspecific binding. This was followed by addition of antibodies labeled with Alexa Fluor 647 at a concentration of 0.4 μg/ml and incubation for 60 min at 37°C. The reaction mixture was then loaded on a sealed chamber coverslip. FCS measurements were performed in a confocal microscope (ISS Q2) with single-molecule detection sensitivity. The excitation source was a Fianium SC-400 supercontinuum laser. An NKT superselect acousto-optic tunable filter (AOTF) was used to select the excitation wavelength of 635 nm, which was reflected by a dichroic mirror to a high-numerical-aperture (NA) water objective (60×; 1.2 NA) and focused on the solution sample. The fluorescence was collected by avalanche photodiodes through a dichroic beam splitter and a band-pass (650- to 720-nm; Chroma) filter, thus eliminating the scattered excitation light and collecting the fluorescence from the Alexa Fluor 647 probes in the region of interest. The data acquisition was enabled by a B&H SPC-150 card operated in a photon time tag time-resolved (TTTR) mode. The ISS VistaVision software was used to analyze the FCS data to assess the in vitro binding of MAbs to HIV-1 virions. We determined the translational diffusion coefficients of Alexa Fluor 647-labeled MAbs and the corresponding complexes with virions using a three-dimensional translational diffusion model. The percentage of total MAb at a given test concentration that shifts into the more slowly diffusing species (i.e., the virion-bound fraction) reflects the relative magnitude of equivalent epitope exposure in the target population of virions. The FCS measurements and analyses were performed similarly to previous reports (56).
Virion capture assay.
The VCA was carried out essentially as described previously (57, 58). Briefly, for each sample, 10 μg of each antibody was conjugated to 30 μl of protein G Dynabeads (Thermo Fisher) for 1 h at room temperature with rotation in 200 μl of PBS containing 0.02% Tween 20. Bead-antibody conjugates were washed three times with 500 μl of PBS containing 0.025% casein and then incubated with HIV-1 particles (20 ng of HIV-1 p24 equivalents) for 2 h at room temperature with rotation. Then, the bead-captured virions were washed three times with 500 μl of PBS containing 0.025% casein. After extraction of viral RNA with a QIAamp viral kit (Qiagen), samples were analyzed by RT-PCR using an AgPath-ID One-Step RT-PCR kit (Thermo Fisher) following the manufacturer’s protocol. The RT step was carried out with random primers. For the PCR step, we used the following primers: forward, 5′-TACTGACGCTCTCGCACC-3′; reverse, 5′-TCTCGACGCAGGACTCG-3′; probe, 5′-6-carboxyfluorescein (FAM)-CTCTCTCCTTCTAGCCTC-3′. The PCR conditions were as follows: 1 cycle at 95°C for 15 min, 40 cycles at 95°C for 15 s, and 60°C for 1 min. Data are expressed as percentages of input HIV-1 (20 ng of HIV-1 p24).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants AI120008 and AI124776 to F.R.; National Institutes of Health grants AI116274 and AI129769 to M.P.; and National Institutes of Health grants GM117836, AI150447, and GM117836-S1 to K.R.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00574-19.
REFERENCES
- 1.Barbeau B, Peloponese JM, Mesnard JM. 2013. Functional comparison of antisense proteins of HTLV-1 and HTLV-2 in viral pathogenesis. Front Microbiol 4:226. doi: 10.3389/fmicb.2013.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma G, Yasunaga J, Matsuoka M. 2016. Multifaceted functions and roles of HBZ in HTLV-1 pathogenesis. Retrovirology 13:16. doi: 10.1186/s12977-016-0249-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM. 2002. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol 76:12813–12822. doi: 10.1128/jvi.76.24.12813-12822.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hivin P, Frédéric M, Arpin-André C, Basbous J, Gay B, Thébault S, Mesnard J-M. 2005. Nuclear localization of HTLV-I bZIP factor (HBZ) is mediated by three distinct motifs. J Cell Sci 118:1355–1362. doi: 10.1242/jcs.01727. [DOI] [PubMed] [Google Scholar]
- 5.Hivin P, Arpin-Andre C, Clerc I, Barbeau B, Mesnard JM. 2006. A modified version of a Fos-associated cluster in HBZ affects Jun transcriptional potency. Nucleic Acids Res 34:2761–2772. doi: 10.1093/nar/gkl375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halin M, Douceron E, Clerc I, Journo C, Ko NL, Landry S, Murphy EL, Gessain A, Lemasson I, Mesnard JM, Barbeau B, Mahieux R. 2009. Human T-cell leukemia virus type 2 produces a spliced antisense transcript encoding a protein that lacks a classic bZIP domain but still inhibits Tax2-mediated transcription. Blood 114:2427–2438. doi: 10.1182/blood-2008-09-179879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbeau B, Mesnard JM. 2011. Making sense out of antisense transcription in human T-cell lymphotropic viruses (HTLVs). Viruses 3:456–468. doi: 10.3390/v3050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Switzer WM, Qari SH, Wolfe ND, Burke DS, Folks TM, Heneine W. 2006. Ancient origin and molecular features of the novel human T-lymphotropic virus type 3 revealed by complete genome analysis. J Virol 80:7427–7438. doi: 10.1128/JVI.00690-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Switzer WM, Salemi M, Qari SH, Jia H, Gray RR, Katzourakis A, Marriott SJ, Pryor KN, Wolfe ND, Burke DS, Folks TM, Heneine W. 2009. Ancient, independent evolution and distinct molecular features of the novel human T-lymphotropic virus type 4. Retrovirology 6:9. doi: 10.1186/1742-4690-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murakami H, Asano S, Uchiyama J, Sato R, Sakaguchi M, Tsukamoto K. 2017. Bovine leukemia virus G4 enhances virus production. Virus Res 238:213–217. doi: 10.1016/j.virusres.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 11.Briquet S, Richardson J, Vanhee-Brossollet C, Vaquero C. 2001. Natural antisense transcripts are detected in different cell lines and tissues of cats infected with feline immunodeficiency virus. Gene 267:157–164. doi: 10.1016/s0378-1119(01)00404-8. [DOI] [PubMed] [Google Scholar]
- 12.Miller RH. 1988. Human immunodeficiency virus may encode a novel protein on the genomic DNA plus strand. Science 239:1420–1422. doi: 10.1126/science.3347840. [DOI] [PubMed] [Google Scholar]
- 13.Clerc I, Laverdure S, Torresilla C, Landry S, Borel S, Vargas A, Arpin-Andre C, Gay B, Briant L, Gross A, Barbeau B, Mesnard JM. 2011. Polarized expression of the membrane ASP protein derived from HIV-1 antisense transcription in T cells. Retrovirology 8:74. doi: 10.1186/1742-4690-8-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cassan E, Arigon-Chifolleau AM, Mesnard JM, Gross A, Gascuel O. 2016. Concomitant emergence of the antisense protein gene of HIV-1 and of the pandemic. Proc Natl Acad Sci U S A 113:11537–11542. doi: 10.1073/pnas.1605739113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vanhee-Brossollet C, Thoreau H, Serpente N, D’Auriol L, Levy JP, Vaquero C. 1995. A natural antisense RNA derived from the HIV-1 env gene encodes a protein which is recognized by circulating antibodies of HIV+ individuals. Virology 206:196–202. doi: 10.1016/S0042-6822(95)80034-4. [DOI] [PubMed] [Google Scholar]
- 16.Champiat S, Raposo RA, Maness NJ, Lehman JL, Purtell SE, Hasenkrug AM, Miller JC, Dean H, Koff WC, Hong MA, Martin JN, Deeks SG, Spotts GE, Pilcher CD, Hecht FM, Kallas EG, Garrison KE, Nixon DF. 2012. Influence of HAART on alternative reading frame immune responses over the course of HIV-1 infection. PLoS One 7:e39311. doi: 10.1371/journal.pone.0039311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bet A, Maze EA, Bansal A, Sterrett S, Gross A, Graff-Dubois S, Samri A, Guihot A, Katlama C, Theodorou I, Mesnard JM, Moris A, Goepfert PA, Cardinaud S. 2015. The HIV-1 antisense protein (ASP) induces CD8 T cell responses during chronic infection. Retrovirology 12:15. doi: 10.1186/s12977-015-0135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tagieva NE, Vaquero C. 1997. Expression of naturally occurring antisense RNA inhibits human immunodeficiency virus type 1 heterologous strain replication. J Gen Virol 78:2503–2511. doi: 10.1099/0022-1317-78-10-2503. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi-Ishihara M, Yamagishi M, Hara T, Matsuda Y, Takahashi R, Miyake A, Nakano K, Yamochi T, Ishida T, Watanabe T. 2012. HIV-1-encoded antisense RNA suppresses viral replication for a prolonged period. Retrovirology 9:38. doi: 10.1186/1742-4690-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saayman S, Ackley A, Turner AM, Famiglietti M, Bosque A, Clemson M, Planelles V, Morris KV. 2014. An HIV-encoded antisense long noncoding RNA epigenetically regulates viral transcription. Mol Ther 22:1164–1175. doi: 10.1038/mt.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michael NL, Vahey MT, d'Arcy L, Ehrenberg PK, Mosca JD, Rappaport J, Redfield RR. 1994. Negative-strand RNA transcripts are produced in human immunodeficiency virus type 1-infected cells and patients by a novel promoter downregulated by Tat. J Virol 68:979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zapata JC, Campilongo F, Barclay RA, DeMarino C, Iglesias-Ussel MD, Kashanchi F, Romerio F. 2017. The human immunodeficiency virus 1 ASP RNA promotes viral latency by recruiting the polycomb repressor complex 2 and promoting nucleosome assembly. Virology 506:34–44. doi: 10.1016/j.virol.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi-Ishihara M, Terahara K, Martinez JP, Yamagishi M, Iwabuchi R, Brander C, Ato M, Watanabe T, Meyerhans A, Tsunetsugu-Yokota Y. 2018. HIV LTR-driven antisense RNA by itself has regulatory function and may curtail virus reactivation from latency. Front Microbiol 9:1066. doi: 10.3389/fmicb.2018.01066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Briquet S, Vaquero C. 2002. Immunolocalization studies of an antisense protein in HIV-1-infected cells and viral particles. Virology 292:177–184. doi: 10.1006/viro.2001.1224. [DOI] [PubMed] [Google Scholar]
- 25.Laverdure S, Gross A, Arpin-Andre C, Clerc I, Beaumelle B, Barbeau B, Mesnard JM. 2012. HIV-1 antisense transcription is preferentially activated in primary monocyte-derived cells. J Virol 86:13785–13789. doi: 10.1128/JVI.01723-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torresilla C, Larocque E, Landry S, Halin M, Coulombe Y, Masson JY, Mesnard JM, Barbeau B. 2013. Detection of the HIV-1 minus-strand-encoded antisense protein and its association with autophagy. J Virol 87:5089–5105. doi: 10.1128/JVI.00225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Folks TM, Justement J, Kinter A, Dinarello CA, Fauci AS. 1987. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 238:800–802. doi: 10.1126/science.3313729. [DOI] [PubMed] [Google Scholar]
- 28.Poli G, Orenstein JM, Kinter A, Folks TM, Fauci AS. 1989. Interferon-alpha but not AZT suppresses HIV expression in chronically infected cell lines. Science 244:575–577. doi: 10.1126/science.2470148. [DOI] [PubMed] [Google Scholar]
- 29.Butera ST, Perez VL, Wu BY, Nabel GJ, Folks TM. 1991. Oscillation of the human immunodeficiency virus surface receptor is regulated by the state of viral activation in a CD4+ cell model of chronic infection. J Virol 65:4645–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perez VL, Rowe T, Justement JS, Butera ST, June CH, Folks TM. 1991. An HIV-1-infected T cell clone defective in IL-2 production and Ca2+ mobilization after CD3 stimulation. J Immunol 147:3145–3148. [PubMed] [Google Scholar]
- 31.Folks TM, Clouse KA, Justement J, Rabson A, Duh E, Kehrl JH, Fauci AS. 1989. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc Natl Acad Sci U S A 86:2365–2368. doi: 10.1073/pnas.86.7.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Folks TM, Powell D, Lightfoote M, Koenig S, Fauci AS, Benn S, Rabson A, Daugherty D, Gendelman HE, Hoggan MD. 1986. Biological and biochemical characterization of a cloned Leu-3- cell surviving infection with the acquired immune deficiency syndrome retrovirus. J Exp Med 164:280–290. doi: 10.1084/jem.164.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Popovic M, Sarngadharan MG, Read E, Gallo RC. 1984. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science 224:497–500. doi: 10.1126/science.6200935. [DOI] [PubMed] [Google Scholar]
- 34.Gallo RC, Sarin PS, Kramarsky B, Salahuddin Z, Markham PD, Popovic M. 1986. First isolation of HTLV-III. Nature 321:119. doi: 10.1038/321119a0.3010136 [DOI] [Google Scholar]
- 35.Gallo RC, Salahuddin SZ, Popovic M, Shearer GM, Kaplan M, Haynes BF, Palker TJ, Redfield R, Oleske J, Safai B. 1984. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 224:500–503. doi: 10.1126/science.6200936. [DOI] [PubMed] [Google Scholar]
- 36.Bentley K, Deacon N, Sonza S, Zeichner S, Churchill M. 2004. Mutational analysis of the HIV-1 LTR as a promoter of negative sense transcription. Arch Virol 149:2277–2294. doi: 10.1007/s00705-004-0386-8. [DOI] [PubMed] [Google Scholar]
- 37.Olins AL, Rhodes G, Welch DB, Zwerger M, Olins DE. 2010. Lamin B receptor: multi-tasking at the nuclear envelope. Nucleus 1:53–70. doi: 10.4161/nucl.1.1.10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lucic B, Lusic M. 2016. Connecting HIV-1 integration and transcription: a step toward new treatments. FEBS Lett 590:1927–1939. doi: 10.1002/1873-3468.12226. [DOI] [PubMed] [Google Scholar]
- 39.Lusic M, Siliciano RF. 2017. Nuclear landscape of HIV-1 infection and integration. Nat Rev Microbiol 15:69–82. doi: 10.1038/nrmicro.2016.162. [DOI] [PubMed] [Google Scholar]
- 40.Berger CT, Llano A, Carlson JM, Brumme ZL, Brockman MA, Cedeno S, Harrigan PR, Kaufmann DE, Heckerman D, Meyerhans A, Brander C. 2015. Immune screening identifies novel T cell targets encoded by antisense reading frames of HIV-1. J Virol 89:4015–4019. doi: 10.1128/JVI.03435-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ludwig LB, Ambrus JL Jr, Krawczyk KA, Sharma S, Brooks S, Hsiao CB, Schwartz SA. 2006. Human immunodeficiency virus type 1 LTR DNA contains an intrinsic gene producing antisense RNA and protein products. Retrovirology 3:80. doi: 10.1186/1742-4690-3-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Torresilla C, Xiao Y, Nguyen PT, Caté C, Barbosa K, Rassart É, Cen S, Bourgault S, Barbeau B. 2018. HIV-1 antisense protein of different clades induces autophagy and associates with the autophagy factor p62. J Virol 93:e01757-18. doi: 10.1128/JVI.01757-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marini B, Kertesz-Farkas A, Ali H, Lucic B, Lisek K, Manganaro L, Pongor S, Luzzati R, Recchia A, Mavilio F, Giacca M, Lusic M. 2015. Nuclear architecture dictates HIV-1 integration site selection. Nature 521:227–231. doi: 10.1038/nature14226. [DOI] [PubMed] [Google Scholar]
- 44.Valecka J, Almeida CR, Su B, Pierre P, Gatti E. 2018. Autophagy and MHC-restricted antigen presentation. Mol Immunol 99:163–170. doi: 10.1016/j.molimm.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Campbell GR, Rawat P, Bruckman RS, Spector SA. 2015. Human immunodeficiency virus type 1 Nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog 11:e1005018. doi: 10.1371/journal.ppat.1005018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, Federico M, Panganiban A, Vergne I, Deretic V. 2009. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 186:255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rancurel C, Khosravi M, Dunker AK, Romero PR, Karlin D. 2009. Overlapping genes produce proteins with unusual sequence properties and offer insight into de novo protein creation. J Virol 83:10719–10736. doi: 10.1128/JVI.00595-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sabath N, Wagner A, Karlin D. 2012. Evolution of viral proteins originated de novo by overprinting. Mol Biol Evol 29:3767–3780. doi: 10.1093/molbev/mss179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pavesi A, Vianelli A, Chirico N, Bao Y, Blinkova O, Belshaw R, Firth A, Karlin D. 2018. Overlapping genes and the proteins they encode differ significantly in their sequence composition from non-overlapping genes. PLoS One 13:e0202513. doi: 10.1371/journal.pone.0202513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pavesi A. 2006. Origin and evolution of overlapping genes in the family Microviridae. J Gen Virol 87:1013–1017. doi: 10.1099/vir.0.81375-0. [DOI] [PubMed] [Google Scholar]
- 51.Pavesi A, Magiorkinis G, Karlin DG. 2013. Viral proteins originated de novo by overprinting can be identified by codon usage: application to the “gene nursery” of Deltaretroviruses. PLoS Comput Biol 9:e1003162. doi: 10.1371/journal.pcbi.1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keese PK, Gibbs A. 1992. Origins of genes: “big bang” or continuous creation? Proc Natl Acad Sci U S A 89:9489–9493. doi: 10.1073/pnas.89.20.9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Korber B, Muldoon M, Theiler J, Gao F, Gupta R, Lapedes A, Hahn BH, Wolinsky S, Bhattacharya T. 2000. Timing the ancestor of the HIV-1 pandemic strains. Science 288:1789–1796. doi: 10.1126/science.288.5472.1789. [DOI] [PubMed] [Google Scholar]
- 54.Worobey M, Gemmel M, Teuwen DE, Haselkorn T, Kunstman K, Bunce M, Muyembe JJ, Kabongo JM, Kalengayi RM, Van Marck E, Gilbert MT, Wolinsky SM. 2008. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature 455:661–664. doi: 10.1038/nature07390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pushker R, Jacque JM, Shields DC. 2010. Meta-analysis to test the association of HIV-1 nef amino acid differences and deletions with disease progression. J Virol 84:3644–3653. doi: 10.1128/JVI.01959-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ray K, Mengistu M, Yu L, Lewis GK, Lakowicz JR, DeVico AL. 2014. Antigenic properties of the HIV envelope on virions in solution. J Virol 88:1795–1808. doi: 10.1128/JVI.03048-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guzzo C, Ichikawa D, Park C, Phillips D, Liu Q, Zhang P, Kwon A, Miao H, Lu J, Rehm C, Arthos J, Cicala C, Cohen MS, Fauci AS, Kehrl JH, Lusso P. 2017. Virion incorporation of integrin alpha4beta7 facilitates HIV-1 infection and intestinal homing. Sci Immunol 2:eaam7341. doi: 10.1126/sciimmunol.aam7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malnati MS, Scarlatti G, Gatto F, Salvatori F, Cassina G, Rutigliano T, Volpi R, Lusso P. 2008. A universal real-time PCR assay for the quantification of group-M HIV-1 proviral load. Nat Protoc 3:1240–1248. doi: 10.1038/nprot.2008.108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.