

IAV can be recognized as a nonself molecular pattern by host immune systems and can cause immune responses. However, the intense immune response induced by influenza virus, known as a “cytokine storm,” can also cause widespread tissue damage (X. Z. J. Guo and P. G. Thomas, Semin Immunopathol 39:541–550, 2017, https://doi.org/10.1007/s00281-017-0636-y; S. Yokota, Nihon Rinsho 61:1953–1958, 2003; I. A. Clark, Immunol Cell Biol 85:271–273, 2007). Meanwhile, the detailed mechanisms involved in the balancing of immune responses in host cells are not well understood. Our studies reveal that, as an IFN-inducible gene, lnc-MxA functions as a negative regulator of the antiviral immune response. We uncovered the mechanism by which lnc-MxA inhibits the activation of IFN-β transcription. Our findings demonstrate that, as an ISG, lnc-MxA plays an important role in the negative-feedback loop involved in maintaining immune homeostasis.
KEYWORDS: lnc-MxA, IFN-β, influenza A virus, antiviral immunity, immune homeostasis
ABSTRACT
Previously, we identified a set of long noncoding RNAs (lncRNAs) that were differentially expressed in influenza A virus (IAV)-infected cells. In this study, we focused on lnc-MxA, which is upregulated during IAV infection. We found that the overexpression of lnc-MxA facilitates the replication of IAV, while the knockdown of lnc-MxA inhibits viral replication. Further studies demonstrated that lnc-MxA is an interferon-stimulated gene. However, lnc-MxA inhibits the Sendai virus (SeV)- and IAV-induced activation of beta interferon (IFN-β). A luciferase assay indicated that lnc-MxA inhibits the activation of the IFN-β reporter upon stimulation with RIG-I, MAVS, TBK1, or active IRF3 (IRF3-5D). These data indicated that lnc-MxA negatively regulates the RIG-I-mediated antiviral immune response. A chromatin immunoprecipitation (ChIP) assay showed that the enrichment of IRF3 and p65 at the IFN-β promoter in lnc-MxA-overexpressing cells was significantly lower than that in control cells, indicating that lnc-MxA interfered with the binding of IRF3 and p65 to the IFN-β promoter. Chromatin isolation by RNA purification (ChIRP), triplex pulldown, and biolayer interferometry assays indicated that lnc-MxA can bind to the IFN-β promoter. Furthermore, an electrophoretic mobility shift assay (EMSA) showed that lnc-MxA can form complexes with the IFN-β promoter fragment. These results demonstrated that lnc-MxA can form a triplex with the IFN-β promoter to interfere with the activation of IFN-β transcription. Using a vesicular stomatitis virus (VSV) infection assay, we confirmed that lnc-MxA can repress the RIG-I-like receptor (RLR)-mediated antiviral immune response and influence the antiviral status of cells. In conclusion, we revealed that lnc-MxA is an interferon-stimulated gene (ISG) that negatively regulates the transcription of IFN-β by forming an RNA-DNA triplex.
IMPORTANCE IAV can be recognized as a nonself molecular pattern by host immune systems and can cause immune responses. However, the intense immune response induced by influenza virus, known as a “cytokine storm,” can also cause widespread tissue damage (X. Z. J. Guo and P. G. Thomas, Semin Immunopathol 39:541–550, 2017, https://doi.org/10.1007/s00281-017-0636-y; S. Yokota, Nihon Rinsho 61:1953–1958, 2003; I. A. Clark, Immunol Cell Biol 85:271–273, 2007). Meanwhile, the detailed mechanisms involved in the balancing of immune responses in host cells are not well understood. Our studies reveal that, as an IFN-inducible gene, lnc-MxA functions as a negative regulator of the antiviral immune response. We uncovered the mechanism by which lnc-MxA inhibits the activation of IFN-β transcription. Our findings demonstrate that, as an ISG, lnc-MxA plays an important role in the negative-feedback loop involved in maintaining immune homeostasis.
INTRODUCTION
Influenza A virus (IAV) belongs to the family Orthomyxoviridae. Its genome consists of eight segments of negative single-stranded RNA, which encodes up to 18 proteins (1, 2). Influenza A virus can also be classified into different subtypes on the basis of 16 kinds of hemagglutinin (HA) and 9 kinds of neuraminidase (NA) (3). Influenza A virus often occurs seasonally and caused several pandemic outbreaks in 1918, 1957, 1968, and 2009, which led to huge economic losses and great damage to health worldwide (4). Avian influenza A virus (H7N9) can also infect human beings and cause death in humans (5).
Influenza virus infection can result in the initiation of the host antiviral immune response. The host can recognize nonself molecular patterns known as pathogen-associated molecular patterns (PAMPs) via pathogen recognition receptors (PRRs) (6). Influenza A virus can be recognized by retinoic acid-inducible protein I (RIG-I) and endosomal Toll-like receptors (TLRs), such as TLR3 and NOD-like receptor family pyrin domain-containing protein 3 (NLRP3) (7). The activated PRRs trigger downstream signaling pathways and induce the expression of inflammatory cytokines and type I interferon (IFN-I) (8). Interferons exert antiviral activity by upregulating a number of interferon-stimulated genes (ISGs), including MxA, ISG15, IFITM3, and ISG56, via the JAK/STAT signaling pathway (9). MxA, an interferon-induced GTPase, can suppress the replication of highly pathogenic influenza A viruses (10). IFITM1, IFITM2, and IFITM3 can inhibit an early step of influenza A virus replication (11). ISG56 can inhibit viral growth via translation-related pathways, as well as other pathways (12). Although IFNs, especially IFN-I, help hosts resist virus invasion, continuous IFN stimulation can cause immune disorders and even lead to autoimmune diseases (13). However, the mechanism involved in the regulation of immunity via negative feedback remains elusive.
Long noncoding RNAs (lncRNAs) are transcripts that are longer than 200 nucleotides (nt) that are not translated into proteins (14). LncRNAs are involved in a variety of biological activities. For example, IFN-inducible lnc-Lsm3b can inactivate RIG-I in the late stage of the innate immune response (15), and lncRNA-ACOD1 promotes viral replication by modulating cellular metabolism (16). In addition, lncRNA-NRAV modulates antiviral responses through the suppression of interferon-stimulated gene transcription (17). Previously, we found that lnc-ISG20 inhibits influenza A virus replication by enhancing ISG20 expression (18). These studies illustrate the importance of lncRNAs in virus infection.
In this study, we focused on the function of the IFN-β-induced lncRNA lnc-MxA. We found that lnc-MxA can promote the replication of IAV. Further studies indicated that lnc-MxA can form triplexes with the IFN-β promoter to interfere with the binding of IRF3 and NF-κB to the promoter and consequently inhibit the transcription of IFN-β. Our studies revealed that lnc-MxA is a negative regulator of the antiviral immune response and may play an important role in maintaining immune homeostasis.
RESULTS
Lnc-MxA is an interferon-stimulated gene.
Previously, we analyzed the differentially expressed lncRNAs in influenza A virus infection by RNA deep sequencing and identified a set of lncRNAs involved in viral replication (18). In this study, we focused on lnc-MxA (NONCODE ID: NONHSAG032905), which was upregulated during influenza A virus infection (Fig. 1A). The gene for lnc-MxA is within the MxA locus (Fig. 1B). It is known that MxA is an important interferon-stimulated gene (19). We performed absolute quantifications of lnc-MxA and MxA mRNAs by real-time quantitative PCR (RT-qPCR). As shown in Fig. 1C, lnc-MxA was more abundant than MxA in influenza virus A/WSN/33 (WSN)-uninfected cells, while both of them were greatly induced in virus-infected cells. We also examined the expression of lnc-MxA during Sendai virus (SeV) infection. The data showed that both lnc-MxA and MxA mRNAs were increased in Sendai virus-infected cells compared with uninfected cells (Fig. 1D). In addition, lnc-MxA was upregulated in poly(I·C)-transfected cells (Fig. 1E). To determine whether lnc-MxA is an interferon-stimulated gene, we treated A549 cells with IFN-β and measured lnc-MxA by RT-qPCR. The data indicated that lnc-MxA was significantly increased in IFN-β-treated cells (Fig. 1F). These results demonstrate that lnc-MxA is an interferon-stimulated gene. We examined the subcellular localization of lnc-MxA by cell fractionation and RT-qPCR. The data showed that lnc-MxA was mainly localized in the nucleus in IAV-infected cells (Fig. 1G). We determined the full length of lnc-MxA by 5′ and 3′ rapid amplification of cDNA ends (RACE) assays. The PCR products of 5′ and 3′ RACE and sequencing data are shown in Fig. 1H.
FIG 1.

Lnc-MxA is an interferon-stimulated gene. (A) A549 cells were infected with influenza virus A/WSN/33 (H1N1) at an MOI of 0.1 for 4 h, 8 h, or 16 h. The relative levels of lnc-MxA in the infected A549 cells were examined by RT-qPCR (n = 3; means and SD; *, P < 0.05; **, P < 0.01). (B) Diagram showing the genomic location of lnc-MxA (red) and its relationship with the MxA gene (blue). The primers for lnc-MxA are indicated. (C) A549 cells were infected with WSN at an MOI of 0.1 for 8 h. Absolute quantification of MxA and lnc-MxA was examined by RT-qPCR (n = 3; means and SD). (D) A549 cells were infected with SeV at an MOI of 0.01 for 6 h. Total RNA was prepared and subjected to RT-qPCR for lnc-MxA with MxA (n = 3; means and SD; **, P < 0.01). (E) A549 cells were induced with poly(I·C) at the indicated doses for 6 h. Total RNA samples were prepared and subjected to RT-qPCR (n = 3; means and SD; **, P < 0.01). (F) A549 cells were stimulated with increasing amounts of IFN-β for 16 h. RT-qPCR was performed to determine the levels of lnc-MxA and MxA (n = 3; means and SD; **, P < 0.01). (G) A549 cells were infected with WSN at an MOI of 0.1 for 8 h (+) or not infected (−). The RNAs from nuclear and cytoplasmic fractions were prepared and subjected to RT-qPCR (n = 3; means and SD). (H) The 5′ and 3′ end sequences of lnc-MxA in A549 cells were determined by 5′ and 3′ RACE. (I) C-terminally FLAG-tagged full-length lnc-MxA in all three reading frames and the longest ORF of lnc-MxA (lnc-MxA-L) was cloned into pcDNA3.1 with a C-terminal FLAG tag. The plasmids were transfected into 293T cells for 48 h. Cell lysates were harvested and subjected to immunoblotting with antibodies against FLAG or GAPDH. FLAG-ISG15 served as a positive control.
To verify that lnc-MxA is indeed a noncoding RNA, we generated plasmids containing C-terminally FLAG-tagged lnc-MxA (full length) in all three reading frames or the longest open reading frame (ORF) of lnc-MxA and transfected them into 293T cells using pCMV-ISG15-FLAG as a positive control. The cell lysates were harvested for immunoblotting assays. The data confirmed that lnc-MxA cannot be translated (Fig. 1I).
Lnc-MxA facilitates the replication of influenza A virus.
Next, we sought to determine whether lnc-MxA can affect the replication of influenza A virus. We transfected the lnc-MxA-expressing plasmids in the indicated amounts into 293T cells for 28 h and then infected the cells with influenza virus A/WSN/33 for 12 h. The relative amounts of lnc-MxA in lnc-MxA-expressing cells and control cells are shown in Fig. 2A. The cell lysates were harvested for immunoblotting with the indicated antibodies. The data showed that the levels of both the NP and the M1 proteins in lnc-MxA-expressing cells were higher than those in control cells (Fig. 2B). The supernatants were collected for plaque assay. As shown in Fig. 2C, the virus titers in the supernatant obtained from lnc-MxA-expressing cells were significantly higher than those in the supernatant obtained from the control cells. These data suggest that the overexpression of lnc-MxA favors the replication of IAV. Then, we generated lnc-MxA knockdown cells (sh-lnc-MxA 1 and 2) (Fig. 2D) and infected the cells with influenza A virus. The data showed that the levels of the NP and the M1 proteins in the lnc-MxA knockdown cells were lower than those in the control cells (Fig. 2E). The virus titer in the supernatant obtained from the lnc-MxA knockdown cells was significantly lower than that in the supernatant obtained from the control cells (Fig. 2F). Overall, these results demonstrate that lnc-MxA facilitates the replication of IAV.
FIG 2.

Lnc-MxA has profound effects on IAV replication. (A to C) 293T cells were seeded into 12 wells and transfected with the indicated amounts of lnc-MxA plasmid or empty vector (EV) for 28 h. (A) The level of lnc-MxA was determined by RT-qPCR. (B and C) The cells were then infected with influenza virus A/WSN/33 at an MOI of 0.5 for 12 h. The cell lysates were harvested for immunoblotting with antibodies against the indicated proteins (B), and the supernatants were collected for a plaque assay (C). (D to F) 293T cells were transfected with control (sh-luc) or lnc-MxA shRNA plasmids. (D) The efficiency of shRNA-based knockdown of lnc-MxA was determined by RT-qPCR. (E and F) The cells were then infected with influenza virus A/WSN/33 at an MOI of 1 for 12 h. The cell lysates were harvested for immunoblotting with antibodies against the indicated proteins (E), and the supernatants were collected for a plaque assay (F). n = 3; means and SD; **, P < 0.01.
Lnc-MxA inhibits the activation of IFN-β.
We wanted to know whether lnc-MxA affects the replication of IAV by interfering with the antiviral immune response. We first examined the effect of lnc-MxA on the activation of IFN-β in Sendai virus-infected cells. The data from the luciferase reporter assay showed that the overexpression of lnc-MxA inhibits the activation of the IFN-β promoter in both Sendai virus-infected cells and poly(I·C)-transfected cells (Fig. 3A and B). Since Sendai virus and poly(I·C) could stimulate the RIG-I signaling pathway, we determined which step lnc-MxA might interfere with. We coexpressed lnc-MxA with RIG-I, active RIG-I (RIG-I N), MAVS, TBK1, or active IRF3 (IRF-5D) and an IFN-β promoter reporter in 293T cells and infected the cells with Sendai virus. The cell lysates were collected for the luciferase assay. The data indicated that the activation of the IFN-β reporter upon stimulation with the above-mentioned components was significantly reduced in lnc-MxA-expressing cells compared with that in control cells (Fig. 3C). We also measured the IFN-β mRNA level in lnc-MxA-expressing cells stimulated by IRF-5D. As shown in Fig. 3D, the level of IFN-β mRNA in lnc-MxA-expressing cells was lower than that in control cells. We also determined whether lnc-MxA affects the levels of protein activators (RIG-I, MAVS, phosphorylated TBK1 [p-TBK1], and IRF3) of IFN-β promoter in SeV-induced or poly(I·C)-transfected cells. The data indicated that lnc-MxA expression did not affect the levels of these proteins (Fig. 3E). Next, we generated the A549-lnc-MxA cell line, which stably expressed lnc-MxA, and the control cell line, A549-ctrl. We infected the cells with Sendai virus and measured the mRNA levels of IFN-β and other ISGs, such as MxA, IFITM1, IFITM3, and ISG15. The data showed that the expression of IFN-β and the ISGs in A549-lnc-MxA cells was significantly lower than that in control cells (Fig. 3F). Consistently, the protein levels of MxA and ISG15 in A549-lnc-MxA cells were obviously lower than those in A549-ctrl cells (Fig. 3G). As lnc-MxA facilitates IAV replication, we examined whether lnc-MxA could affect IFN-β and ISG expression during IAV infection. The data showed that the overexpression of lnc-MxA inhibited IFN-β and ISG expression in IAV-infected cells (Fig. 3H and I).
FIG 3.

Lnc-MxA restrains IFN-β transcription. (A and B) 293T cells were transfected with an IFN-β luciferase reporter along with an empty vector (EV) or an expression vector for lnc-MxA for 28 h. Then, the cells were infected with SeV at an MOI of 1 for 12 h (A) or stimulated with poly(I·C) (2 μg/ml) for 12 h (B). The cell lysates were harvested for a luciferase assay (n = 3; means and SD; *, P < 0.05; **, P < 0.01). (C) 293T cells were transfected with IFN-β luciferase reporter along with the indicated plasmids and either an empty vector or an expression vector for lnc-MxA for 28 h. The cell lysates were harvested for luciferase assay and immunoblotting analysis with the indicated antibodies (n = 3; means and SD; **, P < 0.01). (D) IFN-β mRNA expression was examined by RT-qPCR in 293T cells coexpressing lnc-MxA along with IRF3-5D. (E) 293T cells were transfected with an empty vector or an expression vector for lnc-MxA for 28 h. Then, the cells were infected with SeV or stimulated with poly(I·C). The cell lysates were collected and subjected to immunoblotting with the indicated antibodies. (F to I) A549-lnc-MxA and A549-ctrl cells were infected with SeV at an MOI of 0.1 (F and G) or influenza virus A/WSN/33 (H1N1) at an MOI of 0.1 (H and I) for 12 h. (F and H) Then, the total RNA and protein were prepared. The mRNA levels of the ISGs were determined by RT-qPCR (n = 3; means and SD; *, P < 0.05; **, P < 0.01). (G and I) The proteins were used for immunoblotting with antibodies against the indicated proteins.
Next, we examined the effect of lnc-MxA knockdown on the activation of IFN-β. As shown in Fig. 4A, the mRNA levels of IFN-β and the ISGs (MxA, IFITM1, IFITM3, and ISG15) in lnc-MxA knockdown cells were significantly higher than those in control cells during Sendai virus infection. The efficiency of lnc-MxA knockdown in lnc-MxA knockdown A549 cells is shown in Fig. 4A (first graph). We performed similar experiments by infecting the cells with IAV. Consistently, the mRNA levels of IFN-β and the ISGs in lnc-MxA knockdown cells were significantly higher than those in control cells infected with IAV (Fig. 4B). The protein levels of ISG15 and MxA in lnc-MxA knockdown cells were also obviously higher than those in control cells infected with either Sendai virus or IAV (Fig. 4C and D). These results demonstrate that lnc-MxA negatively regulates the RIG-I-mediated antiviral immune response. To further analyze the mechanism involved in the regulation of the RIG-I signaling pathway by lnc-MxA, we knocked down lnc-MxA in 293T cells (Fig. 4E) and then transfected the cells with MAVS-, TBK1-, or IRF3-5D-expressing plasmids and an IFN-β luciferase reporter. The data showed that the activation of the IFN-β reporter upon stimulation with MAVS, TBK1, or IRF3-5D was significantly higher in lnc-MxA knockdown cells than in control cells (Fig. 4F). These data suggest that lnc-MxA acts downstream of IRF3.
FIG 4.

Knockdown of lnc-MxA promotes the expression of IFN-β. (A to D) lnc-MxA knockdown A549 cells and sh-luc control cells were infected with SeV at an MOI of 0.05 (A and C) or influenza virus A/WSN/33 (H1N1) at an MOI of 0.05 (B and D) for 12 h. (A and B) Then, total RNA and protein were prepared. The mRNA levels of the ISGs and lnc-MxA were determined by RT-qPCR (n = 3; means and SD; *, P < 0.05; **, P < 0.01). (C and D) The proteins were used for immunoblotting with antibodies against the indicated proteins. (E) The efficiency of lnc-MxA knockdown in lnc-MxA knockdown 293T cells was determined by RT-qPCR (n = 3; means and SD; **, P < 0.01). (F) 293T cells were transfected with an IFN-β luciferase reporter along with the indicated plasmids and either a lnc-MxA shRNA vector or a control vector for 36 h. The cell lysates were harvested for luciferase assay and immunoblotting analysis (n = 3; means and SD; *, P < 0.05; **, P < 0.01).
Lnc-MxA binds to the IFN-β promoter by forming RNA-DNA triplexes.
As lnc-MxA inhibits IRF3 transcriptional activity at the IFN-β promoter, we first examined whether it interacts with IRF3 by RNA immunoprecipitation (RIP) assay. As shown in Fig. 5A, lnc-MxA did not interact with either IRF3 or NF-κB subunit p65. Next, we determined whether lnc-MxA affected the association of IRF3 and p65 with the IFN-β promoter using a chromatin immunoprecipitation (ChIP) assay. The data showed that the relative enrichment of IRF3 and p65 at the IFN-β promoter in lnc-MxA-overexpressing cells was significantly lower than that in control cells (Fig. 5B), suggesting that lnc-MxA interfered with the binding of IRF3 and p65 to the IFN-β promoter. By using Triplexator (see below), it was predicted that lnc-MxA might form a triplex with the IFN-β promoter region (Fig. 5C, boxed), which partially overlaps the IRF3 binding site (red) and is near the NF-κB binding site (blue). We generated S1-tagged wild-type (WT) lnc-MxA and an S1-tagged lnc-MxA mutant (described in Materials and Methods) and performed a chromatin isolation by RNA purification (ChIRP) assay. The data indicated that WT lnc-MxA, but not the lnc-MxA mutant, could bind to the IFN-β promoter (Fig. 5D). Furthermore, we carried out an in vitro triplex pulldown assay by using biotin-labeled WT lnc-MxA or its mutant. The results showed that wild-type lnc-MxA could bind to the IFN-β promoter, while the lnc-MxA mutant could not (Fig. 5E). Next, we performed biolayer interferometry (BLI) using biotinylated IFN-β promoter fragments. The data indicated that lnc-MxA can interact with the IFN-β promoter (Fig. 5F). To confirm triplex formation of lnc-MxA on the IFN-β promoter, we carried out an electrophoretic mobility shift assay (EMSA) using a biotinylated IFN-β promoter fragment as the probe. As shown in Fig. 5G and H, wild-type, but not mutant, lnc-MxA could form complexes with biotinylated IFN-β promoter fragments. In addition, the DNA-RNA complexes were not disrupted by RNase H treatment, ruling out the possibility that the band shift occurred due to DNA-RNA heteroduplexes (Fig. 5I). Overall, these results demonstrated that lnc-MxA can form a triplex with an IFN-β promoter and interfere with the activation of IFN-β transcription.
FIG 5.

Lnc-MxA binds to the promoter of IFN-β by forming an RNA-DNA triplex. (A) A549 cells were infected with SeV at an MOI of 0.1 for 12 h. Then, the cells were harvested, and RIP assays were performed using IRF3 antibody and p65 antibody or IgG (n = 3; means and SD; N.S, not significant). (B) lnc-MxA-overexpressing A549 cells were infected with SeV at an MOI of 0.1 for 12 h. Then, the cells were harvested for ChIP-qPCR assay using IRF3 antibody, p65 antibody, or IgG (n = 3; means and SD; **, P < 0.01). (C) Positions of the triplex-forming oligonucleotides (TFO) predicted by Triplexator and their relationship with the binding sites of transcription factors (red, IRF3 binding site; blue, NF-κB binding site) on the IFN-β promoter (boxed). TTS, triplex target sites. (D) 293T cells were transfected with WT lnc-MxA-S1, lnc-MxA-S1 mutant plasmids, or S1 empty vector (EV-S1) as a control for 36 h, followed by infection with SeV at an MOI of 1 for 12 h. Then, the cells were harvested for ChIRP-qPCR assay (n = 3; means and SD; *, P < 0.05). (E) Biotinylated wild-type lnc-MxA or its mutant was incubated with IFN-β promoter fragments (−875/−648) generated by standard PCR for 20 min at room temperature. Upon binding to streptavidin beads, the associated DNA was analyzed by qPCR. The data represent the fold increase in DNA bound to the respective RNAs compared with that measured in assays without RNA (n = 3; means and SD; **, P < 0.01). (F) The ForteBio Octet Red system was used to examine the binding affinities of lnc-MxA for the IFN-β promoter. The vertical and horizontal axes represent the light shift distance (nanometers) and the association/dissociation time(s), respectively. (G) lnc-MxA forms DNA-RNA triplexes with the IFN-β promoter. Increasing amounts (40-, 80-, or 160-fold molar excess) of lnc-MxA were incubated with double-stranded biotinylated IFN-β promoters, and formation of RNA-DNA triplexes was monitored by EMSA. (H) Wild-type lnc-MxA or its mutant was incubated with double-stranded biotinylated IFN-β promoter, and the formation of RNA-DNA triplexes was monitored by EMSA. (I) Reaction mixtures containing labeled IFN-β promoter and an 80-fold molar excess of lnc-MxA were treated with 0.5 U of RNase H or with 0.5 ng RNase A for 30 min at room temperature.
Lnc-MxA negatively regulates antiviral immune responses.
To further understand the function of lnc-MxA in the antiviral immune response, we employed a vesicular stomatitis virus (VSV) infection assay (20). A549 cells with either the overexpression or knockdown of lnc-MxA were transfected with poly(I·C). The conditional medium was collected and applied to A549 cells. Then, the cells were harvested, and the mRNAs were prepared for RT-PCR. As shown in Fig. 6A, the mRNA levels of the ISGs (MxA, ISG15, IFITM3, IFIT1, and IFIT2) in A549 cells treated with conditional medium from lnc-MxA-expressing cells were significantly lower than those in A549 cells treated with conditional medium from the control cells. The levels of the ISG mRNAs in A549 cells treated with conditional medium from A549-sh-lnc-MxA cells were higher than those in A549 cells treated with supernatants from control cells (Fig. 6B). These results suggest that lnc-MxA inhibits type I interferon expression in poly(I·C)-transfected A549 cells. Next, we infected the supernatant-treated A549 cells with green fluorescent protein (GFP)-modified vesicular stomatitis virus (VSV-GFP). Fluorescence-activated cell sorter (FACS) analysis indicated that both the mean fluorescence intensity (MFI) and the percentage of VSV-GFP-positive cells among A549 cells treated with supernatant from lnc-MxA-expressing cells were higher than those in control cells (Fig. 6C and D). Consistently, the MFI and the percentage of VSV-GFP-positive cells among A549 cells treated with supernatant from A549-sh-lnc-MxA cells were lower than those in control cells (Fig. 6E and F). Overall, these results indicate that lnc-MxA negatively regulates the antiviral immune response.
FIG 6.

Lnc-MxA negatively regulates antiviral immune responses. (A and B) Lnc-MxA-overexpressing A549 cells, knockdown cells, or control cells were stimulated with poly(I·C) (0.5 μg/ml) for 6 h. The supernatants were harvested and used to treat fresh A549 cells for 6 h. Then, total RNA was prepared, and the ISGs were examined by RT-qPCR (n = 3; means and SD; *, P < 0.05; **, P < 0.01). (C to F) Lnc-MxA-overexpressing A549 cells, knockdown cells, or control cells were stimulated with poly(I·C) (0.5 μg/ml) for 6 h. The supernatants were harvested and used to treat fresh A549 cells for 6 h. Then, the A549 cells were infected with GFP-tagged VSV for 14 h. The percentage and MFI of VSV-GFP-positive A549 cells were determined by flow cytometry (n = 3; means and SD; *, P < 0.05; **, P < 0.01).
To further verify whether lnc-MxA negatively regulates the antiviral immune response in an IFN-β–dependent manner, we examined whether lnc-MxA could still affect the replication of influenza A virus in 293T IFNAR1 knockout cells. As shown in Fig. 7A and B, lnc-MxA promoted the replication of IAV in WT cells, but not in IFNAR1 knockout cells. These results demonstrate that lnc-MxA affects IAV replication through the IFN-β signaling pathway.
FIG 7.

Lnc-MxA affects IAV replication in an IFN-β-dependent manner. WT and IFNAR1 knockout (IFNAR1 KO) 293T cells were transfected with lnc-MxA or empty-vector plasmid for 28 h. The cells were then infected with influenza virus A/WSN/33 at an MOI of 0.01 for 24 h. The cell lysates were harvested for immunoblotting with antibodies against the indicated proteins (A), and the supernatants were collected for a plaque assay (B) (n = 3; means and SD; **, P < 0.01; N.S, not significant).
In summary, we identified lnc-MxA as a novel interferon-stimulated gene. Our studies show that lnc-MxA inhibits the activation of IFN-β transcription during RNA virus infection and the virus-induced innate immune response. Further studies demonstrated that lnc-MxA inhibits the RIG-I-like receptor (RLR)-mediated signaling pathway by forming a triplex with the IFN-β promoter, which interferes with the binding of IRF3 and NF-κB to the promoter. Consequently, this attenuates the activation of IFN-β transcription and the downstream signaling pathway (Fig. 8). These findings reveal that lnc-MxA is an important negative regulator of antiviral immunity.
FIG 8.
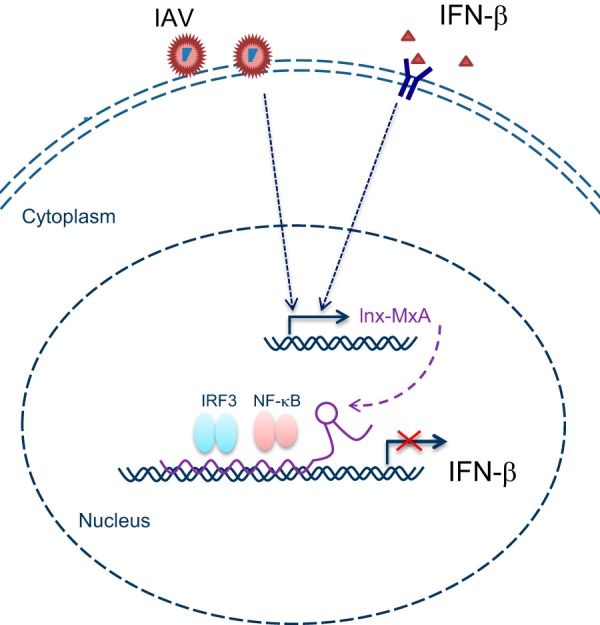
Illustrative model showing the proposed mechanism by which lnc-MxA inhibits the expression of IFN-β via the formation of RNA-DNA triplexes. See the text for details.
DISCUSSION
Influenza virus is a very important human and animal pathogen that can cause extensive morbidity and mortality (21). Type I interferons (IFN-α and IFN-β) are key innate antiviral defense effectors (22). The majority of components involved in controlling the antiviral immune response are proteins, including MxA, IFITM family proteins, PKR, OAS family proteins, and IFIT family proteins. Human MxA protein has been shown to interfere with the intracytoplasmic transport of viral mRNAs, the synthesis of viral proteins, and the translocation of newly synthesized viral proteins to the cell nucleus (23). MxA prevents the viral genome from entering the nucleus to block IAV replication (24). IFITM family proteins inhibit IAV replication at an early stage and restrict the early replication of flaviviruses, including dengue virus and West Nile virus (11). IFIT1 can bind and sequester viral 5′-triphosphate RNAs (5′ ppp-RNAs); when infected with VSV, Ifit1-deficient mice were much less viable than wild-type mice (25–27). Recently, long noncoding RNAs have been found to play important roles in regulating antiviral signaling pathways. For example, the long noncoding RNA NEST binds to WDR5 to alter histone 3 methylation at the IFN-γ locus (28), and lincRNA-THRIL regulates tumor necrosis factor alpha (TNF-α) expression through its interaction with hnRNPL (29). In addition, lincRNA-Cox2 interacts with heterogeneous nuclear ribonucleoproteins A/B and A2/B1 to mediate both the activation and the repression of immune response genes (30), and lnc-NEAT1 increases interleukin 8 (IL-8) transcription by sequestering the repressor SFPQ in the paraspeckle (31). Furthermore, lnc-DC binds directly to STAT3 to control human dendritic cell (DC) differentiation (32). In this study, we found that the IFN-β-induced lncRNA lnc-MxA can inhibit the expression of IFN-β, which plays a role in the negative-feedback loop involved in the antiviral immune response. These findings suggest that lncRNA may contribute to the homeostatic balance in hosts infected by viruses.
In recent studies, it has been demonstrated that lncRNAs perform their functions by forming RNA-DNA triplexes. Nucleolar promoter-associated RNAs (pRNAs) can compete with the transcription factor TTF-I for binding to the rRNA gene promoter by forming RNA-DNA triplexes to mediate transcriptional silencing (33). The lncRNA Khps1 promotes SPHK1 transcription by forming an RNA-DNA triplex structure with the promoter of SPHK1 (34). The lncRNA MEG3 regulates transforming growth factor β (TGF-β) pathway genes through the formation of RNA-DNA triplexes (35). The lncRNA PARTICLE acts as a scaffold for the assembly of gene-silencing machinery in response to irradiation via triplex formation (36). In our study, we found that lnc-MxA forms an RNA-DNA triplex with the purine-rich region of the IFN-β promoter to prevent the binding of transcription factors (IRF3 and p65), which inhibits the activation of IFN-β. Our study contributes to the emerging concept that lncRNAs can influence gene transcription by triplex formation to control cellular processes.
Interferons exert antiviral activity by upregulating a number of ISGs, including MxA, ISG15, IFITM3, and ISG56, via the JAK/STAT signaling pathway (9). Most ISGs encode proteins; however, in recent studies, several groups have found that a variety of lncRNAs are induced by type I, II, and III IFNs (37–40). For example, lnc-ISG15 and lnc-BST2 were upregulated after treatment with different doses of type I IFN-α2 in different types of cells or after treatment with type III IFN-λ (44); the expression of lncRNA-CMPK2, which is a spliced, polyadenylated nuclear transcript, is induced by IFN in diverse cell types in humans and mice, and its induction is dependent on JAK/STAT signaling (37). Our previous study demonstrated that lnc-ISG20 can also be upregulated by IFN-β and inhibit IAV replication (18). LncRNA can also be directly regulated by viral proteins. For example, the lncRNA DREH is downregulated by the HBX protein (41), and lnc-NRON levels are reduced by the early viral accessory protein Nef and increased by the late protein Vpu from HIV (42). In addition, lnc-ACOD1, which is induced by multiple viruses but not by type I interferon, promotes viral replication by modulating cellular metabolism (16). Our studies revealed that lnc-MxA is stimulated by IFN-β and negatively regulates immune responses as a novel ISG. These studies demonstrate that lncRNAs represent another set of ISGs that play important roles in the antiviral immune response.
In summary, we have shown that lnc-MxA can promote the replication of IAV. We demonstrated that lnc-MxA inhibits the transcription of IFN-β via the formation of an RNA-DNA triplex. This research provides more evidence that host lncRNAs are involved in influenza virus replication and immune homeostasis, which will provide new insights for the design of drugs that can treat influenza virus and autoimmune inflammatory diseases.
MATERIALS AND METHODS
Cell lines and viruses.
Madin-Darby canine kidney (MDCK) cells, human embryo kidney 293T cells, and human alveolar epithelial A549 cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, USA). 293T IFNAR1 knockout cells were kindly provided by Liguo Zhang at the Institute of Biophysics, Chinese Academy of Sciences, Beijing, China. Recombinant influenza virus A/WSN/33 was generated by reverse genetics as described previously (43). SeV and VSV-GFP were kindly provided by Wenjun Liu at the Institute of Microbiology, Chinese Academy of Sciences, Beijing, China.
Plasmids and antibodies.
The full-length sequence of lnc-MxA was cloned into the lentiviral pLentiLox3.7 plasmid. To generate pLL3.7-lnc-MxA-S1, a fragment containing a 44-nt-long S1 sequence (5′-ACCGACCAGAATCATGCAAGTGCGTAAGATAGTCGCGGGCCGGG-3′) was cloned into the 3′ end of the plasmid. Short hairpin RNA (shRNA)-based knockdown plasmids were generated with a pSIH-H1-GFP lentiviral vector expressing shRNA. The C-terminally FLAG-tagged full-length lnc-MxA, lnc-MxA-T, lnc-MxA-TT, longest ORF of lnc-MxA, and ISG15 were cloned into pcDNA3.1.
The triplex was formed by a Hoogsteen hydrogen bond or reverse Hoogsteen hydrogen bond between the third base in the RNA and the Watson-Crick base pair in the DNA, which suggested that TA-A or CG-G is critical for the lnc-MxA (118AAAGAGAAGAAGGGA132) and IFN-β triplex. The mutant pLL3.7-lnc-MxA-MUT-S1 was generated by mutating 118AAAGAGAAGAAGG GA132 in pLL3.7-lnc-MxA-WT-S1 to TGTGTTGATGGTCCG.
Mouse anti-M1 monoclonal antibody (MAb) and rabbit anti-NP polyclonal antibody were kindly provided by Wenjun Liu (Institute of Microbiology, Chinese Academy of Sciences). Mouse anti-β-actin MAb (KM9001) and mouse anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) MAb (KM9002) were purchased from Sungene Biotech Co. Rabbit anti-MxA polyclonal antibody (13750-1-AP) was purchased from Proteintech, and mouse anti-ISG15 MAb was purchased from Santa Cruz. Rabbit anti-IFNAR1 polyclonal antibody (ab91466) was purchased from Abcam. Antibodies against RIG-I (3743), MAVS (3993), p-IRF3 (4947), IRF3 (4302), TBK1 (3504), p-TBK1 (5483), p-p65 (3033), and p65 (8242) were from Cell Signaling Technology.
Generation of stable expression and knockdown cell lines.
The lentiviral vectors (pLP1, pLP2, and pLP/VSVG) were cotransfected with pLL-3.7-lnc-MxA or pSIH-lnc-MxA-shRNA into 293T cells. The medium was changed the following day, and the virus-containing supernatant was collected 48 h after transfection and filtered through a 0.45-μm filter. Then, the retrovirus-containing supernatants were used to infect A549 cells, along with Polybrene (8 μg/ml). Infected cells were selected by flow cytometry. The shRNA target sequences were as follows: shRNA-lnc-MxA-1, GCATCTTAAGAAGAGAACAAA; shRNA-lnc-MxA-2, GAACAAATGATCCTACCAATA.
Luciferase reporter assay.
293T cells were cotransfected with the IFN-β promoter luciferase reporter (100 ng) and Renilla luciferase internal control (pRL-TK) (10 ng), along with 100 ng of plasmid encoding FLAG-RIG-I, FLAG-RIG-I (N), FLAG-MAVS, FLAG-TBK1, or FLAG-IRF3 (5D) and plasmid expressing lnc-MxA, using Lipofectamine 2000 (Invitrogen, USA). The empty pLL3.7 vector was used to ensure that equal amounts of DNA were distributed among the wells. Then, the cells were stimulated with poly(I·C) or infected with SeV at the indicated time points. The cells were collected, and luciferase activity was measured with a dual-luciferase assay (Promega, USA) and a Luminoskan Ascent luminometer (Thermo Scientific, USA) according to the manufacturers’ protocols, as previously described (44). Reporter gene activity was determined by the normalization of the firefly luciferase activity to that of Renilla luciferase.
RT-qPCR analysis.
Total RNA was isolated from cells using TRIzol (Invitrogen, USA). Then, cDNA was generated from the total RNA using cDNA Synthesis SuperMix (TransGen Biotech, China). RT-qPCR was performed using SYBR Green real-time PCR master mix (Toyobo, Japan) with specific primers. The following primers were used: lnc-MxA-forward, TCAAATAAATGTATGCCAGGGGTCA, and -reverse, GGAGGCGGATCACTTCTCAC; IFN-β-forward, AACAAGTGTCTCCTCCAAATTGC, and -reverse, GCAGTATTCAA GCCTCCCATTC; MxA-forward, GGTGGTGGTCCCCAGTAATG, and -reverse, ACCACGTCCACAACCTTGTCT; IFIT1-forward, GCCATTTTCTTTGCTTCCCCTA, and -reverse, TGCCCTTTTGTAGCCTCCTTG; IFIT2-forward, CACCTCTGGACTGGCAATAGC, and -reverse, GTCAGGATTCAGCCGAATGG; IFITM1-forward, ACAGGAAGATGGTTGGCGAC, and -reverse, GTAGACTGTCACAGAGCCGAA; IFITM3-forward, GCTGATCTTCCAGGCCTATG, and -reverse, GATACAGGACTCGGCTCCGG; ISG15-forward, ATGGGCTGGGACCTGACGG, and reverse, TTAGCTCCGCCCGCCAGGCT; GAPDH-forward, GTCGGAGTCAACGGATTTGG, and reverse, CGCCCCACTTGATTTTGG.
Absolute quantifications of lnc-MxA and MxA were measured by RT-qPCR using the standard curve generated with the serially diluted PCR products of MxA and lnc-MxA. The number of RNA molecules was then calculated accordingly.
5′ and 3′ RACE.
The 5′ and 3′ RACE analyses were performed using a SMARTer RACE cDNA amplification kit (Clontech). The RACE PCR products were cloned into the pESI-Blunt simple vector (Yeasen, Shanghai, China) and sequenced.
RNA immunoprecipitation.
A549 lnc-MxA-overexpressing cells were seeded and infected with SeV at a multiplicity of infection (MOI) of 1 for 12 h. Then, the cells were cross-linked by UV, as previously described (45). The cells were washed with phosphate-buffered saline (PBS) and lysed with 1 ml radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitor (Roche, Switzerland) and RNase inhibitor (Thermo, USA). The lysates were precleared by incubation with 25 μl protein G magnetic beads (Pierce, USA) for 2 h at 4°C. The precleared lysates were incubated with 2 μg anti-IRF3 antibody, 2 μg anti-p65 antibody, or 2 μg mouse IgG antibody overnight at 4°C. After washing the beads with RIPA buffer and digesting the proteins with proteinase K, the RNA was extracted from the beads and subjected to RT-qPCR to determine the amount of lnc-MxA RNA.
Chromatin immunoprecipitation.
The ChIP assay was performed using an EZ-ChIP kit (Millipore, USA) with anti-IRF3 and anti-p65 antibodies and mouse IgG antibody (Santa Cruz, USA) according to the manufacturer’s instructions. Briefly, A549 cells with stable lnc-MxA expression and control cells were infected with SeV at an MOI of 1 for 12 h. After cross-linking with 1% formaldehyde, the cells were harvested and resuspended in SDS lysis buffer. The lysates were sonicated and then incubated with anti-IRF3, anti-p65, or mouse IgG antibody. The lysates were captured with protein G agarose. The chromatin DNA was eluted from the beads. After treatment with proteinase K, the bound DNA was extracted using phenol-chloroform and precipitated with ethanol. The quantity of DNA was determined by real-time PCR with specific primers (IFN-β-F, TTCCTTTGCTTTCTCCCAAGTC; IFN-β-R, CAGAGGAATTTCCCACTTTCACTT).
Chromatin isolation by RNA purification.
HEK 293T cells were transfected with pLL3.7-lnc-MxA-WT-S1, pLL3.7-lnc-MxA-MUT-S1, or S1 empty vector (EV-S1) for 36 h. Then, the cells were infected with SeV at an MOI of 1 for 12 h. The cells were harvested and cross-linked with 1% glutaraldehyde, followed by sonication. The lysates were subjected to pulldown with streptavidin beads, as previously described (46), and the bound DNA was quantified by RT-qPCR with IFN-β promoter-specific primers.
In vitro triplex pulldown assay.
The IFN-β promoter fragment (−271/+82) was amplified by PCR and digested by exonuclease I (TaKaRa, Japan) for 30 min at 37°C. Then, 100 fmol of the fragment was incubated with 1 pmol of biotin-labeled WT lnc-MxA(5′-biotin-GCUUUUUCAUGGUAAAGAGAAGAAGGGAGCGG-3′) or mutant lnc-MxA (5′-biotin-GCUUUUUCAUGGUCCCUCGCUCUCUGUUGCGG-3′) in 10 mM Tris-HCl (pH 7.5), 20 mM KCl, 10 mM MgCl2, 0.05% Tween 20, and 100 U of RNase inhibitor (Thermo, USA) for 20 min at 25°C. The RNA-DNA complexes were incubated with streptavidin-coated Dynabeads (Life Technologies, USA) for 1 h at 25°C. The beads were washed three times with buffer containing 150 mM KCl, 10 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 0.5% NP-40, and 100 U of RNase inhibitor and were washed once with buffer containing 15 mM KCl, 10 mM Tris-HCl (pH 7.5), and 5 mM MgCl2. The RNA-associated DNA was eluted in 1% SDS, 50 mM Tris-HCl (pH 8.0), and 10 mM EDTA for 5 min at 65°C and digested with RNase A (50 ng/ml) for 30 min at 37°C and with proteinase K (200 ng/ml) for 15 min at 37°C. The recovered DNA was subjected to RT-qPCR using primers specific for the IFN-β promoter.
Electrophoretic mobility shift assay.
One picomole of biotin-labeled IFN-β promoter fragment (−72/−40) was incubated with increasing amounts (40-, 80-, and 160-fold molar excess) of in vitro-transcribed lnc-MxA (+10/+157) in 40 mM Tris-acetate (pH 7.5), 20 mM KCl, 10 mM Mg(CH3COO)2, and 10% glycerol. Triplex formation was monitored by EMSA on 12% polyacrylamide gels containing 40 mM Tris-acetate (pH 7.5) and 10 mM MgCl2, and the gel contents were transferred to a nylon membrane. The biotin-labeled IFN-β fragment and its complexes were detected using a LightShift chemiluminescent EMSA kit (Thermo, USA).
Triplexator.
Triplexator, which is a computational framework for in silico prediction of triplex structures within specific genomic regions, was used to predict whether lnc-MxA could form a triplex with the IFN-β promoter (47). The Triplexator software can be accessed on the Internet (http://bioinformatics.org.au/tools/triplexator/inspector/vm_guide.html).
Biolayer interferometry.
The binding affinity between the IFN-β promoter and lnc-MxA was measured by biolayer interferometry (ForteBio, Inc.). The biotinylated IFN-β promoter was immobilized onto the streptavidin biosensors, and the association and dissociation of lnc-MxA in 200 μl of buffer containing 10 mM Tris-acetate (pH 7.5), 50 mM NaCl, and 1 mM EDTA were monitored. The dissociation constants were calculated with Octet RED software. The sequences of the biotinylated IFN-β promoter were as follows: 5′-TTTTTTTTTTTTTTTAACTGAAAGGGAGAAGTGAAAGTGGGAAAT-3′ and 3′-TTGACTTTCCCTCTTCACTTTCACCCTTTA-5′.
Quantification and statistical analysis.
The data are represented as means and standard deviations (SD) unless otherwise indicated, and Student's t test was used for all statistical analyses, which were performed with GraphPad Prism 5 software. Differences between two groups were considered significant when the P value was ≤0.05.
ACKNOWLEDGMENTS
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB29010000), the Ministry of Science and Technology of China (2015CB910502), and the National Natural Science Foundation of China (81772989 and 31600129). Xin Ye is the principal investigator of the Innovative Research Group of the National Natural Science Foundation of China (81621091).
Xin Ye is the principal investigator of the Innovative Research Group of the National Natural Science Foundation of China (81621091).
REFERENCES
- 1.Yamayoshi S, Watanabe M, Goto H, Kawaoka Y. 2016. Identification of a novel viral protein expressed from the PB2 segment of influenza A virus. J Virol 90:444–456. doi: 10.1128/JVI.02175-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manzoor R, Igarashi M, Takada A. 2017. Influenza A virus M2 protein: roles from ingress to egress. Int J Mol Sci 18:E2649. doi: 10.3390/ijms18122649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stöhr K. 2005. Avian influenza and pandemics—research needs and opportunities. N Engl J Med 352:405–407. doi: 10.1056/NEJMe048344. [DOI] [PubMed] [Google Scholar]
- 4.Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, Gao Q, Andrews SE, Bandyopadhyay S, De Jesus P, Tu BP, Pache L, Shih C, Orth A, Bonamy G, Miraglia L, Ideker T, Garcia-Sastre A, Young JA, Palese P, Shaw ML, Chanda SK. 2010. Human host factors required for influenza virus replication. Nature 463:813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanner WD, Toth DJ, Gundlapalli AV. 2015. The pandemic potential of avian influenza A(H7N9) virus: a review. Epidemiol Infect 143:3359–3374. doi: 10.1017/S0950268815001570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janeway CA Jr, Medzhitov R. 2002. Innate immune recognition. Annu Rev Immunol 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 7.Landeras-Bueno S, Ortín J. 2016. Regulation of influenza virus infection by long non-coding RNAs. Virus Res 212:78–84. doi: 10.1016/j.virusres.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 8.Kell AM, Gale M Jr. 2015. RIG-I in RNA virus recognition. Virology 479–480:110–121. doi: 10.1016/j.virol.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holzinger D, Jorns C, Stertz S, Boisson-Dupuis S, Thimme R, Weidmann M, Casanova JL, Haller O, Kochs G. 2007. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J Virol 81:7776–7785. doi: 10.1128/JVI.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haller O, Staeheli P, Kochs G. 2009. Protective role of interferon-induced Mx GTPases against influenza viruses. Rev Sci Tech 28:219–231. doi: 10.20506/rst.28.1.1867. [DOI] [PubMed] [Google Scholar]
- 11.Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan B, Weyer JL, Weyden LVD, Fikrig E. 2009. IFITM proteins mediate the innate immune response to influenza A H1N1 virus, West Nile virus and dengue virus. Cell 139:1243. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volker F, Sen GC. 2011. The ISG56/IFIT1 gene family. J Interferon Cytokine Res 31:71–78. doi: 10.1089/jir.2010.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao X. 2016. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol 16:35. doi: 10.1038/nri.2015.8. [DOI] [PubMed] [Google Scholar]
- 14.Zhang K, Shi ZM, Chang YN, Hu ZM, Qi HX, Hong W. 2014. The ways of action of long non-coding RNAs in cytoplasm and nucleus. Gene 547:1–9. doi: 10.1016/j.gene.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 15.Jiang M, Zhang S, Yang Z, Lin H, Zhu J, Liu L, Wang W, Liu S, Liu W, Ma Y, Zhang L, Cao X. 2018. Self-recognition of an inducible host lncRNA by RIG-I feedback restricts innate immune response. Cell 173:906–919. doi: 10.1016/j.cell.2018.03.064. [DOI] [PubMed] [Google Scholar]
- 16.Wang P, Xu J, Wang Y, Cao X. 2017. An interferon-independent lncRNA promotes viral replication by modulating cellular metabolism. Science 358:1051–1055. doi: 10.1126/science.aao0409. [DOI] [PubMed] [Google Scholar]
- 17.Ouyang J, Zhu X, Chen Y, Wei H, Chen Q, Chi X, Qi B, Zhang L, Zhao Y, Gao GF, Wang G, Chen J-L. 2014. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe 16:616–626. doi: 10.1016/j.chom.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chai W, Li J, Shangguan Q, Liu Q, Li X, Qi D, Tong X, Liu W, Ye X. 2018. Lnc-ISG20 inhibits influenza A virus replication by enhancing ISG20 expression. J Virol 92:e00539-18. doi: 10.1128/JVI.00539-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Staeheli P, Haller O. 1985. Interferon-induced human protein with homology to protein Mx of influenza virus-resistant mice. Mol Cell Biol 5:2150–2153. doi: 10.1128/mcb.5.8.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi D, Hu L, Jiao T, Zhang T, Tong X, Ye X. 2018. Phosphatase Cdc25A negatively regulates the antiviral immune response by inhibiting TBK1 activity. J Virol 92:e01118-18. doi: 10.1128/JVI.01118-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo XZJ, Thomas PG. 2017. New fronts emerge in the influenza cytokine storm. Semin Immunopathol 39:541–510. doi: 10.1007/s00281-017-0636-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Sastre A, Biron CA. 2006. Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 23.Pavlovic J, Haller O, Staeheli P. 1992. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J Virol 66:2564–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han X, Killip MJ, Peter S, Randall RE, David J. 2013. The human interferon-induced MxA protein inhibits early stages of influenza A virus infection by retaining the incoming viral genome in the cytoplasm. J Virol 87:13053–13058. doi: 10.1128/JVI.02220-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Habjan M, Hubel P, Lacerda L, Benda C, Holze C, Eberl CH, Mann A, Kindler E, Gil-Cruz C, Ziebuhr J, Thiel V, Pichlmair A. 2013. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog 9:e1003663. doi: 10.1371/journal.ppat.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pichlmair A, Lassnig C, Eberle C-A, Górna MW, Baumann CL, Burkard TR, Bürckstümmer T, Stefanovic A, Krieger S, Bennett KL, Rülicke T, Weber F, Colinge J, Müller M, Superti-Furga G. 2011. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat Immunol 12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- 27.Parimal K, Sweeney TR, Skabkin MA, Skabkina OV, Hellen CUT, Pestova TV. 2014. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5’-terminal regions of cap0-, cap1- and 5′ ppp-mRNAs. Nucleic Acids Res 42:3228–3245. doi: 10.1093/nar/gkt1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gomez JA, Wapinski O, Yang Y, Bureau JF, Gopinath S, Monack D, Chang H, Brahic M, Kirkegaard K. 2013. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell 152:743–754. doi: 10.1016/j.cell.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhonghan L, Ti-Chun C, Kung-Yen C, Nianwei L, Patil VS, Chisato S, Head SR, Burns JC, Rana TM. 2014. The long noncoding RNA THRIL regulates TNFα expression through its interaction with hnRNPL. Proc Natl Acad Sci U S A 111:1002–1007. doi: 10.1073/pnas.1313768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Susan C, Daniel A, Atianand MK, Ricci EP, Pallavi G, Hall LL, Meg B, Brian M, Meabh HB, Lawrence JB. 2013. A long noncoding RNA mediates both activation and repression of immune response genes. Science 341:789–792. doi: 10.1126/science.1240925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katsutoshi I, Naoto I, Gen A, Michiko K, Atsushi K, Kyosuke N, Akihisa K, Yasushi K, Hiroki S, Misako Y. 2014. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol Cell 53:393–406. doi: 10.1016/j.molcel.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 32.Pin W, Yiquan X, Yanmei H, Li L, Cong W, Sheng X, Zhengping J, Junfang X, Qiuyan L, Xuetao C. 2014. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science 344:310–313. doi: 10.1126/science.1251456. [DOI] [PubMed] [Google Scholar]
- 33.Schmitz KM, Mayer C, Postepska A, Grummt I. 2010. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev 24:2264–2269. doi: 10.1101/gad.590910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Postepska-Igielska A, Giwojna A, Gasri-Plotnitsky L, Schmitt N, Dold A, Ginsberg D, Grummt I. 2015. LncRNA Khps1 regulates expression of the proto-oncogene SPHK1 via triplex-mediated changes in chromatin structure. Mol Cell 60:626–636. doi: 10.1016/j.molcel.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 35.Mondal T, Subhash S, Vaid R, Enroth S, Uday S, Reinius B, Mitra S, Mohammed A, James AR, Hoberg E, Moustakas A, Gyllensten U, Jones SJ, Gustafsson CM, Sims AH, Westerlund F, Gorab E, Kanduri C. 2015. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat Commun 6:7743. doi: 10.1038/ncomms8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Leary VB, Ovsepian SV, Carrascosa LG, Buske FA, Radulovic V, Niyazi M, Moertl S, Trau M, Atkinson MJ, Anastasov N. 2015. PARTICLE, a triplex-forming long ncRNA, regulates locus-specific methylation in response to low-dose irradiation. Cell Rep 11:474–485. doi: 10.1016/j.celrep.2015.03.043. [DOI] [PubMed] [Google Scholar]
- 37.Hiroto K, Farshad N, Lenche K, Moonka DK, Siegel CT, Post AB, Elena C, Marina B, Puri F, Anthony DD. 2014. Negative regulation of the interferon response by an interferon-induced long non-coding RNA. Nucleic Acids Res 42:10668–10680. doi: 10.1093/nar/gku713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laurence J, Nicolas T, Gralinski LE, Ferris MT, Eisfeld AJ, Green RR, Thomas MJ, Jennifer TG, Schroth GP, Yoshihiro K. 2014. Annotation of long non-coding RNAs expressed in collaborative cross founder mice in response to respiratory virus infection reveals a new class of interferon-stimulated transcripts. RNA Biol 11:875–890. doi: 10.4161/rna.29442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barriocanal M, Carnero E, Segura V, Fortes P. 2014. Long non-coding RNA BST2/BISPR is induced by IFN and regulates the expression of the antiviral factor tetherin. Front Immunol 5:655. doi: 10.3389/fimmu.2014.00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carnero E, Barriocanal M, Segura V, Guruceaga E, Prior C, Börner K, Grimm D, Fortes P. 2014. type I interferon regulates the expression of long non-coding RNAs. Front Immunol 5:548. doi: 10.3389/fimmu.2014.00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang JF, Guo YJ, Zhao CX, Yuan SX, Wang Y, Tang GN, Zhou WP, Sun SH. 2013. Hepatitis B virus X protein (HBx)-related long noncoding RNA (lncRNA) down-regulated expression by HBx (Dreh) inhibits hepatocellular carcinoma metastasis by targeting the intermediate filament protein vimentin. Hepatology 57:1882–1892. doi: 10.1002/hep.26195. [DOI] [PubMed] [Google Scholar]
- 42.Imam H, Bano AS, Patel P, Holla P, Jameel S. 2015. The lncRNA NRON modulates HIV-1 replication in a NFAT-dependent manner and is differentially regulated by early and late viral proteins. Sci Rep 5:8639. doi: 10.1038/srep08639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A 96:9345–9350. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu Y, Jiang L, Lai W, Qin Y, Zhang T, Wang S, Ye X. 2016. MicroRNA-33a disturbs influenza A virus replication by targeting ARCN1 and inhibiting viral ribonucleoprotein activity. J Gen Virol 97:27. doi: 10.1099/jgv.0.000311. [DOI] [PubMed] [Google Scholar]
- 45.Liu N, Liu Q, Yang X, Zhang F, Li X, Ma Y, Guan F, Zhao X, Li Z, Zhang L. 2018. Hepatitis B virus-upregulated lnc-HUR1 promotes cell proliferation and tumorigenesis by blocking p53 activity. Hepatology 68:2130–2144. doi: 10.1002/hep.30098. [DOI] [PubMed] [Google Scholar]
- 46.Zhou J, Yang L, Zhong T, Mueller M, Men Y, Zhang N, Xie J, Giang K, Chung H, Sun X, Lu L, Carmichael GG, Taylor HS, Huang Y. 2015. H19 lncRNA alters DNA methylation genome wide by regulating S-adenosylhomocysteine hydrolase. Nat Commun 6:10221. doi: 10.1038/ncomms10221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buske FA, Bauer DC, Mattick JS, Bailey TL. 2012. Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res 22:1372–1381. doi: 10.1101/gr.130237.111. [DOI] [PMC free article] [PubMed] [Google Scholar]