

The mechanism by which HIV-1 envelope (Env) protein trimers assemble into virus particles is poorly understood but involves an interaction between Env cytoplasmic tails (CTs) and the matrix (MA) domain of the structural precursor Gag (PrGag) proteins. We show here that direct binding of MA to Env CTs correlates with MA trimerization, suggesting models where MA lattices regulate CT interactions and/or MA-CT trimer-trimer associations increase the avidity of MA-CT binding. We also show that MA binding to RNA ligands impairs MA-CT binding, potentially by interfering with MA trimerization and/or directly or allosterically blocking MA-CT binding sites. Rough mapping implicated CT C-terminal helices in MA binding, in agreement with cell culture studies on MA-CT interactions. Our results indicate that targeting HIV-1 MA-CT interactions may be a promising avenue for antiviral therapy.
KEYWORDS: human immunodeficiency virus
ABSTRACT
The matrix (MA) domains of HIV-1 precursor Gag (PrGag) proteins direct PrGag proteins to plasma membrane (PM) assembly sites where envelope (Env) protein trimers are incorporated into virus particles. MA targeting to PM sites is facilitated by its binding to phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2], and MA binding to cellular RNAs appears to serve a chaperone function that prevents MA from associating with intracellular membranes prior to arrival at the PI(4,5)P2-rich PM. Investigations have shown genetic evidence of an interaction between MA and the cytoplasmic tails (CTs) of Env trimers that contributes to Env incorporation into virions, but demonstrations of direct MA-CT interactions have proven more difficult. In direct binding assays, we show here that MA binds to Env CTs. Using MA mutants, matrix-capsid (MACA) proteins, and MA proteins incubated in the presence of inositol polyphosphate, we show a correlation between MA trimerization and CT binding. RNA ligands with high affinities for MA reduced MA-CT binding levels, suggesting that MA-RNA binding interferes with trimerization and/or directly or indirectly blocks MA-CT binding. Rough-mapping studies indicate that C-terminal CT helices are involved in MA binding and are in agreement with cell culture studies with replication-competent viruses. Our results support a model in which full-length HIV-1 Env trimers are captured in assembling PrGag lattices by virtue of their binding to MA trimers.
IMPORTANCE The mechanism by which HIV-1 envelope (Env) protein trimers assemble into virus particles is poorly understood but involves an interaction between Env cytoplasmic tails (CTs) and the matrix (MA) domain of the structural precursor Gag (PrGag) proteins. We show here that direct binding of MA to Env CTs correlates with MA trimerization, suggesting models where MA lattices regulate CT interactions and/or MA-CT trimer-trimer associations increase the avidity of MA-CT binding. We also show that MA binding to RNA ligands impairs MA-CT binding, potentially by interfering with MA trimerization and/or directly or allosterically blocking MA-CT binding sites. Rough mapping implicated CT C-terminal helices in MA binding, in agreement with cell culture studies on MA-CT interactions. Our results indicate that targeting HIV-1 MA-CT interactions may be a promising avenue for antiviral therapy.
INTRODUCTION
Despite many years of investigation, the mechanisms by which HIV-1 envelope (Env) proteins assemble into HIV-1 particles are incompletely elucidated. Evidence indicates that Env proteins arrive at the plasma membranes (PMs) of cells as trimers of associated surface (SU) (gp120) and transmembrane (TM) (gp41) components (1–3). The HIV-1 Env cytoplasmic tails (CTs) are about 150 residues long and include three amphipathic alpha-helical elements, historically designated from the N to C termini as lentivirus lytic peptide 2 (LLP-2), LLP-3, and LLP-1 (1, 4). Investigations have also shown that the N-terminally myristoylated matrix (MA) domains of the HIV-1 precursor Gag (PrGag) proteins trimerize (5–11). MA preferentially associates with cell PMs by virtue of its binding to PM phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] (12–14). Within cells, MA proteins have the capacity to bind RNAs, particularly tRNAs, and this MA-RNA binding appears to serve a chaperone function that keeps MA from associating with intracellular membranes (15–22). Although the organization of MA in virus particles is not known, MA assembles in vitro on PI(4,5)P2-containing membranes as hexamers of trimers (23).
Data indicate genetic evidence for an interaction between HIV-1 MA and Env (8–10, 24–33). Interestingly, nearly complete deletion mutants of MA have been observed to be replication competent if pseudotyped with alternative viral glycoproteins or cytoplasmic tail-deleted (ΔCT) HIV-1 Env proteins, which appear to incorporate into virions in a passive fashion in permissive cells (25, 30). Less drastic MA mutations also perturb Env incorporation. Indeed, a number of single-residue MA mutations reduce both Env assembly into virions and virus infectivity (8–10, 27–29, 32, 33). Similarly, some Env CT mutations reduce Env virus incorporation and infectivity without diminishing Env PM levels (31, 34). Importantly, some suppressors of these Env mutations map to MA and have been observed to suppress the Env incorporation defects of a variety of MA mutations as well (8–10).
Whereas genetic links between HIV-1 Env MA and CT are well established, it has been more difficult to prove direct MA-CT interactions. Nevertheless, a few previous studies, employing different biochemical approaches, have reported direct MA-CT binding (1, 35–37). Based in part on these observations, we recently analyzed MA proteins carrying a 62QR mutation that enhances MA trimerization and suppresses Env incorporation mutations and found that these MA 62QR proteins bound efficiently in vitro to HIV-1 CTs (11). Our new findings demonstrate the importance of MA trimerization to CT binding in three different ways, characterize how MA-RNA binding influences Env interactions, and identify Env elements involved in MA binding. These results provide insights into the nature of MA-CT binding and how this interaction may be targeted for antiviral interference.
RESULTS
Analysis of MA trimerization and CT binding.
When modeled on membranes as a hexamer of MA trimers (23), previously observed single-residue MA mutations that impair Env incorporation (8–10) map to the distal ends or spokes of the trimers (Fig. 1A, red dots). The 62QR mutation at the hubs of trimers (Fig. 1A, green dots) suppresses the effects of these mutations and also suppresses the defects of a CT mutation that impairs Env incorporation (8–10). Previously, we showed that 62QR proteins trimerize and bind CTs more efficiently than wild-type (WT) MA proteins (11). Interestingly, mutation of another amino acid at the trimer hubs, residue 74 (Fig. 1B, red dots), yielded viruses that were defective for replication and Env incorporation (P. Tedbury, M. Novikova, A. Alfadhli, I. Kagiampakis, V. KewalRamani, E. Barklis, and E. O. Freed, unpublished data). Selection for replication-competent revertants of residue 74LG mutations generated second-site mutations at residues 34 and 43 (Fig. 1B, green dots) such that the MA 34VI/43FL/74LG variant restored Env incorporation and replication efficiency (Tedbury et al., unpublished).
FIG 1.

Matrix mutations that affect Env incorporation into virions. Shown are models of membrane-bound HIV-1 MA hexamers of trimers (from PDB accession number 1HIW) viewed perpendicularly to the membrane. In panel A, the red spots indicate locations of MA residues that reduce Env incorporation when mutated (12L, 16E, 30L, 34V, and 98E), and the green spots indicate the location of the 62QR mutation that suppresses Env incorporation defects. In panel B, the red spots indicate the location of the 74LG mutation that reduces Env incorporation, and the green spots indicate the locations of 34VI and 43FL mutations that suppress the Env incorporation defects of 74LG.
We examined the capacity of 74LG and 34VI/43FL/74LG proteins to form trimers in our in vitro UV cross-linking assay (11). As demonstrated in Fig. 2A, the Env incorporation-defective 74LG mutant proteins gave a faint dimer band and little evidence of trimers. In contrast, revertant 34VI/43FL/74LG proteins clearly cross-linked as dimers and trimers (Fig. 2A). The abilities of 74LG and 34VI/43FL/74LG proteins to bind Env CTs were determined using our CT binding assay (11). Briefly, glutathione S-transferase (GST) or GST-CT proteins were prebound to glutathione (GSH) beads and incubated with MA proteins, after which bound and total MA levels were quantified by anti-MA immunoblotting. Not surprisingly, neither MA protein bound to GST beads, and 74LG MA bound poorly to GST-CT beads (Fig. 2B). However, the 34VI/43FL/74LG proteins bound 10 times better to GST-CT than the 74LG proteins. These results recapitulate our observations with the 62QR mutation (11) and substantiate the correlation between MA trimerization and CT binding (8–11).
FIG 2.
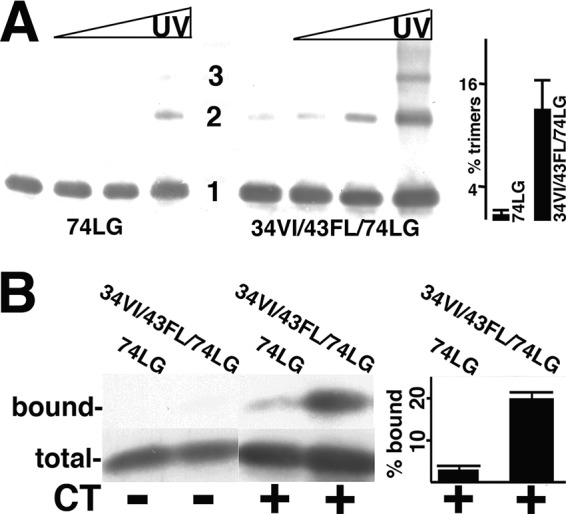
Analysis of MA mutant protein trimerization and CT binding. (A) Purified 74LG or 34VI/43FL/74LG MA proteins at 50 μM concentrations were cross-linked for 0, 1, 3, and 10 min (left to right), separated by electrophoresis, and immunoblotted with an anti-MA antibody. Monomer (1), dimer (2), and trimer (3) sizes were determined by the mobilities of size standards run in parallel. The far-right panel shows the percentage of trimers (relative to monomers) from 10-min cross-linking time points averaged from two separate experiments, with standard deviations as indicated. The P value for the observed difference is 0.0315. (B) The indicated purified MA proteins at 1.5 μM concentrations were incubated with beads coated with glutathione S-transferase (GST) (CT−) or GST-CT (CT+), after which total unbound proteins were collected (total) and beads were washed and eluted to collect bound proteins. Total and bound samples were electrophoretically separated and immunoblotted in parallel for protein detection. Immunoblot bands were quantified densitometrically, and relative binding levels are expressed as percentages of bound versus total MA protein. Calculated percentages are derived from three independent experiments, with standard deviations as indicated. The P value for the observed difference between 74LG and 34VI/43LF/74LG binding to GST-CT is 0.0007.
As reported previously, WT MA inefficiently UV cross-links as trimers (11). These results were reproduced in experiments that showed approximately 2% trimer formation for WT MA (Fig. 3A). Because in vitro Gag assembly experiments have shown that inositol phosphates such as inositol hexakisphosphate (IP6) (phytic acid) might exert effects on HIV-1 MA beyond those documented for CA (38–41), we tested for possible IP6 effects on MA oligomerization. Interestingly, incubation of WT MA with IP6 demonstrated dramatic effects on MA trimerization (Fig. 3A), and we discuss possible mechanisms for this phenomenon in Discussion. However, regardless of the mechanism, this observation afforded us the opportunity to explore a possible correlation between trimerization and CT binding with WT MA. To do so, CT binding analyses were conducted with untreated and IP6-treated WT MA proteins. As seen previously (11), untreated WT MA bound minimally to GST or GST-CT proteins. In contrast, just as IP6 boosted WT MA trimer levels (Fig. 3A), it also increased GST-CT binding about 10-fold (Fig. 3B).
FIG 3.
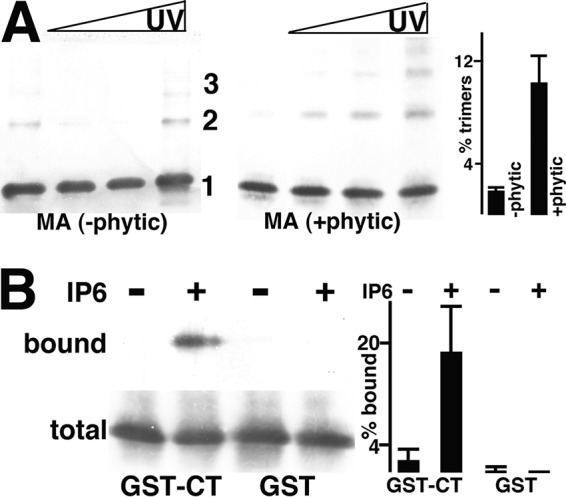
Effects of IP6 on WT MA trimerization and CT binding. (A) WT MA proteins, incubated in either the absence (−phytic) or the presence (+phytic) of IP6 (phytic acid), were UV cross-linked and detected as monomers (1), dimers (2), and trimers (3), as described in the legend to Fig. 2A. The far-right panel shows the percentages of trimers relative to monomers averaged from two independent experiments, with standard deviations as indicated. The P value for the observed difference is 0.0307. (B) Immunoblots of bound and total unbound MA proteins from incubations with GST-CT or GST beads in the absence (−) or presence (+) of IP6. At the far right, relative percentages of bound versus total MA protein levels were derived from either two (GST) or six (GST-CT) independent experiments. The P value for the observed differences in GST-CT binding is 0.0003.
Because, in cells, MA presumably interacts with Env CT in the context of full-length PrGag, we wished to analyze the activities of matrix-capsid (MACA) proteins. Such studies are hampered by the tendency of CA to self-associate (42, 43). Nevertheless, cross-linking experiments for WT HIV-1 MACA gave interpretable results. Indeed, WT MACA UV treatment generated dimers and trimers (Fig. 4A), with trimers representing about 10% of the total MACA levels, similar to our observations with IP6-treated WT MA (Fig. 3A). While WT MACA bound minimally to GST alone (Fig. 4B), it gave a marked improvement in GST-CT binding (Fig. 4B) relative to untreated WT MA (Fig. 3B). Altogether, these results strongly suggest that MA trimerization fosters CT binding.
FIG 4.
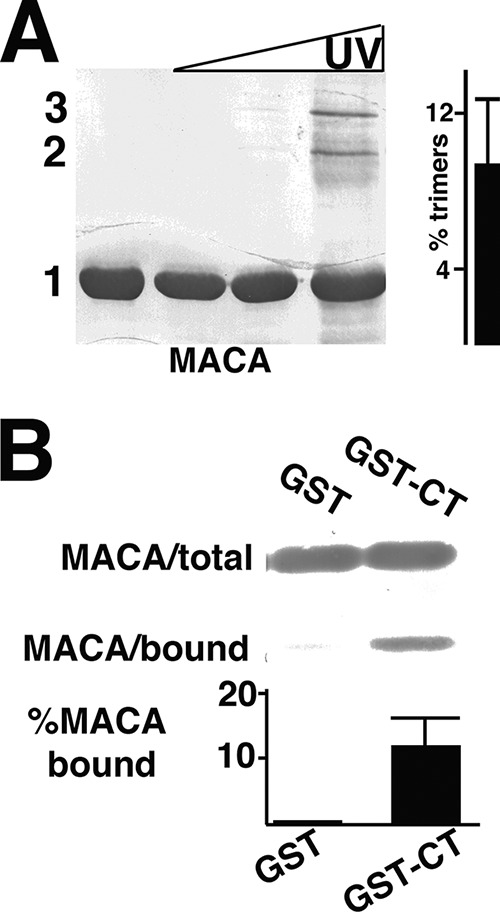
Cross-linking and CT binding of WT MACA proteins. (A) Purified 50 μM WT HIV-1 MACA was UV cross-linked, electrophoretically separated, and detected as described in the legend to Fig. 2A. Monomer (1), dimer (2), and trimer (3) bands were determined by the mobilities of size standards run in parallel. The percentages of trimers relative to monomers are derived from four independent experiments. (B) WT MACA proteins were incubated with GST or GST-CT beads, and bound versus total MACA levels were detected as described in the legend to Fig. 2B. Relative percentages of bound versus total MACA protein levels were calculated from either two (GST) or five (GST-CT) independent experiments. The P value for the observed binding difference is 0.0083.
RNA effects on MA-CT binding.
As noted in the introduction, in addition to interacting with Env, HIV-1 MA binds PI(4,5)P2 and RNA (12–22), and within cells, MA preferentially binds tRNAs rather than viral RNAs (22). Work by our laboratory and others identified an in vitro-selected 25-nucleotide RNA (Sel25), a randomized version of this RNA (Ran25), and tRNAs as ligands with different affinities for MA (11, 17, 19, 22, 44, 45). As a precursor to examining RNA effects in 62QR MA-CT binding assays, where binding signals are relatively high (11), we first examined the affinities of these ligands for 62QR MA in competition fluorescence anisotropy experiments (11, 17, 19). Here, 62QR MA was incubated with fluorescently tagged Sel25 RNA and challenged with increasing concentrations of unlabeled Ran25, Sel25, and Saccharomyces cerevisiae tRNA (Fig. 5A). As illustrated, tRNA competed for binding better than Sel25, while Ran25 competed significantly less well. These results indicate that tRNA, Sel25, and Ran25 respectively bind to 62QR MA with decreasing affinities and are in accordance with expectations (11, 17, 19, 45).
FIG 5.

RNA effects on 62QR MA binding to CT. (A) Fluorescence anisotropy competition binding assays were performed with 1 μM 62QR MA, 10 nM fluorescently tagged Sel25 RNA ligand, and increasing concentrations of untagged Ran25, Sel25, or yeast tRNA ligands to achieve the final indicated concentrations. Competition assays from three separate experiments are plotted as millipolarization-versus-competitor concentration graphs and fitted assuming exponential-decay binding curves. (B) GST-CT binding assays were performed with 62QR MA, and bound versus total 62QR MA levels were monitored as described in the legend to Fig. 2B. Binding assays were performed in the absence of RNA (mock) or in the presence of 5 μM Sel25, Ran25, or yeast tRNA ligand, as indicated. At the far right, levels of 62QR MA binding to GST-CT beads for incubations in the presence of RNAs were normalized to binding levels in the absence of RNA (mock). Results were averaged from five independent experiments. The P values for the observed differences between RNA samples are as follows: 0.007 for Ran25 versus Sel25, 0.0001 for Ran25 versus tRNA, and 0.0301 for Sel25 versus tRNA.
Since RNA promiscuously UV cross-links with protein (46), it was not possible to measure RNA effects on the UV cross-linking of 62QR MA. However, we monitored how our three RNA ligands impacted MA-CT binding. To do so, 62QR MA-CT binding assays were performed in the absence or presence of the different RNA ligands. While Ran25 RNA had little effect on 62QR MA-CT binding, Sel25 RNA reduced CT binding 2.5-fold, and tRNA reduced CT binding 5-fold (Fig. 5B). These observations imply that RNA binding MA may impair trimerization, compete with CT binding, and/or allosterically inhibit CT binding.
Analysis of CT variants.
We undertook rough mapping of CT regions required for MA binding. Thus, in addition to GST and full-length GST-CT (Env residues 705 to 856), we expressed GST fusions to Env residues 705 to 812, 705 to 794, 796 to 856, and 705 to 752 (Fig. 6A). As purified, some of the variants showed minor GST-sized bands, but densitometric analysis indicated that full-length proteins were at least 70% pure (Fig. 6B).
FIG 6.
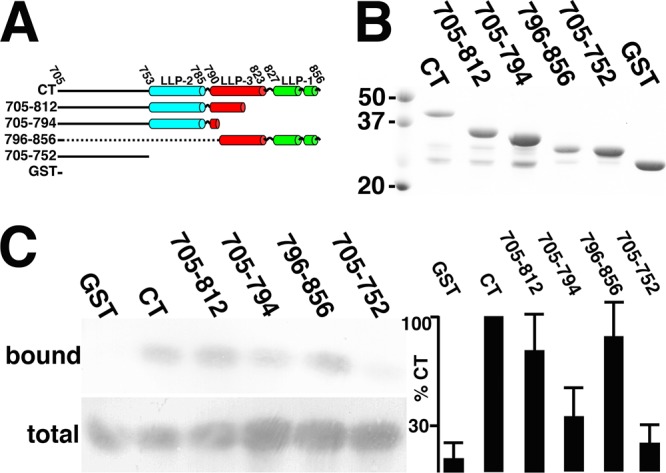
Mapping of CT regions involved in MA binding. (A) N-terminally GST-tagged proteins employed for binding studies are depicted with the borders of the lytic lentiviral peptide (LLP) helical regions LLP-2, LLP-3, and LLP-1, as indicated. (B) GST-CT (Env residues 705 to 856), GST-705-812, GST-705-794, GST-796-856, GST-705-752, and GST proteins, purified as described in Materials and Methods, were separated by electrophoresis in parallel with 50-, 37-, and 20-kDa marker proteins and stained with Coomassie blue for visualization. (C) Binding reactions were performed with 62QR MA and the indicated GST-tagged proteins, and bound and total 62QR MA levels were determined as described in the legend to Fig. 2A. At the far right, binding levels (bound versus total 62QR MA) were normalized to those of GST-CT and are derived from 5 (GST-796-856), 6 (GST-705-812 and GST-705-794), or 10 (GST, GST-CT, and GST-705-752) independent experiments. P values for the observed differences between samples are as follows: 0.65 (not significant) for GST-705-812 versus GST-796-856, 0.0868 (not significant) for GST versus GST-705-752, <0.0001 for GST-796-856 versus GST and GST-705-752, 0.0049 for GST-705-794 versus GST-705-812, 0.0042 for GST-705-794 versus GST-796-856, and 0.0454 for GST-705-794 versus GST-705-752.
We again chose 62QR MA rather than WT MA to measure binding to CT variants, because of its higher levels of binding to full-length GST-CT (11) (Fig. 3B and Fig. 5B). Thus, we performed CT binding assays with 62QR MA and bead-bound GST, GST-CT, GST-705-812 (GST fusion to Env residues 705 to 812), GST-705-794, GST-796-858, and GST-705-752 proteins (Fig. 6C). As observed previously, 62QR MA bound poorly to GST itself but well to GST-CT. Our investigations also showed that the N-terminal CT residues in GST-705-752 were insufficient to produce a good binding signal. In contrast, both GST-705-812 and GST-796-856, each of which includes a significant portion of LLP-3 and either all of LLP-2 (GST-705-812) or LLP-1 (GST-796-856), bound 62QR MA at levels near that of the full-length CT. The GST-705-794 variant provided a noteworthy intermediate case. With several different protein preparations in six independent assays, it consistently gave binding signals above those of GST-705-752 but below those of GST-705-812 and GST-796-856 (Fig. 6C). Interestingly, the residues missing from GST-705-794 but present in GST-705-812 map to mutations that have been observed in cell culture studies to inhibit HIV-1 Env protein incorporation into virions (31, 34).
DISCUSSION
While there exists a wealth of genetic data attesting to an interaction between the HIV-1 MA and CT, it has proven difficult to demonstrate direct MA-CT binding (8–11, 24–37). We have now demonstrated MA-CT binding in vitro with 62QR MA, 62QR MA-green fluorescent protein (GFP), 12LE/62QR MA, 34VI/43FL/74LG MA, IP6-treated WT MA, and WT MACA proteins (11) (Fig. 2 to 6). An obvious consistent observation has been the correlation of MA-CT binding and trimerization (11) (Fig. 2 to 6). One model for the role of MA trimerization (8–10; Tedbury et al., unpublished) is that it may modulate the accessibility of the MA lattice in assembling virions for the accommodation of CTs. A pertinent feature of this lattice modulation model is that it can account for some odd differences in gammaretrovirus Env incorporation and CT proteolytic processing in HIV-1 virions (47, 48), without necessarily invoking a direct gammaretrovirus Env interaction with HIV-1 MA. The lattice model is also consistent with the observation that different membrane-bound MA lattices have been seen in vitro (11, 23, 49) but less readily explains our MA-CT binding assay results; the similarity between WT, 62QR, and 34VI/43FL/74LG MA lattices (albeit at low resolution); or the dearth of evidence for extensive MA lattices in immature or mature HIV-1 particles (11, 50–54).
An alternate model posits that HIV-1 Env incorporation into virions depends on the avidity of a trimer-trimer interaction between CTs of an Env trimer and MA trimers. This conceivably could occur if Env trimers stack directly above MA trimers such that all CTs from one Env trimer bind to the same MA trimer but at different hexamer holes (Fig. 1). By such a model, weakened CT interactions with MA caused by either CT mutations or mutations at the MA spokes (Fig. 1A, red dots) may be compensated for by MA hub mutations that increase the probability of avidity effects. This readily explains how the 62QR mutation might function but less clearly explains observations with WT and 34VI/43FL/74LG MA proteins. In this regard, we hypothesize that the reason why we saw such poor in vitro binding of WT MA to GST-CT beads was due to the low trimerization capacity of WT MA in vitro, while WT MA trimers may be stabilized somewhat in virions due to their associations with capsid lattices. We also speculate that IP6 acted in our in vitro assays as a possible PI(4,5)P2 head group mimic and induced the “flipping out” of myristates, which fosters MA trimerization (7, 55–57). It is relevant to note that we have observed similar, albeit less pronounced, effects on WT MA-CT binding using a short-chain PI(4,5)P2 derivative (data not shown). With respect to 34VI/43FL/74LG MA, we assume that while 74LG destabilizes trimer contacts, 43FL, also located at trimer hubs (Fig. 1B), helps reverse this destabilization. The potential role of 34VI in the stabilization of trimers is less obvious but may also induce myristate exposure or induce longer-range conformational changes (55–57; Tedbury et al., unpublished). As one additional caveat, we also must mention that while the density of CTs on GST-CT-bound beads is adequate enough to permit dimer-dimer or trimer-trimer binding in our system (11), our results could be explained by the specific binding of single CTs to MA trimers.
Our results also have implications concerning the role of MA-RNA binding in the HIV-1 life cycle. We and others have postulated that MA binding to RNA serves a chaperone function, protecting MA (in the context of PrGag) from binding intracellular membranes, prior to arrival at the PM, where high-affinity binding to PI(4,5)P2 can take place (15–22). This model is consistent with our observations that the MA PI(4,5)P2 and RNA binding sites overlap (17–19). In Fig. 5, we show that MA-RNA binding reduced MA-CT binding and that the affinities of RNA ligands for MA correlated with the levels of CT binding reduction. In theory, RNA binding could reduce MA-CT binding by inhibiting MA trimerization and/or occluding the MA-CT recognition site or an allosteric site. Because RNA promiscuously UV cross-links to protein (46), we could not probe for RNA effects in our UV cross-linking assay. However, we previously showed that WT MA and a 15-mer oligonucleotide RNA ligand sedimented as a 1:1 monomeric complex (17), suggesting that RNA binding may affect the oligomerization state of MA. Nonetheless, it would seem that further study on the competition of CT and RNA binding to MA is warranted.
With respect to mapping the MA-CT interaction, our results (Fig. 6) are consistent with the notion that the N-terminal unstructured region of CT is not directly involved and that binding may include multiple elements covering the C-terminal helices (4). The different results obtained with the GST-705-794 and GST-705-812 proteins (Fig. 6B) implicate residues 795 to 812 as contributors to the MA-CT interaction (Fig. 7). These results are in partial agreement with the original observations of Cosson (35), which showed a requirement for the C-terminal LLP-1 region but also for LLP-3, with binding levels dramatically falling off for GST-805-856 versus GST-790-856 proteins (35). Our results are also consistent with those of cell culture-based HIV-1 replication studies (31, 34). In particular, Murakami and Freed (31) clearly showed that deletions 5 to 8 (Fig. 7), including the region spanning residues 787 to 806, resulted in dramatic reductions of the incorporation of Env into virus particles. Similarly, while Bhakta et al. observed effects of LLP-2 mutations on Env incorporation, they also found that double-amino-acid substitutions at residues 795 and 796, 799 and 800, and 802 and 803 (Fig. 7) were defective for replication in CEM and H9 T cell lines (34). Taken together, we believe that these results suggest that targeting the HIV-1 MA-CT interaction will be a viable approach for antiviral interference.
FIG 7.
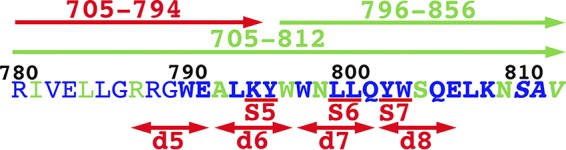
Comparison of CT binding and Env incorporation results. Shown is the sequence of the HIV-1 NL4-3 strain CT region from Env residues 780 to 812. Residues depicted in blue are >90% conserved across 2,351 B clade HIV-1 sequences, while those in green are <90% conserved. Residues in boldface type are part of LLP-3, and the italicized residues (SAV) (residues 810 to 812) are the only ones shown whose codons do not overlap the coding region for Rev exon 3. Above the sequence, the arrows indicate residues included in GST-705-794, which binds poorly to MA relative to the indicated GST-705-812 and GST-796-856 proteins (Fig. 6). Shown below the sequence are the locations of CT deletions d5, d6, d7, and d8, which were observed to impair Env incorporation and HIV-1 replication but did not reduce Gag or cell surface Env levels (31). Also indicated are the locations of double-substitution mutations S5, S6, and S7, which were observed to impair HIV-1 replication in CEM and H9 T cell lines (34).
MATERIALS AND METHODS
Proteins for analysis.
Wild-type (WT) myristoylated MA and MACA C-terminally His-tagged proteins were expressed in Escherichia coli strain BL1(DE3)/pLysS (Novagen) from pET-11a-based vectors, purified, analyzed, and stored as described previously (11, 23, 49). The myristoylated 62QR, 74LG, and 34VI/43FL/74LG MA variants were transferred from the NL4-3 constructs (8–10; Tedbury et al., unpublished) and purified similarly to their WT counterparts.
The Schistosoma japonica glutathione S-transferase (GST) and GST-CT proteins were also expressed and purified from Escherichia coli strain BL21(DE3)/pLysS. GST was expressed from pGEX4T3 (GE Healthcare), and GST-CT was expressed a fusion protein of GST linked at its C terminus with HIV-1 Env CT residues 705 to 856 as described previously (11). We also prepared truncated versions of GST-CT. GST-705-752 ends at Env residue 752 with the sequence GGA TCC TAG GCC CGG G, where the codon for residue 752 is in boldface type and the termination codon is underlined. GST-705-794 ends at Env residue 794 and includes additional codons for WVIS, with the sequence GCC CTC AAA TGG GTA ATT AGC TAG C. GST-705-812 ends at Env residue 812 and includes an additional glycine residue with the sequence AGT GCT GTT GGG TAA TTA GCT AGC. GST-796-856 encodes a fusion protein missing Env residues 705 to 795 and has a GST/CT juncture sequence of CCG AAT TCC CAT TGG TGG AAT, where the codon for Env residue 796 is in boldface type.
GST and GST fusion proteins were purified on glutathione (GSH) columns as described previously by Alfadhli et al. (11) or by a modification of the method described previously by Murphy et al. (4), in which GSH columns were employed, and elutions included 20 mM reduced GSH. Purified proteins were dialyzed in two steps at 4°C with 4 liters of storage buffer (50 mM sodium phosphate [pH 7.0], 150 mM NaCl, 1 mM β-mercaptoethanol [BME]) and analyzed for purity by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (11, 25, 58, 59) followed by staining or immunoblotting (11, 25, 58, 59) with an anti-GST primary antibody (at a 1:1,000 dilution) (catalog number SC-138; Santa Cruz Biotech). GST purifications yielded stocks that were >90% pure and were stored frozen at −80°C in aliquots of 1 to 2 mg/ml. GST fusion proteins yielded protein stocks that corresponded to >70% full-length protein and <30% processed GST based on densitometric analysis of stained gels and were stored frozen as described above.
Biochemical assays.
Fluorescence anisotropy RNA competition binding assays were performed as described previously (11, 17, 19). Briefly, 10 nM fluorescein isothiocyanate (FITC)-labeled Sel25 RNA (11, 17, 19, 44) oligomers (5′-FITC-GGACA GGAAU UAAUA GUAGC UGUCC-3′; Invitrogen) were incubated with 1 μM 62QR MA in 25 mM sodium phosphate (pH 6.0)–50 mM NaCl and challenged with increasing concentrations of untagged Sel25 RNA, a randomized version of Sel25 (Ran25 [5′-GGACA GAAGG AGAUU UAUAC UGUCC-3′), or yeast tRNA (catalog number AM7119; Ambion). Experiments were performed in triplicate, readings were performed in 12- by 75-mm disposable borosilicate glass tubes, and measurements were obtained on a PanVera Beacon 2000 fluorescence polarizer (Invitrogen) at an excitation wavelength of 490 nm. Readings were made at room temperature, polarization values were calculated from emitted light intensities according to the ratio of parallel minus perpendicular to parallel plus perpendicular (11, 17, 19), and binding isotherms were fitted using Prism.
Protein cross-linking analyses were performed by UV light exposure, which has been reported to cross-link proteins at tyrosine, cysteine, and histidine residues (60–62). For UV cross-linking, 20-μl drops of 50 μM protein in a solution containing 50 mM sodium phosphate (pH 7.0), 150 mM NaCl, and 1 mM BME were placed in the center rows of 96-well plates (vinyl; Costar) at 25°C. For cross-linking in the presence of inositol hexakisphosphate (IP6) (phytic acid; Sigma), samples were supplemented with 100 μM IP6. UV cross-linking reactions were performed with a UV Stratalinker system (Stratagene) using the auto-cross-link parameter (1,200 μJ [×100]) for 0, 1, 3, and 10 min. At each time point, samples were removed from the plate and processed for electrophoresis and immunoblotting (11). Monomers, dimers, and trimers were determined by comparison of mobilities with known size standards (catalog number 161-0374; Bio-Rad) run in parallel and quantified by densitometric scanning and analysis with ImageJ software (63). For each 10-min UV reaction, the amounts of trimer signals were normalized to monomer signals to obtain estimates of trimer formation. Standard deviations of percentages of trimers were calculated using Bessel’s sample size correction with Microsoft Excel software. Probability (P) values were calculated from unpaired t tests using GraphPad Prism5 software.
CT binding assays were performed as we described previously (11). GST or GST fusion proteins (1.67 μg) on GSH agarose beads (Sigma, Pierce) (33-μl packed volume) were incubated with 1.5 μM MA or MACA proteins. In some experiments, incubation mixtures were supplemented with 5 μM IP6, Sel25, Ran25, or yeast tRNA. After binding reactions, beads were pelleted, and unbound (total) samples were collected. After washes, bound material was eluted in the presence of 20 mM glutathione. Total (30%) and bound (60%) samples were fractionated by SDS-PAGE and immunoblotted (11) with primary antibodies to MA (catalog number 018-48170, lot number 1429301; Capricorn) (1:2,000) and GST (catalog number SC-138; Santa Cruz Biotech) (1:1,000) for protein detection. Total and bound band levels were quantified by densitometric scanning and analysis with ImageJ software (63) and normalized as relative percent-bound levels. Standard deviations and P values were calculated as described above.
ACKNOWLEDGMENTS
We are grateful to Logan Harper, Andrew Mack, and Christopher Ritchie for advice and assistance.
Eric Barklis, Ayna Alfadhli, and August O. Staubus were supported by NIH grant R01 GM060170. Work in the Freed laboratory was supported by the Intramural Research Program from the Center for Cancer Research, National Cancer Institute, NIH, and by the Intramural AIDS Targeted Antiviral Program.
REFERENCES
- 1.Checkley M, Luttge B, Freed EO. 2011. HIV-1 envelope protein biosynthesis, trafficking and incorporation. J Mol Biol 410:582–608. doi: 10.1016/j.jmb.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lyumkis D, Julien JP, de Val N, Cupo A, Potter CS, Klasse PJ, Burton DR, Sanders RW, Moore JP, Carragher B, Wilson IA, Ward AB. 2013. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 342:1484–1490. doi: 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartesaghi A, Merk A, Borgnia M, Milne J, Subramaniam S. 2013. Prefusion structure of trimeric HIV-1 envelope glycoprotein determined by cryo-electron microscopy. Nat Struct Mol Biol 20:1352–1357. doi: 10.1038/nsmb.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy RE, Samal AB, Vlach J, Saad J. 2017. Solution structure and membrane interaction of the cytoplasmic tail of HIV-1 gp41 protein. Structure 25:1708–1718. doi: 10.1016/j.str.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill C, Worthylake D, Bancroft D, Christensen A, Sundquist W. 1996. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc Natl Acad Sci U S A 93:3099–3104. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morikawa Y, Zhang W, Hockley D, Nermut M, Jones I. 1998. Detection of a trimeric human immunodeficiency virus type 1 Gag intermediate is dependent on sequences in the matrix domain, p17. J Virol 72:7659–7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers M. 2004. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci U S A 101:517–522. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tedbury PR, Ablan SD, Freed EO. 2013. Global rescue of defects in HIV-1 envelope glycoprotein incorporation: implications for matrix structure. PLoS Pathog 9:e1003739. doi: 10.1371/journal.ppat.1003739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tedbury PR, Mercredi PY, Gaines CR, Summers MF, Freed EO. 2015. Elucidating the mechanism by which compensatory mutations rescue an HIV-1 matrix mutant defective for gag membrane targeting and envelope glycoprotein incorporation. J Mol Biol 427(6 Part B):1413–1427. doi: 10.1016/j.jmb.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tedbury P, Freed E. 2014. The role of matrix in HIV-1 envelope glycoprotein incorporation. Trends Microbiol 22:372–378. doi: 10.1016/j.tim.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alfadhli A, Mack A, Ritchie C, Cylinder I, Harper L, Tedbury P, Freed EO, Barklis E. 2016. Trimer enhancement mutation effects on HIV-1 matrix protein binding activities. J Virol 90:5657–5664. doi: 10.1128/JVI.00509-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol(4,5)bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A 101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chukkapalli V, Hogue I, Boyko V, Hu W, Ono A. 2008. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for Gag membrane binding. J Virol 82:2405–2417. doi: 10.1128/JVI.01614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A 103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alfadhli A, Still A, Barklis E. 2009. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J Virol 83:12196–12203. doi: 10.1128/JVI.01197-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chukkapalli V, Oh SJ, Ono A. 2010. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci U S A 107:1600–1605. doi: 10.1073/pnas.0908661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alfadhli A, McNett H, Tsagli S, Bachinger HP, Peyton D, Barklis E. 2011. HIV-1 matrix binding to RNA. J Mol Biol 410:653–666. doi: 10.1016/j.jmb.2011.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones CP, Datta SA, Rein A, Rouzina I, Musier-Forsyth K. 2011. Matrix domain modulates HIV-1 Gag’s nucleic acid chaperone activity via inositol phosphate binding. J Virol 85:1594–1603. doi: 10.1128/JVI.01809-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alfadhli A, McNett H, Eccles J, Tsagli S, Noviello C, Sloan R, Lopez C, Peyton D, Barklis E. 2013. Analysis of small molecule ligands targeting the HIV-1 matrix protein-RNA binding site. J Biol Chem 288:666–676. doi: 10.1074/jbc.M112.399865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chukkapalli V, Inlora J, Todd GC, Ono A. 2013. Evidence in support of RNA-mediated inhibition of phosphatidylserine-dependent HIV-1 Gag membrane binding in cells. J Virol 87:7155–7159. doi: 10.1128/JVI.00075-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inlora J, Collins DR, Trubin ME, Chung JY, Ono A. 2014. Membrane binding and subcellular localization of retroviral Gag proteins are differentially regulated by MA interactions with phosphatidylinositol-(4,5)-bisphosphate and RNA. mBio 5(6):e02202-14. doi: 10.1128/mBio.02202-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kutluay S, Zang T, Blanco-Melo D, Powell C, Jannain D, Errando M, Bieniasz PD. 2014. Global changes in the RNA binding specificity of HIV-1 Gag regulate virion genesis. Cell 159:1096–1109. doi: 10.1016/j.cell.2014.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alfadhli A, Barklis R, Barklis E. 2009. HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)P2. Virology 387:466–472. doi: 10.1016/j.virol.2009.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu X, Yuan X, Matsuda Z, Lee TH, Essex M. 1992. The matrix protein of human immunodeficiency virus type 1 is required for incorporation of viral envelope protein into mature virions. J Virol 66:4966–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang CT, Zhang Y, McDermott J, Barklis E. 1993. Conditional infectivity of a human immunodeficiency virus matrix domain deletion mutant. J Virol 67:7067–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorfman T, Mammano F, Haseltine WA, Gottlinger HG. 1994. Role of the matrix protein in the virion association of the human immunodeficiency virus type 1 envelope glycoprotein. J Virol 68:1689–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freed E, Martin M. 1995. Virion incorporation of envelope glycoproteins with long but not short cytoplasmic tails is blocked by specific, single amino acid substitutions in the human immunodeficiency virus type 1 matrix. J Virol 69:1984–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freed EO, Martin MA. 1996. Domains of the human immunodeficiency virus type 1 matrix and gp41 cytoplasmic tail required for envelope incorporation into virions. J Virol 70:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ono A, Huang M, Freed EO. 1997. Characterization of human immunodeficiency virus type 1 matrix revertants: effects on virus assembly, Gag processing, and Env incorporation into virions. J Virol 71:4409–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reil H, Bukovsky AA, Gelderblom HR, Gottlinger HG. 1998. Efficient HIV-1 replication can occur in the absence of the viral matrix protein. EMBO J 17:2699–2708. doi: 10.1093/emboj/17.9.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murakami T, Freed E. 2000. Genetic evidence for an interaction between human immunodeficiency virus type 1 matrix and alpha-helix 2 of the gp41 cytoplasmic tail. J Virol 74:3548–3554. doi: 10.1128/JVI.74.8.3548-3554.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis MR, Jiang J, Zhou J, Freed EO, Aiken C. 2006. A mutation in the human immunodeficiency virus type 1 Gag protein destabilizes the interaction of the envelope protein subunits gp120 and gp41. J Virol 80:2405–2417. doi: 10.1128/JVI.80.5.2405-2417.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhatia AK, Kaushik R, Campbell NA, Pontow SE, Ratner L. 2009. Mutation of critical serine residues in HIV-1 matrix result in an envelope incorporation defect which can be rescued by truncation of the gp41 cytoplasmic tail. Virology 384:233–241. doi: 10.1016/j.virol.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhakta SJ, Shang L, Prince JL, Claiborne DT, Hunter E. 2011. Mutagenesis of tyrosine and di-leucine motifs in the HIV-1 envelope cytoplasmic domain results in a loss of Env-mediated fusion and infectivity. Retrovirology 8:37. doi: 10.1186/1742-4690-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cosson P. 1996. Direct interaction between the envelope and matrix proteins of HIV-1. EMBO J 15:5783–5788. doi: 10.1002/j.1460-2075.1996.tb00964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hourioux C, Brand D, Sizaret P, Lemiale F, Lebigot S, Barin F, Roingeard P. 2000. Identification of the glycoprotein 41 (TM) cytoplasmic tail domains of HIV-1 that interact with Pr55Gag particles. AIDS Res Hum Retroviruses 16:1141–1147. doi: 10.1089/088922200414983. [DOI] [PubMed] [Google Scholar]
- 37.Wyma D, Jiang J, Shi J, Zhou J, Lineberger J, Miller M, Aiken C. 2004. Coupling of HIV-1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J Virol 78:3429–3435. doi: 10.1128/JVI.78.7.3429-3435.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Datta S, Zhao Z, Clark P, Tarasov S, Alexandratos J, Campbell S, Kvaratskhelia M, Lebowitz J, Rein A. 2007. Interactions between HIV-1 Gag molecules in solution: an inositol phosphate-mediated switch. J Mol Biol 365:799–811. doi: 10.1016/j.jmb.2006.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Datta SA, Curtis JE, Ratcliff W, Clark PK, Crist RM, Lebowitz J, Krueger S, Rein A. 2007. Conformation of the HIV-1 Gag protein in solution. J Mol Biol 365:812–824. doi: 10.1016/j.jmb.2006.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mallery DL, Márquez CL, McEwan WA, Dickson CF, Jacques DA, Anandapadamanaban M, Bichel K, Towers GJ, Saiardi A, Böcking T, James LC. 2018. IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis. Elife 7:e35335. doi: 10.7554/eLife.35335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dick R, Zadrozny K, Yu C, Schur F, Lyddon T, Ricana C, Wagner J, Perilla J, Ganzer-Pornillos B, Johnson M, Pornillos O, Vogt V. 2018. Inositol phosphates are assembly co-factors of HIV-1. Nature 260:509–512. doi: 10.1038/s41586-018-0396-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barklis E, Alfadhli A, McQuaw C, Yalamuri S, Still A, Barklis R, Kukull B, Lopez C. 2009. Characterization of the in vitro HIV-1 capsid assembly pathway. J Mol Biol 387:376–389. doi: 10.1016/j.jmb.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez C, Eccles J, Still A, Sloan R, Barklis R, Tsagli S, Barklis E. 2011. Determinants of the HIV-1 core assembly pathway. Virology 417:137–146. doi: 10.1016/j.virol.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Purohit P, Dupont S, Stevenson M, Green M. 2001. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro selection. RNA 7:576–584. doi: 10.1017/s1355838201002023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaines C, Tkacik E, Rivera-Oven A, Somani P, Achimovich A, Alabi T, Zhu A, Getachew N, Yang A, McDonough M, Hawkins T, Spadaro Z, Summers M. 2018. HIV-1 matrix protein interactions with tRNA: implications for membrane targeting. J Mol Biol 430:2113–2127. doi: 10.1016/j.jmb.2018.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sei E, Conrad N. 2014. UV crosslinking of interacting RNA and proteins in cultured cells. Methods Enzymol 539:53–66. doi: 10.1016/B978-0-12-420120-0.00004-9. [DOI] [PubMed] [Google Scholar]
- 47.Kiernan RE, Freed EO. 1998. Cleavage of the murine leukemia virus transmembrane Env protein by human immunodeficiency virus type 1 protease: transdominant inhibition by matrix mutations. J Virol 72:9621–9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song Y, Olinger G, Janaka S, Johnson M. 2019. Sequence determinants in gammaretroviral Env cytoplasmic tails dictate virus-specific pseudotyping compatibility. J Virol 93:e02172-18. doi: 10.1128/JVI.02172-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alfadhli A, Huseby D, Kapit E, Colman D, Barklis E. 2007. The HIV-1 matrix protein assembles on membranes as a hexamer. J Virol 81:1472–1478. doi: 10.1128/JVI.02122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuller SD, Wilk T, Gowen BE, Krausslich HG, Vogt VM. 1997. Cryo-electron microscopy reveals ordered domains in the immature HIV-1 particle. Curr Biol 7:729–738. doi: 10.1016/S0960-9822(06)00331-9. [DOI] [PubMed] [Google Scholar]
- 51.Wilk T, Gross I, Gowen BE, Rutten T, de Haas F, Welker R, Krausslich HG, Boulanger P, Fuller SD. 2001. Organization of immature human immunodeficiency virus type 1. J Virol 75:759–771. doi: 10.1128/JVI.75.2.759-771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. 2007. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J 26:2218–2226. doi: 10.1038/sj.emboj.7601664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bharat TA, Castillo Menendez LR, Hagen WJ, Lux V, Igonet S, Schorb M, Schur FK, Kräusslich HG, Briggs JA. 2014. Cryo-electron microscopy of tubular arrays of HIV-1 Gag resolves structures essential for immature virus assembly. Proc Natl Acad Sci U S A 111:8233–8238. doi: 10.1073/pnas.1401455111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schur FK, Hagen WJ, Rumlová M, Ruml T, Müller B, Kräusslich HG, Briggs JA. 2015. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature 517:505–508. doi: 10.1038/nature13838. [DOI] [PubMed] [Google Scholar]
- 55.Hermida-Matsumato L, Resh M. 1999. Human immunodeficiency virus type 1 protease triggers a myristoyl switch that modulates membrane binding of Pr55Gag and p17MA. J Virol 73:1902–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Resh M. 2004. A myristoyl switch regulates membrane binding of HIV-1 Gag. Proc Natl Acad Sci U S A 101:417–418. doi: 10.1073/pnas.0308043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saad J, Loeliger E, Luncsford P, Liriano M, Tai J, Kim A, Miller J, Joshi A, Freed E, Summers M. 2007. Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J Mol Biol 366:574–585. doi: 10.1016/j.jmb.2006.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huseby D, Barklis R, Alfadhli A, Barklis E. 2005. Assembly of human immunodeficiency virus precursor Gag proteins. J Biol Chem 280:17664–17670. doi: 10.1074/jbc.M412325200. [DOI] [PubMed] [Google Scholar]
- 59.Ritchie C, Cylinder I, Platt E, Barklis E. 2015. Analysis of HIV-1 Gag protein interactions via biotin ligase tagging. J Virol 89:3988–4001. doi: 10.1128/JVI.03584-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guptasarma P, Balasubramanian D, Matsugo S, Saito I. 1992. Hydroxyl radical mediated damage to proteins, with special reference to the crystallins. Biochemistry 31:4296–4303. doi: 10.1021/bi00132a021. [DOI] [PubMed] [Google Scholar]
- 61.Davies M. 2003. Singlet oxygen-mediated damage to proteins and its consequences. Biochem Biophys Res Commun 305:761–770. doi: 10.1016/S0006-291X(03)00817-9. [DOI] [PubMed] [Google Scholar]
- 62.Leo G, Altucci C, Bourgoin-Voillard S, Gravagnuolo A, Esposito R, Marino G, Costello C, Velotta R, Birolo L. 2013. UV laser-induced cross-linking of proteins. Rapid Commun Mass Spectrom 27:1660–1668. doi: 10.1002/rcm.6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]