

The engagement of antiviral effector responses caused by viral infection is essential when using viruses or virus-like particles (VLPs) as an immunotherapeutic agent. Here, we compare the chemophysical and immunostimulatory properties of wild-type cowpea mosaic virus (CPMV) (RNA containing) and eCPMV (RNA-free VLPs) produced from two expression systems (agrobacterium-based plant expression system and baculovirus-insect cell expression). CPMV and eCPMV could each be developed as novel adjuvants to overcome immunosuppression and thus promote tumor regression in ovarian cancer (and other tumor types). To our knowledge, this is the first study to define the immunotherapeutic differences between CPMV and eCPMV, which is essential for the further development of biomedical applications for plant viruses and the selection of rational combinations of immunomodulatory reagents.
KEYWORDS: empty virus-like nanoparticle, immunotherapy, in situ vaccine, ovarian cancer, plant virus nanoparticle
ABSTRACT
Cowpea mosaic virus (CPMV) is a plant virus that has been developed for multiple biomedical and nanotechnology applications, including immunotherapy. Two key platforms are available: virus nanoparticles (VNPs) based on the complete CMPV virion, including the genomic RNA, and virus-like nanoparticles (VLPs) based on the empty CPMV (eCPMV) virion. It is unclear whether these platforms differ in terms of immunotherapeutic potential. We therefore compared their physicochemical properties and immunomodulatory activities following in situ vaccination of an aggressive ovarian tumor mouse model (ID8-Defb29/Vegf-A). In physicochemical terms, CPMV and eCPMV were very similar, and both significantly increased the survival of tumor-bearing mice and showed promising antitumor efficacy. However, they demonstrated distinct yet overlapping immunostimulatory effects due to the presence of virus RNA in wild-type particles, indicating their suitability for different immunotherapeutic strategies. Specifically, we found that the formulations had similar effects on most secreted cytokines and immune cells, but the RNA-containing CPMV particles were uniquely able to boost populations of potent antigen-presenting cells, such as tumor-infiltrating neutrophils and activated dendritic cells. Our results will facilitate the development of CPMV and eCPMV as immunotherapeutic vaccine platforms with tailored responses.
IMPORTANCE The engagement of antiviral effector responses caused by viral infection is essential when using viruses or virus-like particles (VLPs) as an immunotherapeutic agent. Here, we compare the chemophysical and immunostimulatory properties of wild-type cowpea mosaic virus (CPMV) (RNA containing) and eCPMV (RNA-free VLPs) produced from two expression systems (agrobacterium-based plant expression system and baculovirus-insect cell expression). CPMV and eCPMV could each be developed as novel adjuvants to overcome immunosuppression and thus promote tumor regression in ovarian cancer (and other tumor types). To our knowledge, this is the first study to define the immunotherapeutic differences between CPMV and eCPMV, which is essential for the further development of biomedical applications for plant viruses and the selection of rational combinations of immunomodulatory reagents.
INTRODUCTION
Plant virus nanoparticles (VNPs) are therapeutic reagents based on plant viruses and are useful for vaccine development and immunotherapy because they are noninfectious in mammals, making them safer than mammalian viruses currently used for oncolytic therapy (1). Several expression systems have been used to produce VNPs and their nucleic acid-free derivatives known as virus-like nanoparticles (VLPs), including the bacterium Escherichia coli, yeast, baculovirus-insect cell systems, mammalian cell lines such as Chinese hamster ovary cells, and plants such as Nicotiana benthamiana (2). The use of plants for the large-scale manufacture of plant-based VNPs or VLPs may be particularly attractive based on cost-effectiveness (3, 4).
Among several plant viruses that have been developed as VNPs and/or VLPs, our recent data highlight the potential of cowpea mosaic virus (CPMV) as an in situ vaccine and adjuvant, which is administered directly into a tumor to recruit immune cells and polarize them toward an antitumor immune response. The tumor provides a source of antigens, and effective in situ vaccination induces systemic, durable antitumor immunity against tumor-specific antigens and neoantigens. We previously tested both CPMV-derived VNPs, which contain the RNA genome, and empty CPMV (eCPMV) particles, which are VLPs devoid of genomic RNA (5, 6). In both cases, the administration of the virus achieved potent antitumor efficacy in mouse tumor models (5, 6) and canine patients (7). Wild-type CPMV is a bipartite RNA virus with a 28-nm capsid comprising 60 copies each of the large (L) (42-kDa) and small (S) (24-kDa) coat proteins arranged with pseudo-T=3 icosahedral symmetry. RNA-1 and RNA-2 are encapsidated separately into CPMV particles of identical protein compositions, termed bottom (RNA-1) and middle (RNA-2) components based on their positions after separation on a density gradient; in addition, a small amount of empty CPMV particles can be obtained during infection, and these particles are termed top components (because they appear on top of a density gradient) (8). To exclude the top-component fraction (which essentially is eCPMV) in our CPMV preparation, only RNA-1- and RNA-2-containing particles were collected and used for the following studies. The VNPs derived from this virus can carry cargos of drugs and/or imaging molecules, but because eCPMV lacks the genomic RNA, it has a greater loading capacity for mineral cargo (9), and the inner surface can be conjugated to small-molecule reagents (10). However, the virus RNA fulfills a useful immunostimulatory function because it activates innate immune cells by binding to Toll-like receptor 7 (TLR7). Previously reported in situ vaccination studies using papaya mosaic virus attributed the potency of the VNPs to the presence of the RNA (11, 12).
Our previous work with eCPMV has shown that RNA is not needed to induce antitumor immunity, but it is possible that the RNA may increase the efficacy of in situ vaccination. We therefore carried out a comprehensive comparison of the immunostimulatory properties of CPMV and eCPMV particles, the former produced in a native host (the black-eyed pea Vigna unguiculata subsp. unguiculata, a subspecies of cowpea) and the latter produced in N. benthamiana plants by agroinfiltration or in the baculovirus-insect cell expression system. We compared the immunogenicities of wild-type CPMV and eCPMV using the syngeneic immunocompetent murine orthotopic ovarian cancer model ID8-Defb29/Vegf-A to determine the common and unique immunostimulatory properties of each CPMV platform.
RESULTS
Physicochemical properties of CPMV and eCPMV.
Samples of purified wild-type CPMV, eCPMV produced in agroinfiltrated plants (eCPMV/p), and eCPMV produced in insect cells (eCPMV/i) were denatured and separated by SDS-PAGE (Fig. 1A). The L and S subunits of wild-type CPMV presented as single bands of 42 and 24 kDa, respectively, and both eCPMV preparations yielded the same L and S bands. Nondenaturing agarose gel electrophoresis, through which the nanoparticles travel intact, confirmed the lack of nucleic acids in both eCPMV particles, as indicated by the absence of nucleic acid staining (Fig. 1B). Two bands were observed after electrophoretic separation, and these are due to the presence of two electrophoretic forms for intact eCPMV/CPMV particles, the slow and fast forms; the fast electrophoretic form of CPMV results from a loss of 24 amino acids at the C terminus of the small coat protein, a process that occurs by proteolysis in plants or insect cells (5, 13, 14). Wild-type CPMV traveled farther through the gel than the eCPMV particles, probably reflecting the lack of internal RNA and its strong impact on electrophoretic mobility, which is consistent with our previous observations (15). Interestingly, the eCPMV/i particles were less mobile than the eCPMV/p particles, even though the coat protein composition was identical in both cases (see below). The major difference between the eCPMV/i and eCPMV/p particles was their degree of purity. Insect cell expression was less efficient, and the low yields resulted in a greater quantity of impurities than in the plant-based system, as determined by size exclusion chromatography (SEC) (Fig. 1C). We speculate that residual contaminants in the eCPMV/i formulation led to the formation of aggregates and/or surface-bound molecules that inhibited electrophoretic mobility.
FIG 1.

Characterization of CPMV and eCPMV particles. (A) SDS gels following electrophoresis and staining with Coomassie brilliant blue. (B, top) GelRed nucleic acid gel under UV light; (bottom) the same gel following electrophoresis and staining with Coomassie brilliant blue. Lanes: M, SeeBlue Plus2 molecular weight marker; WT, wild-type CPMV; E/p, empty CPMV from plants (eCPMV/p); E/i, empty CPMV from insect cells (eCPMV/i). The gel images in panels A and B were spliced, as indicated by the gap and yellow arrow. The original gels included additional samples not discussed in this paper; therefore, these lanes were omitted from the figure. (C) Size exclusion chromatography (SEC) analysis of CPMV (top), eCPMV/p (middle), and eCPMV/i (bottom) particles. Blue, 260 nm; red, 280 nm; mAU, milli-absorbance units. (D and E) Sizes of wild-type CPMV (top), eCPMV/p (middle), and eCPMV/i (bottom) measured by dynamic light scattering (D) and transmission electron microscopy (TEM) (E). Bars, 100 nm.
Consistent with the native gels, UV-visible spectroscopy revealed a low A260/A280 ratio (0.7) for the eCPMV particles, indicating the absence of nucleic acids, compared to the typical A260/A280 ratio of 1.8 for wild-type CPMV particles. SEC was carried out to determine the purity and structural integrity of the particles, revealing no significant difference between wild-type CPMV and eCPMV/p particles with the same elution volume of 9.7 ml (Fig. 1C). Although the same elution volume was observed for the eCPMV/i particles, a series of peaks between 25 and 40 ml indicated impurities from the insect cell expression system. Due to low yields, we were unable to purify the samples any further.
Finally, dynamic light scattering (DLS) and transmission electron microscopy (TEM) confirmed the size and icosahedral shape of the CPMV and eCPMV particles (Fig. 1D and E). In agreement with the SEC data, icosahedral particles were found in all samples, but smaller proteins and/or fragmented particles were found in the eCPMV/i preparation. Generally, these data indicated that there were no significant differences between CPMV and eCPMV from different expression systems, except for the lower purity of the eCPMV/i preparation.
Reversed-phase liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was carried out next to identify and quantify the peptide compositions of the 42- and 24-kDa bands revealed by SDS-PAGE. By comparing the data from each sample to the CPMV sequences, 18 target peptide sequences were selected for the L subunit, and 10 were selected for the S subunit (Fig. 2). For all three samples, the intensities of most commonly identified peptides were similar, confirming that the coat protein sequence of both eCPMV formulations is indistinguishable from that of wild-type CPMV.
FIG 2.
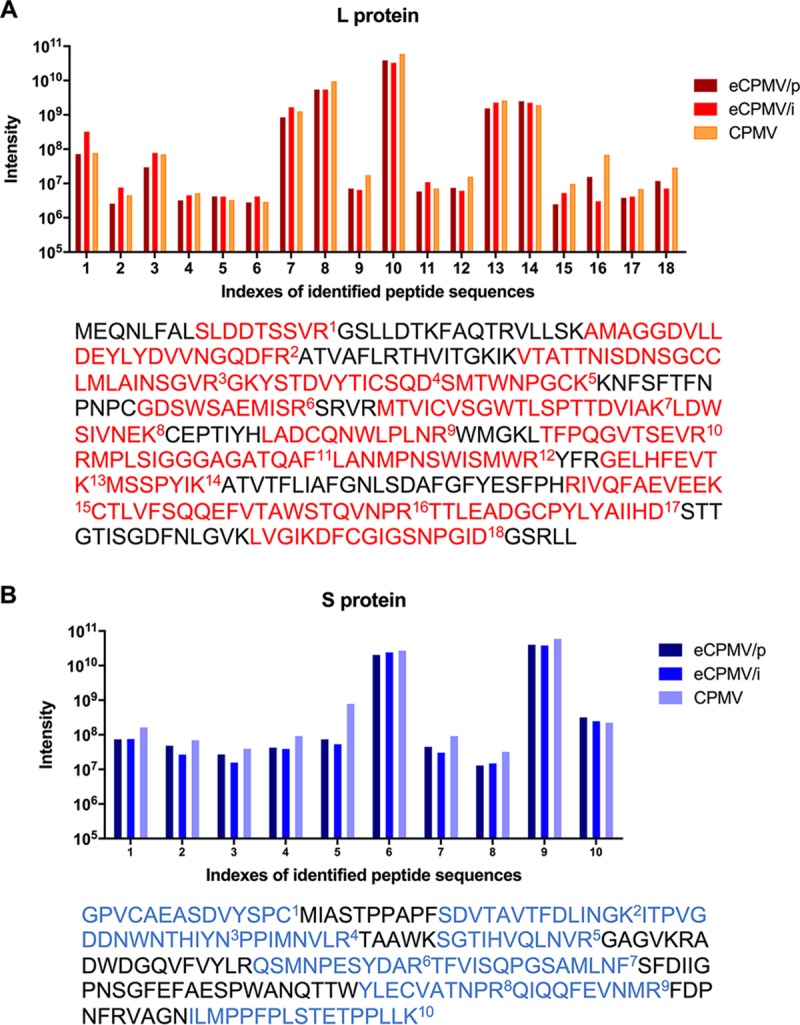
Confirmation of L and S subunit protein sequences in CPMV and eCPMV particles. (A) For the L subunit, 18 target peptides were identified (highlighted in red). (B) For the S subunit, 10 target peptides were identified (highlighted in blue). Each peptide was quantified by label-free proteomic analysis of the excised L and S gel bands. Bars show the intensity of each identified peptide. The position of each peptide in the context of the complete sequence is shown below each graph.
Endotoxin removal.
Endotoxins are lipopolysaccharides (LPSs) that act as pathogen-associated molecular patterns (PAMPs), and they are found in the cell walls of most Gram-negative bacteria, including Agrobacterium tumefaciens, which was used to generate eCPMV in plants. LPS initiates a strong innate immune response when bacteria infect humans (16, 17). The presence of endotoxins can cause adverse reactions, including shock when endotoxin-contaminated reagents are used clinically (18). Therefore, it is essential to remove LPS from recombinant proteins, drugs, and other biological and pharmaceutical products to avoid adverse reactions. Various LPS removal procedures have been reported (19, 20), and we selected a method based on Triton X-114 to extract the LPS from our samples (18, 21).
We also considered the possibility that LPS could be encapsulated within VLPs. Therefore, we heated the eCPMV/p samples to denature the capsids and release any contents. This step was applied before or after the removal of LPS using the Triton X-114 method. We found that endotoxin levels remain unchanged regardless of whether the particles were intact or disassembled (Fig. 3), which indicates that Triton X-114 is sufficient to remove any LPS from the formulation, whether on the particle surface or encapsulated. The data may also suggest that no significant quantities of LPS are enclosed within the capsid.
FIG 3.
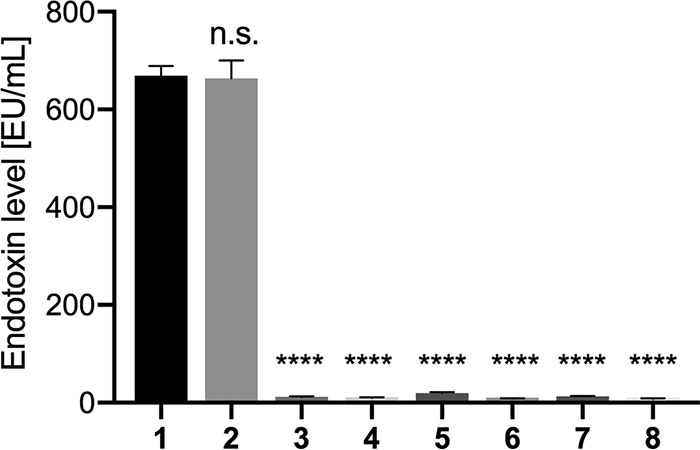
Endotoxin levels of CPMV and eCPMV particles. 1, eCPMV/p directly after purification; 2, eCPMV/p after purification and heat precipitation; 3, eCPMV/p with endotoxins removed; 4, eCPMV with endotoxins removed, followed by heat precipitation; 5, CPMV directly after purification; 6, CPMV after purification and heat precipitation; 7, CPMV with endotoxins removed; 8, CPMV with endotoxins removed, followed by heat precipitation. The significance level was calculated using one-way analysis of variance (ANOVA) and Tukey’s multiple-comparison test. ****, P < 0.0001; n.s., not significant.
Importantly, LPS contamination was apparent only in eCPMV samples produced by agroinfiltration of plants. Wild-type CPMV particles were produced by mechanical inoculation using infectious particles and black-eyed pea plants; these preparations avoid the use of Gram-negative bacteria, such as A. tumefaciens, which are the source of LPS contaminants, therefore highlighting the advantages of the mechanical inoculation method. Obtaining pure particles without LPS contaminants streamlines manufacture and improves yields; any additional postharvest purification steps ultimately reduce yields and require additional steps for characterization, driving up the production costs. For translational applications to avoid the release of a plant pathogen, wild-type CPMV particles could be treated with UV light or chemicals to render them noninfectious toward plants (22) while maintaining the presence of RNA and keeping the manufacture LPS free.
In situ vaccination against ID8-Defb29/Vegf-A ovarian tumor challenge.
In situ vaccines trigger a local innate immune response that leads to the systemic attack of cancer cells carrying the same tumor antigens as a primary tumor. Although ovarian cancer is a devastating disease, metastases are frequently restricted to the peritoneal cavity (where the tumor microenvironment is directly accessible), so local immunotherapy can harness this effect more efficiently (23). To determine whether in situ vaccination with CPMV or eCPMV can increase the survival of tumor-bearing mice and reduce the tumor burden, we inoculated mice intraperitoneally (i.p.) with the luciferase-labeled ID8-Defb29/Vegf-A cell line on day 0. ID8-Defb29/Vegf-A cells were stably transfected with luciferase using a retrovirus as described previously (24) and in Materials and Methods.
Treatments started on day 7 after tumor challenge; treatments (100 μg/200 μl CPMV, eCPMV/p, or eCPMV/i versus the phosphate-buffered saline [PBS] control) were given six times in weekly intervals via i.p. injection (n = 5 per group). Endotoxin levels of all samples were determined using an endotoxin detection assay, and a level of <50 endotoxin units (EU)/mg protein was considered negligible. If necessary, LPS was removed prior to immunization. Tumor growth was monitored by measuring body weight, abdominal circumference, and total bioluminescence, all of which showed a close correlation (Fig. 4A to E). Mice were euthanized when their weight reached 35 g or when moribund. Both eCPMV/p and eCPMV/i reduced the tumor burden significantly in terms of total bioluminescence (P < 0.0001) and prolonged survival (P < 0.01) compared to the control group (Fig. 4D). CPMV prolonged survival even more than the eCPMV formulations, although the differences were not statistically significant. These data confirm that in situ vaccination with either CPMV or eCPMV can be highly efficacious against an aggressive ovarian tumor in mice.
FIG 4.

Tumor growth and survival in mice challenged with ID8-Defb29/Vegf-A tumors and treated weekly with 100 μg of CPMV or eCPMV on days 7, 14, 21, 28, 35, and 42 postinoculation. Green arrows indicate the injection time points. (A to C) Tumor growth was monitored by measuring body weight (A), circumference (B), and luciferase expression in the peritoneal cavity (C). Data are means ± standard errors of the means (SEM) (n = 5). Statistical significance was calculated by two-way ANOVA with Tukey’s test. *, P < 0.05; **, P < 0.01; ***, P < 0.0005; ****, P < 0.0001. (D) Survival rates of treated mice. Statistical significance was calculated using the Mantel-Cox log rank test. **, P < 0.01. (E) Representative live-animal imaging of CPMV- and eCPMV-treated mice with ID8-Defb29/Vegf-A-luc tumors (40 days postinoculation).
CPMV and eCPMV treatment induces immunostimulatory cytokine production and activation of immune cells ex vivo and in vivo.
Next, we compared the effects of CPMV and eCPMV ex vivo and in vivo stimulation on the tumor microenvironment, to determine whether the immunomodulatory activity of eCPMV (i.e., its ability to induce cytokine secretion and the activation of immune cells) is influenced by the lack of virus RNA. We therefore harvested nonadherent peritoneal cavity cells on day 35 from untreated ID8-Defb29/Vegf-A tumor-bearing mice, stimulated these cells ex vivo with 10 μg CPMV or eCPMV, and collected the supernatant 24 h later for cytokine analysis (Fig. 5A). The concentration of the proinflammatory cytokine interleukin-6 (IL-6) increased sharply in cells stimulated by CPMV (P < 0.0005) but not in those stimulated by eCPMV, in each case compared to the unstimulated control (Fig. 5A). However, when we increased the stimulation dose (50 μg), a significant increase of IL-6 was observed in cells stimulated by eCPMV/p, although the level of IL-6 was still 1.8 times lower than the IL-6 level measured after CPMV stimulation (data not shown). The levels of other proinflammatory cytokines (IL-1β, IL-12, interferon beta [IFN-β], and granulocyte-macrophage colony-stimulating factor [GM-CSF]) increased slightly in response to eCPMV but not CPMV (Fig. 5A). Both eCPMV/p and eCPMV/i inhibited the secretion of transforming growth factor β (TGF-β) (P < 0.0001) and IL-10 (P < 0.05 for eCPMV/p and P < 0.01 for eCPMV/i), whereas CPMV inhibited the secretion of only TGF-β (P < 0.0001). All three formulations induced the secretion of IFN-γ (P < 0.05 for CPMV and eCPMV/p and P < 0.01 for eCPMV/i). To further explore the mechanistic differences and similarities between CPMV and eCPMV, cytokine secretion after in vivo stimulation was measured. On day 35 after tumor inoculation, CPMV or eCPMV/p was i.p. administered. After 24 h, peritoneal cavity washes were harvested, and the cytokine levels in the supernatant were measured (Fig. 5B). Different from the ex vivo stimulation results, the level of IL-6 secretion in the CPMV-treated group was similar to the levels in the eCPMV/p- and PBS-treated groups. Besides IL-12, the levels of secretion of other proinflammatory cytokines (IL-1β, TNF-α, IFN-β, and GM-CSF) were increased by both CPMV and eCPMV/p. Moreover, in situ treatment of CPMV as well as eCPMV/p led to reduced TGF-β (P < 0.05 for eCPMV/p and P = 0.0738 for CPMV) and IL-10 (P < 0.05 for eCPMV/p and P < 0.01 for CPMV) secretion. A significant increase in IFN-γ secretion was observed only in CPMV-treated mice (P < 0.001). These data not only highlight the differences between CPMV and eCPMV but also underline the differences between the ex vivo and in vivo assays, the most striking difference being that in vivo, only CPMV stimulated IFN-γ.
FIG 5.

CPMV or eCPMV administration induces cytokine secretion in the tumor microenvironment. (A) Peritoneal cavity wash cells were collected from tumor-bearing mice on day 35 after tumor inoculation. The cells were incubated with CPMV, eCPMV/p, or eCPMV/i (or PBS as a control) for 24 h before measurement of the levels of cytokines in the culture medium. (B) CPMV and eCPMV/p were i.p. injected into the peritoneal cavity of tumor-bearing mice on day 35 after tumor inoculation. After 24 h, the cytokine levels in the peritoneal cavity wash were analyzed by an ELISA. Data are means ± SEM (n = 3). Statistical significance was calculated by one-way ANOVA with a Tukey test. *, P < 0.05; **, P < 0.01; ***, P < 0.0005; ****, P < 0.0001.
We next characterized the phenotype of peritoneal leukocytes after 24 h of stimulation with each formulation (Fig. 6). The gating strategy is shown in Fig. 7. There was no significant difference in monocytic myeloid-derived suppressive cell (M-MDSC) populations between the stimulated groups and the unstimulated control group, while the abundance of granulocytic myeloid-derived suppressive cells (G-MDSCs) was significantly increased by all three formulations (P < 0.0001 for CPMV, P < 0.001 for eCPMV/p, and P < 0.01 for eCPMV/i). The number of tumor-infiltrating neutrophils (TINs) or type 1 neutrophils (N1) was significantly enhanced by CPMV (P < 0.01). Tumor-associated macrophages (TAMs), type 1 TAM (M1) populations (P < 0.01 for CPMV and no significant difference for eCPMV), and type 2 TAM (M2) populations (P < 0.0001 for CPMV and eCPMV/p and P < 0.0005 for eCPMV/i) were increased by all three formulations. The population of NK cells was increased following treatment with eCPMV/p (P < 0.05) and CPMV (no significant difference) stimulation. Furthermore, we observed a significant increase in the population of dendritic cells (DCs) (P < 0.0001) following stimulation with CPMV (but no enhancement of the activated DC subtype). The immune cell profile was also evaluated after in vivo stimulation. Either CPMV or eCPMV/p was i.p. injected into tumor-bearing mice (35 days postinoculation [dpi]), and after 24 h, peritoneal cavity cells were analyzed (Fig. 6B). Consistent with the ex vivo study results, the population of TINs (P < 0.0001) was dramatically enhanced by CPMV in situ treatment; M1 (P < 0.0001 for CPMV and P < 0.05 for eCPMV/p) and NK cell (P < 0.0001) abundances were increased by both CPMV and eCPMV/p treatments. However, different from the ex vivo results, we observed a potent decrease in the G-MDSC population (P < 0.0001) and an increase of M-MDSCs (P < 0.0001) by CPMV and eCPMV/p. CPMV significantly increased the levels of total TAMs and M2, while eCPMV/p reduced the M2 percentages in CD45+ cells. Notably, more activated DCs were found in the tumor microenvironment after CPMV treatment (P < 0.0001).
FIG 6.

CPMV and eCPMV stimulation induces immune cell profile changes in the tumor microenvironment. (A) Peritoneal cavity wash cells were collected from tumor-bearing mice on day 35 after tumor inoculation. The cells were incubated with CPMV or eCPMV (or PBS as a control) for 24 h and then analyzed by flow cytometry. (B) CPMV and eCPMV/p were i.p. injected into the peritoneal cavity of tumor-bearing mice on day 35 after tumor inoculation. The peritoneal cavity wash cells were collected 24 h later and analyzed by flow cytometry. Data are means ± SEM (n = 2 for the eCPMV/i ex vivo stimulation data set; n = 3 for all other data sets). Statistical significance was calculated by one-way ANOVA with a Tukey test. *, P < 0.05; **, P < 0.01; ***, P < 0.0005; ****, P < 0.0001.
FIG 7.

Gating strategy for innate immune cell profiles in the peritoneal cavity of tumor-bearing mice. SSC, side scatter; FSC, forward scatter; FITC, fluorescein isothiocyanate; APC, allophycocyanin; AF-700, Alexa Fluor 700; PE, phycoerythrin.
We therefore conclude that both CPMV and eCPMV can enhance antitumor leukocytes (M1 and NK cell) populations and regulate important cytokines that contribute to an immunostimulatory tumor microenvironment but that the presence of RNA in the CPMV particles has a specific impact, particularly on potent antigen-presenting cells (APCs), such as TINs and activated DCs.
DISCUSSION
The targeting of tumors with plant viruses can activate the innate immune system and instigate the transition to adaptive antitumor immunity. The engagement of antiviral effector responses caused by virus infection is essential when using VNPs or VLPs as immunotherapeutic agents. The repetitive and highly organized structure of coat proteins on VNPs and VLPs is important for the induction of immune responses because the monomeric form of the protein is not recognized as a pathogen-associated molecular pattern (PAMP) by the immune system (1). The VNP and VLP coat proteins may also enhance the costimulation of APCs by TLR signaling. For example, a live-attenuated recombinant poliovirus induces the IFN-dominant activation of dendritic cells and tumor antigen-specific cytotoxic T cells (25). A VLP derived from human papillomavirus was shown to stimulate Th1 responses via MyD88, a key signaling molecule in several TLR pathways (26).
To decipher the immunostimulatory potential of wild-type CPMV and eCPMV, we used a syngeneic immunocompetent murine orthotopic ovarian cancer model (ID8-Defb29/Vegf-A) (27) as the basis for in situ vaccination. Our results show that both CPMV and eCPMV can engage tumors immunologically and induce tumor regression.
Data indicate that CPMV or eCPMV treatment stimulates antitumor immunity through multiple mechanisms of action. Analysis of cytokine secretion in the tumor microenvironment after ex vivo and in vivo stimulation showed that both CPMV and eCPMV can induce broad and similar proinflammatory responses, which would lead to a significant activation of multiple immune cells. Specifically, the immunotherapeutic potential of both formulations involves the induction of type I and II interferons as well as various proinflammatory cytokines while repressing the secretion of TGF-β and IL-10. Type I IFN (IFN-α and IFN-β) is a key component of antigen-specific immunity against viruses, but it has additional effects, such as the upregulation of costimulatory molecules and major histocompatibility complex (MHC) expression, the promotion of dendritic cell maturation and tumor rejection (28), and the direct inhibition of regulatory T (Treg) cell functions (29, 30). IFN-γ plays a key role in the activation and editing of immune responses and is released mainly by cytotoxic T cells and T helper cells to induce a Th1-type immune response (31, 32). TGF-β is an immunosuppressive cytokine that reduces the number and activity of NK cells and cytotoxic T cells while increasing the number of Treg cells (33). In addition, IL-10 and TGF-β also affect myeloid cell functions and polarize TAMs and tumor-associated neutrophils (TANs) toward a protumor phenotype (34, 35). Disrupting the immunosuppressive microenvironment of tumors is one of the key objectives in cancer immunotherapy. The suppression of TGF-β and IL-10 secretion caused by CPMV and eCPMV may convert TANs and TAMs from a protumor to an antitumor phenotype, which restores the cytotoxicity of neutrophils (36) and facilitates the neutrophil-mediated transition from innate to adaptive immunity (37, 38).
Analysis of the cellular components after ex vivo and in vivo stimulation showed that CPMV effectively increased the TIN and M1 populations. We consider that the RNA component in CPMV may play an essential role in recruiting TINs and M1 in the tumor microenvironment by initiating other inflammatory reactions besides those shared with eCPMV. In contrast, eCPMV can only slightly increase the number and intensity of both activated phenotypes in ex vivo and in vivo studies, and the levels of both populations cannot be maintained for a long period.
We observed that CPMV ex vivo stimulation elevated IL-6 secretion and G-MDSCs. In tumor immune responses, IL-6 has a more complex role than other proinflammatory cytokines because tumor-derived IL-6 is associated with disease progression (39, 40). However, IL-6 can also induce anti-inflammatory and antitumor responses to inhibit tumor development (41–43). IL-6 is produced by a variety of different cell types, including fibroblasts, endothelial cells, macrophages, T cells, and polymorphonuclear leukocytes (PMNs) (including neutrophils and G-MDSCs), and it has complex mechanisms of action that include cell type-specific effects (44). IL-6 is predominantly produced by M1 macrophages, which amplify the inflammatory feedback loops in obesity (45). PMNs are the first line of defense against infectious agents or stimuli and can synthesize a variety of proinflammatory cytokines and chemokines, including TNF-α, IL-6, macrophage inflammatory protein alpha (MIP-α), IL-8, and IFN-γ-induced protein 10 (IP-10), which influence the recruitment of inflammatory cells, thus altering the immune response (46–48). Therefore, we hypothesize that IL-6 may be produced by M1 macrophages and PMNs following CPMV stimulation. We recently demonstrated that in vitro CPMV stimulation could activate macrophages and induce strong IL-6 and TNF-α secretion by macrophages (49). Interestingly, the levels of IL-6 and G-MDSCs were reduced after 24 h of in vivo stimulation. A consistent observation was also discussed in our previous study, where IL-6 secretion and G-MDSC numbers were enhanced after repeated in situ CPMV treatments in a short period (6 h) but returned to basal levels in a long period (48 h) (50). We hypothesize that IL-6 may quickly circulate into the system and further recruit/activate more immune cells; the G-MDSCs may differentiate into neutrophils or other immune cells.
The RNA-containing CPMV particles extended the survival of tumor-bearing mice further than the eCPMV particles and also triggered additional cytokine and immune cell responses. Specifically, the CPMV particles induced the proinflammatory cytokine IL-6 and boosted the population of potential APCs by increasing the abundance of TINs, M1, and activated DCs, which should enhance the antigen-processing capacity of APCs in the tumor microenvironment and initiate adaptive immunity and tumor-specific cytotoxic T cells. The different immunomodulatory behaviors of CPMV and eCPMV reflect the encapsulated single-stranded RNA present in CPMV, which can activate TLR7/8. TLR7-driven type I IFN appears to be required for the antitumor potency of RNA-based vaccines (51).
We conclude that CPMV and eCPMV could each be developed as novel adjuvants to overcome immunosuppression and thus promote tumor regression in ovarian cancer. While without further modification, CPMV appears to be more potent, the ability to add a greater secondary payload to eCPMV could enable it to have a unique capability that CPMV could not provide. The interaction between the surface-exposed regions of VNPs and immune cells may trigger the recognition of PAMPs, inducing an efficient immune response in the tumor microenvironment. To our knowledge, this is the first study to define the immunotherapeutic differences between CPMV and eCPMV, which is essential for the further development of biomedical applications for plant viruses and the selection of rational combinations of immunomodulatory reagents.
MATERIALS AND METHODS
Preparation of wild-type CPMV.
CPMV nanoparticles were produced in plants as previously described (10). LPS was quantified using the Endozyme II recombinant factor C endotoxin detection assay (Biovendor), and a level of <50 endotoxin units (EU)/mg protein was considered negligible.
Preparation of eCPMV/p.
N. benthamiana plants were grown in soil for 6 weeks with a 16-h photoperiod (10,000 lux) at 24°C and 60% humidity before transient expression of the CPMV coat proteins using the agroinfiltration system based on A. tumefaciens (52). A preculture of 10 ml lysogeny broth (LB) supplemented with streptomycin and kanamycin was inoculated with 100 μl of an A. tumefaciens LBA4404 glycerol stock carrying the plasmid pEAQ-VP60-24K encoding the L and S coat protein subunits. After overnight cultivation at 28°C and 160 rpm, the preculture was used to inoculate a large culture for vacuum infiltration, which was grown under the same conditions for 48 h. For infiltration, the bacterial suspension was diluted to an optical density (OD) of 1.0 with 2× infiltration medium (100 g/liter sucrose, 4 g/liter glucose, 1 g/liter Jack’s professional 20-10-20 Peat Lite water-soluble fertilizer). To activate the vir genes, we added 200 μM acetosyringone, and the suspension was incubated at room temperature for 1 h.
Prior to infiltration, all plants were sprayed with tap water to facilitate infiltration. Plants were submerged into 2 liters of the bacterial suspension in a bucket in a desiccator. The desiccator lid was replaced, and a vacuum was applied. After pressure equalization, all noninfiltrated leaves were removed, and the plants were incubated under the conditions described above. After 5 days, all leaves were harvested and either directly processed or stored at −80°C.
The harvested plant material was homogenized in a blender with 3 volumes of 0.1 M potassium phosphate (KP) buffer (pH 7.0) and supplemented with 2% (wt/vol) polyvinylpolypyrrolidone (PVPP). After stirring, the material was filtered through two layers of Miracloth and centrifuged at 14,000 × g for 20 min at 4°C before adding 0.25 volumes of polyethylene glycol (PEG) precipitation buffer (1 M NaCl, 20% PEG 6000) to the supernatant and stirring overnight at 4°C. After centrifugation at 14,000 × g for 20 min at 4°C, the supernatant was kept, and the pellet was resuspended in 10 ml 0.01 M KP buffer and centrifuged again at 27,000 × g for 20 min at 4°C. The supernatants from both steps were combined for ultracentrifugation over a 10-ml 30% sucrose cushion at 133,000 × g for 3 h at 4°C. The sucrose fraction was collected and dialyzed against 0.01 M KP buffer for 48 h, with frequent buffer exchange.
Preparation of eCPMV/i.
The eCPMV/i formulation was produced in Sf9 insect cells by Creative Biolabs. The sequences of the CPMV L and S coat protein subunits as well as the 24K protease were codon optimized for insects and introduced into the pFastBac dual-expression vector (Thermo Fisher Scientific) to generate plasmid pFBD-VP60/24K.
Denaturing gel electrophoresis (SDS-PAGE).
CPMV and eCPMV particles were denatured at 100°C for 5 min in 4× lithium dodecyl sulfate (LDS) loading dye (Thermo Fisher Scientific) to obtain a final volume of 20 μl containing 20 μg of particles. The L and S subunits were separated for 40 min at 200 V and 120 mA in 4 to 12% NuPAGE precast gels in 1× morpholinepropanesulfonic acid (MOPS) buffer (Thermo Fisher Scientific). Gels were photographed before and after staining with 0.25% (wt/vol) Coomassie brilliant blue using the AlphaImager imaging system (Protein Simple) under white light.
Agarose gel electrophoresis.
CPMV and eCPMV samples (10 μg in 6× loading dye) were fractionated by 0.8% (wt/vol) agarose gel electrophoresis at 100 V for 30 min in Tris-borate-EDTA (TBE) buffer. Gels were visualized under UV light and after staining with 0.25% (wt/vol) Coomassie brilliant blue.
DLS analysis.
Samples were analyzed at 25°C in plastic disposable cuvettes with a path length of 10 mm using a Wyatt DynaPro NanoStar DLS instrument. The diameter was calculated as the mean of the intensity distribution, and the polydispersity index was determined from the intensity autocorrelation function.
TEM analysis.
TEM was performed using a Zeiss Libra 200EF microscope. Samples were mounted on 400-mesh hexagonal copper grids bearing Formvar support film, stained with 2% uranyl acetate, and allowed to dry for 12 h.
SEC analysis.
One hundred microliters of each sample (1 mg/ml) was injected into a Superose 6 column on an Äkta Explorer chromatography system (GE Healthcare) at a flow rate of 0.5 ml/min in 10 mM KP buffer (pH 7.4), and the absorbance at 260 and 280 nm was recorded.
Mass spectrometry.
L and S gel bands from each sample (20 μg) were excised, cleaned, and digested with lysyl endopeptidase (Wako Chemicals) at an enzyme/substrate ratio of 1:20 for 2 h at 37°C, followed by overnight trypsin digestion using sequencing-grade trypsin (Promega) at an enzyme/substrate ratio of 1:20 at 37°C (53). The digested peptides were analyzed by LC-MS/MS using an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) equipped with a nanoAcquity ultra-high-pressure liquid chromatography system (Waters). Full-scan MS spectra (m/z 380 to 1,800) were acquired at a resolution of 60,000, followed by 20 data-dependent MS/MS scans. MS/MS spectra were generated by collision-induced dissociation of the peptide ions (normalized collision energy of 35%, activation Q of 0.250, and activation time of 20 ms) to generate a series of b and y ions as major fragments. LC-MS/MS raw data were acquired using Xcalibur v2.2 SP1 (Thermo Fisher Scientific). Raw files (one for each sample) were imported into Rosetta Elucidator v3.3.0.1.SP.25 (Rosetta Bio-software) for processing (54). The peak list (.dta) files were searched against the UniProt virus database (550,552 sequences) using Mascot v2.1 (Matrix Science) with the following settings: trypsin enzyme specificity, mass accuracy window for precursor ions of 10 ppm, mass accuracy window for fragment ions of 0.8 Da, carbamidomethylation of cysteines as a fixed modification, oxidation of methionine as a variable modification, and one missed cleavage. The peptide identification criteria were a mass accuracy of ≤10 ppm, expectation P value of <0.05, and estimated false discovery rate (FDR) of <2%. The results were imported into Rosetta Elucidator for the automated differential quantification of peptides (54). Retention time (RT) alignment, feature (RT × m/z surface) identification and peptide ion extraction (peak), and background subtraction and smoothing across both RT and m/z dimensions were performed using PeakTeller. Normalization of signal intensities across samples was achieved using the average signal intensities obtained in each sample. For each peptide, the average intensity for all samples within a group was calculated.
Endotoxin removal.
Endotoxins were removed by treating all particles (CPMV, eCPMV/p, and eCPMV/i) three times with 1% (vol/vol) Triton X-114 (18, 21). After the addition of Triton X-114, samples were incubated on a rolling platform at 4°C for 20 min and then at 37°C for 10 min before centrifugation at 20,000 × g for 10 min at room temperature. The clear layer from the phase separation was collected and transferred to a new tube. After the last step, the samples were centrifuged at 150,000 × g for 1 h at 4°C. The supernatants were discarded, and the pellets were resuspended in 0.1 M potassium phosphate buffer (pH 7.2). Subsequently, Triton was removed using Pierce detergent removal spin columns (Thermo Fisher) with 95% removal efficacy according to manufacturer’s instructions.
Heat precipitation.
We heated 10 μg of CPMV or eCPMV/p particles with or without LPS for 10 min at 90°C, followed by brief centrifugation. To test for endotoxins, 10 μg of CPMV or eCPMV/p with and without LPS was diluted 1:10 in endotoxin-free water and assessed using the Endozyme II recombinant factor C endotoxin detection assay (Biovendor) according to the manufacturer’s instructions.
Animals and in situ vaccination.
Female C57BL/6J mice (6 to 8 weeks old) were purchased from The Jackson Laboratory. All mouse studies were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University. ID8-Defb29/Vegf-A cells were transfected with luciferase as previously described (24). Briefly, 10 μg of plasmids vesicular stomatitis virus G and pBabe-puro Luc was added to the GP2-293 packaging cell line following a 30-min incubation with 60 μl of TransIT (Mirus). GP2-293 cells and plasmids were a generous gift from William P. Schiemann, Case Western Reserve University. Forty-eight hours following the addition of plasmid, medium was collected, filtered to remove cellular debris, and added to ID8-Defb29/Vegf-A cells in the presence of Polybrene (8 μg/ml; Santa Cruz Biotechnology) at a ratio of 50:50 with modified RPMI 1640 medium typically used with this cell line. ID8-Defb29/Vegf-A cells were treated with the retroviral particles for 24 h, allowed to recover in fresh medium for 24 h, and selected for transduction with puromycin for 5 days. We implanted 2 × 106 live cells/200 μl PBS orthotopically into mice by i.p. injection (55). One hundred micrograms of CPMV or eCPMV was administered once a week by i.p. injection in 200 μl PBS for a total of 6 treatments. The mice were monitored weekly for signs of tumor progression, including abdominal distension, weight, circumference, and other morbidity indicators. Tumor growth was also monitored twice weekly by total bioluminescence imaging, based on the i.p. injection of 150 mg/kg of body weight luciferin (Thermo Fisher Scientific), followed by analysis using an Ivis spectrum imaging system (PerkinElmer). Total bioluminescence was determined using Living Image software (PerkinElmer). Regions of interest were quantified as average radiance (photons per second). Mice were euthanized when their weight reached 35 g or when moribund.
Cytokine quantification.
Peritoneal cavity washes and cell culture supernatants were tested by an enzyme-linked immunosorbent assay (ELISA) to detect interleukin-1β (IL-1β), IL-6, IL-10, IL-12, tumor necrosis factor alpha (TNF-α), transforming growth factor β (TGF-β), interferon beta (IFN-β), granulocyte-macrophage colony-stimulating factor (GM-CSF), and IFN-γ (BioLegend) according to the manufacturer’s instructions. For ex vivo stimulation, 105 peritoneal cavity cells from tumor-bearing mice (35 days postinoculation [dpi]) were collected, plated in 96-well plates, and then stimulated with 10 μg CPMV or eCPMV. After 24 h, the supernatant was collected for cytokine quantification; nonadherent cells were washed away with PBS, and the profile of surviving cells was evaluated using flow cytometry. For in vivo stimulation, 100 μg of CPMV or eCPMV/p was i.p. administered to tumor-bearing mice (35 dpi). After 24 h, the peritoneal cavity wash was collected. Cells were spun down by centrifugation (500 × g for 5 min), the supernatant was used for cytokine quantification, and the cells were resuspended and analyzed by flow cytometry.
Flow cytometry.
Cells were washed in cold PBS containing 1 mM EDTA and resuspended in staining buffer (PBS containing 2% fetal bovine serum, 1 mM EDTA, and 0.1% sodium azide). Fc receptors were blocked using anti-mouse CD16/CD32 (BioLegend) for 15 min and then tested with the following fluorescence-labeled antibodies (BioLegend) for 30 min at 4°C: CD45 (30-F11), CD11b (M1/70), CD86 (GL-1), major histocompatibility complex class II (MHCII) (M5/114.15.2), Ly6G (1A8), CD11c (N418 A), F4/80 (BM8), Ly6C (HK1.4), NK1.1 (PK136), and isotype controls. Cells were washed twice and resuspended in staining buffer for data acquisition. Flow cytometry was carried out using a BD LSRII cytometer (BD Biosciences), and the data were analyzed using FlowJo software (TreeStar). OneComp eBeads (eBioscience) were used as compensation controls.
ACKNOWLEDGMENTS
We are grateful to Steven N. Fiering and Chenkai Mao (Dartmouth Geisel School of Medicine) for critical readings of the manuscript.
This work was supported by a grant from the NCI Cancer Nanotechnology Alliance, U01 CA218292 (to N.F.S.).
REFERENCES
- 1.Bachmann MF, Jennings GT. 2010. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol 10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 2.Scotti N, Rybicki EP. 2013. Virus-like particles produced in plants as potential vaccines. Expert Rev Vaccines 12:211–224. doi: 10.1586/erv.12.147. [DOI] [PubMed] [Google Scholar]
- 3.D’Aoust M-A, Couture MMJ, Charland N, Trépanier S, Landry N, Ors F, Vézina L-P. 2010. The production of hemagglutinin-based virus-like particles in plants: a rapid, efficient and safe response to pandemic influenza. Plant Biotechnol J 8:607–619. doi: 10.1111/j.1467-7652.2009.00496.x. [DOI] [PubMed] [Google Scholar]
- 4.Tschofen M, Knopp D, Hood E, Stöger E. 2016. Plant molecular farming: much more than medicines. Annu Rev Anal Chem 9:271–294. doi: 10.1146/annurev-anchem-071015-041706. [DOI] [PubMed] [Google Scholar]
- 5.Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering S. 2016. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat Nanotechnol 11:295–303. doi: 10.1038/nnano.2015.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murray AA, Wang C, Fiering S, Steinmetz NF. 2018. In situ vaccination with cowpea vs tobacco mosaic virus against melanoma. Mol Pharm 15:3700–3716. doi: 10.1021/acs.molpharmaceut.8b00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoopes PJ, Wagner RJ, Duval K, Kang K, Gladstone DJ, Moodie KL, Crary-Burney M, Ariaspulido H, Veliz FA, Steinmetz NF, Fiering SN. 2018. Treatment of canine oral melanoma with nanotechnology-based immunotherapy and radiation. Mol Pharm 15:3717–3722. doi: 10.1021/acs.molpharmaceut.8b00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wellink J. 1998. Comovirus isolation and RNA extraction, p 205–209. In Foster GD, Taylor SC (ed), Plant virology protocols: from virus isolation to transgenic resistance. Humana Press, Totowa, NJ. [Google Scholar]
- 9.Sainsbury F, Cañizares MC, Lomonossoff GP. 2010. Cowpea mosaic virus: the plant virus-based biotechnology workhorse. Annu Rev Phytopathol 48:437–455. doi: 10.1146/annurev-phyto-073009-114242. [DOI] [PubMed] [Google Scholar]
- 10.Wen AM, Shukla S, Saxena P, Aljabali AAA, Yildiz I, Dey S, Mealy JE, Yang AC, Evans DJ, Lomonossoff GP, Steinmetz NF. 2012. Interior engineering of a viral nanoparticle and its tumor homing properties. Biomacromolecules 13:3990–4001. doi: 10.1021/bm301278f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lebel M-È, Chartrand K, Tarrab E, Savard P, Leclerc D, Lamarre A. 2016. Potentiating cancer immunotherapy using papaya mosaic virus-derived nanoparticles. Nano Lett 16:1826–1832. doi: 10.1021/acs.nanolett.5b04877. [DOI] [PubMed] [Google Scholar]
- 12.Carignan D, Herblot S, Laliberté-Gagné M-È, Bolduc M, Duval M, Savard P, Leclerc D. 2018. Activation of innate immunity in primary human cells using a plant virus derived nanoparticle TLR7/8 agonist. Nanomedicine 14:2317–2327. doi: 10.1016/j.nano.2017.10.015. [DOI] [PubMed] [Google Scholar]
- 13.Saunders K, Sainsbury F, Lomonossoff GP. 2009. Efficient generation of cowpea mosaic virus empty virus-like particles by the proteolytic processing of precursors in insect cells and plants. Virology 393:329–337. doi: 10.1016/j.virol.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 14.Sainsbury F, Saunders K, Aljabali AAA, Evans DJ, Lomonossoff GP. 2011. Peptide-controlled access to the interior surface of empty virus nanoparticles. Chembiochem 12:2435–2440. doi: 10.1002/cbic.201100482. [DOI] [PubMed] [Google Scholar]
- 15.Steinmetz NF, Evans DJ, Lomonossoff GP. 2007. Chemical introduction of reactive thiols into a viral nanoscaffold: a method that avoids virus aggregation. Chembiochem 8:1131–1136. doi: 10.1002/cbic.200700126. [DOI] [PubMed] [Google Scholar]
- 16.Raetz CRH, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraker DL, Stovroff MC, Merino MJ, Norton JA. 1988. Tolerance to tumor necrosis factor in rats and the relationship to endotoxin tolerance and toxicity. J Exp Med 168:95–105. doi: 10.1084/jem.168.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu S, Tobias R, McClure S, Styba G, Shi Q, Jackowski G. 1997. Removal of endotoxin from recombinant protein preparations. Clin Biochem 30:455–463. doi: 10.1016/S0009-9120(97)00049-0. [DOI] [PubMed] [Google Scholar]
- 19.Anspach FB. 2001. Endotoxin removal by affinity sorbents. J Biochem Biophys Methods 49:665–681. doi: 10.1016/S0165-022X(01)00228-7. [DOI] [PubMed] [Google Scholar]
- 20.Petsch D, Anspach FB. 2000. Endotoxin removal from protein solutions. J Biotechnol 76:97–119. doi: 10.1016/S0168-1656(99)00185-6. [DOI] [PubMed] [Google Scholar]
- 21.Teodorowicz M, Perdijk O, Verhoek I, Govers C, Savelkoul HFJ, Tang Y, Wichers H, Broersen K. 2017. Optimized Triton X-114 assisted lipopolysaccharide (LPS) removal method reveals the immunomodulatory effect of food proteins. PLoS One 12:e0173778. doi: 10.1371/journal.pone.0173778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rae C, Koudelka KJ, Destito G, Estrada MN, Gonzalez MJ, Manchester M. 2008. Chemical addressability of ultraviolet-inactivated viral nanoparticles (VNPs). PLoS One 3:e3315. doi: 10.1371/journal.pone.0003315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT, Ahonen CL, Conejo-Garcia JR. 2009. In situ stimulation of CD40 and Toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells. Cancer Res 69:7329–7337. doi: 10.1158/0008-5472.CAN-09-0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wallat JD, Czapar AE, Wang C, Wen AM, Wek KS, Yu X, Steinmetz NF, Pokorski JK. 2017. Optical and magnetic resonance imaging using fluorous colloidal nanoparticles. Biomacromolecules 18:103–112. doi: 10.1021/acs.biomac.6b01389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown MC, Holl EK, Boczkowski D, Dobrikova E, Mosaheb M, Chandramohan V, Bigner DD, Gromeier M, Nair SK. 2017. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci Transl Med 9:eaan4220. doi: 10.1126/scitranslmed.aan4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang R, Murillo FM, Cui H, Blosser R, Uematsu S, Takeda K, Akira S, Viscidi RP, Roden RBS. 2004. Papillomavirus-like particles stimulate murine bone marrow-derived dendritic cells to produce alpha interferon and Th1 immune responses via MyD88. J Virol 78:11152–11160. doi: 10.1128/JVI.78.20.11152-11160.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conejo-Garcia JR, Benencia F, Courreges M-C, Kang E, Mohamed-Hadley A, Buckanovich RJ, Holtz DO, Jenkins A, Na H, Zhang L, Wagner DS, Katsaros D, Caroll R, Coukos G. 2004. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med 10:950–958. doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 28.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD. 2011. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bacher N, Raker V, Hofmann C, Graulich E, Schwenk M, Baumgrass R, Bopp T, Zechner U, Merten L, Becker C, Steinbrink K. 2013. Interferon-α suppresses cAMP to disarm human regulatory T cells. Cancer Res 73:5647–5656. doi: 10.1158/0008-5472.CAN-12-3788. [DOI] [PubMed] [Google Scholar]
- 30.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. 2015. Type I interferons in anticancer immunity. Nat Rev Immunol 15:405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 31.Gajewski TF, Fuertes MB, Woo SR. 2012. Innate immune sensing of cancer: clues from an identified role for type I IFNs. Cancer Immunol Immunother 61:1343–1347. doi: 10.1007/s00262-012-1305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. 2011. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J Exp Med 208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, Jay SM, Demento SL, Agawu A, Licona Limon P, Ferrandino AF, Gonzalez D, Habermann A, Flavell RA, Fahmy TM. 2012. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat Mater 11:895–905. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. 2009. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nam J-S, Terabe M, Mamura M, Kang M-J, Chae H, Stuelten C, Kohn E, Tang B, Sabzevari H, Anver MR, Lawrence S, Danielpour D, Lonning S, Berzofsky JA, Wakefield LM. 2008. An anti-transforming growth factor β antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res 68:3835–3843. doi: 10.1158/0008-5472.CAN-08-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C, Cosmi L, Lunardi C, Annunziato F, Romagnani S, Cassatella MA. 2010. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 115:335–343. doi: 10.1182/blood-2009-04-216085. [DOI] [PubMed] [Google Scholar]
- 37.Leliefeld PHC, Koenderman L, Pillay J. 2015. How neutrophils shape adaptive immune responses. Front Immunol 6:471. doi: 10.3389/fimmu.2015.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolaczkowska E, Kubes P. 2013. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 39.Balkwill F, Charles KA, Mantovani A. 2005. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Pollard JW. 2004. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 41.Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, Eppler E, Bouzakri K, Wueest S, Muller YD, Hansen AMK, Reinecke M, Konrad D, Gassmann M, Reimann F, Halban PA, Gromada J, Drucker DJ, Gribble FM, Ehses JA, Donath MY. 2011. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med 17:1481–1489. doi: 10.1038/nm.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White PJ, St-Pierre P, Charbonneau A, Mitchell PL, St-Amand E, Marcotte B, Marette A. 2014. Protectin DX alleviates insulin resistance by activating a myokine-liver glucoregulatory axis. Nat Med 20:664–669. doi: 10.1038/nm.3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilbert LA, Hemann MT. 2012. Context-specific roles for paracrine IL-6 in lymphomagenesis. Genes Dev 26:1758–1768. doi: 10.1101/gad.197590.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mauer J, Denson JL, Brüning JC. 2015. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol 36:92–101. doi: 10.1016/j.it.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 45.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. 2010. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grenier A, Dehoux M, Boutten A, Arce-Vicioso M, Durand G, Gougerot-Pocidalo M-A, Chollet-Martin S. 1999. Oncostatin M production and regulation by human polymorphonuclear neutrophils. Blood 93:1413–1421. [PubMed] [Google Scholar]
- 47.Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL. 2005. Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res 65:8896–8904. doi: 10.1158/0008-5472.CAN-05-1734. [DOI] [PubMed] [Google Scholar]
- 48.Nathan C. 2006. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 49.Wang C, Steinmetz NF. 2019. CD47 blockade and cowpea mosaic virus nanoparticle in situ vaccination triggers phagocytosis and tumor killing. Adv Healthc Mater 8:e1801288. doi: 10.1002/adhm.201801288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang C, Fiering SN, Steinmetz NF. 2019. Cowpea mosaic virus promotes anti-tumor activity and immune memory in a mouse ovarian tumor model. Adv Ther 2:1900003. doi: 10.1002/adtp.201900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, Grunwitz C, Vormehr M, Hüsemann Y, Selmi A, Kuhn AN, Buck J, Derhovanessian E, Rae R, Attig S, Diekmann J, Jabulowsky RA, Heesch S, Hassel J, Langguth P, Grabbe S, Huber C, Türeci Ö, Sahin U. 2016. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534:396–401. doi: 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- 52.Van Larebeke N, Genetello C, Hernalsteens JP, De Picker A, Zaenen I, Messens E, Van Montagu M, Schell J. 1977. Transfer of Ti plasmids between Agrobacterium strains by mobilisation with the conjugative plasmid RP4. Mol Gen Genet 152:119–124. doi: 10.1007/BF00268807. [DOI] [Google Scholar]
- 53.Wiśniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat Methods 6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 54.Azzam S, Schlatzer D, Maxwell S, Li X, Bazdar D, Chen Y, Asaad R, Barnholtz-Sloan J, Chance MR, Sieg SF. 2016. Proteome and protein network analyses of memory T cells find altered translation and cell stress signaling in treated human immunodeficiency virus patients exhibiting poor CD4 recovery. Open Forum Infect Dis 3:ofw037. doi: 10.1093/ofid/ofw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baird JR, Fox BA, Sanders KL, Lizotte PH, Cubillos-Ruiz JR, Scarlett UK, Rutkowski MR, Conejo-Garcia JR, Fiering S, Bzik DJ. 2013. Avirulent Toxoplasma gondii generates therapeutic antitumor immunity by reversing immunosuppression in the ovarian cancer microenvironment. Cancer Res 73:3842–3851. doi: 10.1158/0008-5472.CAN-12-1974. [DOI] [PMC free article] [PubMed] [Google Scholar]