

Type I interferon (IFN-I) plays a critical role in the antiviral innate immune responses that protect the host against virus infection. The negative regulators of IFN-I are important not only for fine-tuning the antiviral responses to pathogens but also for preventing excessive inflammation. Identification of negative regulators and study of their modulation in innate immune responses will lead to new strategies for the control of both viral and inflammatory diseases. Here, we report for the first time that the cell growth-regulating nucleolar protein LYAR behaves as a repressor of host innate immune responses. We demonstrate that LYAR negatively regulates IFN-β-mediated immune responses by inhibiting the DNA binding ability of IFN regulatory factor 3 (IRF3). Our study reveals a common mechanism of LYAR in promoting different virus replication events and improves our knowledge of host negative regulation of innate immune responses.
Keywords: innate immune responses, IFN-β, LYAR, IRF3
ABSTRACT
The innate immune response is vital for host defense and must be tightly controlled, but the mechanisms responsible for its negative regulation are not fully understood. The cell growth-regulating nucleolar protein LYAR was found to promote replication of multiple viruses in our previous study. Here, we report that LYAR acts as a negative regulator of innate immune responses. We found that LYAR expression is induced by beta interferon (IFN-β) during virus infection. Further studies showed that LYAR interacts with phosphorylated IFN regulatory factor 3 (IRF3) to impede the DNA binding capacity of IRF3, thereby suppressing the transcription of IFN-β and downstream IFN-stimulated genes (ISGs). In addition, LYAR inhibits nuclear factor-κB (NF-κB)-mediated expression of proinflammatory cytokines. In summary, our study reveals the mechanism of LYAR in modulating IFN-β-mediated innate immune responses by targeting phosphorylated IRF3, which not only helps us to better understand the mechanisms of LYAR-regulated virus replication but also uncovers a novel role of LYAR in host innate immunity.
IMPORTANCE Type I interferon (IFN-I) plays a critical role in the antiviral innate immune responses that protect the host against virus infection. The negative regulators of IFN-I are important not only for fine-tuning the antiviral responses to pathogens but also for preventing excessive inflammation. Identification of negative regulators and study of their modulation in innate immune responses will lead to new strategies for the control of both viral and inflammatory diseases. Here, we report for the first time that the cell growth-regulating nucleolar protein LYAR behaves as a repressor of host innate immune responses. We demonstrate that LYAR negatively regulates IFN-β-mediated immune responses by inhibiting the DNA binding ability of IFN regulatory factor 3 (IRF3). Our study reveals a common mechanism of LYAR in promoting different virus replication events and improves our knowledge of host negative regulation of innate immune responses.
INTRODUCTION
Viruses must overcome host innate immune responses to establish a productive infection. During infection, the host senses pathogen invasion via the recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs), followed by a series of signal cascades resulting in the transcription of antiviral genes (1–3). Upon viral RNA recognition, PRR retinoic acid-inducible gene I (RIG-I) recruits the adaptor molecule mitochondrial antiviral signaling protein (MAVS) to activate the TANK-binding kinase 1/inhibitor of κB kinase ε (TBK1/IKKε) complex and the inhibitor of κB kinase α/β (IKKα/β) complex, leading to the activation of interferon-regulatory factor 3/7 (IRF3/7) and nuclear factor-κB (NF-κB), which ultimately induces the production of type I interferon (IFN-I) and proinflammatory cytokines (4–6). Following secreted IFN-I (IFN-α, IFN-β) binding to cell surface receptors, the Janus-activated kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway is activated, leading to the phosphorylation of STAT1 and STAT2. The phosphorylated STAT1 and STAT2 associate with IRF9 to form a complex termed interferon-stimulated gene factor 3 (ISGF3), which enters the nucleus. ISGF3 binds to the IFN-stimulated response element (ISRE) to activate the transcription of IFN-stimulated genes (ISGs), which induces a cellular antiviral state (3, 7–9).
Many cellular factors are involved in the IFN-I-inducing pathway, and among them, IRF3 is the major transcription factor which mediates IFN-I production (10). Upon viral infection, cytoplasmic IRF3 is phosphorylated and forms dimers, followed by nuclear translocation, association with CREB-binding protein (CBP)/p300 coactivators to form a complex, and binding to the promoter of the targeted genes, ultimately leading to the transcription of IFN-I and the downstream ISGs (11–14). However, IFN-I must be tightly controlled to avoid inflammatory disorders (15). One of the mechanisms employed by the host to achieve suppression of IFN-I production is inhibition of IRF3 activity. Some negative regulators have been identified and shown to impair IRF3 functions via protein degradation, dephosphorylation, or dimerization inhibition. For instance, Pin 1 (peptidylprolyl cis-trans isomerase NIMA-interacting 1) inhibits innate immune responses via proteasomal degradation of IRF3 (16); INKIT (inhibitor for NF-κB and IRF3) blocks phosphorylation of IRF3 by impairing the recruitment of it to the upstream kinase TBK1/IKKε (17); Rubicon (RUN domain Beclin-1-interacting cysteine-rich domain containing) interacts with IRF3 to inhibit dimerization of IRF3 (18). Regardless, the study of the mechanisms of the negative regulation of IRF3-mediated IFN-I signaling is important for rationally controlling IFN-I to avoid deleterious imbalances in the immune responses.
LYAR, the cell growth-regulating nucleolar protein, which contains two C2HC-type zinc finger motifs and three nuclear localization signal sequences (NLSs) (19), has been identified as an influenza A viral RNP (vRNP)-interacting partner and facilitated virus replication by stimulating vRNP assembly in our previous study (20). In addition, knocking down LYAR can significantly inhibit not only influenza A virus (IAV) but also vesicular stomatitis virus (VSV) and Japanese encephalitis virus (JEV) replication, indicating that LYAR is involved in the regulation of multiple virus replication events and may utilize a common method in these events. In the current study, we show that LYAR participates in antiviral innate immune responses. LYAR represses the expression of IFN-β, ISGs, and proinflammatory cytokines, thus promoting virus replication. Mechanistically, viral infection increases LYAR expression, which in turn associates with phosphorylated IRF3 to impair the DNA binding ability of IRF3. Our findings uncovered a novel role of LYAR as a negative regulator of innate immune responses.
RESULTS
IFN-β increases the expression of LYAR during virus infection.
LYAR was shown to be involved in the replication of multiple viruses, including IAV, VSV, and JEV in our previous study (20). Considering that all these viruses are regulated by IFN-I, LYAR might regulate their replication by modulating IFN-I signal pathways. Moreover, LYAR interacts with influenza A viral RNP (vRNP), and vRNP can recruit host proteins to antagonize innate immune responses during infection (21). Based on these findings, we made a further investigation into the role of LYAR in the virus-triggered innate immune responses.
Since IAV infection enhances LYAR expression, we wondered whether other virus infections have a similar effect. The expression levels of LYAR in Sendai virus (Sev) and VSV-green fluorescent protein (GFP)-infected cells were determined. A549 cells were infected with Sev or VSV-GFP at a multiplicity of infection (MOI) of 1.0 or 10, respectively. The mRNA and protein levels of LYAR and virus replication levels were detected. The data showed that the mRNA and protein levels of LYAR in A549 cells gradually increased until 24 h postinfection after both a high and a low dose of Sev infection, which are positively correlated with virus replication represented by the expression level of Sev M mRNA during the infection course (Fig. 1A and B). It appears that the expression of LYAR responds sensitively to Sev infection, because the expression of LYAR was remarkably induced by Sev during the early infection period, and the high-dose infection did not upregulate LYAR expression to a greater extent than the low-dose infection (Fig. 1A and B). Similarly, both the mRNA and protein levels of LYAR were significantly increased after VSV-GFP infection (Fig. 1C and D). In addition, LYAR expression levels in A549 cells gradually increased with the stimulation of an increasing amount of the viral RNA mimic poly(I·C) (Fig. 1F), which is correlated with the expression of IFN-β upon poly(I·C) stimulation (Fig. 1E). These results collectively illustrate that different virus infections increase the expression of LYAR. A previous study showed that an inducible interaction between oligoadenylate synthetase 3 (OAS3) and LYAR may occur in poly(I·C)-stimulated A549 cells (22). Given that OAS3 is known to be induced by IFNs (23), it would be interesting to see whether the increased expression of LYAR upon virus infection is caused by the virus itself or the virus-induced IFNs. Therefore, IFN-β was used to treat A549 cells to detect the expression of LYAR. As a control, we monitored the expression of Mx1 that is typically induced by IFN-β. We found that the LYAR mRNA and protein levels were upregulated by IFN-β in a dose-dependent manner (Fig. 1G), indicating that IFN-β can induce LYAR expression in A549 cells. To further confirm this result, the Vero cells which lack the functional type I IFN genes (24) were infected with Sev, and then the LYAR expression was detected. Results showed that the expression levels of LYAR were little changed upon Sev infection (Fig. 1H), while when Vero cells were treated with IFN-β, a significant increase in LYAR expression was observed (Fig. 1I), suggesting that the induction of LYAR expression is indeed mediated by IFN-β. Considering that type I IFNs (IFN-α, IFN-β) and type III IFNs (IFN-λ) share similar immune responses and biological activities, both of which can be induced by virus (25). IFN-α and IFN-λ may also induce the expression of LYAR. To test this, the expression levels of LYAR in A549 cells were detected after IFN-α and IFN-λ1 (interleukin 29 [IL-29]) treatment. We observed that the expression of the control ISG, Mx1, a gene that can be induced by type I and III IFNs (25), was effectively induced, whereas the expression levels of LYAR were not affected (Fig. 1J and K), suggesting that IFN-α and IFN-λ1 cannot induce LYAR in A549 cells. Taken together, these results suggest that the virus-induced IFN-β increases the expression of LYAR, indicating a potential role of LYAR in the IFN-β signaling pathway.
FIG 1.

IFN-β increases the expression of LYAR. LYAR expression level in Sev-infected (A and B) and VSV-GFP-infected (C and D) cells. A549 cells were infected with virus at an MOI of 1.0 and 10. Samples were collected at 0, 6, 12, and 24 hpi, followed by qRT-PCR and Western blotting to determine the mRNA (A and C) and protein (B and D) levels of LYAR, respectively. (E and F) LYAR expression level in poly(I·C)-stimulated cells. A549 cells were left untransfected or were transfected with increasing amounts of poly(I·C) (50, 100, and 200 ng) for 6 h. The mRNA levels of IFN-β (E) and both the mRNA and protein levels of LYAR (F) were detected. (G) LYAR expression level in IFN-β-treated A549 cells. Cells were left untreated or were treated with increasing concentrations of IFN-β (500, 1,000, and 2,000 U/ml) for 12 h, and the mRNA level of Mx1 (left) and both mRNA and protein levels of LYAR (right) were detected. (H) LYAR expression level in Sev-infected Vero cells. Cells were infected with Sev at an MOI of 1.0 for the indicated times, and the mRNA level of Sev M segment (left) and mRNA and protein levels of LYAR (right) were detected. (I) LYAR expression level in IFN-β-treated Vero cells. Cells were treated as described for panel G, and the mRNA (left) and protein (right) levels of LYAR were detected. (J and K) LYAR expression level in IFN-α- and IFN-λ1-treated A549 cells. Cells were left untreated or were treated with the indicated concentrations of IFN-α (J) or IFN-λ1 (K) for 12 h, and the mRNA level of Mx1 (left) and both mRNA and protein (right) levels of LYAR were detected. For real-time PCR analysis, the mRNA level was normalized to the GAPDH level. The data are presented as the mean ± SD from three independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001; all by two-tailed Student’s t test). For Western blot analysis, GAPDH was used as a loading control. The band intensities were quantified by ImageJ (NIH), and the relative LYAR levels (LYAR/GAPDH) are shown.
LYAR negatively regulates the antiviral innate immune responses.
To explore the role of LYAR in the innate immune responses, we first examined the effect of LYAR on the expression of some key antiviral genes, which include IFN-β, ISGs, and proinflammatory cytokines. LYAR-overexpressed A549 cells or LYAR-KO A549 cells were stimulated with poly(I·C) for 6 h, and the mRNA levels of the indicated genes were analyzed by reverse transcription-quantitative PCR (qRT-PCR). The data showed that overexpressing LYAR significantly decreased the mRNA levels of IFN-β, the ISGs (Mx1, ISG15, 2-5-oligoadenylate synthetase-like protein [OASL], OAS3, interferon-induced transmembrane protein 3 [IFITM3], and IFN-induced protein with tetratricopeptide repeats 2 [IFIT2]), and the proinflammatory cytokines (IL-6, IL-8, IL-1β, tumor necrosis factor alpha [TNF-α], and CXCL11) (Fig. 2A and B). In contrast, knockout of LYAR showed an opposite effect on these genes’ expression levels (Fig. 2D and E), indicating that LYAR negatively regulates the expression of IFN-β, ISGs, and proinflammatory cytokines. LYAR acts as a transcription factor with transcriptional repression activity (26), which raises a possibility that LYAR inhibits so many genes’ expression due not only to its suppression of IFN-β production but also due to its direct regulation of these genes’ transcription. To demonstrate a specific effect of LYAR on the antiviral responses, we also examined the effect of LYAR on the transcriptions of TGF-β1, vascular endothelial growth factor A (VEGFA), and granulocyte-macrophage colony-stimulating factor (GM-CSF), genes that have not been shown to be regulated by IRF3, NF-κB, or ISGF3. We found that the expression levels of TGF-β1 and VEGFA were hardly affected by LYAR expression, and GM-CSF expression was only slightly increased by LYAR overexpression despite a significant reduction of its expression in LYAR-KO cells (Fig. 2C and F), which indicates that the LYAR suppression of numerous antiviral genes’ expression is more likely to be attributed to its inhibition of IFN-β.
FIG 2.

LYAR suppresses the transcription of IFN-β, ISGs, and cytokines. (A to C) The mRNA levels of IFN-β, ISGs, and cytokines in LYAR-overexpressed A549 cells. Cells were transfected with HA-LYAR (LYAR) or control vector HA (HA). At 24 h posttransfection, cells were left unstimulated with or were stimulated with poly(I·C) (100 ng) for 6 h. The mRNA levels of the indicated target genes were detected using qRT-PCR. (D to F) The mRNA levels of IFN-β, ISGs, and cytokines in LYAR-KO A549 cells. LYAR-KO A549 cells (KO) or wild-type A549 cells (WT) were treated with poly(I·C) as described above. The mRNA levels of the target genes were detected. The mRNA level was normalized to the GAPDH level, and the poly(I·C)-unstimulated HA and WT groups were set up to 1 (mean ± SD from three independent experiments) (*, P < 0.05; **, P < 0.01; *** P < 0.001; all by two-tailed Student’s t test).
To further investigate the role of LYAR in the virus-triggered innate immune signaling pathways, the effect of LYAR on the Sev-induced activation of IFN-β promoter was examined. HEK293T cells were cotransfected with IFN-β-luc, pRL-TK, and si-LYAR or HA-LYAR, and the cells were stimulated with Sev for another 12 h after 24 h posttransfection. The luciferase activity assay was performed to measure the IFN-β promoter activity. The data showed that LYAR silencing significantly enhanced the IFN-β promoter activity (Fig. 3A), while LYAR overexpression reduced the IFN-β promoter activity in a dose-dependent manner (Fig. 3D), suggesting that LYAR inhibits the Sev-induced activation of IFN-β promoter. It is well established that the expression of type I IFN genes is mainly regulated by the transcription factors IRF3 and NF-κB. To examine the effect of LYAR on Sev-induced activation of IRF3 and NF-κB, the promoter activity of ISRE and NF-κB was measured in LYAR-overexpressed and -silenced cells. The data showed that the ISRE (Fig. 3E) and NF-κB (Fig. 3F) promoter activity was gradually reduced along with increasing amounts of LYAR but was significantly increased in the LYAR-silenced cells (Fig. 3B and C), which indicates that LYAR is involved in both IRF3- and NF-κB-mediated IFN-β transcription. IFN-β can induce LYAR, and LYAR in turn inhibits IFN-β expression, which means that when overexpressing LYAR, IFN-β production is inhibited, and then it might affect the endogenous LYAR expression. Here, we conducted an experiment to see whether overexpression of LYAR would affect the endogenous LYAR expression after Sev infection or IFN-β treatment. Here, we used Flag-tagged LYAR instead of HA-LYAR due to an overlap of the endogenous LYAR and HA-tagged LYAR bands. The expression levels of Flag-LYAR and endogenous LYAR in both Sev-infected and IFN-β-treated cells were compared, and the effect of Flag-LYAR on IFN-β promoter activity was also examined to assess whether Flag-LYAR inhibits IFN-β expression like HA-LYAR. Consistently, Flag-LYAR overexpression reduced the IFN-β promoter activity in a dose-dependent manner (Fig. 3G). Upon Sev infection or IFN-β treatment, the expression of endogenous LYAR was significantly enhanced, and the levels in the cells transfected with different doses of Flag-LYAR were very similar, which showed no significant difference compared with the vector control group infected with Sev or treated with IFN-β (Fig. 3G and H). These results indicate that although overexpressing LYAR inhibits IFN-β expression, which may reduce endogenous LYAR expression induced by IFN-β theoretically, the practical effect is negligible. In addition, compared with the expression level of Flag-LYAR, the endogenous LYAR expression level of LYAR in Sev-infected or IFN-β-treated cells was about twice as high as that in the low-dose overexpression group (0.25 μg), similar to that in the middle-dose overexpression group (0.5 μg), and half of that in the high-dose overexpression group (1 μg) (Fig. 3G and H), indicating that the overexpression levels of LYAR are not overloaded under the experimental conditions. Taken together, the above results suggest that overexpressing LYAR with the indicated doses (not overloaded) has little impact in the endogenous LYAR expression.
FIG 3.

LYAR inhibits the Sev-induced activation of IFN-β, ISRE, and NF-κB promoter and facilitates the virus replication. (A to F) Effects of LYAR silencing and overexpression on IFN-β, ISRE, and NF-κB promoter activity. HEK293T cells were transfected with pRL-TK, si-LYAR (A, B, and C), or increasing amounts of HA-LYAR (D, E, and F) together with either IFN-β-luc (A and D), ISRE-luc (B and E), or NF-κB-luc (C and F), followed by Sev infection at 24 h posttransfection. Luciferase activities were measured using luciferase report assay at 12 hpi, and Renilla luciferase was used as an internal control. The expression of LYAR was determined using Western blotting with an anti-LYAR or anti-HA mouse antibody. (G) A comparison of the expression levels of Flag-LYAR and endogenous LYAR in Sev-infected cells. HEK293T cells were transfected with the indicated plasmids as described above, except cells were transfected with Flag-LYAR instead of HA-LYAR. The IFN-β promoter activity was detected (upper panel), and the expression of LYAR was detected using Western blotting with an anti-LYAR antibody (lower panel). (H) A comparison of the expression levels of Flag-LYAR and endogenous LYAR in IFN-β treated cells. HEK293T cells were transfected with Flag-LYAR or Flag, and the cells were left untreated or were treated with IFN-β (1,000 U/ml) at 24 h posttransfection. The expression levels of Flag-LYAR and endogenous LYAR were detected with Western blotting using an anti-LYAR antibody. The relative levels of Flag-LYAR (Flag-LYAR/GAPDH) and endogenous LYAR (Endo-LYAR/GAPDH) and the ratios of endogenous LYAR and Flag-LYAR (Endo-LYAR/Flag-LYAR) are shown. (I and J) The effect of LYAR on Sev replication. LYAR-overexpressed HEK293T cells (left) or LYAR-KO HEK293T cells (right) were infected with Sev at an MOI of 1.0, and cell RNA and supernatants were collected at 12 and 24 hpi. (I) The mRNA levels of Sev M segment were detected by qRT-PCR. (J) The viral titers were determined by TCID50 assay on Vero cells, and the expression levels of LYAR were detected by using an anti-LYAR antibody. (K) The effect of LYAR on VSV replication. LYAR-overexpressed HEK293T cells (left) or LYAR-KO HEK293T cells (right) were infected with VSV-GFP at an MOI of 1.0 for the indicated times. The viral titers were detected by using plaque assay on Vero cells, and the expressions of GFP and LYAR were analyzed using Western blotting. The data are presented as the mean ± SD from three independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001; all by two-tailed Student’s t test).
The influence of LYAR in Sev and VSV replication was further determined to assess its inhibitory effect on antiviral response. Results showed that overexpression and knockout of LYAR significantly promoted and inhibited Sev replication in HEK293T cells, respectively, which was determined by detecting the mRNA levels of Sev M segment by qRT-PCR and the virus titers by 50% tissue culture infective dose (TCID50) assay (Fig. 3I and J). Results from the Western blotting and plaque assay showed that LYAR also enhanced the VSV replication in HEK293T cells (Fig. 3K). Thus, we demonstrated that LYAR negatively regulates the antiviral innate immune responses, thereby extensively promoting virus replication.
LYAR negatively modulates the IFN-β-inducing pathway by targeting IRF3.
Virus can be recognized by RIG-I during infection and activates IFN-I production by signaling transduction from RIG-I, MAVS, and the TBK1/IKKε complex to IRF3. To investigate the mechanism of LYAR regulating IFN-β induction, the effect of LYAR on the IRF3-mediated activation of the IFN-β promoter was examined. HEK293T cells were transfected with IFN-β-luc, pRL-TK, and Flag-tagged signal molecules along with an increasing amount of HA-LYAR, and the luciferase activities were measured at 24 h posttransfection. Results showed that the activation of the IFN-β promoter induced by IRF3-5D (an active phosphomimetic mutant of IRF3) (Fig. 4F), IRF3 (Fig. 4E), and its upstream molecules (Fig. 4A to D) was suppressed by LYAR in a dose-dependent manner, suggesting that LYAR inhibits the induction of IFN-β by targeting IRF3. Considering the structural and functional similarities of IRF3 and IRF7 (27, 28), LYAR might target IRF7 as well. However, we found that LYAR did not affect the IFN-β promoter activity induced by IRF7 (Fig. 4G), indicating that LYAR does not regulate the induction of IFN-β via IRF7. We further examined whether LYAR targets IRF9 to affect the expression of ISGs. The data showed that the ISRE promoter activity induced by IRF9 was not affected by LYAR (Fig. 4H). These results provide evidence that LYAR specifically targets IRF3 to affect the expression of IFN-β and ISGs.
FIG 4.

LYAR inhibits IFN-β promoter activity induced by IRF3-mediated IFN-β signaling molecules. (A to G) The effect of different doses of LYAR on the activation of IFN-β promoter induced by RIG-I (A), MAVS (B), TBK1 (C), IKKε (D), IRF3 (E), IRF3-5D (F), and IRF7 (G). HEK293T cells were transfected with IFN-β-luc, pRL-TK, plus the indicated expression plasmids of signal molecules, along with an increasing amount of HA-LYAR (0, 0.25, 0.5, and 1.0 μg). Luciferase activity was measured at 24 h posttransfection. The expression levels of LYAR and each signal molecule were detected by using Western blotting with an anti-HA antibody and an anti-Flag antibody, respectively. (H) The effect of LYAR on IRF9 induced activation of ISRE promoter. HEK293T cells were transfected with ISRE-luc, pRL-TK, HA-IRF9, and increasing amounts of Flag-LYAR (0, 0.25, 0.5, and 1.0 μg). Luciferase activity was measured at 24 h posttransfection. The expressions of LYAR and IRF9 were detected by using anti-Flag and anti-HA antibodies, respectively. The data are presented as the mean ± SD from three independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001; all by two-tailed Student’s t test).
LYAR interacts with phosphorylated IRF3.
In order to investigate how LYAR represses IFN-β activation via IRF3, the interaction between LYAR and IRF3 was further detected. HEK293T cells were transfected with the indicated plasmids, and the coimmunoprecipitation (Co-IP) experiment was performed by using an anti-Flag or anti-HA antibody. Interestingly, we found that LYAR only coprecipitated with IRF3-5D (the constitutively active form of IRF3), but not with wild-type IRF3 or the negative-control vectors (Fig. 5A), suggesting that LYAR interacts with the phosphorylated form of IRF3. Furthermore, to determine which part of IRF3 mediates the interaction with LYAR, the N-terminal domain (NTD) and C-terminal domain (CTD) of IRF3-5D were used for detection. The data showed that LYAR failed to coprecipitate with the CTD of IRF3-5D (Fig. 5B), suggesting that IRF3 interacts with LYAR via its NTD, which contains the DNA binding domain (29). The interaction between endogenous LYAR and IRF3 was further examined. A549 cells infected with Sev were lysed at 12 hours postinfection (hpi), followed by a Co-IP assay using an anti-LYAR mouse antibody or mouse IgG. The results revealed that the phosphorylated IRF3 was coprecipitated by LYAR, but the negative-control IgG was not (Fig. 5C), indicating a real interaction between LYAR and phosphorylated IRF3 during virus infection. In addition, the colocalization of LYAR and IRF3 was detected using immunofluorescence and confocal microscopy. In HeLa cells, LYAR localized to the nucleoli when expressed alone, and when LYAR coexpressed with IRF3 or IRF3-5D, it colocalized not with IRF3 but with IRF3-5D in the nucleus (Fig. 5D), which further demonstrates that LYAR interacts with the phosphorylated IRF3.
FIG 5.

LYAR interacts with phosphorylated IRF3. (A) The interaction between LYAR and IRF3 in transfected cells. HEK293T cells were cotransfected with the indicated plasmids for 24 h. Co-IP was performed using an anti-Flag (left) or anti-HA (right) antibody, followed by Western blotting to detect the LYAR and IRF3 with the anti-HA and anti-Flag antibodies, respectively. (B) The interactions between LYAR and IRF3-5D or IRF3-5D mutants (N-terminal domain, NTD; amino acids 1 to 197 and C-terminal domain, CTD; amino acids 198 to 427). HEK293T cells were transfected with the indicated plasmids for 24 h. Co-IP was conducted by using an anti-Flag (left) or anti-HA (right) mouse antibody. (C) The interaction between endogenous LYAR and IRF3. A549 cells were infected with Sev for 12 h, and Co-IP was performed using an anti-LYAR mouse antibody or mouse IgG. The endogenous LYAR and coprecipitated IRF3 were detected by using an anti-LYAR antibody and an anti-Ser386 phosphorylated IRF3 antibody, respectively. Mouse IgG served as the negative control, and GAPDH was used as a loading control. (D) Colocalization of LYAR and IRF3. HeLa cells were transfected individually with HA-LYAR or Flag-IRF3 or were cotransfected with HA-LYAR and Flag-IRF3 or Flag-IRF3-5D. Cells were fixed at 24 h posttransfection and stained for LYAR (red) and IRF3 (green) using the anti-HA rabbit antibodies and anti-Flag mouse antibodies, followed by immunostaining with the Alexa Fluor 594-conjugated AffiniPure goat anti-rabbit secondary antibodies and Alexa Fluor 488-conjugated AffiniPure goat anti-mouse antibodies. DAPI was used to stain for the nucleus (blue). The boxed region was enlarged and is shown on the right. Colocalization was quantified using the ZEN Blue software (Zeiss, Germany), and the Pearson’s correlation coefficient (Rr, values from 0.5 to 1.0 indicating colocalization) and overlap coefficient according to Manders (R, values from 0.6 to 1.0 indicating colocalization) are shown below. Images are representative of three independent experiments. Scale bar, 10 μm.
LYAR does not affect the phosphorylation and nuclear import of IRF3.
During virus infection, IRF3 is phosphorylated and subsequently forms dimers to enter the nucleus to bind onto the IFN-β promoter, which activates IFN-β transcription. To determine the effect of LYAR on IRF3 activation, the effect of LYAR on IRF3 phosphorylation was examined. HEK293T cells transfected with HA-LYAR or si-LYAR were stimulated with Sev for 12 h, followed by Western blotting to analyze the expression and phosphorylation levels of IRF3. The data showed that neither the expression nor the phosphorylation levels of IRF3 were visibly changed when LYAR was overexpressed or silenced (Fig. 6A and B), indicating that LYAR does not affect the phosphorylation of IRF3. The effect of LYAR on IRF3 nuclear translocation in Sev-infected cells was further detected. Indirect fluorescent-antibody assay (IFA) results showed that approximately 70% of both the control cells and the LYAR-overexpressed cells displayed IRF3 nuclear localization at 6 hpi (Fig. 6C and D), indicating that IRF3 nuclear import is hardly affected by LYAR expression. Furthermore, the results of Western blotting on nuclear and cytoplasmic fractions of these cells were consistent with the IFA data, i.e., the IRF3 was predominantly in the nuclear fraction of the control cells upon Sev stimulation and so was in the LYAR-overexpressed cells (Fig. 6E), which suggests that LYAR has little effect on the nuclear transportation of IRF3. The above results collectively demonstrate that LYAR regulates IFN-β induction without affecting the phosphorylation and nuclear translocation of IRF3.
FIG 6.

LYAR does not affect the phosphorylation and nuclear import of IRF3. (A and B) Effect of LYAR on IRF3 phosphorylation. HEK293T cells transfected with HA-LYAR (A) or si-LYAR (B) were infected with Sev for 12 h. The protein and phosphorylation levels of IRF3 were detected with Western blotting using an anti-IRF3 rabbit antibody and anti-Ser386 phosphorylated IRF3 rabbit antibody, respectively. GAPDH served as the loading control, and the band intensities were analyzed using the software ImageJ (NIH), and the relative p-IRF3 levels (p-IRF3/GAPDH) are shown below. (C and D) Confocal microscopy analysis of the nucleocytoplasmic distribution of IRF3 in Sev-infected LYAR overexpression cells. A549 cells transfected with HA-LYAR or HA were infected with Sev for 6 h, and endogenous IRF3 and HA-LYAR were detected by IFA using an anti-IRF3 rabbit antibody and anti-HA mouse antibody, followed by immunostaining with 594-conjugated AffiniPure goat anti-rabbit secondary antibodies (red) and Alexa Fluor 488-conjugated AffiniPure goat anti-mouse antibodies (green). Scale bar, 10 μm. Images are representative of three independent experiments. Six images in a random field of view from each sample were scored by the cell counter plugin of ImageJ (NIH). (D) The ratios of IRF3 nuclear localization cells/total cells were analyzed from three independent experiments (means ± SD from three independent experiments). (E) Western blot analysis of the distribution of IRF3 in the cytoplasmic and nuclear fractions in Sev-infected LYAR overexpression cells. A549 cells were treated as described for panel C. Cells were harvested and subjected to nuclear and cytoplasmic fractionation. Western blotting using an anti-IRF3 antibody to determine the IRF3 content of the nuclear (Nuc) and cytoplasmic (Cyt) fractions, respectively. Histone 3.1 was used as a nuclear loading control and marker, and GAPDH, as a cytosolic loading control and marker. The band intensities were quantified using ImageJ, and the relative IRF3 levels (IRF3/GAPDH or histone 3.1) are shown below the images.
LYAR impairs the DNA binding ability of IRF3.
It is known that IRF3 binds to the promoter and IFN-stimulated response element (ISRE) sequence to activate the transcription of target genes (13). Given that IRF3 interacts with LYAR via its DNA binding region, LYAR might interfere with the DNA binding ability of IRF3. To confirm this speculation, the effect of LYAR on IRF3 binding onto ISRE was analyzed. HEK293T cells coexpressed with Flag-IRF3 and HA-LYAR or HA were infected with Sev for 12 h, and a DNA pulldown assay was performed by using a biotinylated ISRE oligonucleotide or an unbiotinylated ISRE oligonucleotide to compete away IRF3-LYAR binding. As shown in Fig. 7A, the IRF3 protein was pulled down by biotinylated ISRE but not by the unbiotinylated ISRE; the amounts of IRF3 associated with IRSE were obviously reduced in the presence of LYAR when only using the biotinylated ISRE; when the biotinylated and unbiotinylated ISRE were used in equal amounts, the amounts of IRF3 bound to ISRE were still visibly reduced in the presence of LYAR, and the amounts of both IRF3 and LYAR pulled down by the biotin-ISRE were also decreased compared to those in the group that only used the biotinylated ISRE; in the group in which much more unbiotinylated ISRE was used, the IRF3 was hardly detected and LYAR was undetected (Fig. 7A). These results indicate that LYAR prevents IRF3 binding onto the ISRE. IRF3 binding to the target genes to activate their transcription requires the interaction with CBP/p300 coactivators. To further investigate whether the interaction of LYAR and IRF3 breaks the association of IRF3 and CBP/p300, the effect of LYAR on the interaction between Flag-IRF3 and endogenous CBP was examined. We found that the endogenous CBP coprecipitated by IRF3 was comparable in the presence and absence of LYAR, indicating that LYAR does not affect the IRF3-CBP interaction (Fig. 7B). Taken together, these results suggest that LYAR does not affect the formation of the IRF3-CBP/p300 complex but attenuates the DNA binding capacity of IRF3, thereby suppressing the transcription of IFN-β and ISGs.
FIG 7.

LYAR impedes IRF3 binding onto ISRE. (A) The effect of LYAR on IRF3 binding onto ISRE. HEK293T cells cotransfected with Flag-IRF3 and HA-LYAR or HA were infected with Sev for 12 h. Biotinylated and unbiotinylated ISRE were mixed with the cell lysates at the indicated ratios and incubated at 4°C for 4 h. The beads were then added and incubated for 2 h at 4°C, followed by washing four times with PBS. Western blotting was done to detect IRF3 and LYAR by using an anti-Flag and anti-HA antibody, respectively. The Flag-IRF3 levels are shown below. (B) The effect of LYAR on the interaction between IRF3 and CBP/p300. HEK293T cells cotransfected with Flag-IRF3 and HA-LYAR or HA were infected with Sev for 12 h. Co-IP assay was performed using an anti-Flag antibody to immunoprecipitate endogenous CBP. The relative precipitated CBP/Flag-IRF3 ratios are shown below.
The lysine-rich region of LYAR plays a dominant role in the inhibition of IFN-β.
To further confirm the suppression of LYAR in IFN-β transcription, the effects of LYAR mutants on virus-induced IFN-β and ISRE promoter activation were examined. The functional domains of LYAR are detailed in the schematic diagram (Fig. 8A). HEK293T cells transfected with pRL-TK and IFN-β-luc or ISRE-luc together with Flag-LYAR or LYAR mutants were infected with Sev, and the luciferase activity was measured at 12 hpi. The data showed that the LYAR wild type (WT) significantly reduced both the IFN-β and ISRE promoter activity, while the N-terminal of LYAR (NTD) slightly increased the promoter activity compared to the vector control (Fig. 8B and C). Deletion of two zinc finger motifs (Δzinc) at the N-terminal of LYAR did not significantly diminish the ability of LYAR to suppress the IFN-β activation, although deletion of zinc finger motif 1 (Δzinc1) led to minor reduction in IFN-β and ISRE promoter activity compared to the WT, whereas zinc finger motif 2 deletion (Δzinc2) resulted in slight increases (Fig. 8B and C). However, the C-terminal of LYAR (CTD) significantly decreased the IFN-β and ISRE promoter activity compared to the WT, and deletion of each individual nuclear localization signal (NLS1, NLS2, or NLS3) at the CTD impaired the inhibitory activity of LYAR in IFN-β activation to various degrees (Fig. 8B and C). Meanwhile, similar results were acquired by using the LYAR-KO HEK293T cells (Fig. 8B and C). Collectively, these results indicate that the lysine-rich region of LYAR plays a leading role in the LYAR inhibited IFN-β activation, and the NLSs also play a role in this process.
FIG 8.

The lysine-rich region of LYAR plays a dominant role in the inhibition of IFN-β. (A) Schematic diagram of LYAR wild type and mutants. (B and C) Effects of LYAR mutants on the IFN-β (B) and ISRE (C) promoter activity. LYAR-KO HEK293T or wild-type HEK293T cells were transfected with pRL-TK, IFN-β-luc, or ISRE-luc together with LYAR WT, LYAR mutants, or control vector Flag. At 24 h posttransfection, cells were mock infected (control) or were infected with Sev for 12 h, and luciferase activities were measured using luciferase report assay. Asterisks indicate the significant difference of the promoter activity in Flag-tagged LYAR WT or mutant transfected cells compared to that in the control vector Flag transfected cells within the same group (HEK-293T group or LYAR-KO group); pentagrams indicate the significant difference of the promoter activity in Flag-tagged LYAR mutant transfected cells compared to that in the Flag-LYAR WT transfected cells within the same group (mean ± SD from three independent experiments) (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ☆, P < 0.05; ☆☆, P < 0.01; ☆☆☆, P < 0.001; all by two-tailed Student’s t test). The blue lines indicate the relative promoter activity in the LYAR WT-overexpressed HEK293T WT or LYAR-KO cells. The expression of endogenous LYAR (endo-LYAR), Flag-tagged LYAR WT, and mutants was determined using Western blotting, and GAPDH was used as a loading control (asterisks indicate specific LYAR or LYAR mutants detected).
DISCUSSION
In this study, LYAR suppressed the transcription of IFN-β and ISGs, including OAS3, OASL, IFITM3, Mx1, ISG15, and IFIT2. ISGs target the key stages of the virus life cycle, or regulate innate immune recognition and cytokine production, thereby establishing the cell’s antiviral status (30). One of the ISGs, OAS3, can be induced by IAV infection and exert antiviral effect together with RNase L by degradation of virus single-stranded RNA (ssRNA) (7, 23). A previous study showed that LYAR might interact with OAS3 in poly(I·C)-stimulated A549 cells (22), and we found that LYAR significantly inhibits the poly(I·C)-induced expression of OAS3 in A549 cells, indicating that LYAR may participate in the OAS3-RNase-L pathway. Other ISGs, including OASL, IFITM3, Mx1, ISG15, and IFIT2, also play important roles in restricting influenza virus replication. LYAR extensively suppresses the expression of these ISGs, thereby inhibiting the antiviral responses. On the other hand, the expression of NF-κB-regulated proinflammatory cytokines, including IL-6, IL-8, IL-1β, and TNF-α are also inhibited by LYAR, and LYAR inhibits the NF-κB promoter activity, suggesting that LYAR may participate in the NF-κB signaling pathway, thus regulating cellular inflammatory responses, and further investigations would benefit for a comprehensive understanding of LYAR’s role in innate immunity. LYAR is a transcription factor, and two zinc finger DNA binding motifs in its N-terminal domain are regarded to be involved in transcriptional regulation (26, 31, 32). This raises the concern that the inhibitory activity of LYAR in antiviral gene expression might be an effect of its transcriptional repression of those genes. However, two pieces of data diminish this possibility. Most importantly, we found that deletion of the two zinc finger motifs retained the ability of LYAR to inhibit the activation of IFN-β and ISRE, and the LYAR inhibition of IFN-β induction was predominantly mediated by its C-terminal region. Second, LYAR did not repress the expression of TGF-β1, VEGFA, and GM-CSF, indicating that genes suppressed by LYAR may be limited to those that are regulated by IRF3, NF-κB, or ISGF3. Thus, it can be speculated that the negative effect of LYAR on the IFN-β-mediated antiviral responses does not require its transcriptional activity.
IRF3 is the key transcription factor which is responsible for the transcription of IFN-I. Phosphorylated IRF3-formed dimers are transported into the nucleus and bind to the promoter of the target genes, resulting in the gene transcription. We found that LYAR inhibits IRF3 and IRF3-5D stimulated IFN-β promoter activity, indicating that LYAR may affect not only IRF3 phosphorylation but also the downstream events. However, LYAR has little effect on the phosphorylation and nuclear import of IRF3, suggesting that LYAR is likely to regulate IRF3 function in the nucleus. In a resting state, IRF3 mainly localizes in the cytoplasm, and it translocates into the nucleus after phosphorylation activation upon virus infection. Of note, LYAR only interacts with the phosphorylated IRF3. Consistent with this observation, LYAR colocalizes with IRF3-5D in the nucleus. In addition, LYAR nuclear localizations play an important role in regulating IFN-β, suggesting a possibility that the association of LYAR with IRF3 occurs in the nucleus. More importantly, the interaction between LYAR and IRF3 is mediated by the DNA binding region of IRF3, which impedes IRF3 binding onto ISRE. However, another important process in the nucleus, IRF3 and CBP/P300 forming a complex to activate the transcription of IFN-β, is not impacted by LYAR. Thus, we propose that LYAR attenuating the DNA binding ability of IRF3 is a major mechanism to impair the IRF3 function. In addition to IRF3, IRF7 is also involved in the RIG-I-like receptor (RLR) signaling pathway to mediate the IFN-β transcription (33), but LYAR did not target IRF7, despite their structural and functional similarities. Another IRF, IRF9, activates the transcription of ISGs by forming the complex ISGF3 together with STAT1 and STAT2 to bind to the ISRE (9), and LYAR did not act on IRF9 to regulate the expression of ISGs. Thus, LYAR specifically regulates the function of IRF3.
LYAR expression is significantly upregulated by Sev and VSV infection as well as poly(I·C) stimulation. Further studies show that LYAR expression is increased in IFN-β-treated cells but is not induced by Sev in the IFN-I deficiency Vero cells, suggesting that LYAR is induced by IFN-β during viral infection. Interestingly, even if IFN-α and IFN-λ1 share similar properties with IFN-β, especially IFN-α, which is very similar to IFN-β in signaling pathways (34), they failed to stimulate LYAR expression in A549 cells, but it is unknown if it is also the case in other types of cells and whether higher concentrations and different time periods of treatment would make a difference. Although many genes induced by type I and III IFNs are shared, the kinetics of induction are distinct, and certain genes may be uniquely regulated by one of the subtypes (35, 36), and it is therefore reasonable that LYAR expression is only induced by IFN-β. However, it is unclear how LYAR is induced by IFN-β and specifically targets the IRF3 during infection. Given that LYAR expression is increased in both mRNA and protein levels, it is plausible that LYAR expression is regulated by the transcription factors in the IFN-β signaling pathways. Further investigation in the mechanism of LYAR induced in virus infection will help us figure out the detailed role of LYAR in the antiviral responses.
Traditional ISGs typically exert antiviral effect, but a small class of IFN-induced genes show proviral activity. ADAR1 (adenosine deaminase acting on RNA 1) is an example of such genes and promotes the replication of several viruses, including VSV, measles virus (MV), human immunodeficiency virus (HIV), and hepatitis C virus (HCV) (37–39). Its role in HIV replication has been well studied. It is reported that ADAR1 can be induced by IFN-α and γ, and it enhances HIV replication by limiting IFN production, by counteracting the activity of another antiviral ISG, PKR (double-stranded RNA [dsRNA]-dependent protein kinase R), and by editing viral RNA (37, 40–43). Other IFN-inducible genes, including LY6E (lymphocyte Ag 6 complex, locus E), Siglec-1 (sialic acid-binding Ig-like lectin 1) (also known as CD169), and IP-10 (IFN-γ-inducible protein 10), positively regulate HIV replication via modulating antiviral immunity (44–48). However, another IFN-I-inducible gene, MCOLN2 (endosomal cation channel mucolipin-2), promotes the replication of diverse RNA virus, including IAV, by increasing virus trafficking efficiency, which is independent of its role in IFN responses (49). These studies suggest that some viruses may have evolved to co-opt IFN effectors for a survival advantage, and those IFN effectors affect virus replication by modulating antiviral immune responses or directly regulating viral life stages. Herein, we characterize LYAR as an IFN-β-inducible gene, which is proposed to have the below double effect on IAV replication. LYAR expression is induced by virus-activated IFN-β, which is in turn hijacked by influenza A viral RNP to facilitate viral RNA synthesis (20). Simultaneously, LYAR targets IRF3 to suppress the host innate immune responses, providing a favorable environment for virus replication.
Based on our findings, we proposed a model for the role of LYAR in the antiviral innate immune responses (Fig. 9). Virus activates IFN-β, which induces the expression of LYAR. LYAR associates with the phosphorylated IRF3 to impede its DNA binding ability, leading to reduced production of IFN-β and its downstream ISGs, which in turn facilitates virus replication. Therefore, for the viruses, LYAR is an inducible factor that is utilized by the virus for replication; for the host, LYAR is induced by IFN-β and regulates the IFN-β expression by negative feedback to avoid excessive immune and inflammatory responses. In summary, our study uncovered an underlying mechanism of how LYAR negatively regulates IFN-β by targeting IRF3, which would increase our understanding of the negative regulation of the host innate immunity and the functions of LYAR in virus replication.
FIG 9.
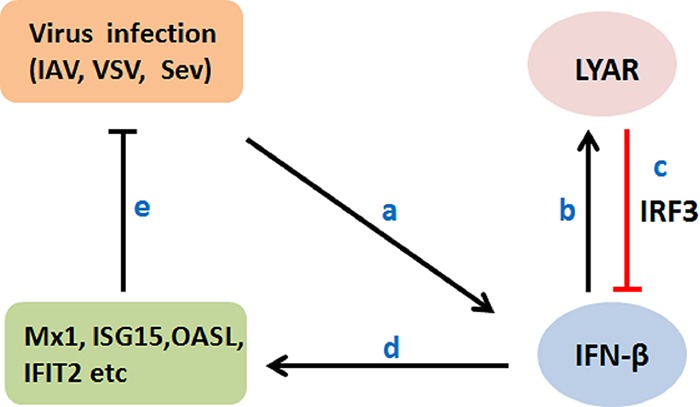
Proposed model for the role of LYAR in the antiviral innate immune responses. Virus-induced IFN-β increases LYAR expression (a and b). In turn, LYAR suppresses IFN-β and ISG production by impeding the DNA binding ability of IRF3, ultimately resulting in enhanced virus replication (c to e).
MATERIALS AND METHODS
Cells and viruses.
Human embryonic kidney 293T cells (HEK293T) and African green monkey kidney (Vero) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, New York, USA). The Henrietta Lacks strain of cancer cells (HeLa) and adenocarcinomic human alveolar basal epithelial cells (A549) were maintained with Park Memorial Institute-1640 medium (RPMI 1640) and F12 medium (HyClone, Beijing, China), respectively. All media were supplemented with 10% fetal bovine serum (FBS) (PAN-Biotech, Germany), and all cells were cultured at 37°C in a 5% CO2 humidified atmosphere. All the cell lines listed above were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). LYAR-KO A549 cells and HEK293T cells were generated as we described previously (20). Sendai virus (Sev) was amplified using 10-day-old embryonic chicken eggs. The recombinant vesicular stomatitis virus encoding green fluorescence protein (VSV-GFP) was a gift from Harbin Veterinary Research Institute (Harbin, China).
Plasmids and small interfering RNAs.
For construction of p3XFlag-CMV-LYAR (Flag-LYAR) and pCAGGS-HA-LYAR (HA-LYAR), the full-length cDNA of LYAR (GenBank accession number NM_017816) amplified by PCR was cloned into vectors p3XFlag-CMV (Flag) and pCAGGS-HA (HA) digested by BglII/XbaI and ECORI/XhoI, respectively. The LYAR mutants (LYAR-Δzinc, LYAR-ΔNLSs, LYAR-NTD, and LYAR-CTD) were constructed by site mutation or overlapping PCR using PrimerSTAR (TaKaRa, Tokyo). Full-length IRF7 (GenBank accession number NM_001572.5) and IRF9 (GenBank accession number NM_006084.4) were inserted into the p3XFlag-CMV and pCAGGS-HA vectors, respectively. PCR primer sequences used in this study are listed in Table 1. Flag-RIG-I, -MAVS, -TBK1, -IKKε, and -IRF3 expression plasmids were kindly provided by Zhengfan Jiang (Peking University, Beijing, China). A luciferase (luc) reporter plasmid for the IFN-β promoter (IFN-β-luc) and Renilla control plasmids pRL-TK were a gift from Ping Qian (Huazhong Agricultural University, Wuhan, China). The Flag-tagged active phosphomimetic mutant of IRF3 (Flag-IRF3-5D) was kindly provided by Yiling Lin (National Defense Medical Center, Taiwan, China). The luciferase (luc) reporter plasmids for the ISRE promoter (ISRE-luc) and the NF-κB promoter (NF-κB-luc) were constructed by our laboratory. Small interfering RNA (siRNA) targeting LYAR (si-LYAR) was used to knock down LYAR, and the sequences were as follows: 5′-CACAGUUUCUGCAAACAGACACAUG-3′ and 5′-CAUGUGUCUGUUUGCAGAAACUGUG-3′; a nontarget siRNA (si-NC) was used as a negative control, and the sequences were as follows: 5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-ACGUGACACGUUCGGAGAATT-3′. All the siRNAs used in this study were synthesized by GenePhama (Shanghai, China).
TABLE 1.
Primers used for PCR
| Plasmid | Forward sequence (5′ to 3′) | Reverse sequence (5′ to 3′) |
|---|---|---|
| Flag-LYAR | GGAAGATCTGATGGTATTTTTTACATGCAATGC | TGCTCTAGATTTCACAAGCTTGACTTTGTCCT |
| HA-LYAR | GGAATTCATGGTATTTTTTACATGCAATGC | CCCTCGAGTCATTTCACAAGCTTGACTTTG |
| LYAR-NTD | GGAAGATCTGATGGTATTTTTTACATGCAATGC | TGCTCTAGAGGTGGAGATTTCTGCATGTGGATTT |
| LYAR-CTD | GGAAGATCTGATGAAGGTTCCAGCCTCCAAAGT | TGCTCTAGATTTCACAAGCTTGACTTTGTCCT |
| LYAR-Δzinc1 | TTACAAGAAACTGTGAATGCCTTT | CAGTTTCTTGTAAAAAATACCATCAGA |
| LYAR-Δzinc2 | GCCTTTCTATAAGTGAAGATCAGA | CACTTATAGAAAGGCATTCACAGT |
| LYAR-Δzinc | GGAAGATCTTGGTGGCAAAGGCTATGAAGGTA | TGCTCTGAGTTTCACAAGCTTGACTTTGTCCT |
| LYAR-ΔNLS1 | AGAATGAACTAAAGTTAGAAAACCA | AGTTCATTCTTCTTCACCTCCCCTT |
| LYAR-ΔNLS2 | AGAAGGGACAGGAGGCTGACC | TGTCCCTTCTGATTCCTTGAGTTTT |
| LYAR-ΔNLS3 | CAGGGATGAAGCTCCCAGAGCAT | TTCATCCCTGCGCCCACGCGTG |
| Flag-IRF7 | CCCAAGCTTATGGCCTTGGCTCCTGAGAG | GCTCTAGAGGCGGGCTGCTCCAGCTCCATA |
| HA-IRF9 | CGGAATTCATGGCATCAGGCAGGGCA | CCGCTCGAGCTACACCAGGGACAGAATGGCT |
Antibodies and reagents.
Antibodies used for Western blotting, immunoprecipitation, and indirect immunofluorescence were anti-Flag M2 mouse monoclonal antibody (F3165; Sigma, USA), anti-LYAR mouse polyclonal antibody (H00055646-B01P; Abnova, China), anti-HA and -GAPDH mouse monoclonal antibodies (PMK013C and PMK043F; PMK Bio, China), anti-histone 3.1 polyclonal rabbit antibody (p30266; Abmart, USA), anti-IRF3 rabbit monoclonal antibody (11904; Cell Signaling Technology, USA), anti-Ser386 phosphorylated IRF3 polyclonal rabbit antibody (AP0091; ABclonal Technology, USA), anti-CBP rabbit monoclonal antibody (D6C5; Cell Signaling Technology, USA), and Alexa Fluor 488-conjugated AffiniPure goat anti-rabbit and Alexa Fluor 594-conjugated AffiniPure goat anti-mouse secondary antibodies (SA00006-2 and SA00006-3; Proteintech, USA). The small-molecule compounds used in this study were DAPI (4′,6′-diamidino-2-phenylindole dihydrochloride; C1002; Beyotime, China) and poly(I·C) [polyinosinic-poly(C) potassium salt; 31852-29-6; Sigma, USA]. The human IFN-α, IFN-β, and IFN-λ1 (IL-29) proteins used in this study were purchased from Sino Biological, Inc. (catalog numbers 12341-H08Y, 10704-HNAS, and 12339-H08H, respectively).
Transfections.
Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Briefly, plasmids, siRNAs, and Lipofectamine were diluted to equal volumes with Opti-MEM and incubated for 5 min at room temperature. The diluted Lipofectamine and the diluted DNA (or RNA) were mixed and incubated for 20 min at room temperature. The mixture was added to cells and incubated for 6 h, and cells were then cultured in fresh medium supplemented with 10% FBS.
Virus titration.
LYAR-overexpressed or LYAR-KO HEK293T cells were infected with VSV-GFP or Sev at an MOI of 1.0. Viral supernatants were harvested at the indicated time points postinfection, and plaque assays were performed on Vero cells to titrate the VSV titer as described previously (50). To detect the Sev virus titer, Vero cells were seeded in 96-well plates before Sev virus infection. Viral supernatants were serially diluted with DMEM and added to each well with eight replicates of each dilution. The 50% tissue culture infective dose (TCID50) was calculated using the Reed-Muench method 48 h after infection.
Co-IP assay.
HEK293T cells were transfected with the indicated plasmids for 24 h, and then cells were washed with cold phosphate-buffered saline (PBS) and lysed in radioimmunoprecipitation assay buffer (V900854; Sigma, USA) containing complete protease inhibitor cocktail (B14001; Biotool, USA) at 24 to 48 h posttransfection. The lysates were pretreated with 20 μl of protein A/G agarose (sc-2003; Santa Cruz, USA) for 1 h at 4°C, and then protein A/G agarose was removed by centrifugation. Before overnight incubation at 4°C, 2 to 3 μg of the indicated antibody was added to the pretreated lysates. Protein A/G agarose was added to the lysates and incubated at 4°C for another 2 h with rotation. The agarose beads were collected using centrifugation and washed four times with lysis buffer. The beads were resuspended in 1× SDS loading buffer, and proteins were resolved by SDS-PAGE, followed by transfer to nitrocellulose and Western blotting.
IFA and confocal microscopy analysis.
Indirect immunofluorescence and confocal microscopy were performed as described previously (51). Briefly, A549 cells were fixed with 4% paraformaldehyde (PFA) for 10 min, treated with 0.2% (vol/vol) Triton X-100 for 10 min, and then incubated with 1% (wt/vol) bovine serum albumin (BSA) for 1 h at room temperature. Samples were then incubated with the indicated primary antibody for 2 h, followed by incubation with the appropriate Alexa Fluor-conjugated secondary antibody, and stained with DAPI to visualize DNA. Images were acquired using a confocal microscope (LSM880; Carl Zeiss, Germany).
Luciferase reporter assay.
HEK293T cells in 24-well plates were transfected with the internal control pRL-TK (5 ng/well) and IFN-β-luc, ISRE-luc, or NF-κB-luc (0.25 μg/well). To stimulate these promoters’ luciferase activity, the RIG-I, MAVS, TBK1, IKKε, IRF3, IRF3-5D, IRF7, or IRF9 expression plasmids (0.25 μg/well) were transfected together with luciferase reporter plasmids, or Sev infection was conducted at 24 h posttransfection. At 24 h posttransfection or 12 h postinfection, cells were lysed in 100 μl of 1× passive lysis buffer (PLB; Promega). The luciferase activity and Renilla activity were measured using a dual-luciferase assay kit (Promega) according to the manufacturer’s instructions. Renilla luciferase activity was used as an internal control and to normalize transfection efficiency. All experiments were performed in triplicate and repeated at least three times.
RNA isolation and qRT-PCR.
For quantitative reverse transcription-PCR (qRT-PCR), cells were lysed with TRIzol reagent (Invitrogen, USA), and total RNA was extracted according to the manufacturer’s instructions. One to two micrograms of RNA was used to generate cDNA using reverse transcriptase (AMV XL; TaKaRa, Tokyo). Real-time PCR (ViiA7; ABI, USA) was performed using FastStart Universal SYBR green master (Roche). The PCR conditions were 2 min at 50°C, 10 min at 95°C, and then 40 cycles for 15 s at 95°C and 1 min at 60°C. The mRNA levels of the target genes were normalized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The sequences of primers used for qRT-PCR are shown in Table 2.
TABLE 2.
Primers used for qRT-PCRa
| Gene | Forward sequence (5′ to 3′) | Reverse sequence (5′ to 3′) |
|---|---|---|
| LYAR | AAGCGGAGGCACTCGGAAGTT | TTCGGATCTGTGATGCTCATCTGTC |
| GAPDH | GCTAAGGCTGTGGGCAAGG | GGAGGAGTGGGTGTCGCTG |
| IFN-β | GCTCCTGTGGCAATTGAATGG | TTGGCCTTCAGGTAATGCAG |
| IL-6 | ACAGCCACTCACCTCTTCAGAAC | GCTCTGGCTTGTTCCTCACTACTC |
| IL-8 | TGCAGCTCTGTGTGAAGGTG | CAGCCCTCTTCAAAAACTTCTCC |
| IL-1β | AGCTACGAATCTCCGACCAC | CGTTATCCCATGTGTCGAAGAA |
| TNF-α | CAGGCGGTGCTTGTTCC | AAGAGGACCTGGGAGTAGATGA |
| OASL | GCCTTCTCTTCCCAACTCCC | AGGCATAGATGGTTAGAAGTTCAAGA |
| MX1 | CCGAGGGAGACAGGACCAT | CGTGGCCTTTCCTTCCTCC |
| CXCL11 | ATTTGCTGCCTTATCTTTCTGACTCTA | TGGCCTTCGATTCTGGATTCA |
| OAS3 | CACAGACCTAAGGGATGGC | TCAGGAAACTGAAGGCTCA |
| ISG15 | TGGACAAATGCGACGAACC | GCCCGCTCACTTGCTGCTT |
| IFITM3 | GGGACAGGAAGATGGTTGG | CACTGGGATGACGATGAGC |
| IFIT2 | GGTCTCTTCAGCATTTATTGGTG | TGCCGTAGGCTGCTCTCCA |
| TGF-β1 | CCCAGCATCTGCAAAGCTC | GTCAATGTACAGCTGCCGCA |
| VEGFA | AGGAGGAGGGCAGAATCATCA | CTCGATTGGATGGCAGTAGCT |
| GM-CSF | TGCTGCTGAGATGAATGAAAC | AAAGGTGATAATCTGGGTTGC |
| Sev-M | GTGATTTGGGCGGCATCT | GATGGCCGGTTGGAACAC |
The gene sequences are downloaded from NCBI, and primers are designed by Primer 5.
Nuclear and cytoplasmic fractionation.
Subcellular fractions were extracted as described previously (52). A total of 106 A549 cells treated accordingly were harvested and lysed with 100 μl of cytoplasmic extraction buffer [10 mM HEPES, 10 mM KCl, 2 mM Mg(Ac)2, 3 mM CaCl2, 340 mM sucrose, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), pH 7.9] on ice for 20 min, followed by the addition of NP-40 (Amresco, Solon, OH, USA) to a final concentration of 0.25% (vol/vol). Samples were then vortexed for 15 s and then centrifuged for 10 min at 3,500 × g at 4°C. Supernatants (the cytoplasmic fraction) were collected and stored at –80°C. Pellets were dissolved in 80 μl nuclear extraction buffer (50 mM HEPES, 500 mM NaCl, 1.5 mM MgCl2, 0.1% [vol/vol] Triton X-100, 1 mM DTT, 1 mM PMSF, pH 7.9), incubated on ice for 10 min, and then centrifuged at 14,000 × g for 10 min at 4°C. Supernatants (the nuclear fraction) were collected and stored at –80°C.
DNA pulldown assay.
DNA probes were synthesized by Tsingke Biological Technology (Wuhan, China). The sequences of triple repeats of the ISG54 ISRE (5′-GGGAAAGTGAAA CTAGGGAAAGTGAAACTAGGGAAAGTGAAACTA-3′) (53, 54), which can be recognized and bound by the activated IRF3, were labeled with biotin and were used in the pulldown experiments, and a cold competitor (unbiotinylated ISRE) was used as a control to compete away IRF3-LYAR binding. Single strains of DNA were thermally annealed to form dsDNA prior to pulldown experiments. HEK293T cells transfected with the indicated plasmids were infected with Sev. At 12 hpi, cells were lysed, and the lysates were then mixed with DNA probes and incubated for 4 h at 4°C. The BeaverBeads streptavidin beads (22307-1; Beaver, China) were washed three times with the buffer (10 mM Tris-HCl, 1 mM EDTA, 1 M NaCl, 0.01% to 0.1% [vol/vol] Tween 20, pH 7.5) and then added to the lysates for 2 h of incubation at 4°C. The beads were collected by magnetic separator and washed four times with cold PBS. Finally, the beads were resuspended in 1× SDS loading buffer, and bound proteins were resolved by SDS-PAGE, followed by immunoblotting with antibody to IRF3 and LYAR.
Statistical analysis.
The data are presented as the means ± standard deviations (SD) from three independent experiments. Statistical significance was determined using two-tailed Student’s t test. A P value of less than 0.05 was considered statistically significant, and a P value of less than 0.01 was considered highly significant.
ACKNOWLEDGMENTS
This work was partially supported by funds from the National Natural Science Foundation of China (31772752, 31761133005, and 31572545), the National Key Research and Development Program (2016YFD0500205 and 2016YFC1200201), and the Outstanding Youth Science Foundation of Hubei Province (2016CFA056).
H.Z. and C.Y. designed the experiments. C.Y., T.C., R.X., Q.G., and F.M. conducted the experiments. C.Y., X.L., M.J., H.C., and H.Z. analyzed the data. C.Y., X.L., and H.Z wrote the paper.
REFERENCES
- 1.Lee B. 2013. HIV provides ample PAMPs for innate immune sensing. Proc Natl Acad Sci U S A 110:19183–19184. doi: 10.1073/pnas.1319118110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kahrstrom CT. 2012. Innate immunity: destructive interference of PRRs. Nat Rev Immunol 12:474. doi: 10.1038/nri3245. [DOI] [PubMed] [Google Scholar]
- 3.Killip MJ, Fodor E, Randall RE. 2015. Influenza virus activation of the interferon system. Virus Res 209:11–22. doi: 10.1016/j.virusres.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 5.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Vallabhapurapu S, Karin M. 2009. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol 27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 7.Iwasaki A, Pillai PS. 2014. Innate immunity to influenza virus infection. Nat Rev Immunol 14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 9.Michalska A, Blaszczyk K, Wesoly J, Bluyssen H. 2018. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front Immunol 9:1135. doi: 10.3389/fimmu.2018.01135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Honda K, Takaoka A, Taniguchi T. 2006. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Lin R, Heylbroeck C, Pitha PM, Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol 18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. 1998. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol Cell 1:507–518. doi: 10.1016/S1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- 13.Weaver BK, Kumar KP, Reich NC. 1998. Interferon regulatory factor 3 and CREB-binding protein/p300 are subunits of double-stranded RNA-activated transcription factor DRAF1. Mol Cell Biol 18:1359–1368. doi: 10.1128/mcb.18.3.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J 17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards KH, Macdonald A. 2011. Putting the brakes on the anti-viral response: negative regulators of type I interferon (IFN) production. Microbes Infect 13:291–302. doi: 10.1016/j.micinf.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 16.Saitoh T, Tun-Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, Akira S, Yamamoto N, Lu KP, Yamaoka S. 2006. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol 7:598–605. doi: 10.1038/ni1347. [DOI] [PubMed] [Google Scholar]
- 17.Lu B, Ren Y, Sun X, Han C, Wang H, Chen Y, Peng Q, Cheng Y, Cheng X, Zhu Q, Li W, Li HL, Du HN, Zhong B, Huang Z. 2017. Induction of INKIT by viral infection negatively regulates antiviral responses through inhibiting phosphorylation of p65 and IRF3. Cell Host Microbe 22:86–98.e84. doi: 10.1016/j.chom.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 18.Kim JH, Kim TH, Lee HC, Nikapitiya C, Uddin MB. 2017. Rubicon modulates antiviral type I interferon (IFN) signaling by targeting IFN regulatory factor 3 dimerization. J Virol 91:e00248-17. doi: 10.1128/JVI.00248-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su L, Hershberger RJ, Weissman IL. 1993. LYAR, a novel nucleolar protein with zinc finger DNA-binding motifs, is involved in cell growth regulation. Genes Dev 7:735–748. doi: 10.1101/gad.7.5.735. [DOI] [PubMed] [Google Scholar]
- 20.Yang C, Liu X, Gao Q, Cheng T, Xiao R, Ming F, Zhang S, Jin M, Chen H, Ma W, Zhou H. 2018. The nucleolar protein LYAR facilitates ribonucleoprotein assembly of influenza A virus. J Virol 92: e01042-18. doi: 10.1128/JVI.01042-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu SF, Su WC, Jeng KS, Lai MM. 2015. A host susceptibility gene, DR1, facilitates influenza A virus replication by suppressing host innate immunity and enhancing viral RNA replication. J Virol 89:3671–3682. doi: 10.1128/JVI.03610-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S, Wang L, Berman M, Kong YY, Dorf ME. 2011. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35:426–440. doi: 10.1016/j.immuni.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Banerjee S, Wang Y, Goldstein SA, Dong B, Gaughan C, Silverman RH, Weiss SR. 2016. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc Natl Acad Sci U S A 113:2241–2246. doi: 10.1073/pnas.1519657113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pauli EK, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG, Ludwig S. 2008. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog 4:e1000196. doi: 10.1371/journal.ppat.1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lazear HM, Schoggins JW, Diamond MS. 2019. Shared and distinct functions of type I and type III INTERFERONS. Immunity 50:907–923. doi: 10.1016/j.immuni.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ju J, Wang Y, Liu R, Zhang Y, Xu Z, Wang Y, Wu Y, Liu M, Cerruti L, Zou F, Ma C, Fang M, Tan R, Jane SM, Zhao Q. 2014. Human fetal globin gene expression is regulated by LYAR. Nucleic Acids Res 42:9740–9752. doi: 10.1093/nar/gku718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 28.Yu Y, Hayward GS. 2010. The ubiquitin E3 ligase RAUL negatively regulates type I interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33:863–877. doi: 10.1016/j.immuni.2010.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Honda K, Taniguchi T. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 30.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun Y, Atmadibrata B, Yu D, Wong M, Liu B, Ho N, Ling D, Tee AE, Wang J, Mungrue IN, Liu PY, Liu T. 2017. Upregulation of LYAR induces neuroblastoma cell proliferation and survival. Cell Death Differ 24:1645–1654. doi: 10.1038/cdd.2017.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y, Liu M, Li Z, Wu XB, Wang Y, Wang Y, Nie M, Huang F, Ju J, Ma C, Tan R, Zen K, Zhang CY, Fu K, Chen YG, Wang MR, Zhao Q. 2015. LYAR promotes colorectal cancer cell mobility by activating galectin-1 expression. Oncotarget 6:32890–32901. doi: 10.18632/oncotarget.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikushima H, Negishi H, Taniguchi T. 2013. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol 78:105–116. doi: 10.1101/sqb.2013.78.020321. [DOI] [PubMed] [Google Scholar]
- 34.Zhou JH, Wang YN, Chang QY, Ma P, Hu Y, Cao X. 2018. Type III interferons in viral infection and antiviral immunity. Cell Physiol Biochem 51:173–185. doi: 10.1159/000495172. [DOI] [PubMed] [Google Scholar]
- 35.Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. 2007. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol 81:7749–7758. doi: 10.1128/JVI.02438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bolen CR, Ding S, Robek MD, Kleinstein SH. 2014. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 59:1262–1272. doi: 10.1002/hep.26657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gelinas JF, Clerzius G, Shaw E, Gatignol A. 2011. Enhancement of replication of RNA viruses by ADAR1 via RNA editing and inhibition of RNA-activated protein kinase. J Virol 85:8460–8466. doi: 10.1128/JVI.00240-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samuel CE. 2011. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 411:180–193. doi: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pujantell M, Franco S, Galván-Femenía I, Badia R, Castellví M, Garcia-Vidal E, Clotet B, de Cid R, Tural C, Martínez MA, Riveira-Muñoz E, Esté JA, Ballana E. 2018. ADAR1 affects HCV infection by modulating innate immune response. Antiviral Res 156:116–127. doi: 10.1016/j.antiviral.2018.05.012. [DOI] [PubMed] [Google Scholar]
- 40.Patterson JB, Thomis DC, Hans SL, Samuel CE. 1995. Mechanism of interferon action: double-stranded RNA-specific adenosine deaminase from human cells is inducible by alpha and gamma interferons. Virology 210:508–511. doi: 10.1006/viro.1995.1370. [DOI] [PubMed] [Google Scholar]
- 41.Yang S, Deng P, Zhu Z, Zhu J, Wang G, Zhang L, Chen AF, Wang T, Sarkar SN, Billiar TR, Wang Q. 2014. Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs. J Immunol 193:3436–3445. doi: 10.4049/jimmunol.1401136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuadrado E, Booiman T, van Hamme JL, Jansen MH, van Dort KA, Vanderver A, Rice GI, Crow YJ, Kootstra NA, Kuijpers TW. 2015. ADAR1 facilitates HIV-1 replication in primary CD4+ T cells. PLoS One 10:e0143613. doi: 10.1371/journal.pone.0143613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pujantell M, Riveira-Munoz E, Badia R, Castellvi M, Garcia-Vidal E, Sirera G, Puig T, Ramirez C, Clotet B, Este JA, Ballana E. 2017. RNA editing by ADAR1 regulates innate and antiviral immune functions in primary macrophages. Sci Rep 7:13339. doi: 10.1038/s41598-017-13580-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu X, Qiu C, Zhu L, Huang J, Li L, Fu W, Zhang L, Wei J, Wang Y, Geng Y, Zhang X, Qiao W, Xu J. 2014. IFN-stimulated gene LY6E in monocytes regulates the CD14/TLR4 pathway but inadequately restrains the hyperactivation of monocytes during chronic HIV-1 infection. J Immunol 193:4125–4136. doi: 10.4049/jimmunol.1401249. [DOI] [PubMed] [Google Scholar]
- 45.Akiyama H, Ramirez NG, Gudheti MV, Gummuluru S. 2015. CD169-mediated trafficking of HIV to plasma membrane invaginations in dendritic cells attenuates efficacy of anti-gp120 broadly neutralizing antibodies. PLoS Pathog 11:e1004751. doi: 10.1371/journal.ppat.1004751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akiyama H, Ramirez NP, Gibson G, Kline C, Watkins S, Ambrose Z, Gummuluru S. 2017. Interferon-inducible CD169/Siglec1 attenuates anti-HIV-1 effects of alpha interferon. J Virol 91:e00972-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Wu T, Ma M, Zhang Z, Fu Y, Liu J, Xu J, Ding H, Han X, Chu Z, Wu Y, Shang H, Jiang Y. 2017. Elevated interferon-gamma-induced protein 10 and its receptor CXCR3 impair NK cell function during HIV infection. J Leukoc Biol 102:163–170. doi: 10.1189/jlb.5A1016-444R. [DOI] [PubMed] [Google Scholar]
- 48.Ramirez LA, Arango TA, Thompson E, Naji M, Tebas P, Boyer JD. 2014. High IP-10 levels decrease T cell function in HIV-1-infected individuals on ART. J Leukoc Biol 96:1055–1063. doi: 10.1189/jlb.3A0414-232RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rinkenberger N, Schoggins JW. 2018. Mucolipin-2 cation channel increases trafficking efficiency of endocytosed viruses. mBio 9:e02314-17. doi: 10.1128/mBio.02314-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xia Z, Xu G, Yang X, Peng N, Zuo Q, Zhu S, Hao H, Liu S, Zhu Y. 2017. Inducible TAP1 negatively regulates the antiviral innate immune response by targeting the TAK1 complex. J Immunol 198:3690–3704. doi: 10.4049/jimmunol.1601588. [DOI] [PubMed] [Google Scholar]
- 51.Monaghan P, Simpson J, Murphy C, Durand S, Quan M, Alexandersen S. 2005. Use of confocal immunofluorescence microscopy to localize viral nonstructural proteins and potential sites of replication in pigs experimentally infected with foot-and-mouth disease virus. J Virol 79:6410–6418. doi: 10.1128/JVI.79.10.6410-6418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Shi M, Lu X, Ma R, Wu C, Guo A, Peng M, Tian W. 2010. A method for protein extraction from different subcellular fractions of laticifer latex in Hevea brasiliensis compatible with 2-DE and MS. Proteome Sci 8:35. doi: 10.1186/1477-5956-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakaya T, Sato M, Hata N, Asagiri M, Suemori H, Noguchi S, Tanaka N, Taniguchi T. 2001. Gene induction pathways mediated by distinct IRFs during viral infection. Biochem Biophys Res Commun 283:1150–1156. doi: 10.1006/bbrc.2001.4913. [DOI] [PubMed] [Google Scholar]
- 54.Meng F, Zhou R, Wu S, Zhang Q, Jin Q, Zhou Y, Plouffe SW, Liu S, Song H, Xia Z, Zhao B, Ye S, Feng XH, Guan KL, Zou J, Xu P. 2016. Mst1 shuts off cytosolic antiviral defense through IRF3 phosphorylation. Genes Dev 30:1086–1100. doi: 10.1101/gad.277533.116. [DOI] [PMC free article] [PubMed] [Google Scholar]