

Periodontal disease is a significant health burden, causing tooth loss and poor oral and overall systemic health. Dysbiosis of the oral biofilm and a dysfunctional immune response drive chronic inflammation, causing destruction of soft tissue and alveolar bone supporting the teeth. Treponema denticola, a spirochete abundant in the plaque biofilm of patients with severe periodontal disease, perturbs neutrophil function by modulating appropriate phosphoinositide (PIP) signaling.
KEYWORDS: PTEN, chemotaxis, neutrophils, periodontal disease, phosphoinositide, spirochetes, Treponema denticola
ABSTRACT
Periodontal disease is a significant health burden, causing tooth loss and poor oral and overall systemic health. Dysbiosis of the oral biofilm and a dysfunctional immune response drive chronic inflammation, causing destruction of soft tissue and alveolar bone supporting the teeth. Treponema denticola, a spirochete abundant in the plaque biofilm of patients with severe periodontal disease, perturbs neutrophil function by modulating appropriate phosphoinositide (PIP) signaling. Through a series of immunoblotting and quantitative PCR (qPCR) experiments, we show that Msp does not alter the gene transcription or protein content of key enzymes responsible for PIP3 signaling: 3′ phosphatase and tensin homolog (PTEN), phosphatidylinositol 3-kinase (PI3K), or 5′ Src homology 2 domain-containing inositol phosphatase 1 (SHIP1). Instead, using immunoblotting and enzyme-linked immunosorbent assays (ELISAs), we found that Msp activates PTEN through dephosphorylation specifically at the S380 site. Msp in intact organisms or outer membrane vesicles also restricts PIP signaling. SHIP1 phosphatase release was assessed using chemical inhibition and immunoprecipitation to show that Msp moderately decreases SHIP1 activity. Msp also prevents secondary activation of the PTEN/PI3K response. We speculate that this result is due to the redirection of the PIP3 substrate away from SHIP1 to PTEN. Immunofluorescence microscopy revealed a redistribution of PTEN from the cytoplasm to the plasma membrane following exposure to Msp, which may contribute to PTEN activation. Mechanisms of how T. denticola modulates and evades the host immune response are still poorly described, and here we provide further mechanistic evidence of how spirochetes modify PIP signaling to dampen neutrophil function. Understanding how oral bacteria evade the immune response to perpetuate the cycle of inflammation and infection is critical for combating periodontal disease to improve overall health outcomes.
INTRODUCTION
Periodontal disease is a significant public health concern worldwide and in the United States, with 47% of the U.S. population suffering from some form of this disease (1, 2). The severity of periodontal disease in the oral cavity is increasingly linked to other chronic diseases and poor health outcomes, including cardiovascular disease (3, 4), diabetes (5), and cancer (6), highlighting the need to more fully understand the relationship between the host immune response and pathogenic bacteria in the oral cavity. Periodontal disease is an inflammatory condition of the periodontium, the tissue which surrounds and supports the tooth, characterized by soft tissue destruction and alveolar bone loss, caused by both dysbiosis of the microbial population and dysregulation of the host immune system, including neutrophils (7–9).
Neutrophils are the most abundant innate immune cells in the oral cavity and play a key role in maintaining healthy gingival tissue, as noted by both their prominence in oral health and increased recruitment during periodontal disease. Furthermore, subjects with severe periodontal disease have impaired neutrophil function, in particular their ability to undergo chemotaxis or migration in a directed fashion toward chemoattractants or bacterial stimuli (10–15). A crucial step in the chemotactic signaling process is the initial polarization of the cell to create a leading edge that dynamically protrudes and retracts through active actin cytoskeleton remodeling (16, 17). To achieve this directed migration as well as a number of other cellular responses, the secondary lipid messenger phosphatidylinositol (3,4,5) triphosphate (PtdIns[(3,4,5)]P3 or PIP3) accumulates asymmetrically at the leading edge of the neutrophil via signaling through G-protein-coupled receptor (GPCR) and receptor tyrosine kinase (RTK) protein signaling (18–22). The PIP3 molecule is produced by the lipid kinase phosphatidylinositol 3-kinase (PI3K) from phosphatidylinositol 4,5-bisphosphate (PtdIns[(4,5)]P2). There are four distinct catalytic isoforms of class I PI3K, termed alpha, beta, delta, and gamma, which are classically associated with cell migration (23, 24). Isoforms alpha and beta are constitutively expressed across all cell types, while gamma and delta appear to play significant roles in immune cells, including neutrophils (23, 25, 26).
PIP3 levels within the cell are further controlled by two lipid phosphatases, the 3′ phosphatase and tensin homolog (PTEN), which localizes to the trailing edge of polarized neutrophils to aid in the localization of PIP3 to the leading edge, and 5′ Src homology 2 (SH2) domain-containing inositol phosphatase 1 (SHIP1) to generate PtdIns[(4,5)]P2 and Ptd[(3,4)]P2, respectively (27, 28). The complex interaction between PI3K, PTEN, and SHIP1 in coordinating neutrophil chemotaxis has been well documented, with PTEN defined as a negative regulator of this system (25, 29–34), while SHIP1 activity has also been shown to be essential for cell polarization and chemotaxis (22, 31, 35).
The anaerobic spirochete Treponema denticola is one of the three hallmark pathogenic bacterial species, along with Porphyromonas gingivalis and Tannerella forsythia, to compose the “red complex,” responsible for the most severe forms of chronic periodontitis (8, 36, 37). Spirochetes are found deep within periodontal pockets closely associated with neutrophils at the biofilm-tissue interface (38, 39). The major outer sheath protein (Msp) of T. denticola is a significant virulence factor for this bacterium (40–44). Msp manipulates signaling through the PI3K pathway, which is essential for an effective host response and bacterial clearance from sites of infection, by altering the PIP balance through the activation of PTEN and inhibition of PI3K, leading to reduced Rac1 and Akt signaling mechanisms and the inhibition of neutrophil chemotaxis (45–48).
While it is clear that Msp manipulates the PIP3 balance in neutrophils, mechanisms for how this signaling disruption begins remain elusive, including how PTEN activity is increased and if PI3K isoform expression is altered by Msp. In addition, alterations in SHIP1 activity in neutrophils exposed to Msp have never been addressed. Here, we have focused on determining how Msp alters the activation state of PTEN and if this activity influences the other key signaling molecules in the signaling pathway, PI3K and SHIP1, to modulate neutrophil signaling and chemotaxis. Understanding the initial mechanisms by which Msp impairs neutrophil function will help provide invaluable knowledge to potentially target Msp for novel treatment and therapy development to improve oral health, which will in turn improve overall health.
RESULTS
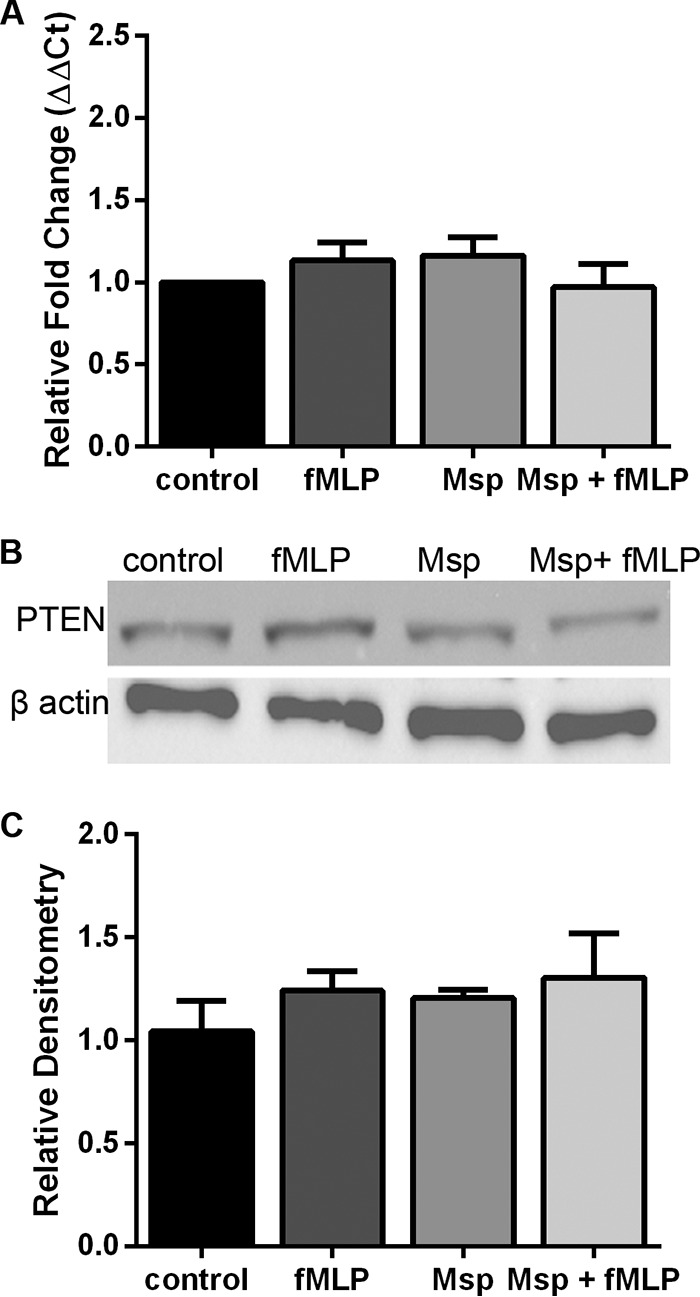
PI3K gene and protein expressions are unaltered by Msp.
We previously reported that Msp prevents appropriate signaling through the PI3K cascade when neutrophils are stimulated with N-formyl-methionine-leucine-phenylalanine (fMLP), resulting in increased PTEN phosphatase activity, reduced phosphorylation of Akt, and reduced Rac1 activity, which culminate in a reduction of neutrophil chemotaxis (45, 46). To determine if this effect is due to a decrease in PI3K gene or protein expression, quantitative PCR (qPCR) and immunoblot assays were performed for all four isoforms of PI3K: alpha, beta, gamma, and delta (Fig. 1). Neutrophils treated with fMLP had a significant fold increase in the gene transcription of all four kinases, alpha (1.5-fold), beta (1.8-fold), gamma (1.9-fold), and delta (1.3-fold), but treatment with Msp for 30 min did not cause any changes in gene expression (Fig. 1A). However, cells treated with Msp and then fMLP were similar to cells alone or cells treated with Msp, suggesting that Msp can block the effects of fMLP stimulation. While there is also no significant difference between gene expression levels in cells treated with fMLP and those treated with Msp and fMLP, the gene expression levels in cells treated with Msp and fMLP never reached those in cells treated with fMLP alone, also suggesting that Msp treatment interferes with or blocks fMLP-stimulated pathways in neutrophils. Cell lysates were collected from the same treatment groups and assayed by immunoblotting for protein expression (Fig. 1B). Similar to the qPCR results, there were no significant changes in P13K protein content with any of the treatments compared to untreated control cells when expression was normalized to β-actin by densitometry (data not shown).
FIG 1.

Msp does not alter PI3K gene or protein expression. (A) Pretreatment of neutrophils with Msp does not change the transcription of any of the four PI3K gene isoforms, alpha, beta, gamma, or delta, when relative fold changes in gene expression compared to GAPDH as the housekeeping control were measured by quantitative RT-PCR. (B) Immunoblots of cell lysates probed with antibodies specific for each PI3K isoform also show that Msp pretreatment does not alter the protein level of any PI3K isoform compared to β-actin as the housekeeping control. Graphs represent the means ± SEM of data from 3 experiments performed in triplicate (*, P < 0.05 by an unpaired t test). Immunoblots are representative images.
T. denticola is known to play a suppressive role in immune cells compared to other members of the red complex which readily activate neutrophils and trigger increases in gene transcription (49–53). Based on this observation, which was supported by the results in Fig. 1, the gene expression level of the transcription factor nuclear factor kappa B (NF-κB) following Msp exposure was measured (Fig. 2). Unsurprisingly, Msp does not alter the expression of this master regulator of the immune response, supporting the results showing no changes in PI3K expression following treatment, which overall suggests minimal cell activation following Msp exposure.
FIG 2.
Msp does not alter NF-κB gene expression. Pretreatment of neutrophils with Msp does not change the gene transcription of NF-κB when relative fold changes in gene expression compared to GAPDH as the housekeeping control were measured by quantitative RT-PCR. Graphs represent the means ± SEM of data from 3 experiments performed in triplicate.
Msp dephosphorylates PTEN specifically at the S380 site.
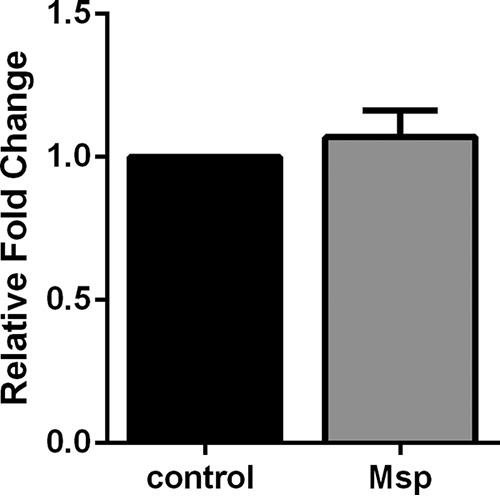
The results presented here (Fig. 1) and our previous findings (45, 46) suggest that the inhibitory effect of Msp on the PIP balance in neutrophils is due to manipulation of PTEN activity. To confirm that the increased activity previously observed following Msp exposure was not due to an increase in gene and protein levels, RNA and cell lysates were assessed for changes in PTEN expression. As expected, Msp treatment with or without fMLP stimulation did not alter PTEN gene transcription (Fig. 3A), nor were there any significant changes in the amounts of PTEN protein present in treated cell lysates compared to neutrophils alone by immunoblotting and densitometry analysis (Fig. 3B and C).
FIG 3.
PTEN expression is unchanged following Msp treatment. (A) Pretreatment of neutrophils with Msp does not change the gene transcription of PTEN when the relative fold change compared to GAPDH as the housekeeping control was measured by quantitative RT-PCR. (B) Immunoblots of cell lysates probed with anti-PTEN also show that Msp pretreatment does not alter PTEN protein levels. (C) Densitometry analysis of immunoblots was performed with ImageJ comparing PTEN to β-actin as the housekeeping control. Results were normalized to the amount of PTEN protein in untreated control cells. Graphs represent the means ± SEM of data from 3 experiments performed in triplicate. Immunoblots are 1 representative image from 3 experiments. No significant results were seen in treatment groups compared to the control in any experiment by a t test.
PTEN regulation occurs at multiple levels, including transcription and posttranscriptional mechanisms such as phosphorylation, oxidation, and localization (reviewed in reference 34). Since Msp does not change the gene or protein levels, we next investigated the phosphorylation state of PTEN (pPTEN) to assess potential changes in activity with Msp exposure using immunoblotting. PTEN has many phosphorylation sites that control activity through conformational changes that modify the substrate binding capacity and recruitment to the plasma membrane (54). When the treated cell lysates were probed with a specific anti-pPTEN S380 antibody, Msp treatment significantly reduced the amount of phosphorylated PTEN compared to that in cells alone by almost one-half (Fig. 4A and B). While fMLP stimulation after Msp exposure slightly restored pPTEN levels, pPTEN never reached the levels found in non-Msp-treated cells, in line with documented Msp inhibition of fMLP stimulation of neutrophils (45, 46). These lysates were also probed with two additional pPTEN antibodies, anti-pPTEN S380/T382/383 and anti-pPTEN T366/S370, but there were no changes in phosphorylation at these sites (see Fig. S2 in the supplemental material; densitometry not shown). To further address increased PTEN activity, a pPTEN S380-specific enzyme-linked immunosorbent assay (ELISA) was performed as an alternative to compare the amount of pPTEN S380 to that of PTEN in the cell lysates. As expected, neutrophils exposed to Msp had significantly less pPTEN S380 than cells alone (Fig. 4C), supporting our hypothesis that PTEN is more active in cells exposed to Msp.
FIG 4.
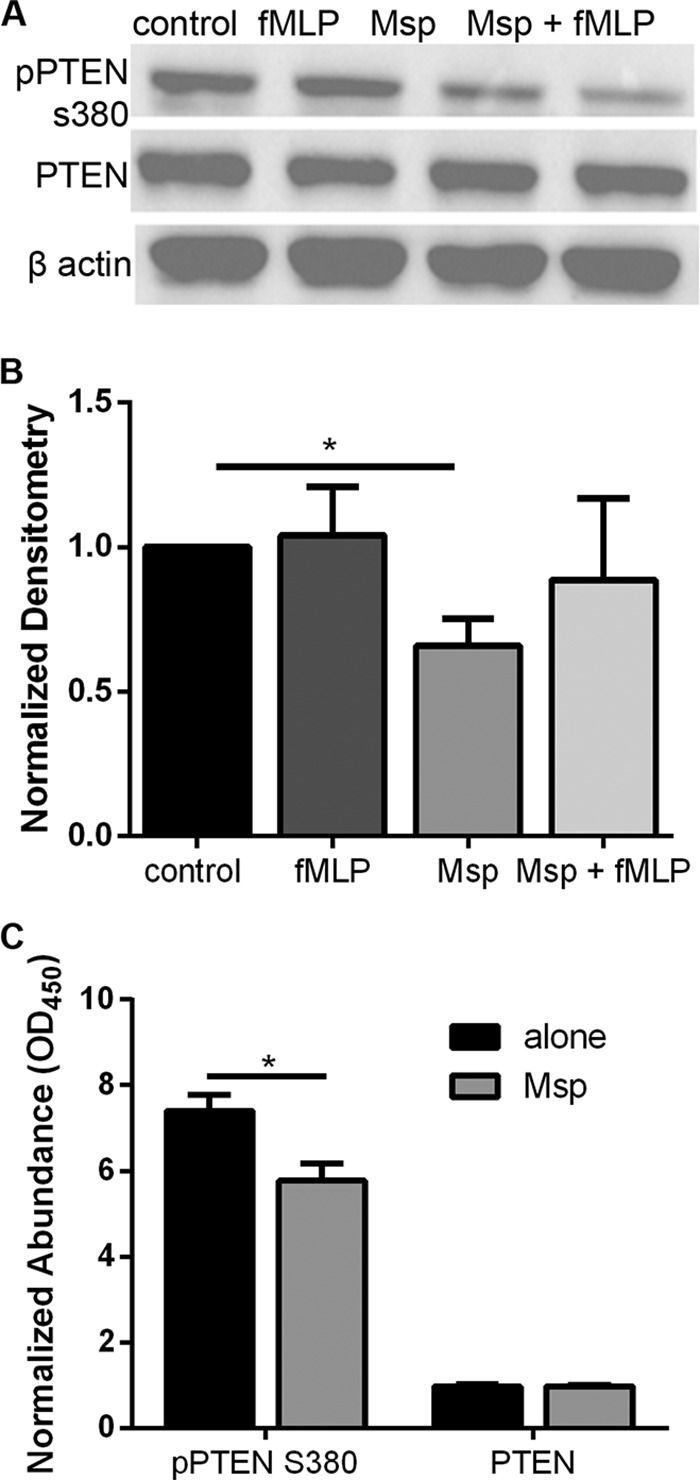
Msp decreases PTEN phosphorylation (pPTEN). (A) Immunoblots probed with anti-pPTEN S380 followed by PTEN and then β-actin show that Msp treatment reduced the phosphorylation of PTEN. (B) Densitometry analysis of pPTEN S380 immunoblots was performed with ImageJ comparing pPTEN to PTEN. Results were normalized to the amount of PTEN protein in untreated control cells. (C) Cell lysates from neutrophils alone or those treated with Msp were used in a pPTEN S380-specific ELISA. PTEN and pPTEN S380 antibodies were used to detect and quantify total PTEN and pPTEN in each sample, where the OD450 value represents the quantity of detectable protein. Graphs represent the means ± SEM of data from 3 experiments. (*, P < 0.05 by an unpaired t test). All immunoblots are 1 representative image from 3 experiments.
Whole bacteria increase PTEN activity via the Msp protein.
To determine if the activation of PTEN by Msp occurs in intact bacteria, phosphate release was measured in neutrophils treated with wild-type (WT) (strain 35405) bacteria or MHE (Msp-deficient mutant). Lysates of cells treated with both strains of bacteria had significantly more free phosphate than untreated cells (Fig. 5A). WT-treated cells had 2.2 times more free phosphate, while those treated with MHE had 1.5 times more phosphate, indicating increased phosphatase activity. While cells treated with MHE did not have statistically different amounts of free phosphate compared to the WT, there was a notable decline, suggesting that Msp is involved in inducing phosphate release from neutrophils.
FIG 5.
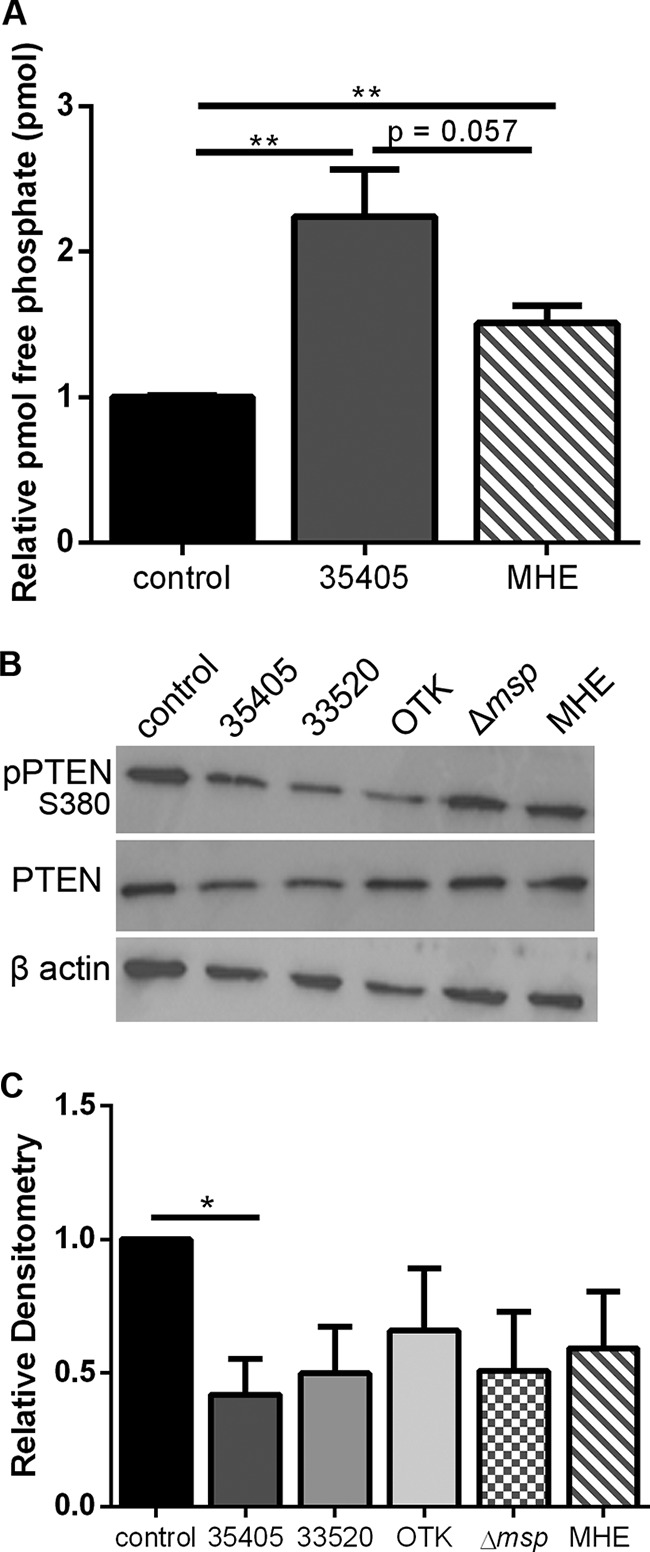
T. denticola activates PTEN by reducing phosphorylation. (A) Phosphate release was measured with a malachite green assay in lysates of neutrophils treated with wild-type strain 35405 or Msp mutant strain MHE bacteria at an MOI of 100. (B) Immunoblots of whole-cell lysates of neutrophils treated with bacteria (MOI of 100) were probed with anti-pPTEN S380. Wild-type strains 35405, 33520, and OTK all had decreased pPTEN levels compared to control cells alone. Mutant strains lacking Msp, Δmsp and MHE, were less effective at dephosphorylating PTEN than WT strains. (C) Densitometry analysis of the pPTEN S380 immunoblot was performed with ImageJ comparing pPTEN to PTEN. Results were normalized to the amount of PTEN protein in untreated control cells. Graphs represent the means ± SEM of data from 3 experiments. (*, P < 0.05, and **, P < 0.01, by an unpaired t test). All immunoblots are 1 representative image from 3 experiments.
Probing the treated cell lysates by immunoblotting for pPTEN S380 revealed that cells treated with WT (35405) bacteria had significantly less phosphorylated PTEN than untreated cells at a relative densitometry value (pPTEN/PTEN ratio) of 0.4 compared to a value of 1 (Fig. 5B and C). MHE-treated lysates had less pPTEN (value of 0.6) than untreated cells and more than cells treated with the WT, but this was not statistically significant. These results suggest that the increased phosphate release is likely due to increased PTEN activity and strongly indicate that Msp is important for this process. In addition to these strains, pPTEN levels in neutrophils treated with two additional WT T. denticola strains, 33520 and OTK, along with an additional Msp mutant strain, Δmsp, were analyzed. Strains 33520 and OTK have differences in the Msp sequence along with differential protease activity compared to 35405 (55–58). All additional WT strains showed reduced pPTEN levels as determined by densitometry analysis, 33520 to 0.5 and OTK to 0.7. Similar to the MHE mutant, the Δmsp mutant-treated cells had more pPTEN (value of 0.5) than the parent WT strain 35405 but still had less than untreated cells.
OMVs activate PTEN and reduce neutrophil chemotaxis.
Outer membrane vesicles (OMVs) are an important virulence factor for many bacteria to disseminate bacterial factors throughout the host (59, 60). T. denticola produces OMVs that contain Msp (61) (Fig. S1), which are likely a mechanism by which neutrophils in the oral cavity may encounter Msp. Neutrophils treated with OMVs have reduced pPTEN (Fig. 6A and B) compared to the amount in untreated cells. While these results are not statistically significant, they suggest that the Msp present in these vesicles is capable of altering PTEN activity in the same manner as purified protein and whole bacteria. To measure PTEN activity directly, PTEN was immunoprecipitated from treated lysates, and phosphate release was quantified using a malachite green assay. There was a slight increase of 14% in the amount of relative phosphate released from OMV-treated cells compared to control cells, indicating increased PTEN activity (Fig. 6C). Furthermore, neutrophils exposed to OMVs followed by fMLP stimulation were significantly inhibited in their ability to migrate (56%) compared to untreated cells (100%) (Fig. 6D). Strikingly, neutrophils exposed to OMVs isolated from strain MHE lacking Msp also displayed reduced directed migration (83%) compared to control cells, but this was significantly greater than for those exposed to OMVs containing Msp (Fig. 6D). These results are consistent with our previous data highlighting a significant role for Msp in manipulating the chemotaxis signaling pathway in neutrophils and demonstrate a novel role for Msp in T. denticola OMVs to cause neutrophil dysfunction.
FIG 6.
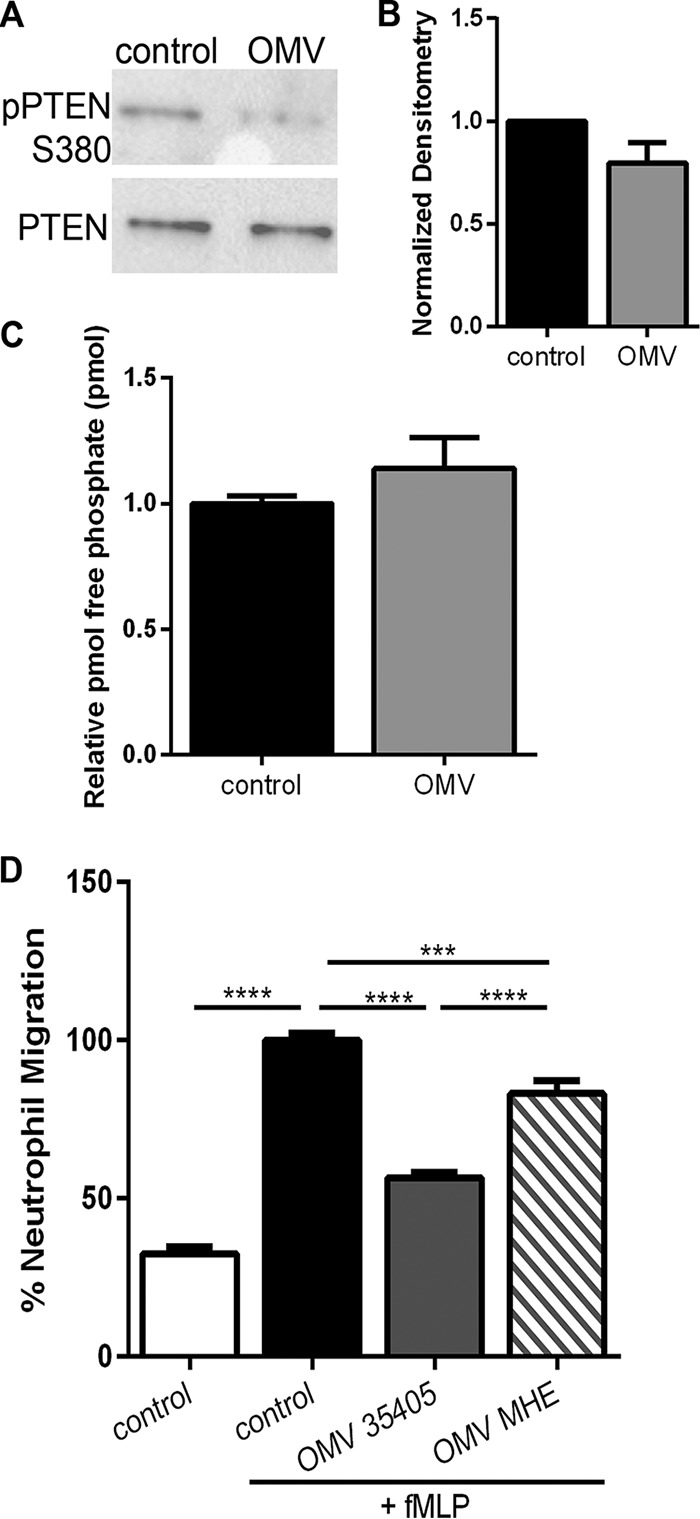
OMVs increase PTEN phosphatase activity and repress chemotaxis. (A) Immunoblots probed with anti-pPTEN S380 followed by PTEN show that OMV (WT) treatment reduced the phosphorylation of PTEN. (B) Densitometry analysis of the pPTEN S380 immunoblot was performed with ImageJ comparing pPTEN to PTEN. Results were normalized to the amount of PTEN protein in untreated control cells. (C) The lysate from neutrophils alone or those treated with OMVs (WT) was incubated with anti-PTEN, followed by immunoprecipitation with protein A-agarose beads. Phosphatase activity was assessed with a malachite green assay. (D) Neutrophils exposed to OMVs from WT T. denticola strain 35405 or the Msp mutant strain MHE prior to stimulation with fMLP in a transwell chemotaxis assay. Untreated cells were used as a positive control. Graphs represent the means ± SEM of data from 3 experiments (***, P < 0.01, and ****, P < 0.001, by an unpaired t test). The immunoblot is 1 representative image from 3 experiments.
Msp suppresses SHIP1 activity.
Since Msp alters the phosphorylation state and therefore the activity of PTEN, we wondered whether Msp treatment would have any effect on SHIP1, another key phosphatase in regulating PIP molecules and signaling in neutrophil chemotaxis (62). Similar to the PTEN experiments, no changes were seen in either gene expression (Fig. 7A) or protein levels (Fig. 7B) of SHIP1 in neutrophils exposed to Msp. To specifically examine the phosphatase activity of SHIP1, SHIP1 was immunoprecipitated, and phosphatase activity was measured by quantifying the amount of free phosphate released by SHIP1 using a malachite green assay. Msp treatment of both cells alone and cells stimulated with fMLP after Msp treatment led to a 14% reduction in relative phosphate release (Fig. 7C). While this was not a significant result, the trend is consistent with Msp altering the phosphatase activity of enzymes involved in PIP3 signaling.
FIG 7.
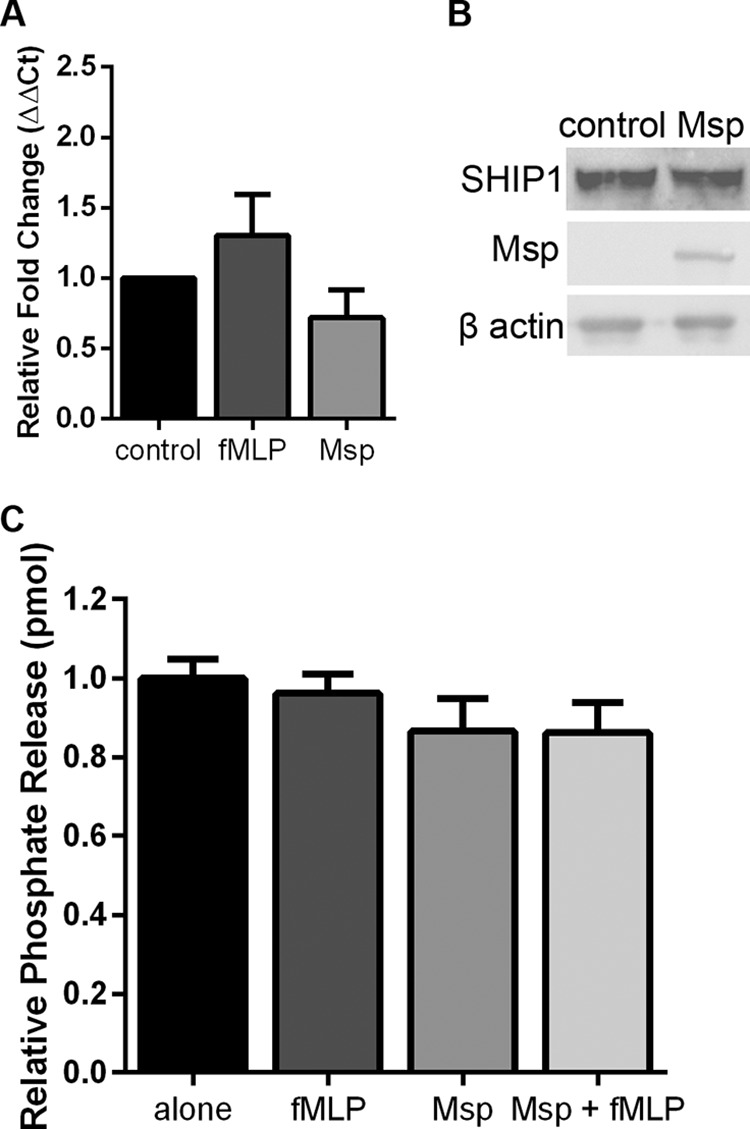
SHIP1 expression and activity are unaltered by Msp. (A) Pretreatment of neutrophils with Msp does not change the gene transcription of SHIP1 when the relative fold change compared to GAPDH as the housekeeping control was measured by quantitative RT-PCR. (B) Immunoblots of cell lysates probed with anti-SHIP1 also show that Msp pretreatment does not alter SHIP1 protein levels. Blots were reprobed for Msp and β-actin as the housekeeping control. (C) Immunoprecipitated SHIP1 from Msp- and fMLP-treated neutrophil cell lysates was used to measure specific SHIP1 activity by incubation with soluble diC8 PIP3, and phosphate release was measured using the malachite green assay. Graphs represent the means ± SEM of data from 3 experiments performed in triplicate. No significant results were seen in treatment groups compared to the control by a t test. Immunoblots are representative images.
Previously, we found that inhibiting PTEN blocks free phosphate release from PTEN activation by Msp (45). Since Msp also appears to inhibit SHIP1 phosphatase activity, we speculated that inhibiting SHIP would lead to additional increased phosphate release with Msp treatment. Neutrophils were treated with a SHIP1 inhibitor, 3-α-aminocholestane (Echelon), followed by the standard Msp treatment used throughout. Cell lysates were then assessed for free phosphate using a modified malachite green assay. Inhibiting SHIP1 alone almost doubled the amount (1.8 times more) of free phosphate compared to the control, while the addition of the SHIP1 inhibitor followed by Msp increased the amount of free phosphate by 2.5 times compared to cells alone (Fig. 8). There was no change in free phosphate release with Msp treatment alone. The additive effect of inhibiting SHIP and Msp supports our hypothesis that the mechanism of action of Msp to modulate neutrophil cell signaling is primarily directed at regulating PTEN activity.
FIG 8.
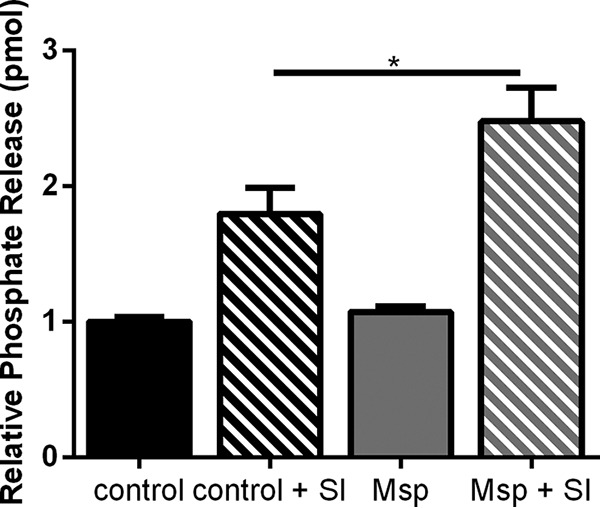
Inhibiting SHIP1 increases phosphate release in response to Msp. Lipid phosphatase activity was measured in partially permeabilized neutrophils treated with the SHIP1 inhibitor 3-α-aminocholestane (SI) prior to Msp incubation using a modified malachite green assay. The graph represents the means ± SEM from 3 experiments performed in triplicate (*, P < 0.05 by an unpaired t test).
Msp and PTEN do not interact directly.
Several reports indicate that the activity of Msp on cell signaling appears to be restricted to the cell surface (63–65), but to our knowledge, no specific Msp interaction at the surface to modulate signaling pathways in neutrophils has been identified. It has been reported that the phosphorylation state of PTEN can be both self-regulated and controlled through direct protein-protein interactions at the C-terminal domain (66, 67). To eliminate the possibility that Msp exerts control over PTEN by a direct interaction, we performed a series of immunoprecipitation assays. Neutrophils alone or treated with Msp were gently lysed, and one-half of each lysate then incubated with anti-Msp rabbit serum or anti-PTEN. Protein-antibody complexes were isolated with protein A-agarose and probed by immunoblotting for Msp and PTEN. Msp and PTEN were successfully immunoprecipitated using their respective antibodies, with no indication of direct binding or interaction between Msp and PTEN proteins under our experimental conditions (Fig. 9). This result suggests that the mechanism by which Msp modulates PTEN activity is likely restricted to an interaction at the plasma membrane of the neutrophil.
FIG 9.
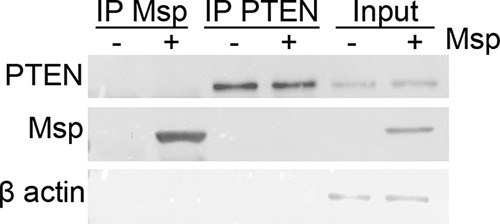
PTEN does not directly interact with Msp. The lysate from neutrophils alone or those treated with Msp was incubated with anti-Msp or anti-PTEN, followed by immunoprecipitation (IP) with protein A-agarose beads. Immunoblots were used to probe the resulting samples for the presence of Msp and PTEN in each lysate. Input samples represent the protein fractions prior to incubation with the immunoprecipitation antibodies. The immunoblot is 1 representative image from 3 independent experiments.
Msp redistributes PTEN localization.
An essential element of neutrophil chemotaxis is the redistribution of PI3K to the leading edge of the cell and PTEN to the trailing edge (19, 20, 68, 69). Similarly, appropriate PTEN activity requires localization at the plasma membrane in association with the lipid substrate (70). Following exposure to Msp, the relative fluorescence from PTEN staining increased to 1.5 × 108 RFU with PTEN localization closer to the plasma membrane compared to untreated cells with cytoplasmic localization and relative fluorescence of only 1.1 × 108 RFU as visualized by fluorescence microscopy (Fig. 10A and B). Moreover, plot profiles of fluorescence intensity across the cell showed fluorescence at the membrane of cells treated with Msp, while untreated cells had an even distribution throughout the cytoplasm, with no visible specific localization to the membrane (Fig. 10C).
FIG 10.
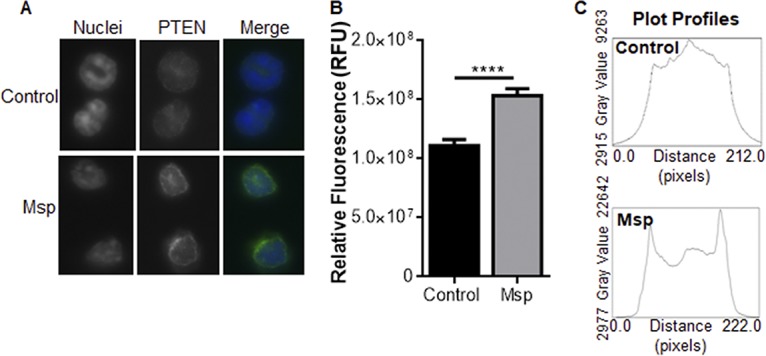
Msp causes PTEN to localize at the cell membrane. (A) Localization of PTEN in neutrophils visualized by indirect immunofluorescence. Nuclei are labeled with DAPI in blue and PTEN antibody in green. (B) Graph representing the mean relative fluorescence (RFU) of PTEN staining throughout the cells (±SEM) (>50 cells/experiment; n = 3) (****, P < 0.0001 by an unpaired t test). (C) Plot profiles representing the fluorescence intensities of representative cells bisected by a line (width of 50; ImageJ), with control cells on top and Msp-treated cells at the bottom.
Msp blocks chemotaxis in response to various stimuli.
As we were unable to show a direct interaction between PTEN and Msp, we wondered if the mechanism of chemotaxis inhibition by Msp is restricted to a specific interaction with the fMLP ligand on the surface of neutrophils or if Msp more broadly limits cell signaling. To address this, neutrophils treated with Msp were then stimulated with other known stimulants of chemotaxis, leukotriene B4 (LTB4) and macrophage inflammatory protein 2 (MIP-2) (CXCL2) (a murine homolog of interleukin-8 [IL-8]). Neutrophils exposed to Msp also failed to respond to these stimuli in a manner similar to that for fMLP (Fig. 11), suggesting that Msp broadly impacts signaling from the cell surface.
FIG 11.
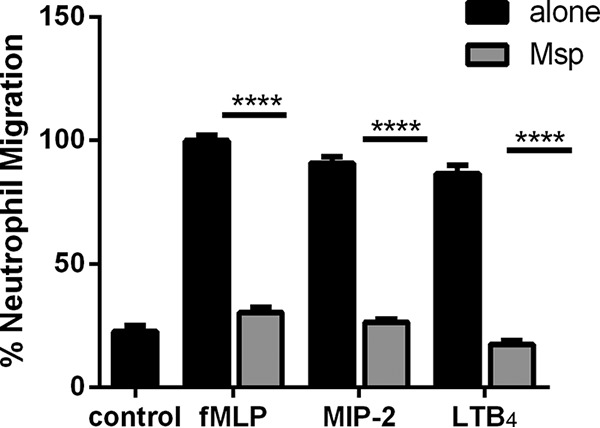
Msp inhibits chemotaxis signaling through multiple pathways. Neutrophils exposed to Msp were stimulated with MIP-2 or LTB4 in a transwell chemotaxis assay. Untreated cells were used as a positive control. The graph represents the means ± SEM of data from 3 experiments performed in triplicate (****, P < 0.001 by an unpaired t test).
DISCUSSION
Neutrophil chemotaxis is driven by a complex series of signaling and regulatory pathways revolving in part around phosphoinositide quantity and distribution at the plasma membrane, with both kinases and phosphatases playing key roles in regulating these processes. Inhibition of fMLP-stimulated chemotaxis by T. denticola Msp involves the activation of PTEN phosphatase activity, along with impaired Akt and Rac1 signaling, likely from the reduction of PIP3 levels due to limitation of PI3K activity or mislocalization at the plasma membrane (45, 46), yet the mechanism of Msp’s impact on PIP3-controlling enzymes is poorly defined. It is unclear if Msp is manipulating multiple steps in this process or altering transcript and/or protein levels to yield the documented effects of increased PTEN activity to limit signaling during neutrophil chemotaxis. A significant finding of this study is that Msp appears to modulate the activity of PTEN by altering phosphorylation specifically at the S380 site, with no direct impact on the transcription or protein content of other key enzymes in this pathway. It is possible that the results observed could be due to the experimental conditions themselves, as an irrelevant protein was not tested in conjunction with purified Msp. However, since similar results were seen with both intact, whole bacteria and outer membrane vesicles (OMVs), we are confident in the interpretations discussed here. Additionally, we provide evidence that this change in PTEN activity alone may be enough to restrict SHIP1 activity, although SHIP1 activity may be modulated by Msp independent of Msp’s role in restricting chemotaxis signaling.
Our previous work on the impact of Msp on neutrophil chemotaxis examined the activity of PTEN and PI3K, and we speculated that the observed inhibition of the PI3K-PTEN axis could be due to changes in total protein or changes in the activation state. We had anticipated seeing an alteration in PI3K gamma or delta, as these are the most significant isoforms to impact neutrophil chemotaxis (26, 30, 71), and T. denticola has been shown to significantly alter gene transcriptional profiles in infected calvarial bone and soft tissue (72). There have been other instances of oral bacteria disrupting gene transcription in immune cells, particularly another member of the red complex, P. gingivalis (8, 50, 73). P. gingivalis increases the activation of the transcription factor nuclear factor kappa B (NF-κB) in neutrophils and monocytes (51, 74, 75) and also differentially regulates gene expression in human gingival epithelial cells (76). Upon investigation, however, Msp did not alter the gene transcription or protein content of either PTEN or SHIP phosphatases, nor were there any changes in any isoform of PI3K (Fig. 1, 3, and 7). Furthermore, there was no change in NF-κB transcription in neutrophils (Fig. 2), which is supported by a study suggesting that T. denticola does not activate NF-κB in gingival epithelial cells (49). These results further support the hypothesis that T. denticola plays a significant role in repressing the host immune response. Interestingly, we observed that exposure to fMLP increased the transcription of all PI3K genes and that prior exposure to Msp appears to limit the response to fMLP (Fig. 1). While these results suggest that Msp does not directly impact transcription, it clearly affects the ability of neutrophils to respond to additional stimuli. Strikingly, we also observed that exposure to Msp prevents further stimulation by fMLP to restore PTEN phosphorylation to normal levels (Fig. 4). This restriction of the neutrophil response to stimuli could have a significant impact on the function of the host immune response in the oral cavity. In line with the new data presented here, our previous studies have also reported similar effects with Msp preventing fMLP stimulation of actin synthesis, dynamics, and migration (47, 48, 77).
PTEN is a complex molecule playing significant roles in the PI3K-Akt pathway in many cell types, with roles in the nucleus and cytoplasm and at the plasma membrane (78, 79). The phosphatase activity responsible for PIP3 processing has been linked to the N-terminal region of the protein, while the C-terminal region is responsible for binding to phospholipid membranes, with the phosphorylation state linked to overall protein stability and function (80). The C-terminal domain is considered responsible for controlling the phosphatase activity of the N-terminal region such that dephosphorylation of this region leads to an increase in PTEN activity (81, 82). It has been demonstrated that PTEN C-terminal phosphorylation keeps the protein in an inactive state and that these changes regulate activity and localization. While there is a wealth of information on the activity and regulation of PTEN, there is still much to be learned about the phosphorylation sites and activity of PTEN in different cell types and the role of the C-terminal domain function (22, 67, 80, 82, 83). Changes in phosphorylation states of PTEN have been documented in many cancers (84), and while knowledge of bacterial manipulation of PTEN phosphorylation and activity specifically is limited, it has been reported that Helicobacter pylori can increase the phosphorylation of PTEN to inactivate this molecule in gastric epithelial cells, with implications for gastric carcinogenesis (85). In this study, we have provided multiple lines of evidence that T. denticola Msp decreases PTEN phosphorylation at the S380 site in the C-terminal domain in neutrophils, which is likely responsible for the increased PTEN activity observed following exposure to Msp (Fig. 4) (45). Many proteins have been documented to modify specific phosphorylation events at the C-terminal region of PTEN (34); for example, PICT-1 is a protein that regulates S380 phosphorylation and turnover of PTEN (66). The upstream Msp-mediated mechanism of this action still requires further investigation.
Direct interactions between bacteria and host cells are one mechanism by which neutrophils may be exposed to Msp, therefore confirming that whole bacteria can modulate PTEN, which was essential to support the results obtained using purified Msp. Immunoblot assays of pPTEN and measurement of phosphate release in cells exposed to intact bacteria confirmed that neutrophils respond to intact bacteria in a manner similar to that of purified protein and that the presence of Msp impacts the neutrophil response to reduce pPTEN and activate phosphatase activity (Fig. 5). The reduced-phosphate-release response to the MHE strain lacking Msp compared to the WT supports a role for Msp in the bacterial membrane to modulate the response of neutrophils to T. denticola. Likewise, wild-type strain 35405 significantly reduced pPTEN levels. The two Msp mutant strains examined, MHE and Δmsp, also reduced pPTEN levels but to a lesser extent than the parental 35405 strain, suggesting a role for Msp but also indicating that other bacterial components contribute to the modulation of PTEN phosphorylation. Other wild-type strains of T. denticola with known variation in the Msp sequence, 33520 and OTK (55, 56), also reduce pPTEN levels but to a lesser extent than 35405. The similarities between these wild-type strains and the 35405 mutant strains strongly suggest that variation in the Msp sequence is an important factor in the response of neutrophils to different strains of T. denticola. This interpretation is supported by our previous work showing that recombinant Msp proteins from strains 35405 and OTK differ significantly in their abilities to block Akt phosphorylation and inhibit neutrophil chemotaxis (46). The overall decrease in pPTEN with all T. denticola strains suggests a potential contribution of other factors in modulating the neutrophil response. For example, the protease dentilisin could contribute to our observed results, as expression and activity vary across strains and the MHE mutant strain demonstrates increased dentilisin activity (58), which may have a significant impact on neutrophil modulation and explain why significant differences in pPTEN levels compared to the parental strain were not seen.
OMVs are a second mechanism by which host cells may encounter bacterial virulence factors such as Msp. OMVs from P. gingivalis, a key pathogen of the red complex associated with severe periodontal disease, alters the growth and function of gingival fibroblasts (86). OMVs from all three oral pathogens of the red complex, T. denticola, P. gingivalis, and Tannerella forsythia, are capable of activating the inflammasome in differentiated macrophages (87). Neutrophils exposed to OMVs reduced pPTEN levels, increased the phosphatase activity of PTEN, and also inhibited the ability of neutrophils to respond appropriately to chemotaxis stimuli (Fig. 6). While these results are not as dramatic as those seen with pure protein, the trend is very consistent and supports our hypothesis of Msp manipulating neutrophil signaling. It is difficult to speculate on how many OMVs host cells may be exposed to at a given time, and we did not correlate the concentration of pure Msp to the amount present in the vesicles. We speculate that if we increased the amount of OMVs, we would see additional increases in PTEN activation.
These results are in line with our previous findings that purified Msp and whole bacteria modulate the chemotaxis signaling pathway through interaction at the plasma membrane (45, 46). Additional studies will be needed to characterize how bacteria and OMVs are interacting with surface ligands or otherwise manipulating the plasma membrane. Other virulence factors, such as the protease dentilisin, also found in OMVs (see Fig. S1 in the supplemental material) (61, 88), are likely contributors to the changes seen even in neutrophils exposed to mutant strains of bacteria lacking Msp. As the Msp sequence varies between strains (55, 56), along with protease expression and activity (58), we are very interested in continuing to compare how host cells respond to different strains of T. denticola, as this could possibly yield interesting correlations with the severity of disease in patients.
Previous work has shown that Msp causes mislocalization of PIP3 and Akt within neutrophils, limiting the ability of cells to properly distribute these molecules to polarize (45). In this study, PTEN localization is also altered following exposure to Msp. In untreated cells, PTEN is evenly dispersed throughout the cytoplasm, without any significant accumulation at the membrane. Upon exposure to Msp, PTEN appears to localize close to the plasma membrane. In activated neutrophils, PTEN is known to localize to the trailing edge of polarized neutrophils and is active there to keep levels of PIP3 low in order to allow the leading edge of the cells to accumulate PIP3 to continue proper signaling for chemotaxis. These results suggest that PTEN in Msp-treated cells localizes to the membrane but not in an organized fashion to a trailing edge (Fig. 10), in line with our previous observations that Msp prevents the ability of neutrophils to properly polarize due to PIP imbalance and repression of appropriate dynamic actin remodeling (47, 77). Dynamic interaction of PTEN at the plasma membrane with localization at its site of lipid activity is required for activity in vivo, and changes in the C-terminal phosphorylation state are crucial for proper function (70, 89). The fact that our immunoblot experiments (Fig. 3) indicate no changes in cellular PTEN expression following Msp exposure while the immunofluorescence data indicate increased intensity (Fig. 10) may appear contradictory. However, we speculate that the increased fluorescence observed with Msp treatment is due to an increased concentration of PTEN at the plasma membrane.
These results are also consistent with another report suggesting that the S380 site plays an important role in PTEN localization in endothelial cells (90). We speculate that having increased activation of PTEN along with redistribution to the membrane would reduce PIP3 levels throughout the cell to prevent the proper localization and signaling of the chemotaxis machinery that have been previously observed (45, 46). Alterations in lipid composition and subsequent membrane surface charge are known to differentially recruit and localize proteins, including Rac isoforms in neutrophils (91). In addition to the role of phosphorylation in PTEN recruitment to the membrane, anionic lipids such as PIP2 and phosphatidylserine play a role through electrostatic interactions (54, 92). Msp-mediated alterations in lipid species such as increased PIP2 (77) and potentially a change in the membrane charge are plausible means for our observed recruitment of PTEN to the membrane, although this requires additional analysis. Exposure to Msp also blocked neutrophil chemotaxis to other stimuli (Fig. 11), which all act through specific G-protein-coupled receptor interactions (93), suggesting that Msp modulates cell signaling through an overall interaction at the surface, possibly disruption of lipid homeostasis, and not by specifically blocking the fMLP ligand. This is also supported by the fact that the other small GTPases Rac1, cdc42, and RhoA are not inhibited following fMLP stimulation of neutrophils (47). Neutrophils possess a broad range of receptor classes that act through complex intracellular signaling pathways, of which the components of many are redundant across receptor engagement. Work is ongoing to examine overall neutrophil receptor profiles following exposure to Msp or intact T. denticola bacteria.
SHIP1 is another key phosphatase in the regulation of PIP3 in many cells, notably including macrophages, T cells, and neutrophils (22, 35, 94), but its role in relation to Msp’s manipulation of PIP3 has never been investigated, and much is still unclear about SHIP1’s role in regulating immune cell signaling (62, 95). Knockout models of SHIP1 suggest that its phosphatase activity on PIP3 plays an important role in neutrophil chemotaxis (35), and zebrafish models of neutrophil chemotaxis also show that SHIP1 is an important regulator of the PI3K signaling pathway (62). While thus far, our data strongly suggest that the main mechanism of Msp to manipulate neutrophil PIP3 signaling is restricted to PTEN activity, we could not rule out possible SHIP1 involvement.
Similar to all the other proteins that we investigated, exposure to Msp does not change gene transcript or protein levels (Fig. 7A and B). Interestingly, in the SHIP1 immunoprecipitation assays, there was a slight, although not significant, decrease in SHIP1 activity after exposure to Msp (Fig. 7C), suggesting that Msp decreases the activity of this phosphatase, but this may appear counterintuitive to our PTEN data, as reduced SHIP1 activity would increase chemotaxis. From the literature, SHIP1 is a multifunctional protein that is controlled by a variety of regulatory mechanisms, including both phosphatase-dependent and -independent manners in immune cells (95). While we suspect that the change in SHIP1 activity is at least partially due to changes in the levels of PIP3, there could be additional responses unrelated to PIP3 levels that are impacting SHIP1, which could explain the modest decrease in SHIP1 activity observed here. Alternatively, we speculate that the decrease in SHIP1 activity may be due to the sequestration of any available PIP3 during incubation with Msp, causing a modification of SHIP1 to decrease its phosphatase activity that could not be altered once we immunoprecipitated it from the cells and exposed it to the PIP3 substrate again.
Inhibiting SHIP1 in cells prior to Msp treatment resulted in an increase in free phosphate release, suggesting that a phosphatase other than SHIP1 had increased activity (Fig. 7). It is likely that the observed elevation in free phosphate is due to increased PTEN activation and more PIP3 substrate being available within the cell for PTEN to act upon since SHIP1 activity was inhibited. Moreover, if Msp decreases SHIP1 activity even modestly, any available PIP3 could be shuttled toward PTEN. These data support this hypothesis, as the largest amount of phosphate release was seen in cells that had both SHIP1 inhibited and Msp treatment. Furthermore, increased PTEN activity resulting in less PIP3 availability as a substrate also explains the decreased PI3K activity and reduced downstream signaling through Rac1 and Akt reported previously (45, 46).
There is still much to be learned about the role of Msp and T. denticola as a significant pathogen in severe periodontitis, but the ability of Msp to manipulate cell signaling in key host defense mechanisms, such as neutrophils, is likely extremely important in promoting bacterial survival and continued dysbiosis of the biofilm community. By limiting the ability of neutrophils to respond to other stimuli and pathogens, we can speculate that T. denticola plays a significant role in the well-characterized restriction of the innate immune response in periodontal disease (96, 97). T. denticola produces OMVs containing Msp (61), and the data presented here indicate that Msp-containing OMVs can activate PTEN to impair neutrophil migration (Fig. 6). Thus, OMVs may be a viable vehicle for Msp action and, combined with the motile nature of T. denticola, may allow for many host cells to be impacted by Msp exposure to reshape the environment in the gingival pocket and progression of periodontal disease. For example, Msp has already been shown to alter actin cytoskeleton rearrangement in fibroblasts (77) and to induce tolerance in macrophages (65).
In summary, we show that the S380 site of PTEN is the key site for Msp’s ability to modulate PIP signaling through the PTEN-PI3K axis and give additional support to the growing evidence that T. denticola functions to repress neutrophil signaling and overall dampen the immune response. Characterizing this mechanism of action of how T. denticola and Msp modulate the innate immune response to perpetuate the cycle of inflammation is an important first step in developing effective treatments to combat the dysregulation of the host response and the dysbiotic biofilm community characteristic of periodontal disease.
MATERIALS AND METHODS
Murine neutrophil isolation.
Murine neutrophil isolation was previously described (45). All procedures were approved by the University at Buffalo Institutional Animal Care and Use Committee. Briefly, C57BL/6J wild-type mice (male, 6 weeks old) were purchased from Jackson Laboratory (Bar Harbor, ME). Femurs and tibias were removed, and cells were isolated from bone marrow by fractionation into discontinuous Percoll (Sigma) gradients (80%, 65%, and 55%). Mature neutrophils were isolated from the 80%–65% interface.
Purification of native Msp protein.
The native Msp complex from strain 35405 was isolated as previously described (Fig. 12) (43, 63). Briefly, T. denticola cultures (2 liters) were grown for 3 days in modified new oral spirochete (NOS) medium (98), followed by centrifugation (10,000 × g for 20 min at 4°C). Cells were washed with cold phosphate-buffered saline (PBS) (pH 7.3), and Msp was highly enriched by sequential detergent extraction steps. Cells were extracted three times overnight with stirring at 4°C with deoxycholate buffer (0.1% with 20 mM Tris [pH 7.5], 1 mM dithiothreitol [DTT], and 2 mM EDTA) (30 ml/liter original culture) and one time with n-octylpolyoxyethylene buffer (1% prepared in 10 mM Tris [pH 8.0]), followed by ultracentrifugation (141,000 × g for 2 h at 4°C) at each step. The enriched preparation was then incubated at 37°C until no peptidase activity could be detected using an N-succinyl-Ala-Ala-Pro-Phe p-nitroanilide (SAAPFNA) protease assay where the molecule SAAPFNA (1.25-μg/μl final concentration) will produce a yellow color when cleaved, measured as the optical density at 405 nm (OD405) after a 1-h incubation at room temperature (46, 99). The time for autoproteolysis of Msp preparations is variable, but our unpublished and previously reported data (63) indicate that this requires from 1 week to 1 month of incubation (Fig. 12E). Following autoproteolysis of the extract, the sample was concentrated by ultrafiltration (Amicon Concentricon Plus 80), washed with 3 liters of 10 mM Tris (pH 8.0) and 1 liter of distilled H2O (dH2O), ultracentrifuged again, extensively dialyzed against distilled H2O, and ultracentrifuged again. Msp preparations were confirmed to be >98% pure by Coomassie- and silver-stained SDS-PAGE gels (Fig. 12A and B) and Western blotting with anti-Msp serum and antidentilisin serum (Fig. 12D). The final purified protein was aliquoted and stored at −80°C until use.
FIG 12.
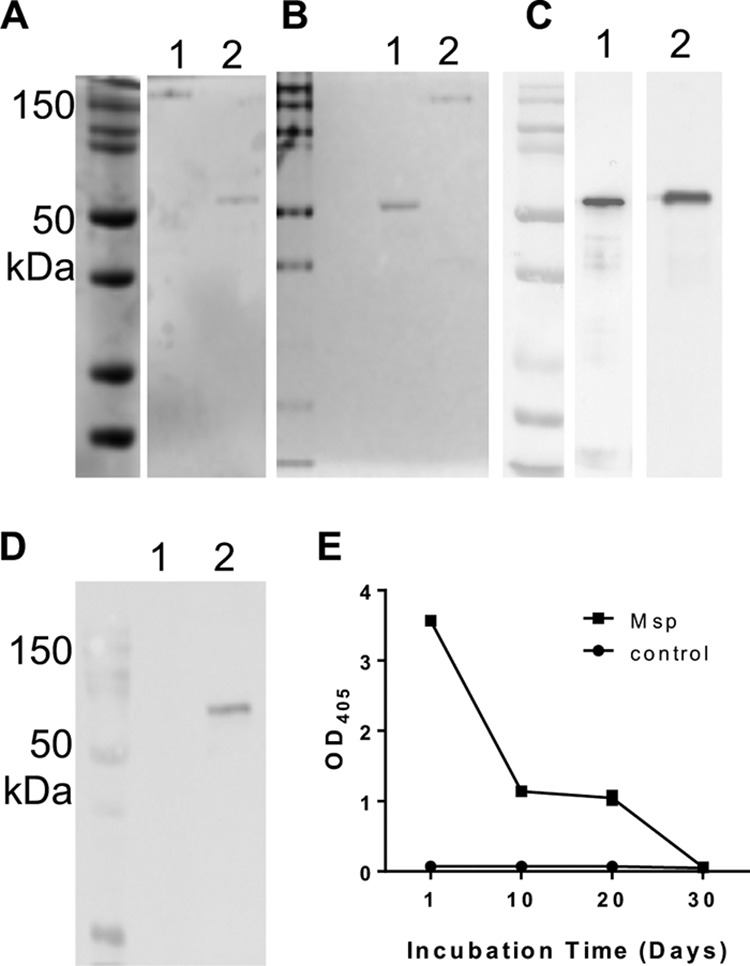
Native Msp protein purity. (A) SDS-PAGE of native Msp showing purity of Msp. Unboiled protein (50 ng) retains its native trimeric conformation at ∼150 kDa (lane 1), while a boiled sample displays the monomeric form at 53 kDa (lane 2). (B) Silver-stained SDS-PAGE gel of boiled (lane 1) and unboiled (lane 2) native Msp. (C) Immunoblot of Msp protein (lane 1) and the WT T. denticola whole-cell lysate (lane 2) probed with anti-Msp. (D) Immunoblot of Msp protein (lane 1) and the whole-cell lysate probed with antidentilisin (72 kDa) showing no protease contamination in the Msp protein preparation. (E) Assessment of proteolytic activity in the Msp preparation using a SAAPFNA assay indicating that at the end of the isolation, there was no detectable proteolytic activity. Data shown represent results of one complete Msp purification.
Purification of outer membrane vesicles.
T. denticola cultures (2 liters) were grown for 3 days in modified NOS medium. Cultures were centrifuged at 10,000 × g at 4°C for 15 min, and supernatants containing outer membrane vesicles (OMVs) were filtered through a 0.2-μm filter (Sigma) and concentrated with sucrose powder (99) in dialysis tubing (3,500-molecular-weight cutoff [MWCO]). The resulting solution was dialyzed overnight in Tris buffer (50 mM; pH 7.2) followed by ultracentrifugation for 2 h at 100,000 × g at 4°C. The pellet of OMVs was washed twice in Tris buffer, centrifuged as described above, resuspended in Tris buffer, and frozen at −80°C until use. Vesicle composition and purity were assessed by Western blotting and scanning electron microscopy (SEM) and transmission electron microscopy (TEM) imaging as described in Fig. S2 in the supplemental material. We speculate the different size of the flagellar protein in the whole-cell lysate compared to that in the vesicles could be due to a different subunit composition or posttranslational modification of the protein (100). Additional experiments to understand this result are ongoing.
Bacterial strains and growth conditions.
Treponema denticola strains used in this study are listed in Table 1. Wild-type strains were routinely grown anaerobically at 37°C in NOS medium, while the Msp mutant strains MHE and Δmsp were grown in NOS medium containing 40 μg/ml erythromycin (101, 102). Cultures were examined for purity and typical morphology and enumerated using dark-field microscopy. For bacterium-neutrophil incubation experiments, strains were grown for 3 days anaerobically and washed 2 times with PBS, followed by counting by dark-field microscopy for use in the assay.
TABLE 1.
Treponema strains
| Strain | Relevant characteristic | Source |
|---|---|---|
| 35405 | Wild-type strain | R. Ellen |
| 33520 | Wild-type strain | J. C. Fenno |
| OTK | Wild-type strain | J. C. Fenno |
| MHE | msp mutant of strain 35405 | J. C. Fenno |
| Δmsp | msp mutant of strain 35405 | K. Nagano |
Standard neutrophil coincubation treatment.
N-Formyl-methionine-leucine-phenylalanine (fMLP) is a bacterial peptide product known to stimulate neutrophil chemotaxis and served as a positive control and stimulus throughout this study. Neutrophils were incubated with Msp (100 nM or 1.8 μg) based on the native trimer form, OMVs (30 μg/ml), or whole bacteria (multiplicity of infection [MOI] of 100) for 30 min at room temperature for all experiments, followed by a 1-min exposure to fMLP (1 μM), as indicated, with appropriate controls of untreated cells and cells stimulated with fMLP alone unless otherwise noted. Previous studies from our group and others support the choice of Msp concentration and incubation times, as these variables are within the range documented to produce observable effects on host cells and also highlighted a dose- and time-dependent effect of Msp (45–47, 77). Msp is highly expressed at both the gene and protein levels in intact organisms in vitro (2 × 105 copies/cell in mid-log phase) (41) as well as in animal models and the human periodontal pocket in vivo (103, 104).
Assessment of protein levels by immunoblotting.
The following antibodies directed toward T. denticola components were obtained: anti-Msp (R. P. Ellen, University of Toronto), antidentilisin (J. C. Fenno, University of Michigan), anti-flagellar protein (J. C. Fenno, University of Michigan), and anti-DnaK (100) (C. Li, Virginia Commonwealth University). For all PI3K and PTEN protein detections, 1 × 106 neutrophils were used, and for SHIP1, 5 × 106 cells were used. All were treated as described above. All samples for immunoblotting were lysed in 2× SDS sample buffer, boiled for 10 min, and separated on 10% SDS-PAGE gels, followed by transfer to nitrocellulose membranes. Membranes were blocked with 5% milk–Tris-buffered saline (TBS)–0.05% Tween and incubated overnight with the following primary antibodies: anti-PI3K alpha, beta, gamma (PI3 kinase antibody sampler kit), and delta (D1Q7R); anti-pPTEN (S380, T366/S370) (GeneTex); anti-pPTEN (S380/T382/383) (44A7); anti-PTEN (1386G); and anti-SHIP1 (D1163). Membranes were incubated with horseradish peroxidase (HRP)-linked secondary antibody followed by detection with Amersham ECL Prime solutions (GE). Membranes were reprobed as appropriate with anti-PTEN and/or anti-β-actin. All antibodies are from Cell Signaling Technology unless otherwise noted. Densitometry analysis was performed using ImageJ software.
Quantitative real-time PCR (qRT-PCR).
Neutrophils were treated with Msp and fMLP as described above. RNA was isolated using E.Z.N.A. Total RNA kit I (Omega), converted to cDNA using iScript (Bio-Rad), and subjected to qPCR using Sso Sybr (Bio-Rad) with an Applied Biosystems 7500 machine. The relative fold change (ΔΔCT) was determined by comparison against the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in the untreated samples (control). Primers are listed in Table 2.
TABLE 2.
Primers for quantitative RT-PCR
| Gene | Sequence |
|---|---|
| PI3K alpha | GCAAAGCATCCATGAAGTCTGGC |
| CAGTTTGGTGTCATCCTGGAAGC | |
| PI3K beta | CAGTTTGGTGTCATCCTGGAAGC |
| TCTGCTCAGCTTCACCGCATTC | |
| PI3K gamma | CTGGAGAGCTTAGAGGACGATG |
| AAGCCACGCTTCAGCAGGAATC | |
| PI3K delta | ACCATCAGTGGCTCTGCGGTTT |
| GTGGTCTTCTGGGAACTCACCT | |
| PTEN | GATTGCAAGTTCCGCCACTGAACA |
| ATTCCCAGTCAGAGGCGCTATGT | |
| SHIP1 | TTCACCCACCTCTTCTGGCTTG |
| TTCCTCTCCAGGAGCAGTTGGT | |
| GAPDH | GCCTTCTCCATGGTGGTGAA |
| GCACAGTCAAGGCCGAGAAT | |
pPTEN ELISA.
The amount of pPTEN was also determined using phospho-PTEN (S380) and total PTEN ELISAs (RayBiotech); all ELISA-specific reagents are included in the kit. Neutrophils (8 × 106 cells under each condition) alone (control) or treated with Msp as described above were lysed and analyzed according to kit instructions, with duplicate wells per sample. Lysate samples were frozen at −80°C until use. All ELISA steps were performed at room temperature with gentle shaking. Briefly, lysates were incubated in anti-PTEN-coated wells, followed by blocking and incubation with either anti-pPTEN S380 or anti-PTEN for 1 h at room temperature. HRP-conjugated solutions were added for 1 h, followed by colorimetric development with a tetramethylbenzidine (TMB) substrate reagent for 30 min, protected from light. After the addition of a stop solution, the plate was immediately read at 450 nm. Levels of pPTEN S380 were normalized to PTEN levels in all samples.
SHIP1 activity detected by immunoprecipitation and a malachite green phosphatase assay.
SHIP1 PIP lipid phosphatase activity was determined by measuring the amount of free phosphate released from a synthetic PIP3 substrate using a modified malachite green phosphatase assay as previously described (45, 77). Neutrophils (5 × 106 under each condition) were treated with Msp, followed by fMLP stimulation of the appropriate samples as described above, and lysed for 15 min at 4°C with agitation in 500 μl of lysis buffer (25 mM Tris, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol), with a protease inhibitor (Halt protease inhibitor; Thermo) added just prior to lysis. Lysates were centrifuged for 10 min at 14,000 × g at 4°C. The collected supernatants were incubated overnight at 4°C with anti-SHIP1 (8 μl), with agitation. The resulting lysates were incubated with 60 μl of 50% protein A-agarose (Thermo) for 2 h at 4°C, followed by 3 washes with reaction buffer (TBS, 10 mM DTT) and final resuspension in 30 μl of reaction buffer. Samples were incubated with 1 mM dioctanoyl phosphatidylinositol 3,4,5-triphospate (diC8) (Echelon) as a substrate for 1 h at 37°C, followed by the addition of 100 μl of a malachite green solution (Echelon) for 30 min at room temperature, protected from light. The absorbance at 620 nm was measured using a microplate reader (FlexStation3 plate reader; Molecular Devices Corporation), and the amount of free phosphate released was calculated using a prepared phosphate standard curve.
SHIP1 inhibitor assay.
Neutrophils (1 × 105/sample) were partially permeabilized with 0.1 volumes of 2% n-octyl-β-glucopyranoside (OG) for 30 s, followed by pretreatment with an SHIP1 inhibitor, 3-α-aminocholestane (1 mM) (Echelon) for 15 min at room temperature in Hanks’ balanced salt solution (HBSS) without phosphate. Cells were then divided appropriately and treated with Msp followed by fMLP, as indicated. Samples (25 μl/well) were then incubated with a malachite green solution, and free phosphate release was measured as described above. The ratio of free phosphate in cells with and without treatment was normalized to the value for untreated control cells.
Protein-protein interaction immunoprecipitation.
For the PTEN-Msp interaction, neutrophils were treated and lysed as described above for SHIP1 immunoprecipitations. After collection of an input lysate sample, untreated and Msp-treated lysates were equally divided and incubated with anti-PTEN (8 μl) or anti-Msp (8 μl of complete rabbit serum) overnight with agitation at 4°C. The resulting lysates were then incubated with 60 μl of 50% protein A-agarose (Thermo) for 2 h at 4°C, followed by 3 washes with PBS. Beads were resuspend in 60 μl of 2× sample buffer and boiled. The resulting prepared samples were separated by SDS-PAGE and immunoblotted for PTEN and Msp, along with β-actin as a control.
PTEN immunofluorescence.
Neutrophils treated as indicated and as described above were allowed to attach to 5% bovine serum albumin (BSA)-coated coverslips. Cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.5% Triton X-100 for 10 min, and blocked with 5% normal goat serum (NGS) for 30 min. Anti-PTEN was diluted 1:200 in 5% NGS and applied for 1 h at room temperature, followed by Alexa 488 goat anti-rabbit secondary antibody (1:100; Invitrogen) and nucleus staining with 4′,6-diamidino-2-phenylindole (DAPI; Roche). Cells were examined by fluorescence microscopy (Nikon Eclipse TE2000-U), and images were obtained using SPOT Advanced software (SPOT Imaging). Quantification of images was performed with ImageJ software.
Neutrophil transwell chemotaxis assay.
Analysis of neutrophil chemotaxis using a transwell assay was previously described (46). Briefly, medium with stimulants, fMLP (1 μM), LTB4 (25 ng/μg), and MIP-2 (CKCL2) (20 ng/μg), was placed in the bottom of the plates. MIP-2 is a murine homolog of IL-8. Neutrophils (0.5 × 106) were incubated with or without Msp protein (100 nM) for 30 min at room temperature prior to placement in chemotaxis chambers at 37°C for 1 h. Membranes were fixed in 4% paraformaldehyde, washed, and stained with crystal violet. After subsequent washing with dH2O to remove excess dye, cells fixed in the membrane were counted using an inverted microscope. The cells in 5 different areas of each membrane were counted, with duplicate transwells under each condition. All data were normalized to the control with fMLP alone.
Statistical analysis.
Comparisons between two groups were performed by a t test using PRISM software. Results are based on at least 3 independent experiments. Statistical significance was defined as a P value of <0.05. Error bars represent the standard errors of the means (SEM).
Supplementary Material
ACKNOWLEDGMENTS
We declare no conflict of interest.
We thank Rikin Patel and Robert Ferguson for their assistance with completing the immunoblot experiments and Peter Bush (UB South Campus Instrument Center) for assistance with the electron microscopy analysis.
This research was supported by funding from the NIH/NIDCR under grants F32DE027612 (M.M.J.), R01 DE027073, and R03DE024769 (M.B.V.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00553-19.
REFERENCES
- 1.Eke PI, Dye BA, Wei L, Slade GD, Thornton-Evans GO, Borgnakke WS, Taylor GW, Page RC, Beck JD, Genco RJ. 2015. Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol 86:611–622. doi: 10.1902/jop.2015.140520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersen PE, Ogawa H. 2012. The global burden of periodontal disease: towards integration with chronic disease prevention and control. Periodontol 2000 60:15–39. doi: 10.1111/j.1600-0757.2011.00425.x. [DOI] [PubMed] [Google Scholar]
- 3.Scannapieco FA, Bush RB, Paju S. 2003. Associations between periodontal disease and risk for atherosclerosis, cardiovascular disease, and stroke. A systematic review. Ann Periodontol 8:38–53. doi: 10.1902/annals.2003.8.1.38. [DOI] [PubMed] [Google Scholar]
- 4.Leishman SJ, Do HL, Ford PJ. 2010. Cardiovascular disease and the role of oral bacteria. J Oral Microbiol 2:5781. doi: 10.3402/jom.v2i0.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Preshaw PM, Alba AL, Herrera D, Jepsen S, Konstantinidis A, Makrilakis K, Taylor R. 2012. Periodontitis and diabetes: a two-way relationship. Diabetologia 55:21–31. doi: 10.1007/s00125-011-2342-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeng X-T, Xia L-Y, Zhang Y-G, Li S, Leng W-D, Kwong JSW. 2016. Periodontal disease and incident lung cancer risk: a meta-analysis of cohort studies. J Periodontol 87:1158–1164. doi: 10.1902/jop.2016.150597. [DOI] [PubMed] [Google Scholar]
- 7.Darveau RP. 2010. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol 8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 8.Hajishengallis G, Lamont RJ. 2012. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol 27:409–419. doi: 10.1111/j.2041-1014.2012.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hajishengallis G, Chavakis T, Hajishengallis E, Lambris JD. 2015. Neutrophil homeostasis and inflammation: novel paradigms from studying periodontitis. J Leukoc Biol 98:539–548. doi: 10.1189/jlb.3VMR1014-468R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delima AJ, Van Dyke TE. 2003. Origin and function of the cellular components in gingival crevice fluid. Periodontol 2000 31:55–76. doi: 10.1034/j.1600-0757.2003.03105.x. [DOI] [PubMed] [Google Scholar]
- 11.Ryder MI. 2010. Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontol 2000 53:124–137. doi: 10.1111/j.1600-0757.2009.00327.x. [DOI] [PubMed] [Google Scholar]
- 12.Landzberg M, Doering H, Aboodi GM, Tenenbaum HC, Glogauer M. 2015. Quantifying oral inflammatory load: oral neutrophil counts in periodontal health and disease. J Periodontal Res 50:330–336. doi: 10.1111/jre.12211. [DOI] [PubMed] [Google Scholar]
- 13.Kumar RS, Prakash S. 2012. Impaired neutrophil and monocyte chemotaxis in chronic and aggressive periodontitis and effects of periodontal therapy. Indian J Dent Res 23:69–74. doi: 10.4103/0970-9290.99042. [DOI] [PubMed] [Google Scholar]
- 14.Roberts HM, Ling MR, Insall R, Kalna G, Spengler J, Grant MM, Chapple IL. 2015. Impaired neutrophil directional chemotactic accuracy in chronic periodontitis patients. J Clin Periodontol 42:1–11. doi: 10.1111/jcpe.12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fine N, Hassanpour S, Borenstein A, Sima C, Oveisi M, Scholey J, Cherney D, Glogauer M. 2016. Distinct oral neutrophil subsets define health and periodontal disease states. J Dent Res 95:931–938. doi: 10.1177/0022034516645564. [DOI] [PubMed] [Google Scholar]
- 16.Fenteany G, Glogauer M. 2004. Cytoskeletal remodeling in leukocyte function. Curr Opin Hematol 11:15–24. doi: 10.1097/00062752-200401000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Pollard TD, Borisy GG. 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell 112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 18.Wang F. 2009. The signaling mechanisms underlying cell polarity and chemotaxis. Cold Spring Harb Perspect Biol 1:a002980. doi: 10.1101/cshperspect.a002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuiper JW, Sun C, Magalhaes MA, Glogauer M. 2011. Rac regulates PtdInsP(3) signaling and the chemotactic compass through a redox-mediated feedback loop. Blood 118:6164–6171. doi: 10.1182/blood-2010-09-310383. [DOI] [PubMed] [Google Scholar]
- 20.Wang F, Herzmark P, Weiner OD, Srinivasan S, Servant G, Bourne HR. 2002. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Biol 4:513–518. doi: 10.1038/ncb810. [DOI] [PubMed] [Google Scholar]
- 21.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. 2001. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem 70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 22.Luo HR, Mondal S. 2015. Molecular control of PtdIns(3,4,5)P3 signaling in neutrophils. EMBO Rep 16:149–163. doi: 10.15252/embr.201439466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jean S, Kiger AA. 2014. Classes of phosphoinositide 3-kinases at a glance. J Cell Sci 127:923–928. doi: 10.1242/jcs.093773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawkins PT, Stephens LR, Suire S, Wilson M. 2010. PI3K signaling in neutrophils. Curr Top Microbiol Immunol 346:183–202. doi: 10.1007/82_2010_40. [DOI] [PubMed] [Google Scholar]
- 25.Sadhu C, Masinovsky B, Dick K, Sowell CG, Staunton DE. 2003. Essential role of phosphoinositide 3-kinase δ in neutrophil directional movement. J Immunol 170:2647–2654. doi: 10.4049/jimmunol.170.5.2647. [DOI] [PubMed] [Google Scholar]
- 26.Kok K, Nock GE, Verrall EAG, Mitchell MP, Hommes DW, Peppelenbosch MP, Vanhaesebroeck B. 2009. Regulation of p110δ PI 3-kinase gene expression. PLoS One 4:e5145. doi: 10.1371/journal.pone.0005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, Krystal G. 1996. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc Natl Acad Sci U S A 93:1689–1693. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maehama T, Dixon JE. 1998. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 29.Heit B, Robbins SM, Downey CM, Guan Z, Colarusso P, Miller BJ, Jirik FR, Kubes P. 2008. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat Immunol 9:743–752. doi: 10.1038/ni.1623. [DOI] [PubMed] [Google Scholar]
- 30.Hannigan M, Zhan L, Li Z, Ai Y, Wu D, Huang CK. 2002. Neutrophils lacking phosphoinositide 3-kinase gamma show loss of directionality during N-formyl-Met-Leu-Phe-induced chemotaxis. Proc Natl Acad Sci U S A 99:3603–3608. doi: 10.1073/pnas.052010699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mondal S, Subramanian KK, Sakai J, Bajrami B, Luo HR. 2012. Phosphoinositide lipid phosphatase SHIP1 and PTEN coordinate to regulate cell migration and adhesion. Mol Biol Cell 23:1219–1230. doi: 10.1091/mbc.E11-10-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Subramanian KK, Jia Y, Zhu D, Simms BT, Jo H, Hattori H, You J, Mizgerd JP, Luo HR. 2007. Tumor suppressor PTEN is a physiologic suppressor of chemoattractant-mediated neutrophil functions. Blood 109:4028–4037. doi: 10.1182/blood-2006-10-055319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gambardella L, Vermeren S. 2013. Molecular players in neutrophil chemotaxis—focus on PI3K and small GTPases. J Leukoc Biol 94:603–612. doi: 10.1189/jlb.1112564. [DOI] [PubMed] [Google Scholar]
- 34.Song MS, Salmena L, Pandolfi PP. 2012. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 35.Nishio M, Watanabe K, Sasaki J, Taya C, Takasuga S, Iizuka R, Balla T, Yamazaki M, Watanabe H, Itoh R, Kuroda S, Horie Y, Forster I, Mak TW, Yonekawa H, Penninger JM, Kanaho Y, Suzuki A, Sasaki T. 2007. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat Cell Biol 9:36–44. doi: 10.1038/ncb1515. [DOI] [PubMed] [Google Scholar]
- 36.Ellen RP, Galimanas VB. 2005. Spirochetes at the forefront of periodontal infections. Periodontol 2000 38:13–32. doi: 10.1111/j.1600-0757.2005.00108.x. [DOI] [PubMed] [Google Scholar]
- 37.Sela MN. 2001. Role of Treponema denticola in periodontal diseases. Crit Rev Oral Biol Med 12:399–413. doi: 10.1177/10454411010120050301. [DOI] [PubMed] [Google Scholar]
- 38.Visser MB, Ellen RP. 2011. New insights into the emerging role of oral spirochaetes in periodontal disease. Clin Microbiol Infect 17:502–512. doi: 10.1111/j.1469-0691.2011.03460.x. [DOI] [PubMed] [Google Scholar]
- 39.Uriarte SM, Edmisson JS, Jimenez-Flores E. 2016. Human neutrophils and oral microbiota: a constant tug-of-war between a harmonious and a discordant coexistence. Immunol Rev 273:282–298. doi: 10.1111/imr.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nuzzi PA, Lokuta MA, Huttenlocher A. 2007. Analysis of neutrophil chemotaxis, p 23–35. In Coutts AS. (ed), Adhesion protein protocols. Methods in molecular biology, vol 370 Humana Press, Totowa, NJ. doi: 10.1007/978-1-59745-353-0_3. [DOI] [PubMed] [Google Scholar]
- 41.Anand A, Luthra A, Edmond ME, Ledoyt M, Caimano MJ, Radolf JD. 2013. The major outer sheath protein (Msp) of Treponema denticola has a bipartite domain architecture and exists as periplasmic and outer membrane-spanning conformers. J Bacteriol 195:2060–2071. doi: 10.1128/JB.00078-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haapasalo M, Muller KH, Uitto VJ, Leung WK, McBride BC. 1992. Characterization, cloning, and binding properties of the major 53-kilodalton Treponema denticola surface antigen. Infect Immun 60:2058–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Egli C, Leung WK, Muller KH, Hancock RE, McBride BC. 1993. Pore-forming properties of the major 53-kilodalton surface antigen from the outer sheath of Treponema denticola. Infect Immun 61:1694–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fenno JC, Muller KH, McBride BC. 1996. Sequence analysis, expression, and binding activity of recombinant major outer sheath protein (Msp) of Treponema denticola. J Bacteriol 178:2489–2497. doi: 10.1128/jb.178.9.2489-2497.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Visser MB, Sun CX, Koh A, Ellen RP, Glogauer M. 2013. Treponema denticola major outer sheath protein impairs the cellular phosphoinositide balance that regulates neutrophil chemotaxis. PLoS One 8:e66209. doi: 10.1371/journal.pone.0066209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones MM, Vanyo ST, Visser MB. 2017. The C-terminal region of the major outer sheath protein (Msp) of Treponema denticola inhibits neutrophil chemotaxis. Mol Oral Microbiol 32:375–389. doi: 10.1111/omi.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Magalhaes MAO, Sun CX, Glogauer M, Ellen RP. 2008. The major outer sheath protein of Treponema denticola selectively inhibits Rac1 activation in murine neutrophils. Cell Microbiol 10:344–354. doi: 10.1111/j.1462-5822.2007.01045.x. [DOI] [PubMed] [Google Scholar]
- 48.Puthengady Thomas B, Sun CX, Bajenova E, Ellen RP, Glogauer M. 2006. Modulation of human neutrophil functions in vitro by Treponema denticola major outer sheath protein. Infect Immun 74:1954–1957. doi: 10.1128/IAI.74.3.1954-1957.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brissette CA, Pham TTT, Coats SR, Darveau RP, Lukehart SA. 2008. Treponema denticola does not induce production of common innate immune mediators from primary gingival epithelial cells. Oral Microbiol Immunol 23:474–481. doi: 10.1111/j.1399-302X.2008.00452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holt SC, Ebersole JL. 2005. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the ‘red complex’, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000 38:72–122. doi: 10.1111/j.1600-0757.2005.00113.x. [DOI] [PubMed] [Google Scholar]
- 51.Sochalska M, Potempa J. 2017. Manipulation of neutrophils by Porphyromonas gingivalis in the development of periodontitis. Front Cell Infect Microbiol 7:197. doi: 10.3389/fcimb.2017.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ji S, Kim Y, Min B-M, Han SH, Choi Y. 2007. Innate immune responses of gingival epithelial cells to nonperiodontopathic and periodontopathic bacteria. J Periodontal Res 42:503–510. doi: 10.1111/j.1600-0765.2007.00974.x. [DOI] [PubMed] [Google Scholar]
- 53.Shin JE, Choi Y. 2010. Treponema denticola suppresses expression of human β-defensin-2 in gingival epithelial cells through inhibition of TNFα production and TLR2 activation. Mol Cells 29:407–412. doi: 10.1007/s10059-010-0048-5. [DOI] [PubMed] [Google Scholar]
- 54.Das S, Dixon JE, Cho W. 2003. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci U S A 100:7491–7496. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaibani P, Pellegrino MT, Rossini G, Alvisi G, Miragliotta L, Prati C, Sambri V. 2010. The central region of the msp gene of Treponema denticola has sequence heterogeneity among clinical samples, obtained from patients with periodontitis. BMC Infect Dis 10:345. doi: 10.1186/1471-2334-10-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fenno JC, Wong GW, Hannam PM, Müller KH, Leung WK, McBride BC. 1997. Conservation of msp, the gene encoding the major outer membrane protein of oral Treponema spp. J Bacteriol 179:1082–1089. doi: 10.1128/jb.179.4.1082-1089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Godovikova V, Goetting-Minesky MP, Fenno JC. 2011. Composition and localization of Treponema denticola outer membrane complexes. Infect Immun 79:4868–4875. doi: 10.1128/IAI.05701-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goetting-Minesky MP, Godovikova V, Li JJ, Seshadrinathan S, Timm JC, Kamodia SS, Fenno JC. 2013. Conservation and revised annotation of the Treponema denticola prcB-prcA-prtP locus encoding the dentilisin (CTLP) protease complex. Mol Oral Microbiol 28:181–191. doi: 10.1111/omi.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kulp A, Kuehn MJ. 2010. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol 64:163–184. doi: 10.1146/annurev.micro.091208.073413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schwechheimer C, Kuehn MJ. 2015. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol 13:605–619. doi: 10.1038/nrmicro3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Veith PD, Glew MD, Gorasia DG, Chen D, O’Brien-Simpson NM, Reynolds EC. 2019. Localization of outer membrane proteins in Treponema denticola by quantitative proteome analyses of outer membrane vesicles and cellular fractions. J Proteome Res 18:1567–1581. doi: 10.1021/acs.jproteome.8b00860. [DOI] [PubMed] [Google Scholar]
- 62.Lam P, Yoo SK, Green JM, Huttenlocher A. 2012. The SH2-domain-containing inositol 5-phosphatase (SHIP) limits the motility of neutrophils and their recruitment to wounds in zebrafish. J Cell Sci 125:4973–4978. doi: 10.1242/jcs.106625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Q, Ko KS, Kapus A, McCulloch CA, Ellen RP. 2001. A spirochete surface protein uncouples store-operated calcium channels in fibroblasts: a novel cytotoxic mechanism. J Biol Chem 276:23056–23064. doi: 10.1074/jbc.M011735200. [DOI] [PubMed] [Google Scholar]
- 64.Mathers DA, Leung WK, Fenno JC, Hong Y, McBride BC. 1996. The major surface protein complex of Treponema denticola depolarizes and induces ion channels in HeLa cell membranes. Infect Immun 64:2904–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nussbaum G, Ben-Adi S, Genzler T, Sela M, Rosen G. 2009. Involvement of Toll-like receptors 2 and 4 in the innate immune response to Treponema denticola and its outer sheath components. Infect Immun 77:3939–3947. doi: 10.1128/IAI.00488-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Okahara F, Ikawa H, Kanaho Y, Maehama T. 2004. Regulation of PTEN phosphorylation and stability by a tumor suppressor candidate protein. J Biol Chem 279:45300–45303. doi: 10.1074/jbc.C400377200. [DOI] [PubMed] [Google Scholar]
- 67.Zhang XC, Piccini A, Myers MP, Van Aelst L, Tonks NK. 2012. Functional analysis of the protein phosphatase activity of PTEN. Biochem J 444:457–464. doi: 10.1042/BJ20120098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Funamoto S, Meili R, Lee S, Parry L, Firtel RA. 2002. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell 109:611–623. doi: 10.1016/s0092-8674(02)00755-9. [DOI] [PubMed] [Google Scholar]
- 69.Billadeau DD. 2008. PTEN gives neutrophils direction. Nat Immunol 9:716–718. doi: 10.1038/ni0708-716. [DOI] [PubMed] [Google Scholar]
- 70.Vazquez F, Matsuoka S, Sellers WR, Yanagida T, Ueda M, Devreotes PN. 2006. Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc Natl Acad Sci U S A 103:3633–3638. doi: 10.1073/pnas.0510570103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parent CA. 2004. Making all the right moves: chemotaxis in neutrophils and Dictyostelium. Curr Opin Cell Biol 16:4–13. doi: 10.1016/j.ceb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 72.Bakthavatchalu V, Meka A, Sathishkumar S, Lopez MC, Verma RK, Wallet SM, Bhattacharyya I, Boyce BF, Mans JJ, Lamont RJ, Baker HV, Ebersole JL, Kesavalu L. 2010. Molecular characterization of Treponema denticola infection-induced bone and soft tissue transcriptional profiles. Mol Oral Microbiol 25:260–274. doi: 10.1111/j.2041-1014.2010.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL Jr.. 1998. Microbial complexes in subgingival plaque. J Clin Periodontol 25:134–144. doi: 10.1111/j.1600-051X.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 74.Sugita N, Kimura A, Matsuki Y, Yamamoto T, Yoshie H, Hara K. 1998. Activation of transcription factors and IL-8 expression in neutrophils stimulated with lipopolysaccharide from Porphyromonas gingivalis. Inflammation 22:253–267. doi: 10.1023/A:1022344031223. [DOI] [PubMed] [Google Scholar]
- 75.Grenier D, Morin M-P, Fournier-Larente J, Chen H. 2016. Vitamin D inhibits the growth of and virulence factor gene expression by Porphyromonas gingivalis and blocks activation of the nuclear factor kappa B transcription factor in monocytes. J Periodontal Res 51:359–365. doi: 10.1111/jre.12315. [DOI] [PubMed] [Google Scholar]
- 76.Handfield M, Mans JJ, Zheng G, Lopez MC, Mao S, Progulske-Fox A, Narasimhan G, Baker HV, Lamont RJ. 2005. Distinct transcriptional profiles characterize oral epithelium-microbiota interactions. Cell Microbiol 7:811–823. doi: 10.1111/j.1462-5822.2005.00513.x. [DOI] [PubMed] [Google Scholar]
- 77.Visser MB, Koh A, Glogauer M, Ellen RP. 2011. Treponema denticola major outer sheath protein induces actin assembly at free barbed ends by a PIP2-dependent uncapping mechanism in fibroblasts. PLoS One 6:e23736. doi: 10.1371/journal.pone.0023736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Georgescu M-M. 2010. PTEN tumor suppressor network in PI3K-Akt pathway control. Genes Cancer 1:1170–1177. doi: 10.1177/1947601911407325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bononi A, Pinton P. 2015. Study of PTEN subcellular localization. Methods 77–78:92–103. doi: 10.1016/j.ymeth.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. 2000. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol 20:5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sun Z, Huang C, He J, Lamb KL, Kang X, Gu T, Shen WH, Yin Y. 2014. PTEN C-terminal deletion causes genomic instability and tumor development. Cell Rep 6:844–854. doi: 10.1016/j.celrep.2014.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gericke A, Munson M, Ross AH. 2006. Regulation of the PTEN phosphatase. Gene 374:1–9. doi: 10.1016/j.gene.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 83.Lee Y-R, Chen M, Pandolfi PP. 2018. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol 19:547–562. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- 84.Bunney TD, Katan M. 2010. Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat Rev Cancer 10:342–352. doi: 10.1038/nrc2842. [DOI] [PubMed] [Google Scholar]
- 85.Xu W, Huang Y, Yang Z, Hu Y, Shu X, Xie C, He C, Zhu Y, Lu N. 2018. Helicobacter pylori promotes gastric epithelial cell survival through the PLK1/PI3K/Akt pathway. Onco Targets Ther 11:5703–5713. doi: 10.2147/OTT.S164749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bartruff JB, Yukna RA, Layman DL. 2005. Outer membrane vesicles from Porphyromonas gingivalis affect the growth and function of cultured human gingival fibroblasts and umbilical vein endothelial cells. J Periodontol 76:972–979. doi: 10.1902/jop.2005.76.6.972. [DOI] [PubMed] [Google Scholar]
- 87.Cecil JD, O’Brien-Simpson NM, Lenzo JC, Holden JA, Singleton W, Perez-Gonzalez A, Mansell A, Reynolds EC. 2017. Outer membrane vesicles prime and activate macrophage inflammasomes and cytokine secretion in vitro and in vivo. Front Immunol 8:1017. doi: 10.3389/fimmu.2017.01017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rosen G, Naor R, Rahamim E, Yishai R, Sela MN. 1995. Proteases of Treponema denticola outer sheath and extracellular vesicles. Infect Immun 63:3973–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nguyen HN, Afkari Y, Senoo H, Sesaki H, Devreotes PN, Iijima M. 2014. Mechanism of human PTEN localization revealed by heterologous expression in Dictyostelium. Oncogene 33:5688–5696. doi: 10.1038/onc.2013.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu Y, Zhou H, Wu K, Lee S, Li R, Liu X. 2014. PTEN phosphorylation and nuclear export mediate free fatty acid-induced oxidative stress. Antioxid Redox Signal 20:1382–1395. doi: 10.1089/ars.2013.5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Magalhaes MAO, Glogauer M. 2010. Pivotal advance: phospholipids determine net membrane surface charge resulting in differential localization of active Rac1 and Rac2. J Leukoc Biol 87:545–555. doi: 10.1189/jlb.0609390. [DOI] [PubMed] [Google Scholar]
- 92.Yasui M, Matsuoka S, Ueda M. 2014. PTEN hopping on the cell membrane is regulated via a positively-charged C2 domain. PLoS Comput Biol 10:e1003817. doi: 10.1371/journal.pcbi.1003817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Futosi K, Fodor S, Mocsai A. 2013. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol 17:638–650. doi: 10.1016/j.intimp.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rauh MJ, Sly LM, Kalesnikoff J, Hughes MR, Cao L-P, Lam V, Krystal G. 2004. The role of SHIP1 in macrophage programming and activation. Biochem Soc Trans 32:785–788. doi: 10.1042/BST0320785. [DOI] [PubMed] [Google Scholar]
- 95.Pauls SD, Marshall AJ. 2017. Regulation of immune cell signaling by SHIP1: a phosphatase, scaffold protein, and potential therapeutic target. Eur J Immunol 47:932–945. doi: 10.1002/eji.201646795. [DOI] [PubMed] [Google Scholar]
- 96.Rosales C, Uribe‐Querol E. 2017. Chapter 4: Neutrophil role in periodontal disease In Khajah MA. (ed), Role of neutrophils in disease pathogenesis. IntechOpen, Rijeka, Croatia. doi: 10.5772/67789. [DOI] [Google Scholar]
- 97.Cortes-Vieyra R, Rosales C, Uribe-Querol E. 2016. Neutrophil functions in periodontal homeostasis. J Immunol Res 2016:1396106. doi: 10.1155/2016/1396106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dawson JR, Ellen RP. 1990. Tip-oriented adherence of Treponema denticola to fibronectin. Infect Immun 58:3924–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Horowitz NH, Fling M. 1962. A method for concentrating dilute protein solutions. Fungal Genet Rep 2:18. doi: 10.4148/1941-4765.1057. [DOI] [Google Scholar]
- 100.Kurniyati K, Kelly JF, Vinogradov E, Robotham A, Tu Y, Wang J, Liu J, Logan SM, Li C. 2017. A novel glycan modifies the flagellar filament proteins of the oral bacterium Treponema denticola. Mol Microbiol 103:67–85. doi: 10.1111/mmi.13544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Abiko Y, Nagano K, Yoshida Y, Yoshimura F. 2014. Characterization of Treponema denticola mutants defective in the major antigenic proteins, Msp and TmpC. PLoS One 9:e113565. doi: 10.1371/journal.pone.0113565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fenno JC, Wong GW, Hannam PM, McBride BC. 1998. Mutagenesis of outer membrane virulence determinants of the oral spirochete Treponema denticola. FEMS Microbiol Lett 163:209–215. doi: 10.1111/j.1574-6968.1998.tb13047.x. [DOI] [PubMed] [Google Scholar]
- 103.Veith PD, Dashper SG, O’Brien-Simpson NM, Paolini RA, Orth R, Walsh KA, Reynolds EC. 2009. Major proteins and antigens of Treponema denticola. Biochim Biophys Acta 1794:1421–1432. doi: 10.1016/j.bbapap.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 104.Deng Z-L, Sztajer H, Jarek M, Bhuju S, Wagner-Döbler I. 2018. Worlds apart—transcriptome profiles of key oral microbes in the periodontal pocket compared to single laboratory culture reflect synergistic interactions. Front Microbiol 9:124. doi: 10.3389/fmicb.2018.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.