

Fibrosis is a significant complication of intestinal disorders associated with microbial dysbiosis and pathobiont expansion, notably Crohn’s disease (CD). Mechanisms that favor fibrosis are not well understood, and therapeutic strategies are limited. Here we demonstrate that colitis-susceptible Il10-deficient mice develop inflammation-associated fibrosis when monoassociated with adherent/invasive Escherichia coli (AIEC) that harbors the yersiniabactin (Ybt) pathogenicity island.
Keywords: fibrosis, AIEC, Crohn’s disease, colitis, microbiome, yersiniabactin, intestinal inflammation, microbiota
ABSTRACT
Fibrosis is a significant complication of intestinal disorders associated with microbial dysbiosis and pathobiont expansion, notably Crohn’s disease (CD). Mechanisms that favor fibrosis are not well understood, and therapeutic strategies are limited. Here we demonstrate that colitis-susceptible Il10-deficient mice develop inflammation-associated fibrosis when monoassociated with adherent/invasive Escherichia coli (AIEC) that harbors the yersiniabactin (Ybt) pathogenicity island. Inactivation of Ybt siderophore production in AIEC nearly abrogated fibrosis development in inflamed mice. In contrast, inactivation of Ybt import through its cognate receptor FyuA enhanced fibrosis severity. This corresponded with increased colonic expression of profibrogenic genes prior to the development of histological disease, therefore suggesting causality. fyuA-deficient AIEC also exhibited greater localization within subepithelial tissues and fibrotic lesions that was dependent on Ybt biosynthesis and corresponded with increased fibroblast activation in vitro. Together, these findings suggest that Ybt establishes a profibrotic environment in the host in the absence of binding to its cognate receptor and indicate a direct link between intestinal AIEC and the induction of inflammation-associated fibrosis.
INTRODUCTION
Inflammatory bowel diseases (IBD), including Crohn’s disease (CD), are characterized by chronic intestinal inflammation that develops as a result of prolonged and inappropriate mucosal immune responses to luminal antigens in genetically susceptible individuals (1). The chronic and relapsing nature of IBD, in conjunction with the lack of curative therapies for many patients, enhances risk for inflammation-associated comorbidities, including intestinal fibrosis (2). Approximately 30% of CD patients develop fibrotic disease that can result in intestinal strictures and bowel obstructions (2–4). Current treatments for intestinal fibrosis are inadequate and rely on anti-inflammatory therapies (which are often ineffective) and surgical interventions (3). Fibrosis is recurrent in large proportions of the population with CD (4), thus necessitating the development of specific antifibrotic therapeutics.
Fibrosis is characterized by excess accumulation of extracellular matrix (ECM) components that results in the pathological remodeling of tissues and consequent organ dysfunction. Mesenchymal cells such as fibroblasts, myofibroblasts, and smooth muscle cells become highly activated in response to transmural injury or inflammation and hypersecrete ECM components and profibrogenic factors that further propagate fibrotic processes. The tissue microenvironment also plays an important role in modulating the activity of mesenchymal cells, where host-derived signals such as cytokines and growth factors serve as additional fibrogenic or antifibrotic mediators (3, 4). Activation of mesenchymal cells is also subject to regulation by microbial factors (5, 6). Fibrosis can occur in bacterium-induced models of acute colitis, including mice chronically colonized with the enteric pathogen Salmonella enterica or with a CD-associated Escherichia coli pathobiont (7, 8). Importantly, progression from intestinal inflammation to inflammation-associated fibrosis is incompletely penetrant in bacterium-induced colitis models and in clinical populations with microbe-driven diseases like IBD. It remains unclear which microbiota-derived signals favor the establishment of a profibrogenic microenvironment.
Members of the intestinal microbiota are key modulators of mucosal immunity under homeostatic conditions and in numerous inflammatory pathologies, including IBD (1). A subset of resident intestinal E. coli organisms known as adherent/invasive E. coli (AIEC) is enriched in CD patients (9–11). AIEC breaches the intestinal epithelium and induces inflammation in various rodent models of experimental colitis (12–15). Colonization of germfree, inflammation-prone Il10−/− mice with AIEC induces aggressive, transmural intestinal inflammation driven by bacterial antigen-specific T-helper 1 (Th1) and Th17 immune responses (13, 16). Studies with germfree Il10−/− mice individually colonized with AIEC have led to the identification of several bacterial factors that augment or diminish the colitis-inducing and procarcinogenic capabilities of AIEC (17–20).
Comparative phylogenetic studies have demonstrated that the yersiniabactin (Ybt) high-pathogenicity island (HPI) is overrepresented in human, canine, and murine AIEC strains (21). The Ybt HPI encodes enzymatic machinery required for the biosynthesis of the siderophore Ybt (22). Once Ybt is released from bacterial cells, it sequesters extracellular metals, including iron, zinc, and copper. The Ybt-metal chelate is subsequently imported through its cognate outer membrane receptor FyuA for bacterial use (22–24). The Ybt HPI is harbored by numerous Enterobacteriaceae pathogens and contributes to in vivo fitness, niche formation, and virulence (25–27). However, the contribution of the Ybt HPI to the proinflammatory potential of resident intestinal E. coli such as AIEC has not been explored, despite its prevalence in this population. We therefore utilized our gnotobiotic Il10−/− mouse model to investigate whether inactivation of the Ybt system in AIEC modulates immune-mediated colitis. While abrogation of Ybt biosynthesis in AIEC delayed colitis onset, colonization of mice with Ybt-positive AIEC was associated with the development of inflammation-associated fibrosis. Severity of fibrosis was enhanced in mice colonized with the Ybt-positive transport mutant (ΔfyuA), which corresponded with increased profibrogenic gene signatures in the colon and in cultured fibroblasts and enhanced AIEC subepithelial localization within fibrotic lesions. Abrogation of Ybt biosynthesis in the ΔfyuA mutant attenuated fibrosis in inflamed mice, restored AIEC localization to the epithelium, and reduced fibroblast activation. Collectively, our findings introduce a noncanonical role for Ybt in mediating fibrosis development independent of its established function in delivering iron to bacteria through FyuA. More broadly, we introduced a novel microbe-driven, immune-mediated model of inflammation-associated fibrosis that recapitulates key histopathological features of fibrotic disease in human CD.
RESULTS
Inactivation of Ybt biosynthesis, but not Ybt transport, in AIEC delays progression of colitis.
The siderophore Ybt and its cognate receptor FyuA mediate bacterial metal acquisition in pathogenic Enterobacteriaceae. Because the Ybt HPI is also harbored by many IBD-associated AIEC strains, we hypothesized that like its pathogenic counterparts, the Ybt HPI enhances the proinflammatory potential of AIEC. To determine whether an intact Ybt siderophore system in AIEC contributes to colitis development, we inactivated Ybt biosynthesis or import by creating isogenic mutants unable to import Ybt-metal chelates (ΔfyuA) or unable to synthesize Ybt (Δirp1) in the AIEC strain NC101 (which also harbors the enterobactin and salmochelin siderophore systems). We colonized germfree, inflammation-susceptible Il10−/− mice with NC101 or the ΔfyuA or Δirp1 mutant and compared the severities of colitis induction. At 5 weeks, colitis histopathology was significantly attenuated in mice colonized with the Δirp1 mutant compared with Ybt+ NC101 and the ΔfyuA mutant (Fig. 1A to E), an attenuation that was no longer apparent by 10 weeks (see Fig. S1 in the supplemental material). In contrast, colitis development did not differ in mice colonized with NC101 versus the ΔfyuA mutant. Colitis scores differences did not correlate with altered expression of proinflammatory cytokines known to correlate with disease in this model (Fig. S1) (13, 16). The reduced colitis potential of the Δirp1 mutant did not correspond with diminished luminal growth in the gut (Fig. 1G to I) or in vitro growth defects under iron-replete or -limiting conditions (Fig. S2). While the ΔfyuA mutant exhibited a growth defect at 5 weeks, its attenuated growth was not sustained throughout colitis development and did not correlate with colitis severity (Fig. 1G to I). Together, these findings demonstrate that Ybt enhances the proinflammatory potential of AIEC in gnotobiotc, inflammation-susceptible hosts.
FIG 1.

Yersiniabactin enhances the proinflammatory potential of AIEC in gnotobiotic Il10−/− mice. Germfree Il10−/− mice were monoassociated with the AIEC strain E. coli NC101 (NC) or the ΔfyuA or Δirp1 mutant for 5 weeks. (A to D) Composite (A) and regional (B to D) histopathology colitis scores. (E) Representative H&E histology of the colon. Scale bar, 50 μm. (F) Composite histopathology colitis scores of Il10−/− mice colonized with yersiniabactin (Ybt)-positive or Ybt-deficient NC101. Lines are at the medians. P values were determined by Kruskal-Wallis or Mann-Whitney test. (G to I) Quantitative bacterial culture from feces at 1 week (G), 5 weeks (H), or 10 weeks (I) postcolonization. Lines are at the means. P values were determined by one-way analysis of variance (ANOVA). Each symbol represents an individual mouse (n = 8 to 14). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Ybt-positive AIEC promotes fibrosis development in inflamed Il10−/− mice.
In a subset of NC101- and ΔfyuA mutant-colonized inflamed Il10−/− mice, but rarely in Δirp1 mutant-colonized Il10−/− mice, pathological remodeling of the colonic submucosa was observed in hematoxylin and eosin (H&E)-stained colon sections (Fig. 2 and Fig. S3). Histological features consistent with fibrosis, including marked expansion of the submucosa with excessive deposition of lightly eosinophilic, fibrillar substances, characterized the pathology. Positive staining with Masson’s trichrome and Sirius red confirmed the presence of collagen fibers as part of the expanded ECM in fibrotic mice (Fig. 2B). Lamina propria collagen localization was also altered in fibrotic mice, exhibiting a basal predilection. In contrast, in nonfibrotic AIEC-colonized Il10−/− mice, the submucosal ECM was structured and organized and stained collagen fibrils in the lamina propria exhibited an apical propensity (Fig. 2A). Taken together, our results show that a subset of AIEC-colonized Il10−/− mice developed histopathological lesions that are consistent with fibrosis.
FIG 2.

Ybt+ AIEC promotes fibrosis development in colitic Il10−/− mice. Germfree Il10−/− mice were monoassociated with the Ybt+ AIEC strain NC and the ΔfyuA mutant or the Ybt− Δirp1 strain for 10 weeks. (A and B) Representative colonic histology of gnotobiotic Il10−/− mice colonized with NC (A) or the ΔfyuA mutant (B). Colon sections were stained with H&E, Sirius red/fast green, or Masson’s trichrome. Regions of Sirius red binding are indicated by white arrowheads in the submucosa and red arrowheads in the lamina propria. (C to F) Composite (C) and regional (D to F) fibrosis histology scores. Each symbol represents an individual mouse (n = 11 to 29). Lines are at the medians. P values were determined by Kruskal-Wallis test. *, P < 0.05. (G) Representative colonic histology from gnotobiotic Il10−/− mice colonized with the ΔfyuA mutant for 5 weeks. Colonic sections were stained with antibodies against the established macrophage cell surface markers CD206, CD11b, and F4/80 and were counterstained with the DNA stain 4′,6-diamidino-2-phenylindole (DAPI). Scale bars, 200 μm. SM, submucosa; LP, lamina propria.
Because fibrosis incidences seemed to differ between NC101-, ΔfyuA mutant-, and Δirp1 mutant-colonized Il10−/− mice, we next utilized a fibrosis pathology scoring system to determine whether the Ybt system in AIEC impacts inflammation-associated fibrosis (28, 29) (see Materials and Methods). The most severe fibrosis pathology in all regions of the colon were observed in ΔfyuA mutant-colonized Il10−/− mice, which corresponded with higher incidence of severe disease. In contrast, moderate to severe fibrosis in NC101-colonized mice was mostly restricted to the proximal colon (Fig. 2C to F). These differences in fibrosis severity and incidence were associated with altered cellular populations infiltrating the submucosa, with immunologically defined macrophages (CD206+, CD11b+, and/or F4/80+ cells) observed in ΔfyuA mutant-colonized fibrotic mice (Fig. 2G) versus the inflammatory lymphocytes consistently observed in NC101-colonized, nonfibrotic mice (20). Inflamed Il10−/− mice colonized with the Ybt-deficient Δirp1 mutant did not develop moderate to severe fibrotic lesions and rarely exhibited mild disease (Fig. 2A to D), suggesting a role for Ybt in inducing and exacerbating this pathology. To validate that the histopathology in our mouse model is consistent with inflammation-associated fibrosis in human CD, we evaluated H&E and Sirius red staining of full-thickness colon resection tissues from fibrotic CD, ulcerative colitis, diverticulitis, and healthy margins of colorectal cancer resections (Fig. 3 and Fig. S5). Fibrotic CD tissues exhibited remarkable similarity to our mouse model, with transmural inflammation, expansion of the submucosa, thick collagen fibrils, and disruption of the muscularis by collagen and infiltrating cells (Fig. 3). Fibrosis was not evident by H&E, Sirius red, or Masson’s trichrome or at 5 weeks in Il10−/− mice (data not shown). Collectively, these observations demonstrate that Ybt+ AIEC promotes the development of fibrotic disease in an experimental model of pathobiont-induced colitis.
FIG 3.
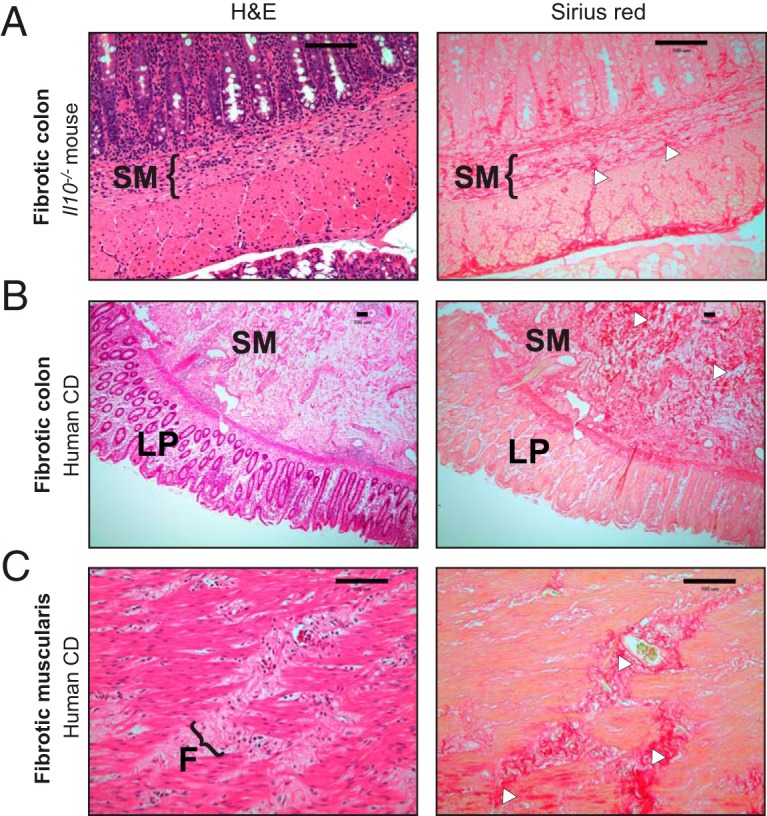
Fibrosis development in AIEC-colonized Il10−/− mice recapitulates histopathological features of fibrosis in human Crohn’s disease. (A) Representative colonic histology of ΔfyuA mutant-colonized fibrotic Il10−/− mice. (B and C) Representative histology of full-thickness colon cross sections from fibrotic Crohn’s disease patients, representative of 3 per group. (C) Magnification of the muscularis serosa. Colon sections were stained with H&E or Sirius red. Regions of Sirius red binding are indicated with white arrowheads. F, fibrotic lesion. Scale bars, 100 μm.
Because fibrosis occurs in response to tissue injury instigated by inflammation, we next determined whether fibrosis severity positively correlates with inflammation. Linear regression analysis revealed a significant negative correlation between fibrosis and colitis histopathology in the middle colon and no significant correlations in the proximal and distal colon (Fig. S4). Moreover, NC101- and ΔfyuA mutant-colonized mice exhibited similar levels of colitis histopathology despite the exacerbated fibrosis observed in ΔfyuA mutant-colonized mice (Fig. 1 and 2). Nonetheless, as previously reported (4), inflammation is required for the profibrotic activities of NC101 and ΔfyuA given that fibrosis was not observed in uninflamed wild-type (WT) mice colonized with either strain (Fig. S3). These results demonstrate that while inflammation is required for fibrosis development, Ybt+ AIEC exacerbates inflammation-associated fibrosis independent of effects on the proinflammatory potential of AIEC.
Fibrosis development corresponds with enhanced subepithelial invasion of fyuA-deficient AIEC.
We next determined whether the profibrogenic potential of Ybt+ AIEC corresponds with altered bacterial localization within the intestines. While colonic mucus colonization did not differ between the strains, colonic tissue loads of AIEC were significantly increased in ΔfyuA mutant-colonized mice at 10 weeks (Fig. 4A and B). In contrast, colonic tissue colonization did not differ between NC101 and the Δirp1 mutant. Colonic mucus and tissue loads were also comparable at 5 weeks (Fig. S6A and B). Because AIEC is functionally characterized by epithelial invasiveness, intramacrophagic survival, and robust biofilm formation, we performed standard in vitro assays commonly utilized to distinguish AIEC strains (9). While iron availability altered AIEC epithelial invasion, no differences in epithelial adherence or invasion were observed between NC101 and the Δirp1 and ΔfyuA mutants under iron-replete or -limiting conditions (Fig. S6C and D). Similarly, genetic ablation of Ybt transport did not alter macrophage phagocytosis or intracellular survival of AIEC (Fig. S6E to G) and had no effect on AIEC biofilm formation (Fig. S6H). Thus, while Ybt transport and biosynthesis did not impact defining in vitro characteristics of AIEC, deletion of fyuA enhanced AIEC colonic tissue colonization, suggesting that FyuA may be important in modulating bacterial localization within the intestines.
FIG 4.

Inactivation of yersiniabactin transport enhances AIEC mucosal invasion. Germfree Il10−/− mice were monoassociated with NC or the ΔfyuA or Δirp1 mutant for 10 weeks. (A and B) Quantitative bacterial cultures of colonic mucus (A) and colonic (B) tissues. Each symbol represents an individual mouse (n = 11 to 15). Lines are at the medians. P values were determined by Kruskal-Wallis test. (C) FISH analysis of proximal colons (n = 4 to 8). P values were determined by Kruskal-Wallis test. (D to F) Representative FISH images of the proximal colon. Red, E. coli; blue, DAPI. Arrowheads in panel F indicate E. coli localized within the lamina propria (white) and submucosa (yellow). E, epithelium. Scale bars, 200 μm. (G) Subepithelial AIEC invasion scores as assessed by FISH in panel C. Red squares represent fibrotic mice as assessed by histopathology. Lines are at the medians. P values were determined by Kruskal-Wallis test. (H) Linear regression analysis of quantitative bacterial culture versus FISH score from colonic tissues. Red squares represent fibrotic mice as assessed by histopathology. *, P < 0.05; **, P < 0.01.
To further assess how FyuA impacts AIEC localization in the gut, we employed a more sensitive approach—E. coli 16S fluorescence in situ hybridization (FISH)—to visualize tissue-associated AIEC. FISH analysis revealed an overall increase in the tissue-associated ΔfyuA mutant relative to NC101 and the Δirp1 mutant (Fig. 4C). This difference was primarly driven by enhanced subepithelial (lamina propria and submucosa) localization of ΔfyuA (Fig. 4D to G). Moreover, ΔfyuA was observed within submucosal fibrotic lesions, demonstrating its colocalization with diseased tissue (Fig. 4F, arrowheads). Importantly, tissue bacteria loads assessed by quantitative bacterial culture and FISH analysis were positively correlated (Fig. 4H). Together, these results suggest that inactivation of fyuA enhances the subepithelial localization of AIEC, which may contribute to its profibrogenic potential.
Inactivation of Ybt-mediated metal acquisition does not alter AIEC iron sensing.
The canonical function of Ybt is to scavenge extracellular metals for bacterial use (22, 30). Because the most severe fibrosis occurred in mice colonized with the ΔfyuA mutant, we first assessed whether Ybt functionality was altered in this mutant. The extents of Ybt secretion were comparable between NC101 and the ΔfyuA mutant, and as expected, Ybt secretion was not detected in the Δirp1 Ybt biosynthesis mutant (Fig. S7a). We next confirmed the functionality of Ybt produced by NC101 and the ΔfyuA mutant. To accomplish this, we assessed whether Ybt produced by these strains can restore the growth of siderophore-deficient Klebsiella pneumoniae (ΔentB irp1) cultivated under iron-limiting conditions. In contrast to the Δirp1 mutant, both Ybt+ NC101 and the ΔfyuA mutant rescued K. pneumoniae ΔentB irp1 mutant growth (Fig. S7b). Taken together, these data suggest that altered Ybt functionality does not correspond with the increased profibrogenic potential of the ΔfyuA mutant.
Mutants lacking FyuA are unable to import Ybt-iron chelates and may therefore be unable to satisfy their iron requirements. Thus, the enhanced profibrogenic potential of the ΔfyuA mutant may be the result of altered bacterial function mediated through disrupted bacterial iron homeostasis. To test this idea, we first compared in vivo expressions of iron-responsive genes in NC101 and the ΔfyuA and Δirp1 mutants. Transcript levels of several iron-responsive genes did not differ between strains (Fig. S8A and B), suggesting that NC101 iron homeostasis is not perturbed upon inactivation of Ybt transport or biosynthesis in the intestines. Similarly, in vitro iron depletion with the iron chelator 2,2′-bipyridyl (BPD) did not alter transcription of iron-responsive genes (Fig. S8C).
To corroborate these results, we performed transcriptional reporter assays utilizing vectors harboring gfp fused to the iron-responsive promoter PtonB. To first validate this approach, the NC101 reporter strain was cultivated under iron-replete and -limiting conditions, and as expected, iron depletion enhanced gfp activity driven by the tonB promoter (Fig. S9a). We next assessed whether NC101, ΔfyuA, and Δirp1 iron-sensing reporters respond differently to iron depletion. In agreement with our transcriptional results, levels of gfp expression were comparable between NC101 and the ΔfyuA mutant (Fig. S9a), suggesting that inactivation of FyuA does not impact AIEC iron sensing. Because Ybt can also bind other metals, including zinc (22, 31; our own observations) and copper (our own observations) (23, 24; our own observations), we performed similar assays with the zinc-responsive promoter PznuA and the copper-responsive promoter PcusC. As with the iron-sensing reporters, altering zinc and copper availability did not alter sensing of the respective metals in the ΔfyuA mutant relative to that in NC101 (Fig. S9b and c). In contrast, the activities of iron- and zinc-responsive promoters were significantly increased in the Δirp1 mutant (Fig. S9a and b), suggesting that metal starvation is enhanced in this mutant under iron- and zinc-limiting conditions. Taken together, these data suggest that while metal sensing in AIEC is not altered with disruption of Ybt transport, metal homeostasis appears to be disrupted in the Ybt-negative Δirp1 mutant.
Deletion of fyuA in AIEC promotes the establishment of a profibrotic colonic environment that precedes fibrosis development.
The increased incidence of fibrosis in ΔfyuA mutant-colonized Il10−/− mice may in part be driven by differential host responses to fyuA-expressing versus fyuA-deficient AIEC. To test this idea, we utilized high-throughput RNA sequencing (RNA-seq) to determine whether global differences are apparent in the colonic transcriptomes of inflamed Il10−/− mice and noninflamed WT mice colonized with NC101 or ΔfyuA. Principal-coordinate analysis (PCoA) revealed significant differences in the colonic transcriptomes of NC101-colonized nonfibrotic versus ΔfyuA mutant-colonized fibrotic Il10−/− mice at 10 weeks, when fibrosis is apparent (Fig. 5A). This corresponded with 2,692 genes and 71 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that were differentially expressed between ΔfyuA mutant- versus NC101-colonized Il10−/− mice (Tables S3 and S4). In contrast, the transcriptomes of NC101- versus ΔfyuA mutant-colonized WT mice clustered together (Fig. 5A), suggesting that differences in the host transcriptional responses to either strain predominantly occur in Il10−/− mice. To determine whether the differing host responses precede histological evidence of fibrosis, we also compared the colonic transcriptomes of NC101-colonized versus ΔfyuA mutant-colonized Il10−/− mice at 5 weeks. RNA-seq analysis revealed that 169 genes and 116 KEGG pathways were differentially expressed in NC101-colonized versus ΔfyuA mutant-colonized Il10−/− mice (Tables S1 and S2), many of which were differentially regulated at both 5 and 10 weeks. However, testing overall community composition did not show statistical significance after false-discovery rate (FDR) correction (P < 0.084) (Fig. 5B). Thus, the presence of fyuA in AIEC significantly altered host transcriptional responses in the inflamed colon prior to and throughout the development of fibrosis.
FIG 5.

Deletion of fyuA in AIEC promotes transcriptome-wide changes in the colons of Il10−/− mice. Shown are the results of principal-component analysis of transcriptome-wide changes in the colons of NC- versus ΔfyuA mutant-colonized WT or Il10−/− mice after 10 weeks (A), NC- versus ΔfyuA mutant-colonized Il10−/− mice after 5 weeks (B), and NC- versus ΔfyuA mutant-colonized Il10−/− mice after 5 and 10 weeks (C).
Transcriptomic analysis of a prospectively followed inception cohort of pediatric CD patients revealed high expression of profibrogentic genes and pathways prior to the development of stricturing fibrotic disease (32). This included ECM structural constituents and collagen binding pathways (32). In agreement with these results, the ECM-receptor interaction KEGG pathway is significantly upregulated in ΔfyuA mutant-colonized mice during (10 weeks) and prior to (5 weeks) histological evidence of fibrosis (Tables S2 and S4). We generated a heat map to visualize expression of individual genes in this KEGG pathway between individual NC101- versus ΔfyuA mutant-colonized Il10−/− mice (Fig. 6A). Phylogenetic clustering of the 5-week samples demonstrated that three of the ΔfyuA mutant-colonized mice clustered together and exhibited increased expression of numerous ECM genes, including type I, IV, and VI collagens and fibronectin (arrowheads, Fig. 6A). Careful histological observation by a pathologist blinded to the treatment groups revealed early evidence of fibrosis in these three ΔfyuA mutant-colonized mice but not in the remaining ΔfyuA mutant-colonized mice that clustered with the NC101-colonized mice and exhibited lower expression of ECM genes. These unbiased molecular findings are consistent with our observation that a subset, and not 100%, of ΔfyuA mutant-colonized mice develop fibrosis. These findings were confirmed by targeted quantitative PCR analysis, in which transcript levels of col1a2 (type 1 collagen) and fn1 (fibronectin) were significantly increased in fyuA mutant- versus NC101-colonized Il10−/− mice (Fig. 6B and C). This corresponded with increased positivity of α-SMA (smooth muscle actin), a common feature of fibrosis, in ΔfyuA mutant-colonized Il10−/− mice (Fig. 6E and F). Taken together, these findings demonstrate that ECM components are upregulated in prefibrotic Il10−/− mice colonized with the ΔfyuA mutant prior to the development of fibrotic disease.
FIG 6.

Deletion of fyuA in AIEC promotes profibrotic host responses preceding fibrosis development. (A) Heat map of log2 normalized counts of genes in the ECM-receptor interaction KEGG pathway in NC- and ΔfyuA mutant-colonized Il10−/− mice at 5 weeks. (B to D) Relative colonic transcript levels of Col1a1 (B), Fn1 (C), and Tgfbr2 (D) in Il10−/− mice monoassociated with NC or the ΔfyuA mutant for 5 weeks. Each symbol represents an individual mouse (n = 10 to 13). Lines are at the medians. P values were determined by Mann-Whitney test. *, P < 0.05; **, P < 0.01. (E and F) Proximal colons from Il10−/− mice colonized with NC (E) or the ΔfyuA mutant (F) for 10 weeks were stained with α-SMA (red), CD31 (green), or DAPI. MS, muscularis serosa. Scale bars, 50 μm.
Transforming growth factor β (TGF-β) signaling represents the canonical profibrotic activation pathway. Therefore, to further confirm the presence of a profibrotic gene signature in fibrotic mice, we evaluated the expression of genes within the TGF-β pathway. RNA-seq analysis detected colonic expression of the three TGF-β and TGF-β receptor isoforms. Prefibrotic (5 weeks) ΔfyuA mutant- versus NC101-colonized Il10−/− mice trended toward elevated expression of TGF-β1 and TGF-β3 and TGF-β receptor isoforms 1 and 2 (Fig. S10A). Increased expression of the TGF-β2 receptor was confirmed by quantitative PCR (Fig. 6D). Similarly, a significant increase in TGF-β1 to -β3 and TGF-β receptor isoform 2 and 3 expression was observed at 10 weeks in fibrotic ΔfyuA mutant-colonized Il10−/− mice (Fig. S10b). Together, these data further support our hypothesis that deletion of fyuA in AIEC promotes a profibrogenic environment in inflammation-susceptible hosts, which occurs at an early phase of the inflammatory response prior to onset of fibrosis.
Ybt-dependent fibrosis is not associated with altered host systemic iron homeostasis.
Membrane-permeative siderophores like Ybt disrupt host iron homeostasis and modulate iron-sensitive host responses, which includes the induction of Ndrg1 (33, 34). Because deletion of fyuA does not alter Ybt secretion, colonization with the ΔfyuA mutant may instead increase Ybt internalization by host cells in the absence of bacterial import and alter host iron homeostasis to promote fibrosis. To address this possibility, we determined whether colonization with the ΔfyuA mutant versus NC101 or the Δirp1 mutant alters systemic iron homeostasis in Il10−/− mice. At 2 weeks (prior to histological inflammation or fibrosis) and at 10 weeks (when colitis and fibrosis are evident in affected animals), plasma hemoglobin levels did not differ (Fig. S11A and B). Similarly, Prussian blue staining did not reveal differences in splenic iron stores at 10 weeks (Fig. S11C). To determine whether local iron homeostasis was altered in the colon, we utilized our RNA-seq data to assess whether established host iron-responsive genes were differentially expressed in mice colonized with NC101 or the ΔfyuA mutant (Table S5) (35–38). Of the 15 canonical iron-responsive genes investigated, three were differentially regulated in Il10−/− mice, including Ndrg1 and Tfrc (transferrin receptor), and two were differentially regulated in WT mice, including Tfrc, at 10 weeks. At 5 weeks, Epas1 was the only iron-responsive gene that was altered between NC101- versus ΔfyuA mutant-colonized Il10−/− mice, a change not observed at 10 weeks. Together, these findings suggest that the ΔfyuA mutant does not profoundly alter systemic or colonic iron homeostasis in the host and may not be a driving factor for fibrosis induction.
Yersiniabactin biosynthesis is required for AIEC-mediated fibrosis induction.
Abrogation of Ybt transport in AIEC had opposing effects on fibrosis induction in Il10−/− mice compared to the inactivation of Ybt biosynthesis (Fig. 2). Because fibrosis development was minimal in Il10−/− mice colonized with the Δirp1 mutant, we next determined whether Ybt biosynthesis is required for the fibrosis-inducing potential of the ΔfyuA mutant. Genetic inactivation of Ybt biosynthesis in the ΔfyuA mutant (ΔfyuA irp1) significantly reduced fibrosis incidence in Il10−/− mice (Fig. 7A to C). Moreover, when comparing fibrosis incidence in mice colonized with Ybt-positive versus Ybt-negative AIEC, we noted that 22 out of 51 mice colonized with Ybt-positive AIEC developed fibrotic disease, whereas 3 out of 26 mice colonized with Ybt-deficient AIEC exhibited histological evidence of fibrosis (Fig. 7B). Inactivation of Ybt production in the ΔfyuA mutant also reduced its subepithelial invasiveness, resulting in a pattern of tissue localization similar to that of NC101 (Fig. 7D). This further reinforces the link between increased mucosal invasiveness and the enhanced profibrogenic potential of the ΔfyuA mutant. Importantly, colitis severities at 10 weeks were comparable between the ΔfyuA and ΔfyuA irp1 mutants (Fig. S12), suggesting that differences in inflammation were not driving fibrosis severity.
FIG 7.

Yersiniabactin biosynthesis promotes fibrosis in AIEC-driven colitis. Germfree Il10−/− mice were monoassociated with the AIEC strain NC or the ΔfyuA, Δirp1, or ΔfyuA irp1 mutant for 10 weeks. (A) Composite fibrosis histology scores. Each symbol represents an individual mouse (n = 10 to 29). Lines are at the medians. P values were determined by Kruskal-Wallis test. (B) Composite histopathology colitis scores of Il10−/− mice colonized with Ybt-positive or Ybt-deficient AIEC. Lines are at the medians. P values were determined by Mann-Whitney test. (C) Fibrosis incidence rates of Il10−/− mice colonized with Ybt-positive or Ybt-deficient AIEC as assessed by H&E histology. P values were determined by Fisher’s exact test. (D) FISH analysis of proximal colons (n = 4 to 8). P values were determined by one-way ANOVA. (E) Swiss 3T3 fibroblasts were cocultured with NC or the Δirp1, ΔfyuA, ΔfyuA chromosomally complemented with fyuA under its native promoter, or ΔfyuA irp1 mutant. Fibroblasts stimulated with TGF-β served as a positive control (Pos). Data are represented as the means ± SEMs. P values were determined by Kruskal-Wallis test. (F) Working model. Data for NC-, ΔfyuA mutant-, and Δirp1 mutant-colonized mice are also presented in Fig. 2 and 4. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further demonstrate the profibrogenic potential of the ΔfyuA mutant, we next determined whether inactivation of Ybt transport in AIEC enhances the activation of cultured fibroblasts in vitro. Fibroblasts that were cultured with the ΔfyuA mutant expressed significantly higher levels of the fibroblast activation marker Fn1 than fibroblasts cultured with the parental and Ybt-deficient strains (Fig. 7E). This corresponded with our in vivo observation that Fn1 transcripts were elevated in ΔfyuA mutant-colonized Il10−/− mice (Fig. 6C). Together, these results demonstrate that inactivation of the Ybt siderophore system in AIEC in two distinct manners (i.e., Ybt transport versus Ybt biosynthesis) does not have similar effects on colitis induction and fibrosis development in genetically susceptible hosts. More broadly, in addition to its role in bacterial iron acquisition, our findings collectively introduce a novel, noncanonical role of Ybt in establishing a profibrogenic microenvironment in inflammation-susceptible hosts.
DISCUSSION
Siderophore biosynthetic gene clusters are abundant in the gut microbiota, with 232 putative clusters identified from metagenomes in the Human Microbiome Project study (39). Given that IBD-associated AIEC strains also harbor many of these siderophore systems (21), it is conceivable their siderophores may contribute to AIEC-associated intestinal disease. Indeed, here we introduce the siderophore Ybt as a novel bacterial factor that promotes profibrogenic host responses in the inflamed intestinal environment. Our findings demonstrate that AIEC is profibrogenic and that inactivation of Ybt transport in a colitogenic AIEC strain enhances fibrosis development in inflammation-susceptible mice. Inactivation of Ybt biosynthesis in both the Ybt transport mutant and the parental strain abrogates their fibrosis-inducing potential, suggesting that Ybt promotes fibrosis development even in the absence of uptake through its canonical receptor. Profibrogenic transcriptional signatures are evident in the colon prior to histological presentation of disease, suggesting a causative role for Ybt-mediated induction of fibrosis. Together, our findings introduce a specific microbiota-derived factor that promotes the development of inflammation-associated fibrosis.
The canonical function of the Ybt siderophore system is to import extracellular iron sequestered by Ybt through FyuA for bacterial use. Thus, inactivation of FyuA may enhance the profibrogenic potential of AIEC by perturbing bacterial iron homeostasis and subsequently modulating bacterial function. However, levels of luminal expression of highly sensitive, iron-responsive genes (30, 40) were comparable between the NC101 parental strain, the ΔfyuA transport mutant, and the Δirp1 Ybt-deficient mutant. This indicates a lack of strain-specific differences in iron sensing. Similarly, while the functional outcome of iron starvation in bacteria is a fitness disadvantage (30, 41), we observed no prolonged differences in luminal colonization between NC101 and the ΔfyuA and Δirp1 mutants in the noninflamed intestines or during the course of inflammation and fibrosis development. This is likely the result of additional iron scavenging systems in NC101 that play compensatory roles in the Ybt-deficient and transport mutants (21, 42). Indeed, in other Enterobacteriaceae strains, inactivation of multiple iron acquisition systems is required to attenuate in vivo fitness (25, 30). Most importantly, if the enhanced profibrogenic potential of the ΔfyuA mutant was the result of dysregulated bacterial iron homeostasis and consequent effects on Ybt-independent functions, we would expect levels of fibrosis induction mediated by the Δirp1 and ΔfyuA mutants to be comparable, as both mutants cannot scavenge iron through Ybt (43). Instead, fibrosis induction was further attenuated in mice colonized with Ybt-deficient AIEC strains. Collectively, our findings support a model in which Ybt stimulates host profibrogenic responses through a mechanism independent of its role in importing iron through FyuA.
While the Ybt system did not impact overall AIEC intestinal fitness, inactivation of Ybt transport altered the distribution of AIEC colonization within colonic tissues. This may contribute to AIEC-driven fibrosis by activating myofibroblasts and mesenchymal cells either directly via bacterial recognition receptors (i.e., Toll-like receptors [TLRs]) or indirectly by activating intestinal immune cells that modulate profibrogenic cellular responses (5, 6). The ΔfyuA mutant was more abundant than the parental strain within the colonic subepithelium and colocalized with fibrotic lesions in Il10−/− mice at 10 weeks. In contrast, inactivation of Ybt biosynthesis did not alter tissue localization of AIEC, further uncoupling the effects of Ybt biosynthesis and Ybt transport on bacterial function. Instead, inactivation of Ybt biosynthesis in the ΔfyuA mutant restored tissue colonization patterns exhibited by the parental strain, suggesting that Ybt mediates the mislocalization of the ΔfyuA mutant to the subepithelium independent of its role in importing iron through FyuA. Consistent with our findings, several studies have also reported altered tissue localization of Enterobacteriaceae pathogens with inactivation of the Ybt system in extraintestinal mucosal environments (26, 44, 45). Finally, it should be noted that while we observed a statistically significant decrease in fecal colonization of the ΔfyuA mutant at 5 weeks, it remains unclear whether a <0.5-log difference in bacterial burdens in a monocolonized mouse can impart any meaningful effects on the host, especially as this decrease was also not observed at 1 or 10 weeks postcolonization. Collectively, these findings highlight one putative noncanonical function of Ybt that may enhance the profibrogenic potential of AIEC. The precise mechanisms by which inactivation of FyuA enhances fibrosis development in susceptible hosts will be the subject of future studies.
Because Ybt is a secreted bacterial product that permeates mammalian membranes, Ybt may also promote profibrogenic host responses by perturbing cellular iron homeostasis in the host. Indeed, the membrane-permeative siderophores enterobactin and yersiniabactin stimulate epithelial proinflammatory responses by decreasing intracellular iron pools, an effect that is reversed with the addition of iron (33, 34). Ybt disruption of local iron homeostasis may similarly drive fibrosis development by stimulating profibrogenic responses in epithelial, mesenchymal, and immune cells. While our host transcriptomics analyses demonstrated similar colonic expression profiles of numerous iron responsive host genes in NC101- versus ΔfyuA mutant-colonized mice (Table S5), differences in the canonical iron response genes Ndrg1 and Tfrc were uniquely observed in Il10−/− mice. This suggests that local and/or cell-specific alterations in host iron homeostasis may contribute to the progression of fibrosis. However, as these changes were observed at 10 weeks but not 5 weeks postcolonization, the initiation of fibrosis and early profibrotic gene signatures cannot be attributed to major alterations in host iron homeostasis.
In addition to iron, Ybt is capable of binding other metals, including nickel, cobalt, chromium, gallium, and copper (46). This raises the possibility that its profibrogenic potential is the result of interactions with other metals present in the colonic environment. For example, when complexed with copper, Ybt acts to limit the lethal effects of macrophage reactive oxygen species (47). Ybt-copper chelates have been detected in urine samples from patients infected with uropathogenic E. coli (UPEC), demonstrating that Ybt binds copper in vivo (23). Bacterial cells can also import Ybt-copper chelates through FyuA (24, 31). Together, these findings introduce two putative mechanisms by which Ybt promotes fibrosis development: (i) through chelation of host sources of metals other than iron and/or (ii) by modulating the transcriptome and metabolome of AIEC by altering the flux of micronutrients into the bacterial cell. Finally, because the Ybt enzymatic machinery produces additional secreted metabolites that remain uncharacterized (48), it is intriguing to speculate that these Ybt precursors and Ybt-like molecules may also play a role in inflammation-associated fibrosis.
Fibrosis complicates many inflammatory intestinal disorders associated with microbial dysbiosis; however, profibrotic mechanisms remain incompletely understood and limit therapeutic strategies. This has been hampered by the lack of rodent models that recapitulate the complex interactions between host genetics and microbial factors important for inflammation and fibrosis development. Here we introduce a new model for inflammation-associated fibrosis driven by a pathobiont-derived small molecule produced from the Ybt pathogenicity island. Consistent with human CD (32), profibrogenic pathways are upregulated prior to histological presentation of fibrosis and mirror the incidence rate of fibrotic disease in our model. Moreover, our model recapitulates key histological and transcriptomic aspects of fibrotic disease in human CD. More broadly, our findings demonstrate that manipulating the same pathogenicity island in different ways can result in distinct consequences for disease development. This highlights an important difference in targeting siderophore biosynthesis versus the cognate receptors as putative bacterial targets in microbe-driven diseases such as CD. Furthermore, other siderophore and metallophore systems of the gut microbiota may induce similar responses and contribute to fibrosis. Given the prevalence of AIEC among the population with CD, the presence of the Ybt siderophore system could serve as a useful prognostic tool in identifying patient subsets susceptible to fibrotic disease.
MATERIALS AND METHODS
Bacterial strains.
The fecal isolate E. coli NC101 was isolated from WT mice (13). The λ-red recombinase system was utilized to generate mutants (49) (Table S7). Bacterial strains and plasmids are listed in Table S6.
Mice.
Germfree Il10−/− and WT 129S6/SvEV mice were maintained at the National Gnotobiotic Rodent Resource Center at the University of North Carolina at Chapel Hill (UNC-CH). Absence of isolator contamination was confirmed by Gram stain and fecal culture. Eight- to 12-week-old mice were inoculated via oral and rectal swabs with E. coli following overnight growth in LB broth (13). Colonization was confirmed by fecal plating. Five cohorts of Il10−/− mice were colonized with the NC101 WT or the ΔfyuA mutant, and two cohorts of Il10−/− mice were colonized with the Δirp1 or ΔfyuA irp1 mutant. Animal protocols were approved by the UNC-CH Institutional Animal Care and Use Committee.
Quantification of bacteria.
E. coli CFU in feces were quantified by serial dilutions and plating on LB plates. Mucus- and tissue-associated bacteria were enumerated as described previously (10).
Colitis histopathology.
At necropsy, tissues were fixed in 10% neutral buffered formalin. Colon sections were stained with H&E, Masson’s trichrome, or Sirius red; spleens were stained with Prussian blue. Colitis scores (0 to 12) of Swiss-rolled colons were blindly assessed as described previously (13, 20). Composite scores (0 to 36) are the sums of proximal, middle, and distal colon scores.
Fibrosis histopathology.
Fibrosis was blindly assessed on colonic H&E sections and validated by Sirius red. Severity of fibrosis (0 to 5) was evaluated using a validated scoring system (28, 29) assessing the extent of submucosal involvement: no fibrosis (score of 0) or fibrosis in <25% (score of 1), 26 to 50% (score of 2), 51 to 75% (score of 3), or 76 to 100% (score of 4) of the colon section. One point was added for lamina propria involvement. Composite scores (0 to 15) are the sums of the proximal, middle, and distal colon scores. A score of 0 was considered nonfibrotic, 1 to 3 represented mild fibrosis, and 4+ represented moderate/severe fibrosis. Histopathology was blindly confirmed by a small-animal veterinarian specializing in gastrointestinal histopathology.
Human intestinal samples.
Formalin-fixed, paraffin-embedded tissue blocks from routine diagnostic surgical resections were transferred to UNC-CH under an approved Institutional Review Board protocol of the Cleveland Clinic. Sections were H&E and Sirius red stained from three individuals per disease category: CD, ulcerative colitis (UC), diverticulitis, and noninflamed controls (healthy margins of colorectal cancer patients).
FISH.
Colons were washed in phosphate-buffered saline (PBS) to remove contents and loosely adhered bacteria. Formalin-fixed, paraffin-embedded sections were mounted on charged glass slides and incubated with an oligonucleotide probe directed against E. coli (Cy3-E. coli/Shigella probe) and an antisense probe (6-carboxyfluorescein [FAM]-non-EUB338) (50). Hybridized samples were washed in PBS, air dried, and mounted with ProLong antifade (Molecular Probes Inc.). Sections were examined on a BX51 epifluorescence microscope with an Olympus DP-7 camera. Fluorescent in situ hybridization (FISH) analysis was performed in a blind fashion by two independent investigators as follows: to assess bacterial colonization, we enumerated individual bacterial cells adhered to epithelial cells (epithelial attachment), localized within epithelial cells (epithelial invasion), and translocated across the epithelium (subepithelial invasion). The quantity of bacteria per colon Swiss roll was converted to a FISH score of 0 to 4 (Table 1).
TABLE 1.
FISH scoring
| FISH score | No. of bacteria demonstrating: |
||
|---|---|---|---|
| Epithelial attachment | Epithelial invasion | Subepithelial invasion | |
| 0 | None | None | None |
| 1 | 1–50 | 1–10 | 1–5 |
| 2 | 51–150 | 11–20 | 6–10 |
| 3 | 151–250 | 21–30 | 11–15 |
| 4 | 251+ | 31+ | 16+ |
RNA-seq analysis.
RNA-seq reads were quality filtered at Q20 and trimmed to remove remaining adaptors using Trimmomatic (51) version 0.35. Resulting reads were aligned to the Illumina iGenome Mus musculus GRCm38 reference genome using Tophat (52) version 2.1.0 utilizing Bowtie2 (53) version 2.2.5. Resulting alignments were processed using Cufflinks (54) version 2.2.1 along with the Illumina iGenome Mus musculus GRCm38 Gene transfer format file, after masking rRNA features as described previously (55). Transcripts were quantified using cuffquant, and gene counts were exported to text files and then imported to edger (56) version 3.12.1 (running inside R version 3.2.3) for detecting differentially expressed genes. A gene was considered for differential-expression testing if present in at least three samples. We considered a gene differentially expressed if its edgeR FDR-adjusted P value was <0.05. Parallel analysis using featureCounts (57) from the subread package version 1.4.6 for transcript quantification showed similar results.
Pathway analysis was conducted using GAGE (58) version 2.20.1 using Mus musculus (mmu) Kyoto Encyclopedia of Genes and Genomes (KEGG) (59) pathways, and genes were mapped to KEGG pathways using Pathview version 1.10.1 (60). Pathways were considered significant if the GAGE q-value was <0.05. ECM-receptor interaction pathway genes (Fig. 6) are based on KEGG pathway mmu04512. We tested the effect of sequencing run and lane on the clustering of the samples and found both to be insignificant (P value > 0.05) for PC1 and PC2 in Fig. 5.
Statistical analysis.
P values were calculated using the nonparametric Mann-Whitney test when 2 experimental groups were compared or Kruskal-Wallis test with Dunn’s posttest when ≥3 experimental groups were compared. Data from quantitative bacterial cultures were log transformed for normalization. P values of <0.05 were considered significant.
Additional methods are described in the supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the Histology and Gnotobiotic Cores at the UNC Center for Gastrointestinal Biology and Disease (CGIBD; supported by NIH grant P30DK34987), the Translational Pathology Laboratory at the UNC Lineberger Comprehensive Cancer Center (supported by NCI grant P30-CA016086-40), NIEHS (P30ES010126-15A1), UCRF, and NCBT (2015-IDG-1007) and the National Gnotobiotic Rodent Resource Center (supported by NIH grant P40 OD01995). We thank the University of North Carolina’s Department of Chemistry Mass Spectrometry Core Laboratory, especially Brandie Ehrmann, for their assistance with mass spectrometry analysis, supported by the NSF (CHE-1726291) and the UNC-CH School of Medicine Office of Research. This work was supported by the following grants: NIH/NIDDK grant K01 DK103952 (J.C.A.), Kenneth Rainin Foundation Innovator Award (J.C.A.), pilot feasibility grant from the CGIBD to J.C.A. (NIH grant P30DK34987), American Gastroenterological Association Augustyn Award in Digestive Cancer (J.C.A.), Lineberger Comprehensive Cancer Center Pilot Grant and UCRF (J.C.A.), North Carolina Translational and Clinical Sciences Institute (NC TraCS) pilot grant (J.C.A.), American Cancer Society postdoctoral fellowship (J.C.A.), UNC dissertation completion fellowship (M.E.), Microbiome Initiative (R.B.S. and K.W.S.) and Gnotobiotic Facility (R.B.S.) grants from the Crohn’s and Colitis Foundation of America (CCFA), a Student Research Fellowship Award from the CCFA (L.N.M.), and pilot grant P30DK097948 and K08DK110415 (F.R.).
We acknowledge Taylor Tibbs and Lacey Lopez for assistance with histological stains and Elise Sloey for assistance with green fluorescent protein reporter assays.
Author contributions were as follows: conceptualization, M.E., K.W.S., R.B.S., and J.C.A.; methodology, M.E. and J.C.A.; software, R.Z.G.; validation, A.R.; formal analysis, M.E., R.Z.G., B.D., and J.C.A.; investigation, M.E., L.F., B.D., L.N.M., C.A.B., L.R.L., A.M.R., C.R.B., J.W.H., I.O.G., and J.C.A.; resources, C.B., K.W.S., C.J., I.O.G., F.R., R.B.S., and J.C.A.; data curation, R.Z.G.; writing of original draft, M.E. and J.C.A.; review and editing, M.E., J.C.A., and R.B.S.; visualization, M.E.; supervision, J.C.A.; and funding acquisition, J.C.A. and R.B.S.
We declare no competing interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00587-19.
REFERENCES
- 1.Sartor RB, Wu GD. 2017. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology 152:327–339.e4. doi: 10.1053/j.gastro.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peyrin-Biroulet L, Loftus EV, Colombel JF, Sandborn WJ. 2010. The natural history of adult Crohn’s disease in population-based cohorts. Am J Gastroenterol 105:289–297. doi: 10.1038/ajg.2009.579. [DOI] [PubMed] [Google Scholar]
- 3.Latella G, Rieder F. 2017. Intestinal fibrosis: ready to be reversed. Curr Opin Gastroenterol 33:239–245. doi: 10.1097/MOG.0000000000000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rieder F, Fiocchi C, Rogler G. 2017. Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology 152:340–350.e6. doi: 10.1053/j.gastro.2016.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Tol EA, Holt L, Li FL, Kong FM, Rippe R, Yamauchi M, Pucilowska J, Lund PK, Sartor RB. 1999. Bacterial cell wall polymers promote intestinal fibrosis by direct stimulation of myofibroblasts. Am J Physiol 277:G245–G255. doi: 10.1152/ajpgi.1999.277.1.G245. [DOI] [PubMed] [Google Scholar]
- 6.Otte J-M, Rosenberg IM, Podolsky DK. 2003. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology 124:1866–1878. doi: 10.1016/s0016-5085(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 7.Grassl GA, Valdez Y, Bergstrom KSB, Vallance BA, Finlay BB. 2008. Chronic enteric salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology 134:768–780. doi: 10.1053/j.gastro.2007.12.043. [DOI] [PubMed] [Google Scholar]
- 8.Small C-L, Reid-Yu SA, McPhee JB, Coombes BK. 2013. Persistent infection with Crohn’s disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat Commun 4:1957. doi: 10.1038/ncomms2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser A-L, Barnich N, Bringer M-A, Swidsinski A, Beaugerie L, Colombel J-F. 2004. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 10.Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. 2004. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 11.Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R, Orsi RH, Wiedmann M, Mcdonough P, Kim SG, Berg D, Schukken Y, Scherl E, Simpson KW. 2007. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J 1:403–418. doi: 10.1038/ismej.2007.52. [DOI] [PubMed] [Google Scholar]
- 12.Boudeau J, Glasser AL, Masseret E, Joly B, Darfeuille-Michaud A. 1999. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect Immun 67:4499–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, Huycke MM, Sartor RB. 2005. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Carvalho FA, Koren O, Goodrich JK, Johansson MEV, Nalbantoglu I, Aitken JD, Su Y, Chassaing B, Walters WA, González A, Clemente JC, Cullender TC, Barnich N, Darfeuille-Michaud A, Vijay-Kumar M, Knight R, Ley RE, Gewirtz AT. 2012. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe 12:139–152. doi: 10.1016/j.chom.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez-Medina M, Denizot J, Dreux N, Robin F, Billard E, Bonnet R, Darfeuille-Michaud A, Barnich N. 2014. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 63:116–124. doi: 10.1136/gutjnl-2012-304119. [DOI] [PubMed] [Google Scholar]
- 16.Kim SC, Tonkonogy SL, Karrasch T, Jobin C, Sartor RB. 2007. Dual-association of gnotobiotic IL-10-/- mice with 2 nonpathogenic commensal bacteria induces aggressive pancolitis. Inflamm Bowel Dis 13:1457–1466. doi: 10.1002/ibd.20246. [DOI] [PubMed] [Google Scholar]
- 17.Patwa LG, Fan T-J, Tchaptchet S, Liu Y, Lussier YA, Sartor RB, Hansen JJ. 2011. Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology 141:1842–1851.e10. doi: 10.1053/j.gastro.2011.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tchaptchet S, Fan T-J, Goeser L, Schoenborn A, Gulati AS, Sartor RB, Hansen JJ. 2013. Inflammation-induced acid tolerance genes gadAB in luminal commensal Escherichia coli attenuate experimental colitis. Infect Immun 81:3662–3671. doi: 10.1128/IAI.00355-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellermann M, Huh EY, Liu B, Carroll IM, Tamayo R, Sartor RB. 2015. Adherent-invasive Escherichia coli production of cellulose influences iron-induced bacterial aggregation, phagocytosis, and induction of colitis. Infect Immun 83:4068–4080. doi: 10.1128/IAI.00904-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan T-J, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C. 2012. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dogan B, Suzuki H, Herlekar D, Sartor RB, Campbell BJ, Roberts CL, Stewart K, Scherl EJ, Araz Y, Bitar PP, Lefébure T, Chandler B, Schukken YH, Stanhope MJ, Simpson KW. 2014. Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm Bowel Dis 20:1919–1932. doi: 10.1097/MIB.0000000000000183. [DOI] [PubMed] [Google Scholar]
- 22.Perry RD, Fetherston JD. 2011. Yersiniabactin iron uptake: mechanisms and role in Yersinia pestis pathogenesis. Microbes Infect 13:808–817. doi: 10.1016/j.micinf.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaturvedi KS, Hung CS, Crowley JR, Stapleton AE, Henderson JP. 2012. The siderophore yersiniabactin binds copper to protect pathogens during infection. Nat Chem Biol 8:731–736. doi: 10.1038/nchembio.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koh EI, Robinson AE, Bandara N, Rogers BE, Henderson JP. 2017. Copper import in Escherichia coli by the yersiniabactin metallophore system. Nat Chem Biol 13:1016–1021. doi: 10.1038/nchembio.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bachman MA, Oyler JE, Burns SH, Caza M, Lépine F, Dozois CM, Weiser JN. 2011. Klebsiella pneumoniae yersiniabactin promotes respiratory tract infection through evasion of lipocalin 2. Infect Immun 79:3309–3316. doi: 10.1128/IAI.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia EC, Brumbaugh AR, Mobley H. 2011. Redundancy and specificity of Escherichia coli iron acquisition systems during urinary tract infection. Infect Immun 79:1225–1235. doi: 10.1128/IAI.01222-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rakin A, Schneider L, Podladchikova O. 2012. Hunger for iron: the alternative siderophore iron scavenging systems in highly virulent Yersinia. Front Cell Infect Microbiol 2:151. doi: 10.3389/fcimb.2012.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rigby RJ, Hunt MR, Scull BP, Simmons JG, Speck KE, Helmrath MA, Lund PK. 2009. A new animal model of postsurgical bowel inflammation and fibrosis: the effect of commensal microflora. Gut 58:1104–1112. doi: 10.1136/gut.2008.157636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordon IO, Agrawal N, Willis E, Goldblum JR, Lopez R, Allende D, Liu X, Patil DY, Yerian L, El-Khider F, Fiocchi C, Rieder F. 2018. Fibrosis in ulcerative colitis is directly linked to severity and chronicity of mucosal inflammation. Aliment Pharmacol Ther 47:922–939. doi: 10.1111/apt.14526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deriu E, Liu JZ, Pezeshki M, Edwards RA, Ochoa RJ, Contreras H, Libby SJ, Fang FC, Raffatellu M. 2013. Probiotic bacteria reduce salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe 14:26–37. doi: 10.1016/j.chom.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bobrov AG, Kirillina O, Fetherston JD, Miller MC, Burlison JA, Perry RD. 2014. The Yersinia pestis siderophore, yersiniabactin, and the ZnuABC system both contribute to zinc acquisition and the development of lethal septicaemic plague in mice. Mol Microbiol 93:759–775. doi: 10.1111/mmi.12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kugathasan S, Denson LA, Walters TD, Kim M-O, Marigorta UM, Schirmer M, Mondal K, Liu C, Griffiths A, Noe JD, Crandall WV, Snapper S, Rabizadeh S, Rosh JR, Shapiro JM, Guthery S, Mack DR, Kellermayer R, Kappelman MD, Steiner S, Moulton DE, Keljo D, Cohen S, Oliva-Hemker M, Heyman MB, Otley AR, Baker SS, Evans JS, Kirschner BS, Patel AS, Ziring D, Trapnell BC, Sylvester FA, Stephens MC, Baldassano RN, Markowitz JF, Cho J, Xavier RJ, Huttenhower C, Aronow BJ, Gibson G, Hyams JS, Dubinsky M-C. 2017. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. Lancet 389:1710–1718. doi: 10.1016/S0140-6736(17)30317-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson AL, Ratner AJ, Barasch J, Weiser JN. 2007. Interleukin-8 secretion in response to aferric enterobactin is potentiated by siderocalin. Infect Immun 75:3160–3168. doi: 10.1128/IAI.01719-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holden VI, Lenio S, Kuick R, Ramakrishnan SK, Shah YM, Bachman MA. 2014. Bacterial siderophores that evade or overwhelm lipocalin 2 induce hypoxia inducible factor 1α and proinflammatory cytokine secretion in cultured respiratory epithelial cells. Infect Immun 82:3826–3836. doi: 10.1128/IAI.01849-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez M, Galy B, Dandekar T, Bengert P, Vainshtein Y, Stolte J, Muckenthaler MU, Hentze MW. 2006. Iron regulation and the cell cycle: identification of an iron-responsive element in the 3’-untranslated region of human cell division cycle 14A mRNA by a refined microarray-based screening strategy. J Biol Chem 281:22865–22874. doi: 10.1074/jbc.M603876200. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez M, Galy B, Schwanhaeusser B, Blake J, Bahr-Ivacevic T, Benes V, Selbach M, Muckenthaler MU, Hentze MW. 2011. Iron regulatory protein-1 and -2: transcriptome-wide definition of binding mRNAs and shaping of the cellular proteome by iron regulatory proteins. Blood 118:e168–e179. doi: 10.1182/blood-2011-04-343541. [DOI] [PubMed] [Google Scholar]
- 37.Anderson CP, Shen M, Eisenstein RS, Leibold EA. 2012. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta 1823:1468–1483. doi: 10.1016/j.bbamcr.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holden VI, Bachman MA. 2015. Diverging roles of bacterial siderophores during infection. Metallomics 7:986–995. doi: 10.1039/c4mt00333k. [DOI] [PubMed] [Google Scholar]
- 39.Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, Clardy J, Linington RG, Fischbach MA. 2014. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158:1402–1414. doi: 10.1016/j.cell.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McHugh JP, Rodríguez-Quinoñes F, Abdul-Tehrani H, Svistunenko DA, Poole RK, Cooper CE, Andrews SC. 2003. Global iron-dependent gene regulation in Escherichia coli. A new mechanism for iron homeostasis. J Biol Chem 278:29478–29486. doi: 10.1074/jbc.M303381200. [DOI] [PubMed] [Google Scholar]
- 41.Stojiljkovic I, Cobeljic M, Hantke K. 1993. Escherichia coli K-12 ferrous iron uptake mutants are impaired in their ability to colonize the mouse intestine. FEMS Microbiol Lett 108:111–115. doi: 10.1111/j.1574-6968.1993.tb06082.x. [DOI] [PubMed] [Google Scholar]
- 42.Arthur JC, Gharaibeh RZ, Mühlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, Fodor AA, Jobin C. 2014. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat Commun 5:4724. doi: 10.1038/ncomms5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bearden SW, Fetherston JD, Perry RD. 1997. Genetic organization of the yersiniabactin biosynthetic region and construction of avirulent mutants in Yersinia pestis. Infect Immun 65:1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bachman MA, Lenio S, Schmidt L, Oyler JE, Weiser JN. 2012. Interaction of lipocalin 2, transferrin, and siderophores determines the replicative niche of Klebsiella pneumoniae during pneumonia. mBio 3:e00224-11. doi: 10.1128/mBio.00224-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brumbaugh AR, Smith SN, Subashchandrabose S, Himpsl SD, Hazen TH, Rasko DA, Mobley H. 2015. Blocking yersiniabactin import attenuates extraintestinal pathogenic Escherichia coli in cystitis and pyelonephritis and represents a novel target to prevent urinary tract infection. Infect Immun 83:1443–1450. doi: 10.1128/IAI.02904-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koh E-I, Hung CS, Parker KS, Crowley JR, Giblin DE, Henderson JP. 2015. Metal selectivity by the virulence-associated yersiniabactin metallophore system. Metallomics 7:1011–1022. doi: 10.1039/c4mt00341a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaturvedi KS, Hung CS, Giblin DE, Urushidani S, Austin AM, Dinauer MC, Henderson JP. 2014. Cupric yersiniabactin is a virulence-associated superoxide dismutase mimic. ACS Chem Biol 9:551–561. doi: 10.1021/cb400658k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohlemacher SI, Giblin DE, d’Avignon DA, Stapleton AE, Trautner BW, Henderson JP. 2017. Enterobacteria secrete an inhibitor of Pseudomonas virulence during clinical bacteriuria. J Clin Invest 127:4018–4030. doi: 10.1172/JCI92464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simpson KW, Dogan B, Rishniw M, Goldstein RE, Klaessig S, McDonough PL, German AJ, Yates RM, Russell DG, Johnson SE, Berg DE, Harel J, Bruant G, McDonough SP, Schukken YH. 2006. Adherent and invasive Escherichia coli is associated with granulomatous colitis in boxer dogs. Infect Immun 74:4778–4792. doi: 10.1128/IAI.00067-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gilad Y, Mizrahi-Man O. 2015. A reanalysis of mouse ENCODE comparative gene expression data. F1000Res 4:121. doi: 10.12688/f1000research.6536.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 58.Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ. 2009. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10:161. doi: 10.1186/1471-2105-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luo W, Brouwer C. 2013. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 29:1830–1831. doi: 10.1093/bioinformatics/btt285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.