

Abstract
Aim:
The p53 cancer mutation Y220C creates a conformationally unstable protein with a unique elongated surface crevice that can be targeted by molecular chaperones. We report the structure-guided optimization of the carbazole-based stabilizer PK083.
Materials & methods:
Biophysical, cellular and x-ray crystallographic techniques have been employed to elucidate the mode of action of the carbazole scaffolds.
Results:
Targeting an unoccupied subsite of the surface crevice with heterocycle-substituted PK083 analogs resulted in a 70-fold affinity increase to single-digit micromolar levels, increased thermal stability and decreased rate of aggregation of the mutant protein. PK9318, one of the most potent binders, restored p53 signaling in the liver cancer cell line HUH-7 with homozygous Y220C mutation.
Conclusion:
The p53-Y220C mutant is an excellent paradigm for the development of mutant p53 rescue drugs via protein stabilization. Similar rescue strategies may be applicable to other cavity-creating p53 cancer mutations.
Keywords: : cancer mutations, cancer therapy, CRISPR/Cas9 p53 knockout, molecular chaperones, p53, protein stabilization, structure-based drug design, tumor suppression
The transcription factor p53 plays a key control in the cell cycle. It regulates diverse cellular processes ranging from DNA repair, apoptosis, metabolic processes and autophagy to stem cell biology and epigenetic control of gene expression [1–3]. The p53 protein is arguably the most important tumor suppressor in humans and is inactivated in virtually every cancer, making it a prime target for cancer therapy [1,4]. In about half of all human cancers, p53 is directly inactivated by mutation, and in the remainder, p53 activity is abolished through aberrations in regulatory pathways, for example, overexpression of the E3 ubiquitin ligase MDM2 and its homolog MDMX [1,4]. Most p53 cancer mutations are missense mutations in the DNA-binding domain (DBD) of the protein [5,6]. Although there is a great heterogeneity among common p53 cancer mutants, they can be roughly subdivided into DNA-contact and structural mutations [7]. DNA-contact mutations impair p53-DNA binding by eliminating essential protein-DNA contacts, for example, mutation of Arg248 or Arg273 [8,9]. Structural mutations destabilize the DBD, which has a relatively low intrinsic kinetic and thermodynamic stability, so that it rapidly unfolds at body temperature, followed by aggregation [10,11]. An estimated 30% of human p53 cancer mutants are temperature sensitive: the protein is denatured and inactive at body temperature but shows transcriptional activity at subphysiological temperature where it adopts a folded, wild-type-like conformation [7,12,13]. In principle, small molecules that bind to the folded but not to the unfolded state of these temperature-sensitive mutants should shift the equilibrium toward the active, folded state and, hence, restore p53 function in tumor cells. Current strategies to develop such molecular chaperones are targeting a transiently open pocket in the L1/S3 region of the DBD [14] or mutant-specific surface lesions [15,16]. In addition, alkylating agents, such as PRIMA-1Met/APR-246 and 2-sulfonylpryrimidines, that modify exposed cysteine residues have been shown to induce cell death in p53-compromised cells through a combination of p53 protein stabilization and an increase in reactive oxygen species levels via alkylation of other cellular targets [17–19]. Another rescue strategy targets the zinc-binding deficient R175H mutant with metallochaperones [20], and more recently, multifunctional compounds combining a small-molecule stabilizer with a metallochaperone have been suggested for targeting thermolabile mutants [21].
We have shown that the destabilizing Y220C mutation, a large-to-small substitution that is found in approximately 100,000 new cancer cases per year, creates a druggable crevice on the surface of the protein [1,15]. Conveniently, this binding site is distant from the DNA-binding surface of the protein, so that no inhibitory effects from binding should arise. Using in silico methods and fragment-based screening, followed by structure-based design, we have identified several classes of lead structures that bind to the Y220C pocket, stabilize the protein and slow down its rate of aggregation [16,22–28]. Initial biological testing of these compounds showed that they are biologically active in cancer cell lines with homozygous Y220C mutation. PK7088, for example, induced a mutant-specific increase in the amount of correctly folded protein and upregulation of p53 target genes p21 and NOXA [25]. Despite these encouraging preliminary data, most of the current molecules have a relatively low affinity and some of them exhibit general toxicity, which hampers extensive biological evaluation. There is therefore a need for more potent binders to fully exploit the potential of this rescue strategy in cancer cell lines with the Y220C mutation.
In-depth analyses of Y220C-ligand structures, combined with molecular dynamics simulations, have provided valuable insight into the structural plasticity of the mutation-induced surface crevice, revealing, for example, a transiently open subpocket and, importantly, pinpointing key interactions for the development of molecules with increased potency [16]. Here, we have revisited our carbazole-based fragment-lead structure PK083 [22], which binds in the Y220C pocket with a dissociation constant of ca. 125 μM. Interestingly, a recent paper described related carbazoles as a new class of anthelmintic agent [29]. A structure-based design strategy has led to the synthesis of carbazole derivatives targeting an unoccupied subpocket of the mutation-induced crevice with appropriately substituted five-membered heterocycles, which resulted in an affinity increase by almost two orders of magnitude. These next-generation carbazoles showed increased thermostabilization of the Y220C mutant and were efficient in slowing down aggregation of the mutant protein. Most importantly, large-scale gene expression analysis revealed that one of these compounds induces upregulation of a series of p53 target genes in the p53-Y220C liver cancer cell line HUH-7 compared with an isogenic knockout cell line created by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9.
Materials & methods
Compound synthesis
A detailed description of compound synthesis, including analytical data, is provided in the supplementary information. Briefly, in many instances, the bromo-substituted carbazole PK9284 was subjected to a range of palladium- or copper-mediated coupling reactions to further adorn this position with heteroatom or heterocyclic substituents to explore the structure–activity relationship (SAR) tolerance in the subsite II position.
Differential scanning fluorimetry
Differential scanning fluorimetry (DSF) measurements were performed as described [27], using 8-μM protein (stabilized p53-Y220C DBD) and 10 × SYPRO Orange (Life Technologies) in a 25-mM KPi (pH 7.2), 150-mM NaCl and 1-mM TCEP assay buffer at a final DMSO concentration of 5% (v/v). ΔTm values were calculated as ΔTm = Tm (p53 mutant + ligand) − Tm (p53 mutant). All samples were measured in triplicate.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) was performed as described [27]. Briefly, the cell unit contained 50–100 μM protein in a 25-mM KPi (pH 7.2), 150-mM NaCl, 1-mM TCEP and 5% (v/v) DMSO assay buffer. The syringe contained 2−5 mM compound in the same buffer. For poorly soluble compounds (e.g., PK9318-9328), a reverse titration was performed in the same assay buffer using 190-μM protein for the syringe and 15-μM compound for the cell.
Light scattering assay
Aggregation kinetics of the p53 Y220C DBD (94-312) were measured and analyzed as previously described [30]. Briefly, light scattering was recorded at 37°C using 500 nm as excitation and emission wavelengths. Experiments were performed in standard phosphate buffer (as described above) with 3-μM protein using a Horiba FluoroMax-3 spectrophotometer. The initial aggregation rates V0 were calculated from the slopes of the initial linear part of the graph, plotting light scattering against t2 (t being time).
Generation of Y220C-p53 knockout cell line
A p53 knockout clone of liver cancer cell line HUH-7 was generated using the CRISPR/cas9 technique [31]. A detailed description is provided in the Supplementary Information.
Cell culture & viability
Cells were cultured as described in Bauer et al. [19]. Cell viability was measured as previously described [11,19].
Real-time PCR & p53 target gene array
HUH-7 and HUH-7 p53 KO cell lines were grown in a humidified incubator at 37°C with 5% (v/v) CO2 in Dulbecco's modified Eagle's medium (DMEM) containing 10% (v/v) Fetal calf serum (FCS) and 1% antibiotic stock mix (10 mg/ml penicillin, 10 mg/ml streptomycin). Cells were treated in 6-well plates for 18 h with a DMSO content of 0.5% for both compound and control samples. Total RNA extraction and purification was done using the RNeasy Mini Kit (Qiagen), according to the manufacturer's instructions. cDNA synthesis was performed with a RT2 First Strand Kit (Qiagen), using 800-ng RNA per reaction. After transferring RT2 SYBR Green ROX FAST Mastermix (Qiagen) and cDNA (22.5 × dilution of cDNA synthesis reaction as final concentration) into a rotor-disc ring containing the RT2 Profiler PCR Array Human p53 Signaling Pathway (Qiagen), real-time PCR was performed according to the manufacturer's instructions using the Rotor-Gene 6000 (Corbett Life Science) PCR cycler. mRNA fold changes were quantified with the ΔΔCt method using the housekeeping gene β2-microglobulin as a reference.
Protein purification & crystallography
A stabilized version of the human p53 DBD (residues 94–312; M133L/V203A/N239Y/N268D) with an additional Y220C mutation was expressed and purified as described [22]. Crystals were grown using the sitting drop vapor diffusion technique at 18°C following a published protocol [15]. They were soaked for 3–4 h in a saturated solution of compound (<30 mM) in cryo buffer (19% [w/v] polyethylene glycol 4000, 20% [v/v] glycerol, 100-mM Tris, pH 7.2, 10-mM sodium phosphate, pH 7.2, 150-mM NaCl) and then flash frozen in liquid nitrogen. X-ray datasets were collected at 100 K at beamlines I02, I03 and I04 of the Diamond Light Source, Oxford, UK. The diffraction data were integrated using XDS [32] and scaled with SCALA [33], which is implemented in the CCP4 package [34]. The structures were solved by difference Fourier analysis in PHENIX [35], using Protein Data Bank (PDB) entry 2J1X as a starting model with initial rigid body refinement. The structures of the Y220C-ligand complexes were then refined using iterative cycles of manual model building in COOT [36] and refinement in PHENIX. Ligand dictionary files for refinement were generated using the Grade Web Server (http://grade.globalphasing.org). Structural figures were prepared using PyMOL (www.pymol.org).
Results & discussion
Compound design, synthesis & biophysical characterization
The mutation-induced surface crevice in the Y220C mutant consists of a central pocket leading into subsites 1 and 2 and a transiently open subsite 3 at the bottom of the central pocket that is modulated by the conformational state of Cys220 [16]. The carbazole-based compound PK083 binds to this surface crevice with a dissociation constant, KD. of ca. 125 μM. In the crystal structures of the Y220C mutant in complex with PK083 [22] and fluoroalkyl analogs [27], subsite 2 of the binding pocket is not occupied by these ligands, which provides an excellent opportunity for targeting this site to improve the binding affinity of the carbazole-based lead structure (Figure 1). However, this is not without challenges, as this site is relatively small and very hydrophobic. In the first step of our synthesis strategy, we introduced a bromine at C7 (facing subsite 2) of the carbazole ring system to probe this subpocket and to provide a synthetic handle for further lead optimization via Suzuki–Miyaura coupling chemistry. In the subsequent synthesis round, we substituted the bromine by various aromatic heterocycles. We focused on directly bound five-membered rings because modeling studies had suggested that they fit better into this subsite than 6-membered rings or systems with, for example, amide or ether linkages, which were predicted to cause steric clashes (confirmed by SAR studies also, vide infra).
Figure 1. . Structure-based design strategy.

(A) Molecular surface representation of the p53 mutant Y220C in complex with the carbazole-based molecule PK083 (Protein Data Bank ID: 2VUK), highlighting different subsites of the binding pocket. Subsite 2 is not occupied by the ligand. The side chain of Cys220 blocks access to subsite 3 in the complex with PK083. Two different orientations are shown: cross-section through the binding site (top) and top view (bottom). (B) Scheme showing the design strategy for improving the binding affinity of PK083 by targeting subsite 2. See text for further details.
The effect of the synthesized compounds on the thermostability of the Y220C mutant DBD was measured by differential scanning fluorimetry, and dissociation constants were determined by ITC. Data for selected compounds are shown in Table 1 and Supplementary Figure 1. Gratifyingly, the introduction of a bromine in the first step already significantly increased binding affinity ca. tenfold, from 125 to 14 μM, for the bromocarbazole PK9284. Substitution of the bromine with a five-membered heterocycle further improved affinity. The highest affinity in this second-generation series was observed with a C2-linked furan (PK9320) or thiophene (PK9318) moiety, with KD. of 4.0 and 2.6 μM, respectively. Improved affinities were accompanied by a substantial increase in protein stabilization. The same two compounds, increased the Tm of the DBD by almost 4°C at a ligand concentration of 250 μM compared with a stabilization of only about 1°C for to the starting scaffold, PK083, at the same ligand concentration. Among the five-membered heterocycles, C4-linked rings with two neighboring heteroatoms (PK9324 and PK9326) resulted in significantly weaker thermostabilization. Introduction of a pyrimidine moiety in PK9305 or an O-linked phenyl group in PK9296 almost completely abolished thermostabilization, validating our initial modeling studies that six-membered rings cause steric clashes in subsite 2.
Table 1. . Thermostabilization and dissociation constants of carbazole-based Y220C binders.
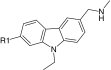 | |||||||
|---|---|---|---|---|---|---|---|
| Compound ID | R1† | DSF ΔTM (°C)‡ | ITC KD (μM) | Compound ID | R1† | DSF ΔTM (°C)‡ | ITC KD (μM) |
| PK083 | H | 1.0 ± 0.1 | 125 ± 10§ | PK9321 | 3.2 ± 0.1 | 5.6 ± 1.1¶ | |
| PK9284 | Br | 3.0 ± 0.1 | 14.2 ± 2.0¶ | PK9322 | 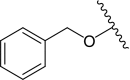 |
3.0 ± 0.1 | 8.3 ± 2.6¶ |
| PK9295 | 1.3 ± 0.3 | 62 ± 6.5¶ | PK9323 | 3.4 ± 0.2 | 5.3 ± 1.4# | ||
| PK9296 | 1.9 ± 0.0 | – | PK9324 | 1.8 ± 0.1 | – | ||
| PK9304 | -0.6 ± 0.0 | – | PK9326 | 0.8 ± 0.1 | – | ||
| PK9305 | 0.6 ± 0.0 | – | PK9331 | 1.7 ± 0.1 | – | ||
| PK9318 | 3.6 ± 0.2 | 2.6 ± 0.4# | PK9327 | 3.1 ± 0.2 | – | ||
| PK9319 | 0.5 ± 0.1 | – | PK9328 | 3.3 ± 0.0 | 1.7 ± 0.2# | ||
| PK9320 | 3.7 ± 0.0 | 4.1 ± 0.2# | PK9329 | 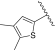 |
0.1 ± 0.0 | – | |
All compounds were synthesized as the amine-hydrochloride salt to aid aqueous solubility.
Measured at a compound concentration of 250 μM. Mean values of quadruplicate measurements are shown.
Value taken from [22].
Single measurement with ITC fit error.
Mean value of at least three independent measurements with SEM.
DSF: Differential scanning fluorimetry; ITC: Isothermal titration calorimetry; SEM: Standard error of the mean.
Next, we performed a methyl scan of the thiophene moiety in PK9318 to explore affinity optimization and potential derivatization. The scan revealed that methylation was well tolerated at C4 and C5 of the heterocycle, however, not at C3. A methyl substituent at C4 actually further improved the KD to 1.7 μM in PK9328, which is the most potent Y220C binder reported to date. Interestingly, double methylation at C4 and C5 completely abrogated binding (PK9329) and no stabilization of the Y220C mutant was observed. Overall, targeting subsite 2 with appropriately substituted five-membered heterocyclic rings gave a more than 70-fold affinity increase compared with the parent carbazole PK083.
Inhibition of Y220C mutant aggregation
Conformational mutants of p53 not only unfold at physiological temperatures but also quickly aggregate due to unfolding-induced exposure of aggregation-prone sequences [11]. Accordingly, aggregation of the Y220C mutant should, in theory, be inhibited upon stabilization. Coaggregation with other cellular proteins, including the p53 family members p63 and p73, is considered to be one of the main reasons for the so-called oncogenic gain-of-function of mutant p53 [37–40]. We used light scattering at 500 nm to monitor aggregation of the Y220C mutant DBD at 37°C with and without our carbazole-based stabilizers (Figure 2). PK9318 significantly slowed down aggregation in a concentration-dependent manner, with an approximately sixfold lower initial aggregation rate at a compound concentration of 30 μM, corresponding to a 1:10 protein–ligand ratio (Supplementary Table 1). PK9327 and PK9328 slowed down the initial rate of Y220C aggregation (nucleation phase) even more strongly than PK9318 (by a factor of two) at a concentration of 15 μM (Supplementary Table 2). However, PK9318 inhibited more strongly the formation of larger aggregates (Figure 2).
Figure 2. . Inhibition of protein aggregation.

The effect of Y220C mutant stabilizers on the rate of aggregation of the mutant protein was measured at 37°C by monitoring light scattering at 500 nm. (A) Concentration-dependence of aggregation inhibition by PK9318. (B) Aggregation of the Y220C mutant at 15 μM PK9318 (green), PK9327 (red) or PK9328 (orange), and without compound (black).
Structural analysis of ligand-binding modes & its implications for ligand optimization
We determined the high-resolution crystal structures of the Y220C mutant in complex with selected compounds in order to rationalize the measured affinities and guide future compound design (Table 2). As anticipated, the structure of the Y220C-PK9284 complex shows that adding a bromine results in new hydrophobic interactions in subsite two while retaining the overall binding mode of the carbazole (Figure 3A). The central carbazole ring system is sandwiched between several prolines on both sides of the binding pocket, with the N-ethyl substituent serving as a hydrophobic anchor pointing toward subsite 3. The secondary amine is solvent-exposed and forms a hydrogen bond with the backbone oxygen of Asp228 in subsite 1. The bromine-targeting subsite two is in close contact to Thr150, Pro151 and Pro222 on both sides of the binding pocket as well as the sulfur atom of Cys220 at the bottom of the pocket. It is also close to the backbone oxygen of Cys220 (3.6 Å), but at a C-Br··O angle of about 125°, no energy gain from halogen bonding is expected, which would require a close to linear arrangement for favorable overlap of the lone electron pair of the oxygen with the sigma hole of the bromine [41].
Table 2. . X-ray data collection and refinement statistics of p53-Y220C ligand structures.
| Compound | PK9284 | PK9318 | PK9320 | PK9324 | PK9327 | PK9328 |
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| a (Å) | 65.11 | 64.83 | 64.89 | 64.89 | 65.08 | 65.11 |
| b (Å) | 71.26 | 71.18 | 71.17 | 71.25 | 71.22 | 71.21 |
| c (Å) | 105.31 | 104.98 | 105.02 | 105.06 | 105.27 | 105.35 |
| Molecules/AU | 2 | 2 | 2 | 2 | 2 | 2 |
| Resolution (Å)† | 29.6–1.55 (1.63–1.55) | 29.5–1.32 (1.39–1.32) | 29.5–1.24 (1.31–1.24) | 29.5–1.40 (1.48–1.40) | 29.6–1.25 (1.32–1.25) | 29.6–1.32 (1.39–1.32) |
| Unique reflections | 71,228 | 112,690 | 137,227 | 96,032 | 129,162 | 114,821 |
| Completeness (%)† | 99.8 (99.0) | 98.6 (97.1) | 99.3 (98.3) | 99.6 (99.9) | 95.5 (93.0) | 99.5 (99.3) |
| Multiplicity† | 4.4 (4.3) | 5.5 (5.4) | 4.6 (4.6) | 4.7 (4.8) | 5.3 (5.4) | 5.1 (4.8) |
| Rmerge (%)† | 3.6 (46.8) | 6.9 (50.8) | 3.8 (51.3) | 5.0 (52.3) | 5.4 (58.1) | 4.9 (55.0) |
| Mean I/σ(I)† | 17.9 (2.9) | 12.7 (3.2) | 17.4 (3.3) | 14.5 (2.9) | 13.8 (2.8) | 16.0 (3.3) |
| Wilson B value (Å2) | 20.7 | 10.8 | 10.5 | 14.0 | 10.7 | 11.6 |
| Refinement | ||||||
| Rwork (%)‡ | 18.0 | 14.2 | 15.0 | 14.9 | 15.1 | 14.9 |
| Rfree (%)‡ | 20.0 | 16.1 | 16.6 | 17.0 | 17.1 | 17.1 |
| No. of atoms | ||||||
| Protein§ | 3072 | 3106 | 3112 | 3107 | 3104 | 3114 |
| Zinc | 2 | 2 | 2 | 2 | 2 | 2 |
| Water | 374 | 507 | 443 | 364 | 482 | 454 |
| Ligands | 38 | 71 | 60 | 52 | 48 | 54 |
| RMSD bonds (Å) | 0.006 | 0.004 | 0.005 | 0.005 | 0.005 | 0.005 |
| RMSD angles (°) | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 |
| Mean B (Å2) | 27.0 | 19.0 | 18.0 | 22.0 | 20.2 | 20.0 |
| PDB entry | 6GGA | 6GGB | 6GGC | 6GGD | 6GGE | 6GGF |
Values in parentheses are for the highest resolution shell.
Rwork and Rfree.#x00A0;= ∑||Fobs. - |Fcalc.|/∑|Fobs., where Rfree was calculated with 5% of the reflections chosen at random and not used in the refinement.
Number includes alternative conformations.
PDB: Protein Data Bank; RMSD: Root-mean-square deviation.
Figure 3. . Structures of p53-Y220C with subsite 2-targeting carbazole derivatives.

(A) Structure of the Y220C mutant bound to PK9284 (Protein Data Bank [PDB] ID 6GGA). The protein is shown as a gray surface representation, with the sulfur of Cys220 highlighted in yellow. The ligand is shown as a stick model. (B) Structure of the Y220C-PK9318 complex (PDB ID 6GGB). A cross-section of the binding pocket is shown. (C) Superposition of the binding modes of PK083 (gray carbon atoms; PDB ID 2VUK), PK9318 (green carbon atoms; PDB ID 6GGB), PK9320 (pink carbon atoms; PDB ID 6GGC) and PK9324 (cyan carbon atoms; PDB ID 6GGD) upon binding to the Y220C mutant. (D) Structure of the Y220C-PK9320 complex (PDB ID 6GGC). Orientation and color codes are the same as in panel B. In all four panels, chain B of the asymmetric unit is shown.
The five-membered heterocyclic rings in compounds PK9318 and PK9320 (thiophene and furan) occupy subsite 2 almost fully, with the heteroatom facing Cys220 in both cases (Figure 3B & D). The distance between the sulfur atom of the thiophene ring and that of Cys220 is 3.7 Å. Interestingly, the thiophene sulfur atom is in close proximity (3.2 Å, slightly below the sum of the van der Waals radii) to the Cys220 backbone carbonyl oxygen at a C-S··O angle of 156°. This orientation indicates that there are attractive interactions between: the sulfur sigma hole of the thiophene sulfur (analogous to halogen sigma holes) and the Cys220 carbonyl oxygen; and the polarized thiophene Cα hydrogen atom and the Cys220 carbonyl oxygen (weak hydrogen bonding) [42].
The subsite 2 rings are almost coplanar with the carbazole scaffold. They form additional hydrophobic packing interactions with Pro153 at the far end of the pocket, which explains the significantly improved affinity of these compounds. Conversely, PK9324, which places two polar atoms at this position, induced a much lower thermostabilization, indicative of weaker binding, despite the same space filling and overall binding mode (Figure 3C). Presumably, there is a higher desolvation penalty associated with binding because polar atoms are buried in a hydrophobic pocket. Accommodation of the thiophene or furan ring in subsite 2 induced a small shift in the position of the carbazole scaffold by up to 0.7 Å compared with the parent molecule PK083 (Figure 3C).
Methyl scanning of the thiophene moiety in PK9318 showed that methylation at either C4 or C5 of the ring system is well tolerated, resulting in an appreciable affinity increase, whereas double methylation abrogated binding. Again, this is in agreement with the structural observations (Figure 4A–C). Addition of a methyl group at C4 of the thiophene moiety improves hydrophobic packing against Pro153. Interestingly, the structure of the Y220C-PK9328 complex also reveals that methylation at C4 induces a rotation of the thiophene ring by about 16° relative to its close to coplanar orientation with the carbazole moiety in the unsubstituted ring in order to accommodate the ligand. Such a rotation of the thiophene moiety would result in steric clashes with a methyl group present at C5, which explains the observed affinity loss for PK9329. The location of the C4 methyl substituent at the entrance of subsite 2 means that further extension at this position would lead into the solvent, providing an opportunity for modulating the biophysical and pharmacokinetic properties of future compounds without interfering with binding. The methyl substituent in PK9327 sits close to protein-backbone hydrogen-bond donors and acceptors (Gly154 N and Cys220 O), which can be exploited for future ligand optimization (Figure 4D).
Figure 4. . Binding mode of methylated thiophenes in subsite 2.

(A) Crystal structure of the Y220C-PK9328 complex (Protein Data Bank [PDB] ID 6GGF). Cross-section of the binding pocket. The Y220C mutant protein is shown as a surface representation. Selected side chains and the ligand are shown as stick models. 2Fo–Fc electron density for the ligand is shown in blue at a contour level of 1.5 σ. (B) Structure of Y220C-PK9328 (green) superimposed onto Y220C-PK9318 (gray; PDB ID 6GGB). Shown are Cα traces and selected side chains plus ligands as stick models. (C) Superposition of the binding modes of PK9328 (green stick model) and PK9318 (magenta stick model) shown in a different orientation, highlighting the rotation of the thiophene moiety upon methylation. The protein chain of the Y220C-PK9328 complex is shown in gray, with selected residues in the binding pocket displayed as stick models. (D) Structure of Y220C-PK9327 (pink; PDB ID 6GGE) superimposed onto Y220C-PK9318 (gray). Shown are Cα traces and selected side chains plus ligands as stick models. In all four panels, chain B of the asymmetric unit is shown.
In the wild-type structure, the tyrosine side chain of Tyr220 effectively blocks the binding site of the central carbazole moiety of our ligands (Supplementary Figure 2). Accordingly, neither the starting scaffold PK083 nor the second-generation carbazole PK9323 did show any stabilization of the wild type in DSF experiments under the same conditions as in Table 1, confirming that the compound-mediated stabilization of the DBD is due to binding to the Y220C pocket.
Selective viability reduction in Y220C cancer cell lines
We selected the most effective compounds for Y220C stabilization and aggregation inhibition (PK9318 and PK9328) to study the effects of our Y220C stabilizers on the viability of liver cancer cell line HUH-7, which has a homozygous p53-Y220C mutation. For control experiments, we generated an isogenic p53 knockout cell line, HUH7-F1, using the CRISPR/Cas9 technique [31] (detailed description of generation and validation in the Supplementary Information). This cell line contains a p53 frameshift mutation at codon 124 of one allele and a deletion of amino acids 125–223 on the other allele that results in a truncated, transcriptionally inactivated p53 protein (Supplementary Figures 3 & 4). Cell viability assays were performed at concentrations ranging from 0.6 to 15 μM after 72 h incubation (Figure 5).
Figure 5. . Second-generation carbazole-based binders reduce the viability of liver cancer cell line HUH-7.

p53-Y220C-dependent cell viability reduction was observed for PK9318 (A) and PK9328 (B) after 72 h. PK9329 did not show any p53-Y220C-dependent viability reduction (C), consistent with its inability to stabilize the mutant. Cell viability was measured in quadruplicate and normalized against the values of blank (viability = 1) and no cell (viability = 0) controls. Data were measured in triplicates and are shown as mean ± SEM (paired t-test to test for significance in differences in HUH-7 and HUH-7 p53 KO viability reduction.
*p < 0.05; **p < 0.01; ***p < 0.001.
KO: Knockout; SEM: Standard error of the mean.
Both PK9318 and PK9328 induced viability loss in HUH-7 cells at concentrations ≥3.75 mM. Viability was also reduced in the isogenic knockout cell line, HUH-7 p53 KO, but not as strongly as in HUH-7, and onset of viability loss occurred at higher concentration. For example, cell viability with PK9318, at a concentration of 7.5 μM, was 58% in HUH-7 and 88% in the isogenic p53 knockout. Increasing the compound concentration to 15 μM resulted in complete viability loss in HUH-7, whereas the viability of the knockout cell lines was still at 40%. Similarly, PK9328, at a concentration of 7.5 μM, resulted in complete viability loss for HUH-7 but more than 50% viability for the p53 knockout cells. The control compound PK9329, which has a similar structure but impaired binding to the Y220C mutant due to dimethylation of the thiophene ring, did not have a significant effect on the viability of either HUH-7 or the knockout cell line at the concentrations tested. Taken together, these data show a Y220C-mutant-specific viability reduction for PK9318 and PK3328, combined with general toxicity at higher compound concentrations. The same trend of preferential viability reduction in p53-Y220C cells was also observed when treating gastric cancer cell lines NUGC-3 (Y220C) and NUGC-4 (wild-type p53) with compounds PK9318, PK9320, PK9323 and PK9328 (Supplementary Figures 5). Collectively, these data indicate that our second-generation carbazoles preferentially kill cancer cells with Y220C-mutated p53, albeit within a relatively narrow concentration window (slightly above the in vitro dissociation constants of the compounds) due to the onset of Y220C-independent toxicity at higher compound concentrations.
Restoration of p53 transcriptional activity in p53-Y220C cancer cell lines
To monitor the effect of our compounds on restoring p53 transcriptional activity in cancer cells, we determined relative changes in mRNA levels for 84 genes related to p53-mediated signaling in the liver cancer cell line HUH-7 (Y220C mutation). After 9-h incubation with 10-μM PK9318, mRNA levels in both HUH-7 and HUH7-F1 cells were measured via real-time PCR (Figure 6). Many well-established p53 target genes [43] were upregulated in HUH-7 cells, including BAX, PUMA (BBC3), BTG2, GML, ESR1 SESN2 and GADD45A, which are involved in apoptosis, cell-cycle arrest or DNA repair, the negative regulator MDM2 as well as brain-specific angiogenesis inhibitor 1 (BAI1/ADGRB1). p53 levels were also upregulated upon PK9318 treatment. In addition, we observed significant upregulation of several apoptosis-related p53 downstream targets (e.g., BCL2 and CASP2), and MYOD1, which has been shown to regulate PUMA expression [44]. p21 (CDKN1A) levels remained virtually the same in HUH-7 after PK9318 treatment. Only a few of the investigated genes were upregulated in the p53 knockout cell line, HUH7-F1, but many were downregulated. We also observed a Y220C-independent upregulation of FASLG, a member of the tumor necrosis factor superfamily, which was much more pronounced in the p53 knockout cell line. Overall, the mRNA level analysis shows that PK9318 activates p53 signaling in Y220C mutant cancer cells and leads to an increased expression of a whole series of p53 target genes, in agreement with the observed in vitro thermostabilization of the Y220C mutant by this compound.
Figure 6. . Heatmap of mRNA fold changes in p53 signaling after treatment with 10 μM PK9318 for 9 h of HUH-7 and the isogenic HUH-7 p53 knockout cell line.

The qPCR array comprises 84 genes related to p53-mediated signal transduction, classified into subgroups. Changes in mRNA levels were calculated using the ΔΔCt method. A value of 1 indicates no change in relative transcript levels between control- and PK9318-treated samples (values between 0.66 and 1.5 are shown in white). Increased mRNA levels are shown in green, starting from 1.5 (light green) to 4 (dark green), and decreased mRNA levels are shown in red, ranging from 0.66 (light red) to 0 (dark red). For ADGRB1 in HUH-7 p53 KO cells and TP73, WT1 in HUH-7 cells, no reliable ΔΔCt values could be obtained (shown as NA). Especially p53-target genes that are involved in apoptotic signaling (e.g., PUMA [BBC3], BTG2, ESR1, EGFR) and cell cycle modulation (GML, MDM2, SESN2) were selectively upregulated in HUH-7 (p53-Y220C) cells after PK9318 treatment, indicating Y220C-dependent induction of apoptosis and cell-cycle arrest in this cell line.
Conclusion
We have shown that restoring the function of mutant p53 with small-molecule stabilizers is a promising therapeutic approach. Structure-guided optimization of the carbazole-based small-molecule PK083 by targeting an unoccupied subsite of the binding pocket with suitably substituted five-membered heterocycles resulted in a ca. 70-fold increase in binding affinity. This has led to PK083 analogs with the highest Y220C-binding affinities so far (low single-digit micromolar) that increase the thermostability of the conformationally unstable Y220C mutant by up to 4°C, the largest Y220C stabilization so far, and efficiently slow down its rate of aggregation in biophysical studies. Importantly, one of best binders induced upregulation of a series of p53 target genes in a gene transcription assay in liver cancer cell line HUH-7 with homozygous Y220C mutation, which was not seen in a CRISPR/cas9-generated p53-Y220C knockout, providing the proof-of-concept that our second-generation carbazoles have the potential to restore p53 tumor-suppressor function in cancer cells.
Future perspective
The presented structural and biophysical data will guide efforts to further increase the affinity and reduce the general cytotoxicity of our Y220C binders, which will aid the development of potent mutant-specific p53 rescue drugs for use in personalized cancer therapy. Additionally, the generated mutant p53 knockout cell line provides a valuable control for future biological testing. We further envision molecules with added functionalities at solvent-exposed positions, for example, fluorophores, to be used as chemical probes for studying p53 function and cellular trafficking. A similar rescue strategy may be applied to other thermolabile p53 cancer mutants or, more generally, misfolding disorders where disease-related mutants display druggable pockets in their folded state.
Summary points.
Destabilized p53 cancer mutant Y220C has a unique druggable surface crevice.
Structure-guided growing of the carbazole-based compound PK083 into an unoccupied subsite of the surface crevice yielded a 70-fold affinity increase.
Second-generation carbazole binders increased the thermostability of the Y220C mutant and inhibited its aggregation.
Design strategy validated by high-resolution crystal structures of Y220C-ligand complexes.
Second-generation carbazoles reduced viability of cancer cell lines with Y220C mutation.
Best binders restored p53 signaling in liver cancer cells with homozygous Y220C mutation.
Generation of a CRISPR/Cas9 p53-Y220C knockout HUH-7 cell line as a tool for mutant p53 drug discovery.
Proof of concept that the Y220C mutant is an excellent paradigm for the development of mutant p53 rescue drugs via protein stabilization.
Similar rescue strategies may be applicable to other cavity-creating p53 cancer mutations.
Supplementary Material
Acknowledgments
The authors thank the staff at beamlines I02, I03 and I04 of the Diamond Light Source, Oxford, UK, for their assistance during data collection. The authors also thank Sara Gomes for assistance in the generation of the CRISPR/Cas9 knockout cell line.
Footnotes
Open access
This article is distributed under the terms of the Creative Commons Attribution License 4.0 which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/full/10.2217/fmc-2019-0181
Data availability
The atomic coordinates and structure factors of the Y220C-ligand complexes have been deposited in the Protein Data Bank, www.pdb.org. PDB codes: 6GGA (Y220C-PK9284) 6GGB (Y220C-PK9318), 6GGC (Y220C-PK9320), 6GGD (Y220C-PK9324), 6GGE (Y220C-PK9327) and 6GGF (Y220C-PK9328).
Financial & competing interests disclosure
This work was funded by Worldwide Cancer Research (grants 14-1002, 18-0043), Sussex University (RN Jones), German Research Foundation (DFG) grant JO 1473/1-1 (AC Joerger) and ERC advanced grant 268506 (AR Fersht). The authors are grateful for support by the SGC, a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute, Innovative Medicines Initiative (EU/EFPIA) (ULTRA-DD grant no. 115766), Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: •• of considerable interest
- 1.Joerger AC, Fersht AR. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 85, 375–404 (2016). [DOI] [PubMed] [Google Scholar]; •• Comprehensive review of p53 drug discovery and structural biology as well as of the evolutionary history of p53 family proteins.
- 2.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell 137, 413–431 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 13, 217–236 (2014). [DOI] [PubMed] [Google Scholar]; •• Excellent review of different p53 drug discovery strategies.
- 5.Bouaoun L, Sonkin D, Ardin M. et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum. Mutat. 37, 865–876 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Leroy B, Fournier JL, Ishioka C. et al. The TP53 website: an integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 41, D962–D969 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joerger AC, Fersht AR. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene 26, 2226–2242 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 265, 346–355 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Eldar A, Rozenberg H, Diskin-Posner Y, Rohs R, Shakked Z. Structural studies of p53 inactivation by DNA-contact mutations and its rescue by suppressor mutations via alternative protein-DNA interactions. Nucleic Acids Res. 41, 8748–8759 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19, 1245–1256 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Wang G, Fersht AR. Multisite aggregation of p53 and implications for drug rescue. Proc. Natl Acad. Sci. USA 114, E2634–E2643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Como CJ, Prives C. Human tumor-derived p53 proteins exhibit binding site selectivity and temperature sensitivity for transactivation in a yeast-based assay. Oncogene 16, 2527–2539 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Dearth LR, Qian H, Wang T. et al. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis 28, 289–298 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Wassman CD, Baronio R, Demir O. et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 4, 1407 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joerger AC, Ang HC, Fersht AR. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl Acad. Sci. USA 103, 15056–15061 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• First paper describing a druggable pocket in the p53-Y220C mutant.
- 16.Joerger AC, Bauer MR, Wilcken R. et al. Exploiting transient protein states for the design of small-molecule stabilizers of mutant p53. Structure 23, 2246–2255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambert JM, Gorzov P, Veprintsev DB. et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 15, 376–388 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Bykov VJ, Zhang Q, Zhang M, Ceder S, Abrahmsen L, Wiman KG. Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front. Oncol. 6, 21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bauer MR, Joerger AC, Fersht AR. 2-Sulfonylpyrimidines: mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl Acad. Sci. USA 113, E5271–E5280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanden AR, Yu X, Wolfe AJ. et al. Synthetic metallochaperone ZMC1 rescues mutant p53 conformation by transporting zinc into cells as an ionophore. Mol. Pharmacol. 87, 825–831 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller JJ, Orvain C, Jozi S. et al. Multifunctional compounds for activation of the p53-Y220C mutant in cancer. Chemistry 24, 17734–17742 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl Acad. Sci. USA 105, 10360–10365 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basse N, Kaar JL, Settanni G, Joerger AC, Rutherford TJ, Fersht AR. Toward the rational design of p53-stabilizing drugs: probing the surface of the oncogenic Y220C mutant. Chem. Biol. 17, 46–56 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Wilcken R, Liu X, Zimmermann MO. et al. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J. Am. Chem. Soc. 134, 6810–6818 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Wilcken R, Joerger AC. et al. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 41, 6034–6044 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilcken R, Zimmermann MO, Bauer MR. et al. Experimental and theoretical evaluation of the ethynyl moiety as a halogen bioisostere. ACS Chem. Biol. 10, 2725–2732 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauer MR, Jones RN, Baud MG. et al. Harnessing fluorine-sulfur contacts and multipolar interactions for the design of p53 mutant Y220C rescue drugs. ACS Chem. Biol. 11, 2265–2274 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baud MGJ, Bauer MR, Verduci L. et al. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 152, 101–114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rennison D, Gueret SM, Laita O. et al. Substituted carbazoles – a new class of anthelmintic agent. Aust. J. Chem. 69, 1268–1276 (2016). [Google Scholar]
- 30.Wilcken R, Wang G, Boeckler FM, Fersht AR. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc. Natl Acad. Sci. USA 109, 13584–13589 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, La Russa M, Qi LS. CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem. 85, 227–264 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W. XDS. Acta Crystallogr. D 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans P. Scaling and assessment of data quality. Acta Crystallogr. D 62, 72–82 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Winn MD, Ballard CC, Cowtan KD. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkoczi G. et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr. D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu J, Reumers J, Couceiro JR. et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 7, 285–295 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Wang G, Fersht AR. Propagation of aggregated p53: cross-reaction and coaggregation vs. seeding. Proc. Natl Acad. Sci. USA 112, 2443–2448 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aschauer L, Muller PA. Novel targets and interaction partners of mutant p53 gain-of-function. Biochem. Soc. Trans. 44, 460–466 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Kehrloesser S, Osterburg C, Tuppi M, Schafer B, Vousden KH, Dotsch V. Intrinsic aggregation propensity of the p63 and p73 TI domains correlates with p53R175H interaction and suggests further significance of aggregation events in the p53 family. Cell Death Differ. 23, 1952–1960 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 56, 1363–1388 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Zhang X, Gong Z, Li J, Lu T. Intermolecular sulfur…oxygen interactions: theoretical and statistical investigations. J. Chem. Inf. Model. 55, 2138–2153 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Fischer M. Census and evaluation of p53 target genes. Oncogene 36, 3943–3956 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harford TJ, Kliment G, Shukla GC, Weyman CM. The muscle regulatory transcription factor MyoD participates with p53 to directly increase the expression of the pro-apoptotic Bcl2 family member PUMA. Apoptosis 22, 1532–1542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.