

Abstract
Mutations in at least 12 genes are responsible for a group of congenital skeletal muscle diseases known as nemaline myopathies (NMs). NMs are associated with a range of clinical symptoms and pathological changes often including the presence of cytoplasmic rod-like structures (nemaline bodies) and myofiber hypotrophy. Our recent work has identified a variable degree of behavioral benefit when treating 2 NM mouse models due to mutations in Acta1 with myostatin inhibition. This study is focused on the effects of delivering ActRIIB-mFc (Acceleron; a myostatin inhibitor) to the nebulin conditional knockout KO (Neb cKO) mouse model of NM. Treatment of Neb cKO mice with ActRIIB-mFc did not produce increases in weight gain, strength, myofiber size, or hypertrophic pathway signaling. Overall, our studies demonstrate a lack of response in Neb cKO mice to myostatin inhibition, which differs from the response observed when treating other NM models.
Keywords: Myofiber, Myopathies, Myostatin inhibition, Nemaline
INTRODUCTION
Nemaline myopathy (NM) is a clinically and genetically heterogeneous disease that is associated with mutations in at least 12 genes (NEB, ACTA1, TPM3, TPM2, TNNT1, CFL2, KBTBD13, KLHL40, KLHL41, LMOD3, MYPN, and MYO18B) (1–8). While there is enormous variation in disease course even within genetic groups, aggregates of nemaline rods made of α-actinin and other sarcomeric proteins (9) are present in muscle biopsies from all cases. Disease severity, however, does not correlate well with the abundance, size, or distribution of the nemaline rods (10, 11).
Mutations in the nebulin (NEB, 50% of NM cases) and skeletal muscle actin (ACTA1, 30% of NM cases) genes constitute genetic abnormality in the vast majority of NM patients (1, 12–14). Nebulin acts as a thin filament stabilizer that regulates the length of the sarcomeric thin filament (15–17) and is thought to increase the force-generating cross-bridge population (18, 19). In the context of nebulin deficiency, abnormalities of thin filament length and cross bridge cycling kinetics produce impaired contractile function and muscle weakness (20). Murine models with a complete loss of nebulin exhibit severe muscle weakness and die soon after birth (21–23), but complete loss of nebulin is only observed very rarely in human patients (24). As the vast majority of patients with NEB mutations express 10%–20% of normal nebulin levels with a considerably milder phenotype (17), the nebulin conditional knockout (Neb cKO) mouse was developed in hopes of better modeling the human disease state (25). While the conditional knockout strategy does not mimic the genetic defects seen in NM patients with NEB mutations (NEB-NM patients), this mouse displays low levels of nebulin expression, nemaline rod formation, moderate weakness, and considerable mortality in the first few months of life. These features make the Neb cKO model a better candidate system for therapeutic testing and development in NEB-NM in comparison to models that have complete nebulin deficiency from birth.
Similar to many congenital myopathies, myofiber smallness is observed in many cases of NM despite the fact that NM-associated genes are not known to be associated with muscle growth pathways. Recent work in our laboratory has focused on myostatin inhibitors as potential treatments using a variety of NM models, based on the hypothesis that increasing the effective contractile area of muscle would be of therapeutic benefit. Myostatin inhibits myogenesis by binding to activin type IIB receptors (ActRIIB), thereby activating the TGF-β pathway to inhibit cell cycle progression and signaling processes involved in myofiber hypertrophy (26, 27). We have previously used myostatin inhibition to pharmacologically induce myofiber hypertrophy in mouse models of X-linked myotubular myopathy (XLMTM) (28, 29) and in the Acta1H40Y (30) and TgActa1D286G (31) mouse models of NM. There was an encouraging impact on muscle growth and function observed in wild-type (WT) mice in all of these studies. Additionally, myostatin inhibition increased muscle size in both mouse models of ACTA1-NM, suggesting that the induction of myofiber hypertrophy was possible in this disorder and that it could potentially increase muscle strength and be of symptomatic benefit in NM. As myofiber smallness and inadequate or variable sarcomeres contribute to weakness in NM and occur to an even greater extent in the Neb cKO mouse, we predicted that myostatin inhibition would show considerable benefit in this mouse model.
The current study evaluates myostatin inhibition using ActRIIB-mFc in the Neb cKO mouse, based on the hypothesis that ActRIIB-mFc treatment would increase muscle size and strength in WT and Neb cKO mice. In contrast to our prior studies of myostatin inhibition in Acta1H40Y (30) and TgActa1D286G mice (31), treatment with ActRIIB-mFc did not improve any of these parameters in Neb cKO mice. To investigate the lack of treatment efficacy in Neb cKO mice, key components of myostatin signaling, hypertrophic pathway signaling, and the expression of myogenic factors were investigated. While these studies identified interesting differences between WT and Neb cKO mice with respect to some specific signaling molecules and myogenic markers, none of these differences explain the inability to grow muscle in response to ActRIIB-mFc treatment. Overall, our findings in this study indicate that myostatin inhibition does not improve disease in Neb cKO mice. As we have observed more positive therapeutic responses with myostatin inhibition in our prior work, this suggests that specific mutations or degrees of disease severity will impact the usefulness of this therapeutic strategy in NM.
MATERIALS AND METHODS
Live Animal Studies
All studies were performed with approval from the IACUC at The Medical College of Wisconsin. Genotyping of the Neb cKO mice was performed using a modification of previously described methods (Supplementary Data Fig. 1 [25]). This modification differed from the original genotyping strategy by placing both the WT and KO primers in the same Mastermix tube (as opposed to one mix for WT and one mix for KO). The MCK-Cre Mastermix was made separately from the WT and KO mix. Solutions were aliquoted into PCR tubes and 1 µL of each animal’s tail mix was added to the tube. Tubes were then placed into a Bio-Rad C1000 Touch ThermoCycler and samples run on a 1.5%–2% agarose gel in 1× Tris acetate-EDTA buffer with SYBR Safe DNA Gel Stain (Invitrogen, Carlsbad, CA). Gels were imaged on a Bio-Rad ChemiDoc MP and all PCR results were run twice to confirm genotypes. Male mice that were homozygous for the floxed Cre-positive nemaline allele (Neb cKO) and mice that were homozygous for the Cre-positive WT allele (control animals, WT) were used for dosing experiments. Additionally, 1 heterozygous animal was used for Western blot experiments due to a genotyping error that led us to initially group the sample in the Neb cKO group.
Beginning at 14 days of life, all mice were given intraperitoneal injections twice per week with either a soluble activin type IIB receptor (ActRIIB-mFc, also termed RAP-031, Acceleron Pharma, Cambridge, MA) at a dose of 10 mg/kg or an equivalent volume of tris-buffered saline ([TBS], referred to as “vehicle”) as previously described (28). Beginning when the first injection was given, animals were weighed 5 days per week and weaned at 3 weeks of age. After weaning, forelimb grip strength was performed weekly using a grip strength meter (Columbus Instruments, Columbus, OH) and open field tests were performed monthly as previously described (30, 31). Animals were killed at 9 weeks of life or when they reached an “end-stage” phenotype, which was characterized by body weight loss of more than 20%, paralysis of the hind limbs, or urinary obstruction, based on the characteristics of mice with severe muscle disease in our prior treatment studies. Neb cKO mice began to physically decline at a faster rate than anticipated, thus an earlier time point was chosen than in previous studies. Only a small proportion of animals survived until the 9-week terminal time point.
Pathological Evaluation and Tissue Collection
Animals were killed by CO2 and cervical dislocation. Photographs of skinned animals were taken and the internal organs and brain were placed in zinc-buffered formalin. Quadriceps, gastrocnemius, triceps, and diaphragm muscles were removed, weighed, and frozen in liquid nitrogen-cooled isopentane.
Muscle Histology
Hematoxylin and eosin (H&E) and Gomori trichrome stains were performed on quadriceps samples, which were cross-sectioned at 8 µm and mounted on slides. Images were taken using an Olympus BX53 microscope with an Olympus DP72 camera and cellSens Standard software (Olympus, Center Valley, PA). Frozen 8-µm quadriceps cross-sections were also double-stained with rabbit antidystrophin antibodies (ab15277, Abcam, Cambridge, MA, 1:100) and mouse monoclonal antibodies against myosin heavy chain type 2b (clone BF-F3, Developmental Studies Hybridoma Bank [DSHB], Iowa City, IA; 1:50) to determine myofiber size. Additional fiber typing experiments were performed using monoclonal antibodies against developmental myosin heavy chain (NCL-MHCd, Leica Biosystems, Buffalo Grove, IL, 1:25), fast myosin (Leica NCL-MHCf, 1:25), and neonatal myosin (Leica NCL-MHCn, 1:10). Secondary antibodies used were Alexa488 conjugated antimouse IgM (Sigma Aldrich, St. Louis, MO, 1:400) and AlexaFluor-conjugated antirabbit IgG (Molecular Probes, Carlsbad, CA, 1:200). Satellite cell number was evaluated using immunostaining for Pax7 (DSHB, 1:10) and quantified by manually counting the number of positive cells and the number of fibers in 2–5 fields photographed at 200× magnification (ranging from 466 to 1447 fibers assessed per animal), as previously described (32).
Fiber size was quantified through measurements of cross-sectional area and MinFeret diameter of myofibers from the quadriceps muscle. As in our recent studies (30, 31), quantitation was performed by evaluating the type 2b myosin-positive (glycolytic) and type 2b myosin-negative (oxidative) populations on a whole slide scan of 1 quadriceps muscle from 6 vehicle-injected WT mice, 6 ActRIIB-mFc WT mice, 6 vehicle-injected Neb cKO mice, and 6 ActRIIB-mFc Neb cKO mice. MinFeret diameter was evaluated using a novel automated technique developed at Cytoinformatics LLC by Dr Lin Yang.
Organ Histology
Major organs from 5 animals per treatment group were paraffin-embedded at the Children’s Hospital of Wisconsin Research Institute Histology Core, as previously described and stained using H&E. Organ histology was reviewed by a board certified anatomic- and neuro-pathologist (M.W.L.) using H&E-stained slides.
Western Blots
Muscle tissues from the gastrocnemius muscle were frozen in liquid nitrogen-cooled isopentane at the time of necropsy. Frozen muscles were sliced into 8-μm sections and homogenized using RIPA lysis buffer (EMD Millipore, Temecula, CA) containing protease inhibitor (Roche, Basel, Switzerland) and phosphatase inhibitor (Roche). Western blot procedures were performed as previously described (29, 30, 33). Following transfer, the PVDF membranes were probed with antibodies against Akt (4691), phospho-Akt (Ser473; 4060), p70-S6K (2708), phospho-p70-S6K (p-p70S6K) (Thr421/Ser424; 9204), S6 Ribosomal Protein (rps6) (2217), phospho-S6 Ribosomal Protein (p-rps6) (Ser240/244; 5364), phospho-eEF2k (p-eEF2K) (Ser366; 3691), phospho-4E-BP1(p-4EBP1) (Thr37/46; 2855), MTOR (2983) (Cell Signaling, Danvers, MA), ActRIIB (ab76940) (Abcam), MYF5 (SAB4501943), MYOD1 (M6190), GAPDH (G8795, Sigma Aldrich), myostatin (MAB788, R&D Systems, Minneapolis, MN), and myogenin (MYF4). Antibodies were visualized using ECL (Bio-Rad Laboratories, Hercules, CA), a chemiluminescent HRP antibody detection reagent. Quantification of protein expression was done using Image Lab Software (Bio-Rad Laboratories). All values were normalized to GAPDH. Three to 4 animals per treatment group were used for analysis. Blots that showed acceptable signal/noise and that passed appropriate quality control parameters (even loading controls, no disruption of bands to be assessed, appropriate transfer on Ponceau S staining) were used for the figure and data analysis. Additional replicates of these blots were not pursued due to the very limited amount of tissue available in Neb cKO animals and the large number of potential proteins of interest.
Statistical Analysis
Prism 7.0 software (GraphPad, Inc., La Jolla, CA) was used to perform ANOVAs and Tukey post-tests on animal weight, forelimb grip strength, open field and individual muscle weights. Survival data were analyzed using the Wilcoxon rank sum test using SPSS software (IBM, Armonk, NY). For average myofiber diameter measurements and Western blot data, t-tests were used to compare protein expression between different genotypes or treatments.
RESULTS
Whole Animal Studies
Vehicle-injected WT animals differentiated from vehicle-injected Neb cKO mice on the basis of animal weight by 21 days of life (DOL, p < 0.0332, Fig. 1A), and similar differences were observed when comparing individual muscle weights in vehicle-injected mice (quadriceps: 40.3 mg vs 172.2 mg, p < 0.0001; triceps: 28.7 mg vs 103.7 g, p < 0.0002; gastrocnemius: 44.4 mg vs 136.1 g, p < 0.0001; Fig. 1B). With respect to treatment, significant whole animal weight differences between WT mice ActRIIB-mFc-treated and vehicle-injected mice were short lived (from 55 to 58 days of life; 32.0 vs 27.7 g, p < 0. 0332) and did not regain significance by the end of the study (63 days). However, significant differences in individual muscle weight were observed when comparing vehicle-injected WT mice with ActRIIB-mFc-treated WT mice (quadriceps: 172.2 g vs 252.1 mg, p < 0.0001; triceps: 103.7 mg vs 159.5 g, p < 0.0002; gastrocnemius: 136.1 mg vs 206.6 g, p < 0.0001). No significant differences in animal weight or muscle weight were observed when comparing vehicle-injected Neb cKO mice to Neb cKO mice injected with ActRIIB-mFc.
FIGURE 1.

Behavioral impact of ActRIIB-mFc treatment in Neb cKO mice. (A) Total bodyweight of VEH- and ActRIIB-mFc-treated mice. Significant differences in body mass were observed when comparing genotypes, and for a very short period during the treatment of WT mice. (B) Individual muscle weights from VEH- and ActRIIB-mFc-treated mice. Treatment of WT mice with ActRIIB-mFc produced significant increases in triceps, quadriceps, and gastrocnemius muscle weights. (C) Forelimb grip force of VEH- and ActRIIB-mFc-treated mice. Treatment of WT and Neb cKO mice with ActRIIB-mFc had no effect on forelimb grip strength. (D) Open field analysis revealed no impact of treatment in either WT or Neb cKO mice. (E) Survival curve of VEH- and ActRIIB-mFc -treated mice. Regardless of treatment, Neb cKO mice did not survive as long as WT animals. Bars and asterisk denote significant (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001) differences between treatment groups with relevant comparisons. Values are based on measurements made on 9–13 animals per group for each assay.
To evaluate muscle weakness and stamina, forelimb grip strength and open field activity were assessed. Vehicle-injected Neb cKO mice did show weakness on grip strength testing compared with vehicle-injected WT mice (p < 0.0001; Fig. 1C) beginning with their first grip strength at 21 DOL and continuing until the end of the study. Treatment of WT mice with ActRIIB-mFc did not significantly increase forelimb grip strength over the course of the study compared with what was observed in vehicle-injected WT animals. ActRIIB-mFc-treated Neb cKO mice did not show a significant increase in forelimb grip strength over vehicle-injected Neb cKO mice. Interestingly, no differences were seen between WT and Neb cKO vehicle-injected animals when distance traveled was assessed in the open field (Fig. 1D), although a trend suggesting decreasing activity in Neb cKO mice was present. Treatment with ActRIIB-mFc did not affect open field outcomes. Some behavioral assessments pursued in our prior studies were not performed in this study either due to the lack of significant findings between WT and diseased Neb cKO mice (inverted screen and treadmill tests) or the shorter-than-expected duration of the study (terminal rotarod and voluntary running wheel). Overall, Neb cKO mice has a significantly reduced lifespan compared with WT mice (p = 0.001). Neb cKO ActRIIB-mFc-treated animals began dying or reaching end-stage criteria at 28 days of life with 53.8% of the animals continuing until the end of the study. Vehicle-treated Neb cKO animals also experienced an earlier mortality (39 days), with only 43.5% of the animals surviving until the end of the study. Treatment had no effect on the survival of the WT animals with both groups losing an animal at 53 days and an approximately 90% survival rate until the end of the study (WT Veh: 90.0%, WT ActRIIB-mFc: 91.7%; Fig. 1E).
Pathological Studies
Histological evaluation of mouse quadriceps muscle revealed appropriate morphology in WT mice, while Neb cKO mice displayed dramatic myopathic features (Fig. 2A). A subpopulation of fibers in Neb cKO muscle (approximately 30%) displayed myopathic features including markedly decreased fiber size, round myofiber contours, centrally located nuclei, and cytoplasmic basophilia. Notably, the majority of small myofibers in the quadriceps muscle were located along one edge of the muscle. Another significant subpopulation of fibers displayed myofiber hypertrophy with minimal central nucleation or other myopathic findings. Neither the large nor small fiber populations were restricted to a single fiber type (Fig. 2B). Nemaline rods were not clearly identifiable at the light microscopic level. Following treatment with ActRIIB-mFc, WT quadriceps muscles displayed modest myofiber enlargement (hypertrophy) without other histological abnormalities. None of the pathological findings observed in the Neb cKO mice were significantly impacted by ActRIIB-mFc treatment. In order to quantify observations of differences in myofiber size, automatic quantitative analysis of muscle histology was performed (Fig. 2B–E). Myofiber size of vehicle-injected Neb cKO mice was significantly decreased according to analysis of both cross-sectional areas (WT: 2161 vs Neb cKO: 898.2 μm2, p = 0.0007; Fig. 2C) and MinFeret diameter (WT: 40.95, Neb cKO: 23.58, p < 0.0001; Fig. 2D). Treatment with ActRIIB-mFC significantly increased fiber size in WT muscles (p = 0.0113) but had no effect in Neb cKO mice.
FIGURE 2.
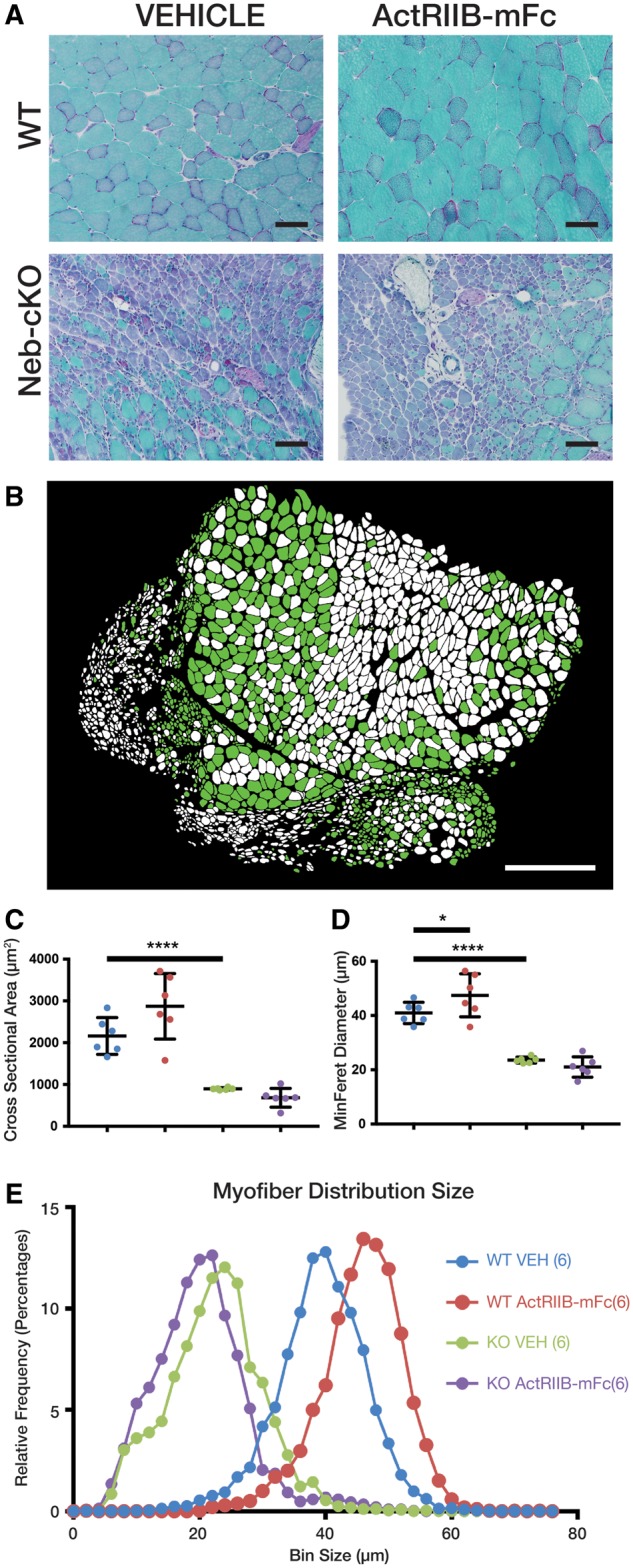
Pathology and myofiber size following ActRIIB-mFc treatment in Neb cKO mice. (A) Representative areas of transversely sectioned quadriceps muscle after sectioning and Gomori trichrome staining, representative of 7–10 animals per group. (B) Representative image of Neb cKO type 2b myosin-positive (glycolytic, green) and type 2b myosin-negative (oxidative, white) populations on a whole slide scan of one quadriceps muscle, using the Cytoinformatics automated quantification algorithm. Tissue from 6 to 7 animals per group was assessed. (C) Quadriceps myofiber cross-sectional area. (D) Quadriceps myofiber min ferret diameter. Bars and asterisk denote significant (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001) differences between treatment groups with relevant comparisons. (E) Frequency histogram depicting myofiber size distributions by minFeret diameter, with a rightward shift in curves as consistent with myofiber growth in treated WT mice.
As the presence of basophilic fibers raised the possibility of muscle regeneration, myofiber types were assessed by immunofluorescence for the expression of fast, slow, developmental, and embryonic myosin isoforms (Fig. 3). As previously described (25), Neb cKO animals had significant abnormalities in relation to the proportion of fiber types, with many more oxidative (type I and type IIA) fibers than WT animals and marked myofiber smallness particularly seen in the type IIB fiber population. These 3 fiber types accounted for the vast majority of the myofibers in all of the Neb cKO specimens, with only rare fibers positive for embryonic myosin and essentially no positivity for developmental myosin (not shown).
FIGURE 3.

Expression of myosin isoforms in Neb cKO quadriceps muscle. The expression of myosin isoforms was assessed to identify the identity of small and basophilic myofibers in the sampled tissues. Staining patterns of similar fields, serially sectioned and stained with dystrophin (red) and one of several myosin heavy chains (I, IIa, IIb, embryonic) are shown to demonstrate differences in fiber typing between WT and Neb cKO quadriceps muscles. Additionally, it is evident that very few fibers are positive for embryonic myosin and developmental myosin was negative in all samples (not shown). Bar=200 μm.
Organ histology was evaluated to assess potential off-target effects of the treatment. Histological evaluation of organs revealed no structural abnormalities in any of the treatment groups (data not shown). An in-depth evaluation of urinary tracts did not reveal urinary obstructions, which have previously been reported in other NM mouse models (30).
Hypertrophic Pathway Studies
The activation of hypertrophic pathways in WT and Neb cKO mice was investigated using Western blots. Most signaling molecules were similar when comparing between genotypes and treatment groups; however, specific molecules did show genotype- or treatment-related changes (Fig. 4). Vehicle-injected Neb cKO animals showed 136% upregulation of rpS6 as compared with the vehicle-injected WT animals (p < 0.05). While myostatin expression in WT vehicle-injected mice was undetectable, the expression was markedly increased in the Neb cKO vehicle group as compared with the WT vehicle group (p < 0.0001). Expression of p70S6K and p-p70S6K was elevated, 19% and 193%, respectively, when comparing ActRIIB-mFc-treated WT mice to vehicle-injected WT mice (p < 0.05 and p < 0.01). Phosphorylated and nonphosphorylated constituents of the Akt signaling pathway, including AKT, p-AKT, P70/S6K, p-p70/S6K, rps6, p-rps6, p-4EBP1, p-eEf2k ActRIIB, myostatin, and mTOR did not show significant differences between vehicle and ActRIIB-mFc-injected Neb cKO mice.
FIGURE 4.

Expression of hypertrophic signaling proteins in Neb cKO mice. (A) Western blot analysis of the quadriceps muscle reveals the similarities and differences between genotypes and treatment groups of 9-week-old mice. (B) A graphical representation of protein expression normalized to GAPDH is shown. Statistical analysis indicates significant relationships most notably between WT vehicle and WT drug for p70/S6K and p-p70/S6K and between WT vehicle and Neb cKO vehicle for rpS6 (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001). Note that the orange boxes identify a lane that was initially identified as a Neb cKO animal but was later found to be a heterozygote. This lane was not included in the statistical analysis of any group.
Assessment of Myogenic Precursor Populations
Immunohistochemical and Western blot studies were also performed to assess whether Neb cKO mice were deficient in populations of myogenic precursor cells. Satellite cell populations were assessed using immunohistochemistry for Pax7 and normalizing cell counts to the number of fibers within the assessed area (Fig. 5). These assessments revealed significantly higher Pax7-positive cells/myofiber when comparing vehicle-injected WT and Neb cKO mice (0.04 vs 0.10, respectively, p < 0.001) and higher Pax7-positive cells/myofiber when comparing vehicle-injected WT mice and ActRIIB-mFc-treated WT mice (0.04 vs 0.07, respective, p < 0.01). ActRIIB-mFc treatment of Neb cKO mice did not affect the number of Pax7-positive cells/myofiber. Additional markers of satellite cell differentiation assessed by Western blot included MyoD, myogenin, and Myf5 (Fig. 5). With respect to all of these markers, some differences were observed with respect to genotype but not with respect to treatment. Expression of myogenin and Myf5 in vehicle-injected WT mouse muscle was 35% and 29%, respectively, of the expression observed in vehicle-injected Neb cKO mice (p < 0.01). Expression of MyoD was not significantly different when comparing vehicle-injected WT and vehicle-injected Neb cKO mice, although this was partially due to considerable variation in Neb cKO animals while WT animals consistently displayed low expression levels. With respect to treatment, there were no significant differences in myogenin, MYF5, and MyoD due to treatment with ActRIIB.
FIGURE 5.

Expression of myogenic markers in Neb cKO mice and following treatment. (A) Pax7 immunostaining of quadriceps muscle tissue reveals increased in satellite cell number in Neb cKO mice and ActRIIB-mFc-treated WT mice in comparison to vehicle-injected WT mice (quantified in B). Treatment of Neb cKO mice did not significantly affect satellite cell number. Counts were normalized to the number of myofibers in the field due to the large difference in fiber size between genotypes. (C) Western blot analysis of the gastrocnemius muscle reveals the similarities and differences between genotypes and treatment groups of 9-week-old mice with respect to other myogenic markers. (D) A graphical representation of protein expression normalized to GAPDH is shown (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001). Note that the orange boxes identify a lane that was initially identified as a Neb cKO animal but was later found to be a heterozygote. This lane was not included in the statistical analysis of any group.
DISCUSSION
In this report, we examined the effects of myostatin inhibition in the Neb cKO mouse model of NM. The rationale for these studies was based on the presence of myofiber smallness in some NM patients and animal models, and the hypothesis that increasing fiber size pharmacologically would lead to useful gains in strength. We previously used myostatin inhibition to induce myofiber hypertrophy in 2 murine models of NM. Treatment of Acta1H40Y mice with ActRIIB-mFc produced significant increases in body mass, muscle mass, quadriceps myofiber size, and survival, but did not improve forelimb grip strength or ex vivo measurements of contractile function (30). Further investigation in the Acta1 TgActa1D286G mouse model using the second-generation myostatin inhibitor, mRK35 (Pfizer, New York, NY) demonstrated treatment-related increases in animal size and muscle strength (31). Therefore, we anticipated myostatin inhibition to have an even greater effect in the Neb cKO mouse model because hypotrophic fibers are a major pathological abnormality in this mouse model. Despite these expectations, ActRIIB-mFc treatment had no effect on animal size, muscle size, muscle strength, or disease histopathology in Neb cKO mice and thus did not provide any improvement in the disease process in this model.
When comparing the findings in the current trial to our prior, more promising studies of myostatin inhibition in the Acta1H40Y and Acta1D286G NM mouse models, several features of the Neb cKO model may have contributed to the lack of treatment response. Of the 3 models, Neb cKO mice are the most severely affected, with an earlier age of onset, lower body weight, a greater degree of muscle hypotrophy, and a shorter lifespan than the previously tested NM models. It is possible that the disruption of muscle biology imposed by the Neb cKO model is too severe to allow muscle growth to occur. Findings of myostatin inhibition in murine spinal muscular atrophy (SMA) suggest an inverse relationship between disease severity and treatment responsiveness. Treatment had no effect in a model of severe SMA (34) but improved muscle mass and function in a model of mild disease (35). While strain-related differences could have potentially contributed to differences in therapeutic response, it should be noted that the Neb cKO mouse is on a C57/BL6 background and the WT C57/BL6 littermates in this study showed positive effects of treatment. Additionally, several of our prior studies of myostatin inhibition in TgActa1D286G (31) NM mice and 2 models of XLMTM (28, 29) were conducted on this background and showed excellent muscle growth in treated WT animals.
In evaluating possible explanations for the lack of response to ActRIIB-mFc in Neb cKO mice, we investigated specific issues including (1) the expression of myostatin and its receptor, (2) protein expression in major hypertrophic signaling pathways, and (3) the possibility of a deficiency of myogenic precursors. In some diseases, myostatin is downregulated and greater downregulation increases with disease severity (36), but the Western blot data in our study clearly demonstrates higher levels of myostatin in Neb cKO mice. With respect to the myostatin receptor (ActRIIB), WT and Neb cKO mice displayed a similar level of expression. Thus, it would appear that signaling through the myostatin pathway should be occurring in Neb cKO mice, and it remains surprising that myostatin inhibition has no impact on myofiber size in these animals.
Western blots were used to determine how the Neb cKO genotype impacts the hypertrophic signaling pathway and to identify the effects of ActRIIB-mFc on the hypertrophic signaling in these animals. In our prior work, such studies have been helpful in identifying signaling abnormalities that correlated with limited treatment efficacy (29), but evaluation of this signaling system did not offer an explanation for a lack of treatment efficacy in this study. The expression of proteins associated with hypertrophic signaling was variable between animals within the same treatment group in this study, with only a few significant differences observed between treatment groups. The baseline levels of nonphosphorylated rpS6 in vehicle-injected Neb cKO mice were significantly higher than WT animals; but it is difficult to imagine how this could impact treatment efficacy without similar differences in phosphorylated rpS6. In contrast, treated WT mice had increased level of p-p70S6K compared with vehicle-injected WT mice, consistent with an activation of the Akt pathway by ActRIIB-mFc in WT animals. We have not observed this pathway activation in our prior studies of myostatin inhibition; however, our previous studies evaluated protein expression in tissue at a later time point (29–31). Overall, however, these studies do not identify any evidence of impaired hypertrophic signaling in Neb cKO mice, and so the explanation for the small fiber size and the inability to pharmacologically induce hypertrophy in Neb cKO mice remains unclear.
An additional mechanism by which muscle growth can be impaired is through abnormalities or deficiencies of critical myogenic components. Immunohistochemical studies revealed that the Pax7-positive satellite cell population was actually more numerous in Neb cKO mice in comparison to vehicle-injected WT mice, which demonstrates that satellite cell populations are intact. Interestingly, we also observed an increase in Pax7-positive cells with ActRIIB-mFc treatment of WT mice, whereas there was no significant impact on satellite cell number when comparing vehicle-injected to ActRIIB-mFc-treated Neb cKO mice. Western blots were performed to evaluate the expression of several other key myogenic signaling molecules (MyoD, myogenin, Myf5) across the 4 treatment groups in this study. While expression levels once again showed considerable variation between animals, there was no evidence of a deficiency in myogenic signaling in Neb cKO mice, and no significant impact of treatment on the expression of these proteins in either genotype. Thus, it would appear that myogenic cell populations are present in Neb cKO mice at a level that should be appropriate for muscle growth.
Given the extensive genetic heterogeneity in NM and the paucity of information on how NM mouse models relate to human patients, it is also important to consider whether ACTA-NM and NEB-NM truly represent the same disease. This study hypothesized that myostatin inhibition, which showed positive results in 2 mouse models of ACTA-NM (30, 31), would similarly produce benefits in a mouse model of NEB NM. The lack of treatment efficacy observed in the Neb cKO mouse suggests that nebulin deficiency impairs muscle growth in different fashion than can be observed in Acta1-mutant muscle, although there is no clear mechanism for this when comparing hypertrophic signaling studies across the 3 models. Alternatively, it is possible that the Neb cKO model produces abnormalities of skeletal muscle biology that are dissimilar to NEB-NM patients. The Neb cKO mouse was designed to provide postnatal nebulin deficiency as a means of improving upon mouse models devoid of nebulin expression, as those displayed death early in life and did not recapitulate many pathological aspects of NEB-NM. The Neb cKO mouse displays a gradual decrease in nebulin expression that models progressive nebulin deficiency, while most NEB-NM patients have persistently low nebulin levels since birth. Further study to identify the degree of similarity between disease progression in the Neb cKO mouse in comparison to NEB-NM patients is necessary to more fully understand the implication of our findings to the treatment of NEB-NM.
In summary, treatment with ActRIIB-mFc did not significantly improve structural or functional phenotypes in the Neb cKO mouse model of NM. While our prior work still suggests a role for myostatin inhibition in the treatment of a subset of NM, this study clearly demonstrates that some models of NM will not improve with this treatment strategy. The mechanism responsible for the failure of treatment here is unclear, but there was no evidence of abnormalities in myostatin signaling, hypertrophic signaling, or the expression of myogenic markers to explain our findings. Additional work is required to understand the appropriateness of using the Neb cKO model in treatment studies of NEB-NM and to understand how the findings of our murine treatment studies can be most effectively applied to the NM patient population.
Supplementary Material
{kind=link}
ACKNOWLEDGMENTS
ActRIIB-mFc was provided by R. Scott Pearsall, PhD at Acceleron (Cambridge, MA). Behavioral testing was performed at the Neuroscience Research Center’s Behavioral Core Facility at the Medical College of Wisconsin, which is funded by the Research and Education Initiative Fund, a component of the Advancing a Healthier Wisconsin Endowment at the Medical College of Wisconsin. Histology and imaging work was performed (in part) using the Histology and Imaging Core Facilities at the Children’s Hospital of Wisconsin Research Institute. The Myosin Heavy Chain Type IIB monoclonal antibody developed by S. Schiaffino, Universita degli Studi di Padova, was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA.
This work was supported by funding from the Muscular Dystrophy Association (MDA 295195; Improving Muscle Function in Nebulin-based Nemaline Myopathy), A Foundation Building Strength (AFBS) and NIH R01AR053897.
Disclosure: M.W.L. is a member of advisory boards for Audentes Therapeutics, Solid Biosciences, and Ichorion Therapeutics and is a consultant for Wave Life Sciences, Valerion Therapeutics, and Dynacure. The remaining authors have no relevant disclosures.
Supplementary Data can be found at academic.oup.com/jnen.
REFERENCES
- 1. Wallgren-Pettersson C, Sewry CA, Nowak KJ, et al. Nemaline myopathies. Sem Ped Neurol 2011;18:230–8 (Review) [DOI] [PubMed] [Google Scholar]
- 2. Gupta VA, Ravenscroft G, Shaheen R, et al. Identification of KLHL41 mutations implicates BTB-Kelch-mediated ubiquitination as an alternate pathway to myofibrillar disruption in nemaline myopathy. Am J Hum Genet 2013;93:1108–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ravenscroft G, Miyatake S, Lehtokari VL, et al. Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am J Hum Genet 2013;93:6–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yuen M, Sandaradura SA, Dowling JJ, et al. Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest 2015;125:456–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miyatake S, Mitsuhashi S, Hayashi YK, et al. Biallelic mutations in MYPN, encoding myopalladin, are associated with childhood-onset, slowly progressive nemaline myopathy. Am J Hum Genet 2017;100:169–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lornage X, Malfatti E, Cheraud C, et al. Recessive MYPN mutations cause cap myopathy with occasional nemaline rods. Ann Neurol 2017;81:467–73 [DOI] [PubMed] [Google Scholar]
- 7. Malfatti E, Bohm J, Lacene E, et al. A premature stop codon in MYO18B is associated with severe nemaline myopathy with cardiomyopathy. J Neuromuscul Dis 2015;2:219–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alazami AM, Kentab AY, Faqeih E, et al. A novel syndrome of Klippel-Feil anomaly, myopathy, and characteristic facies is linked to a null mutation in MYO18B. J Med Genet 2015;52:400–4 [DOI] [PubMed] [Google Scholar]
- 9. Dubowitz V, Sewry C, Oldfors A.. Congenital myopathies and related disorders In: Dubowitz V, Sewry C, Oldfors A, eds. Muscle Biopsy: A Practical Approach. China: Saunders Elsevier; 2013:358–405 [Google Scholar]
- 10. Imoto C, Nonaka I.. The significance of type 1 fiber atrophy (hypotrophy) in childhood neuromuscular disorders. Brain Dev 2001;23:298–302 [DOI] [PubMed] [Google Scholar]
- 11. Ilkovski B, Cooper ST, Nowak K, et al. Nemaline myopathy caused by mutations in the muscle alpha-skeletal-actin gene. Am J Hum Genet 2001;68:1333–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pelin K, Wallgren-Pettersson C.. Nebulin—A giant chameleon. Adv Exp Med Biol 2008;642:28–39 [PubMed] [Google Scholar]
- 13. Pelin K, Hilpela P, Donner K, et al. Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci USA 1999;96:2305–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pelin K, Donner K, Holmberg M, et al. Nebulin mutations in autosomal recessive nemaline myopathy: An update. Neuromuscul Disord 2002;12:680–6 [DOI] [PubMed] [Google Scholar]
- 15. Labeit S, Gibson T, Lakey A, et al. Evidence that nebulin is a protein-ruler in muscle thin filaments. FEBS Lett 1991;282:313–6 [DOI] [PubMed] [Google Scholar]
- 16. Gokhin DS, Bang ML, Zhang J, et al. Reduced thin filament length in nebulin-knockout skeletal muscle alters isometric contractile properties. Am J Physiol Cell Physiol 2009;296:C1123–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ottenheijm CA, Witt CC, Stienen GJ, et al. Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum Mol Genet 2009;18:2359–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chandra M, Mamidi R, Ford S, et al. Nebulin alters cross-bridge cycling kinetics and increases thin filament activation: A novel mechanism for increasing tension and reducing tension cost. J Biol Chem 2009;284:30889–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ottenheijm CA, Hooijman P, DeChene ET, et al. Altered myofilament function depresses force generation in patients with nebulin-based nemaline myopathy (NEM2). J Struct Biol 2010;170:334–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Labeit S, Ottenheijm CA, Granzier H.. Nebulin, a major player in muscle health and disease. FASEB J 2011;25:822–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Witt CC, Burkart C, Labeit D, et al. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO J 2006;25:3843–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bang ML, Li X, Littlefield R, et al. Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J Cell Biol 2006;173:905–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ottenheijm CA, Buck D, de Winter JM, et al. Deleting exon 55 from the nebulin gene induces severe muscle weakness in a mouse model for nemaline myopathy. Brain 2013;136:1718–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lawlor MW, Ottenheijm CA, Lehtokari VL, et al. Novel mutations in NEB cause abnormal nebulin expression and markedly impaired muscle force generation in severe nemaline myopathy. Skelet Muscle 2011;1:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li F, Buck D, De Winter J, et al. Nebulin deficiency in adult muscle causes sarcomere defects and muscle-type-dependent changes in trophicity: Novel insights in nemaline myopathy. Hum Mol Genet 2015;24:5219–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCroskery S, Thomas M, Maxwell L, et al. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol 2003;162:1135–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Joulia-Ekaza D, Dominique J-E, Cabello G, Gérard C.. Myostatin regulation of muscle development: Molecular basis, natural mutations, physiopathological aspects. Exp Cell Res 2006;312:2401–14 [DOI] [PubMed] [Google Scholar]
- 28. Lawlor MW, Read BP, Edelstein R, et al. Inhibition of activin receptor type IIb increases strength and lifespan in myotubularin-deficient mice. Am J Pathol 2011;178:784–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lawlor MW, Viola MG, Meng H, et al. Differential muscle hypertrophy is associated with satellite cell numbers and Akt pathway activation following activin type IIB receptor inhibition in Mtm1 p.R69C mice. Am J Pathol 2014;184:1831–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tinklenberg J, Meng H, Yang L, et al. Treatment with ActRIIB-mFc produces myofiber growth and improves lifespan in the Acta1 H40Y murine model of nemaline myopathy. Am J Pathol 2016;186:1568–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tinklenberg JA, Siebers EM, Beatka MJ, et al. Myostatin inhibition using mRK35 produces skeletal muscle growth and tubular aggregate formation in wild type and TgACTA1D286G nemaline myopathy mice. Hum Mol Genet 2018;27:638–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lawlor MW, Alexander MS, Viola MG, et al. Myotubularin-deficient myoblasts display increased apoptosis, delayed proliferation, and poor cell engraftment. Am J Pathol 2012;181:961–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wattanasirichaigoon D, Swoboda KJ, Takada F, et al. Mutations of the slow muscle alpha-tropomyosin gene, TPM3, are a rare cause of nemaline myopathy. Neurology 2002;59:613–7 [DOI] [PubMed] [Google Scholar]
- 34. Sumner CJ, Wee CD, Warsing LC, et al. Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Hum Mol Genet 2009;18:3145–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu M, Hammers DW, Barton ER, et al. Activin receptor type IIB inhibition improves muscle phenotype and function in a mouse model of spinal muscular atrophy. PLoS One 2016;11:e0166803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mariot V, Joubert R, Hourde C, et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat Commun 2017;8:1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.