

Introduction
Chordomas are rare primary bone tumors that originate from the notochord remnants. This embryonic structure is a cord of cells that retrogrades, thereby putting pressure on the spine and causing malformation. Chordomas are observed on the axial axis, mainly in the sacrococcygeal region. They are slow-growing tumors with a tendency to recur, presenting an aggressive clinical course frequently associated with metastasis.1, 2 Skin and subcutaneous tissue involvement is unusual but may eventually occur as a result of local infiltration and/or metastasis.3, 4
Here, we present the case of a 60-year-old man diagnosed with chordoma in the gluteal region associated with bone, lung, and skin metastases.
Case report
A 60-year-old man with metastatic chordoma affecting the lungs, bones, and skin was referred to the dermatology department for evaluation of the nodules growing on his scalp and right arm. Five years earlier, he had been diagnosed with chordoma of the right gluteus. The patient had extensive surgical resection of the initial gluteal tumor, followed by adjuvant radiation and chemotherapy. One year later, metastases developed in the bones, lungs, and, subsequently, skin.
On initial examination, the patient presented with multiple firm, pink, translucid, widely distributed nodules, mainly on the gluteus and right arm, followed by the scalp, neck, face, and back (Fig 1).
Fig 1.
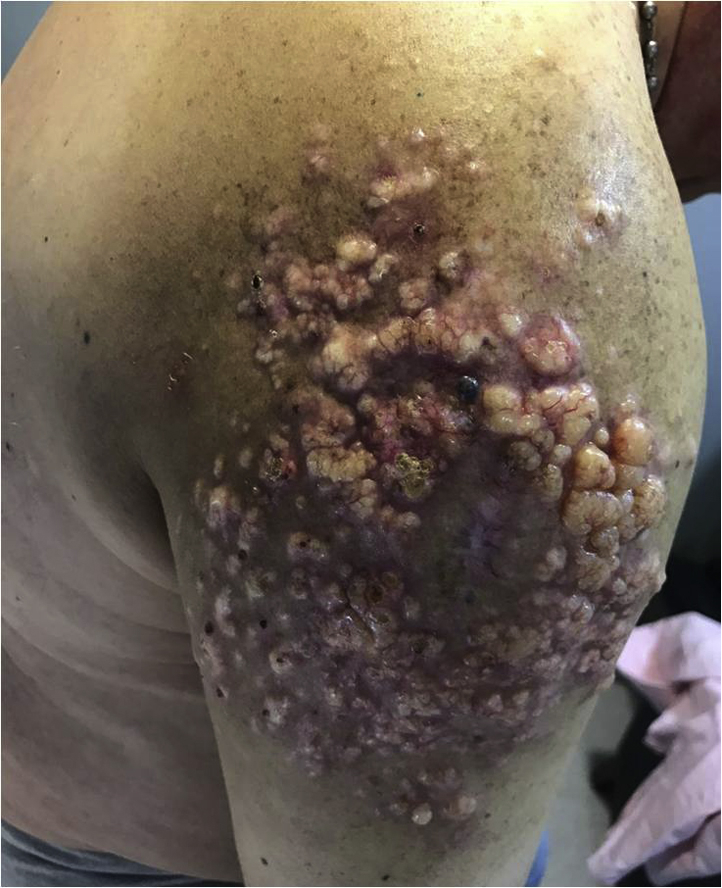
Multiple firm, translucent, nodular tumors in the right arm.
Dermoscopic examination showed multiple tumors with branched vessels over a yellow-white background (Fig 2).
Fig 2.

Dermoscopy showed branched vessels on a yellow-white background.
Histologic assessment showed that the lesions were composed of large neoplastic pleomorphic cells (physaliphorous cells), with vacuolated periodic acid–Schiff–positive cytoplasm and prominent vesicular nuclei with mild atypia. The neoplastic cells seemed to be organized in cords and lobules separated by fibrous septae within a mucoid intercellular matrix. Tumor cells were positive for cytokeratin AE1/AE3 and EMA (Fig 3).
Fig 3.

Eosinophilic pale polygonal cells with variable vacuolar presence, most centrally located. Characteristic finding: physaliphorous cells. H&E; original magnification, ×20.
After metastases developed, treatment with sunitinib was initiated, with partial response. However, after 35 months of treatment, the progression of brain metastasis caused the patient's death.
Discussion
Chordoma is an extremely rare aggressive tumor. Half of these tumors are located in the sacrococcygeal area, although they can also be found in the sphenooccipital region of the skull and along the spine.1
They generally appear between the ages of 40 and 70 years and are more common in men than women. In childhood, they are rare, but they tend to be more aggressive if they do occur.1, 2
Metastasis frequency varies between 3% and 48% and usually occurs several years after the initial diagnosis. Usual metastasis sites include the lungs (52%), liver (23%), lymph nodes (20%), and bone (16%). The skin as a primary tumor location not typical, usually occurring only by contiguity or metastasis, both of which are termed chordoma cutis in the literature.1, 3
Clinical manifestations are nonspecific, typically presenting as mucoid translucid nodules with a firm consistency and a pale, reddish brown or bluish color,5 as we observed in our patient. With dermoscopy, we found focused linear vessels on a translucent yellow-white background.
On microscopic examination, we observed a multinodular organization, analogous to cartilaginous tumors. Tumor cells are cohesive, organized in networks, cords, or nests, often surrounded by abundant mucin. Physaliphorous (“bubbly”) or signet-ring cells, which are characteristic and strongly suggestive of chordoma, are occasionally identified.2
Regarding immunohistochemical profile, chordomas express cytokeratins AE1/AE3, CK8/18, and CK19. EMA and S100 protein are almost always expressed. Brachyury is currently the most useful marker for the diagnosis of chordoma.6 In our case, brachyury was not performed; however, positive markers for cytokeratins AE1/AE3 and EMA confirmed the clinical suspicion and allowed differential diagnosis from other tumors with similar histologic features.
Surgery is the treatment of choice, but it may be complicated by vessel and nerve involvement. Therefore, aggressive surgical excision followed by radiation represents the best long-term therapeutic option.1
Cytotoxic chemotherapy is not recommended because it has not yet proven beneficial.7
Potentially relevant therapeutic targets have been identified for chordoma, including platelet-derived growth factor receptor inhibitor (imatinib), epidermal growth factor receptor inhibitors (cetuximab, gefitinib, erlotinib), mechanistic target of rapamycin inhibitor (sirolimus), and multitarget inhibitor (sunitinib). However, the inhibition of several of these targets has shown a partial response, and the 5-year survival rate for chordoma is 51%1, 8, 9; hence, new treatment strategies are needed for these patients.
Multiple distant cutaneous metastases, as seen in our patient, are relatively infrequent. Taking this into consideration, new cutaneous lesions appearing in patients with a history of chordoma should lead to a high index of suspicion of cutaneous metastasis; dermoscopy can be a beneficial tool in the early diagnosis of these tumors, thereby affecting the outcomes for these patients.
Footnotes
Funding sources: None.
Conflicts of interest: None disclosed.
References
- 1.D'Amuri A., Brunelli M., Floccari F. On a rare cutaneous metastasis from a sacrococcygeal chordoma. Case Rep Pathol. 2017;2017:5281239. doi: 10.1155/2017/5281239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Su W.P., Louback J.B., Gagne E.J., Scheithauer B.W. Chordoma cutis: a report of nineteen patients with cutaneous involvement of chordoma. J Am Acad Dermatol. 1993;29(1):63–66. doi: 10.1016/0190-9622(93)70153-k. [DOI] [PubMed] [Google Scholar]
- 3.Delteil C., Malissen N., Appay R. Chordoma cutis, an unusual clinical presentation of a rare neoplasm: chordoma. Ann Pathol. 2018;38(2):126–130. doi: 10.1016/j.annpat.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Riesco-Martínez M.C., Parrilla-Rubio L., Enguita-Valls A.B., Delgado-Márquez A.M., Ruste S., López-Martin J.A. A unique case of distant skin metastasis from chondroid chordoma. JAAD Case Rep. 2016;2(1):63–66. doi: 10.1016/j.jdcr.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubin A., Bagel J., Niedt G. Chordoma cutis. JAAD Case Rep. 2005;52(5):s105–s108. doi: 10.1016/j.jaad.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 6.Miettinen M., Wang Z., Lasota J., Heery C., Schlom J., Palena C. Nuclear brachyury expression is consistent in chordoma, common in germ cell tumors and small cell carcinomas, and rare in other carcinomas and sarcomas: an immunohistochemical study of 5229 cases. Am J Surg Pathol. 2015;39(10):1305–1312. doi: 10.1097/PAS.0000000000000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stacchiotti S., Sommer J. Building a global consensus approach to chordoma. Lancet Oncol. 2015;16(2):e71–e830. doi: 10.1016/S1470-2045(14)71190-8. [DOI] [PubMed] [Google Scholar]
- 8.George B., Bresson D., Herman P. Chordoma: a review. Neurosurg Clin N Am. 2015;26(3):437–452. doi: 10.1016/j.nec.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Liplaa A., Dijkstra S., Gelderblom H. Efficacy of pazopanib and sunitinib in advanced axial chordoma: a single reference centre case series. Clin Sarcoma Res. 2016;6:19. doi: 10.1186/s13569-016-0059-x. [DOI] [PMC free article] [PubMed] [Google Scholar]