

Abstract
Artemisinin and its derivatives (ARTs) were reported to display heme-dependent antitumor activity. On the other hand, histone deacetylase inhibitors (HDACi) were known to be able to promote heme synthesis in erythroid cells. Nevertheless, the effect of HDACi on heme homeostasis in non-erythrocytes remains unknown. We envisioned that the combination of HDACi and artesunate (ARS) might have synergistic antitumor activity through modulating heme synthesis. In vitro studies revealed that combination of ARS and HDACi exerted synergistic tumor inhibition by inducing cell death. Moreover, this combination exhibited more effective antitumor activity than either ARS or HDACi monotherapy in xenograft models without apparent toxicity. Importantly, mechanistic studies revealed that HDACi coordinated with ARS to increase 5-aminolevulinate synthase (ALAS1) expression, and subsequent heme production, leading to enhanced cytotoxicity of ARS. Notably, knocking down ALAS1 significantly blunted the synergistic effect of ARS and HDACi on tumor inhibition, indicating a critical role of ALAS1 upregulation in mediating ARS cytotoxicity. Collectively, our study revealed the mechanism of synergistic antitumor action of ARS and HDACi. This finding indicates that modulation of heme synthesis pathway by the combination based on ARTs and other heme synthesis modulators represents a promising therapeutic approach to solid tumors.
Keywords: Artesunate, HDAC inhibitor, Heme, ALAS1, Antitumor
Abbreviations: ALA, 5-aminolevulinic acid; ALAD, 5-aminolevulinate dehydratase; ALAS, 5-aminolevulinate synthase; ARS, artesunate; ART, artemisinin; CCK-8, cell counting kit 8; CI, combination index; CMCNa, carboxymethyl cellulose; DHA, dihydroartemisinin; DMAB, (dimethylamino)benzaldehyde; FECH, ferrochelatase; GSDME, gasdermin E; HDAC, histone deacetylase; HDACi, HDAC inhibitor; HMBS, hydroxymethylbilane synthase; KD, knockdown; KO, knockout; LBH589, panobinostat; PDT, photodynamic therapy; PI, propidium iodide; PpIX, protoporphyrin IX; ROS, reactive oxygen species; SA, succinyl acetone; SAHA, vorinostat; sgRNA, single guide RNA; WT, wild-type
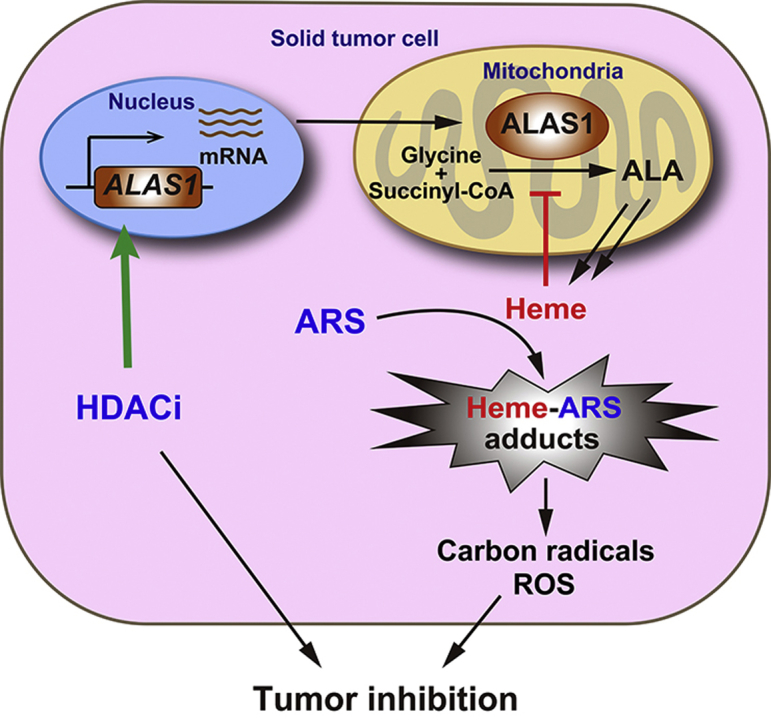
Graphical abstract
HDAC inhibitors (HDACi) cooperate with artesunate (ARS) to synergistically induce tumor cell death through promoting ALAS1 expression and subsequent heme synthesis, leading to enhanced cytotoxicity of ARS. This finding demonstrates a promising therapeutic approach to solid tumors based on modulating heme synthesis by the combination of artemisinin derivatives with HDACi.
1. Introduction
Heme, a complex of ferrous iron (Fe2+) and protoporphyrin IX (PpIX), is an essential molecule to all aerobic organisms. In mammals, heme synthesis is conducted by eight enzymes localized in the mitochondrion or cytosol. The key enzyme, 5-aminolevulinate synthase (ALAS), is the first and rate-limiting enzyme that mediates biosynthesis of 5-aminolevulinic acid (ALA) from glycine and succinyl CoA. Two isoforms of ALAS with distinct tissue distribution have been identified. ALAS1, also known as non-specific ALAS (ALAS-N), is ubiquitously expressed in both erythrocytes and non-erythrocytes while ALAS2 (also called as ALAS-E) is specifically expressed in erythroid cells1. Notably, excessive heme or heme precursors is toxic to cells and tissues2. Thus, the biosynthesis of heme is strictly controlled in non-erythrocytes by negative feedback mechanisms via ALAS1 repression by excessive heme through reduction of transcription and translation, destabilization of mRNA, inhibition of mitochondrial transport of precursor protein, and degradation3, 4. In erythroid cells, the regulation of ALAS2 is much different from that of ALAS1, as a huge amount of heme is needed for hemoglobin production5.
In tumor cells, the ability of heme biosynthesis seems to be higher than that in normal cells6, 7. Notably, heme precursor ALA has been in clinical use to produce the photosensitizer PpIX allowing for photodynamic therapy (PDT) for cancers8, 9. The antimalarial drugs, artemisinin (ART) and its derivatives (ARTs) have been reported to exhibit heme-dependent antitumor activity10, 11, 12, 13, 14. The mechanism of antitumor action of ARTs is considered to be similar to that of their antimalarial action. That is, heme or none-heme Fe2+ triggers the cleavage of endoperoxide bridge of ARTs, producing carbon centered radicals that alkylate multiple proteins, lipids and DNA, leading to oxidative stress, apoptosis, ferroptosis, necrosis, arrest of cell cycle, and inhibition of angiogenesis15, 16, 17. On the other hand, histone deacetylases inhibitors (HDACi) have been reported to promote erythroid differentiation with increased ALAS2 expression and heme synthesis18, 19, 20. Nevertheless, the effect of HDACi on heme synthesis and homeostasis in non-erythrocytes is still unclear.
Combination therapy using two or more therapeutic agents, is progressively emerging as a cornerstone of cancer therapy. This approach exhibits enhanced efficacy21, 22, 23, 24, 25, 26 in an additive or synergistic manner, potentially also reducing drug resistance27 and adverse effects28, 29. In an earlier study, Zhang et al30. found that HDACi facilitated dihydroartemisinin (DHA)-induced apoptosis in hepatocellular cancer cells, and proposed a mechanism involving altered ERK phosphorylation and MCL-1 expression. In this study, we verified a novel mechanism involving the synergistic modulation of heme synthesis by the combination of HDACi and ARTs to combat against solid tumors. We first confirmed the synergistic antitumor effect of artesunate (ARS) and pan-HDACi (SAHA and LBH589) as well as isoform specific HDACi (romidepsin) in several cancer cell lines. Then, the results of in vivo study showed that the combination treatment exhibited a greater anti-tumor effect on xenograft tumor in mice than the single-agent treatment group with no obvious toxicity. Mechanistic studies revealed that HDACi synergized with ARS to sustainably upregulate ALAS1 expression and thus promote heme synthesis, which in turn enhanced antitumor action of ARS. While this paper was under review, Lee et al31. reported that hemin (oxidized version of heme with Fe3+) in combination with metformin could suppress tumor growth.
2. Materials and methods
2.1. Reagents
ARS, succinyl acetone (SA), ALA, hemin, (dimethylamino)benzaldehyde (DMAB) and perchloric acid were bought from Sigma–Aldrich (St. Louis, MO, USA). SAHA, LBH589, romidepsin, CI994 and tubastatin A were purchased from Selleckchem (Houston, TX, USA). PpIX was obtained from Aladdin (Shanghai, China). A 50 mmol/L stock solution of ARS or SAHA dissolved in DMSO was prepared and stored at −20 °C and refreshed monthly. A 100 mol/L stock solution of LBH589 was prepared using DMSO and stored at −20 °C. Primary antibodies against ALAS1 (Cat#ab154860), ALAD (Cat#ab151697), HMBS (Cat#ab129092), FECH (Cat#ab137042) and ALAS2 (Cat#ab184964) were purchased from Abcam (Cambridge, UK, USA).
2.2. Cell cultures and growth conditions
Huh-7, Hep3B, HCT116 and PANC-1 cells were purchased from the Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). All these cells were verified by STR analysis, provided by the Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences and reconfirmed by Guangzhou Cellcook Biotech Co., Ltd. (Guangzhou, China). Huh-7 and Hep3B cell were not contaminated by mycoplasma, provided by Guangzhou Cellcook Biotech Co., Ltd. (Guangzhou, China). Mycoplasma testing on HCT116 and PANC-1 cells were not performed, as cell morphology, in vitro growth properties and tumor formation in nude mice provide evidence of health. The Huh-7 cells and PANC-1 cells were maintained in DMEM medium, Hep3B cells were maintained in MEM medium, HCT116 cells were maintained in McCoy's 5A medium supplemented with 10% fetal bovine serum (FBS), 100 μg/mL penicillin and 100 U/mL streptomycin in a 5% CO2 humidified incubator at 37 °C.
2.3. Cell viability assays
Cells were seeded at about 3000 cells/well in 96-well plates within medium supplemented with 10% FBS for 24 h, and subsequently treated with compounds for 48 or 72 h as indicated. Cell viability were determined by MTT (3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay or cell counting kit 8 (CCK-8) assay as previously described32, 33.
2.4. Flow cytometry
Apoptosis levels were analyzed after staining with annexin V and propidium iodide (PI) by flow cytometry (BD Accuri C6, BD Biosciences, Franklin Lakes, NJ, USA) as previously described32, 33, 34.
2.5. Construct ALAD knockout cells
ALAD knockouts were generated using the CRISPR-Cas9 technology as described35, 36. Briefly, ALAD single guide RNA (sgRNA) was ligated into the lentiCRISPR V2 vector (Focus Bioscience Co., Ltd., Nanchang, China); sgRNA sequence: 5ʹ-AGCGGCTGGAAGAGATGCTG-3ʹ. LentiCRISPR-sgRNA-ALAD plasmid was co-transfected with pMD2.G and psPAX2 into HEK-293T cells for 48 h. The recombinant viruses were collected and added to Hep3B cells cultured with 8 μg/mL polybrene for 24 h. After puromycin selection, we performed single-cell cloning by seeding survival cells into 96-well plates at ratio of 0.6 cell per well. Individual clones were screened by Western blot for ALAD protein expression and further identify null alleles or frame-shift mutation by sequence of TA cloning.
2.6. Protein sample preparation and Western blot assay
After treated with the compounds for the indicated times, cells were lysed by RIPA buffer (150 mmol/L NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mmol/L Tris-HCl, pH 7.5) containing protease and phosphatase inhibitor cocktail (Merck Millipore, Billerica, MA, USA) with freshly added 1 mmol/L of PMSF. After centrifugation at 12,000 rpm (MicroCL 17R, Thermo Fisher Scientific, Waltham, MA, USA) at 4 °C for 20 min, supernatants were collected and equal amounts of protein were subjected to Western blot assay as described before37, 38. After being visualized using an electrochemiluminescence kit (Sudgen, China), luminescence was assessed by chemiluminescent imaging system (Tanon, Shanghai, China).
2.7. Real-time reverse transcription–PCR
Total RNA from cultured tumor cells was extracted using TRIzol (Tiangen, Beijing, China) as previously described39, 40. 500 ng of total RNA of each sample were used for reverse transcription (Takara, Tokyo, Japan). Specific primer pairs for real-time PCR are as follows: ALAS1, GGC AGC ACA GAT GAA TCA GAG AG (for) and TTC AGC AAC CTC TTT CCT CAC GG (rev); ALAD, TTC CAC CCA CTA CTT CGG (for) and GCT ACC ACC TGA CAT CCT G (rev); HMBS, TGC AAC GGC GGA AGA AAA (for) and ACG AGG CTT TCA ATG TTG CC (rev); ALAS2, GCA GCA CTC AAC AGC AAG (for) and ACA GGA CGG CGA CAG AAA (rev) and GAPDH, CGG AGT CAA CGG ATT TGG TCG TAT (for) and AGC CTT CTC CAT GGT GGT GAA GAC (rev).
2.8. Whole cell heme measurement
Tumor cells cultured in 10 cm dish and were treated with compounds for 14 h. After washed twice with PBS, cells were lysed in extraction solution (100 mmol/L NaCl, 2.5% Triton X-100, 50 mmol/L Tris-HCl, pH 7.5), mixed with vortex and centrifuged at 5000×g for 10 min. Equal volume of the supernatant were mixed with 1.5 mol/L oxalic acid and heated at 100 °C for 30 min to release iron from heme, generating fluorescent PpIX. After cooling, fluorescence was assessed by Enspire (PerkinElmer, Waltham, MA, USA) at Ex 405 nm/Em 600 nm and were normalized by protein content. Meanwhile, samples diluted with PBS, without heating were performed to determine the endogenous fluorescence. Standards made by hemin were used to quantify heme values in all the samples. Alternatively, cells cultured in 12-well plate were refreshed with serum free medium containing ALA or PpIX for another 4 h and then subjected to heme measurement same as above.
2.9. Transfections of siRNAs
Duplexes of siRNA were synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China). The three siRNA sequences targeting human ALAS1 are as follows: si ALAS1#1, ACA ACA GAG AUU UGC CUG CTT; si ALAS1#2, AGG UAG UCA UUA CUG CAC CTT; si ALAS1#3, ACG UAG AUG UUA UGU CUG CTT. siRNA sequence for negative control is UUC UCC GAA CGU GUC ACG UTT. Hep3B and Huh-7 cells cultured in 96-well plates or 12-well plates were transfected with a concentration of 30 nmol/L of siRNA duplexes using Lipofectamine™ RNAiMAX Reagent (Invitrogen, Carlsbad, CA, USA). Afterwards, the cells were performed as described in the figure legends.
2.10. ALA measurement
Tumor cells cultured in 15 cm dish were washed with PBS and lysed in 1 mL of 1% Triton X-100 after treatment with compounds. After 15 min centrifuging at 12,000 rpm (MicroCL 17R, Thermo Fisher, Waltham, MA, USA) at 4 °C, 500 μL of the supernatant of each sample was mixed with equal volume of sodium acetate buffer (pH 4.6) and 100 μL of ethyl acetoacetate, and heated at 100 °C for 10 min. After cooling, 200 μL of ethyl acetate was added and mixed thoroughly, later layered by centrifuging at 12,000 rpm (MicroCL 17R, Thermo Fisher, Waltham, MA, USA) for 5 min. 100 μL of the upper layer was transferred to react with Ehrlich reagent and was determined colorimetrically at 553 nm. Ehrlic reagent was prepared as follows: 1 g of DMAB is dissolved in 30 mL of glacial acetic acid, then 5 mL of 70% perchloric acid are added, followed by 5 mL H2O and the solution is diluted to 50 mL with glacial acetic acid41. This reagent should be used freshly since it is unstable.
2.11. Intracellular reactive oxygen species (ROS) detection
Huh-7 cells were seeded at 5 × 105 cells/well in 6-well plates and allowed to adhere overnight, followed by treatment with ARS, SAHA, or the combination for 48 h. Then, cells were probed with DCFH (2.5 μmol/L) for 30 min in the dark. After being washed with PBS for at least three times, cells were trypsin digested and collected. Rinsed cells were transferred at about 3 × 104 cells/well into black with clear bottom 96-well plates (Corning 3603, Corning, NY, USA) and ROS was measured by Enspire (PerkinElmer, Waltham, MA, USA) at Ex 488 nm/Em 525 nm and were normalized by protein content.
2.12. Subcutaneous xenograft model
Female nude mice (5–6 weeks) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). A total of 5 × 106 HCT116 and Hep3B cells were subcutaneously inoculated to nude mice. When the tumors reached a mean group size of about 100 mm3, mice were randomly divided into four groups receiving sodium carboxymethyl cellulose (CMCNa) as vehicle, ARS (50 mg/kg), SAHA (50 mg/kg), and combination (50 mg/kg ARS plus 50 mg/kg SAHA) daily by intragastric administration for 3 weeks. Tumor volume was calculated as (length × width2)/2.
2.13. Statistical analysis
Data are presented as mean ± SD. The statistical significance of the differences was assessed by unpaired Student's t-test.
3. Results
3.1. Synergistic antitumor activity of ARS and HDAC inhibitors in vitro
We first examined the therapeutic potential of the combination of ARS and pan-HDACi, vorinostat (SAHA) on the cell viability of various human cancer cell lines, including hepatocellular cancer cells (Huh-7 and Hep3B), colorectal cancer cell (HCT116), non-small cell lung cancer (A549) and pancreatic cancer cell (PANC-1). As shown in Fig. 1A and Supporting Information Fig. S1A, co-treatment with ARS and SAHA decreased cell viability more potently than treatment with each drug alone. Combination index (CI) values were calculated to determine the interaction between ARS and SAHA by using Compusyn software (version 1.0). CI values < 0.9 indicate synergism, CI values < 0.5 indicate strong synergism, CI values of 0.9–1.1 indicate an additive effect, and CI values > 1.1 indicate antagonism. Combination index analysis showed that there was a synergistic or strong synergistic effect between ARS and SAHA at most of the tested concentrations (Fig. 1A).
Figure 1.

ARS and pan-HDACi synergistically induce cell death in solid cancer cells. (A)–(B) Effects of ARS, SAHA (A) or LBH-589 (B) and combinations of the two compounds on the cell viability of Huh-7 and Hep3B hepatocellular cancer cells, HCT116 colorectal cancer cell, A549 lung cancer cells and PANC-1 pancreatic cancer cell, as measured by MTT assay after 72 h treatment. Error bars represent SD of triplicate experiments. CI values were calculated using Compusyn software for the ARS and HDACi concentrations used in the MTT assay. (C)–(D) Representative microphotographs of Huh-7 and Hep3B cells after receiving with ARS (20 μmol/L), SAHA (2 μmol/L) (C) or LBH-589 (25 nmol/L) (D) and combination treatment for 48 h respectively. Extensive cell death, swollen cells with large bubbles commonly seen after treatment with both agents. Scale bars = 40 μm. (E)–(F) Flow cytometric analysis of Hep3B cells treated with vehicle control, ARS (20 μmol/L), SAHA (2 μmol/L) (E) or LBH-589 (25 nmol/L) (F) and the combination as indicated for 48 h. Cell death was analyzed by annexin V/PI assay.
To further confirm the effect of SAHA was due to its HDAC inhibitory activity, another pan-HDACi, panobinostat (LBH589) was used. Similarly, the results showed that ARS in combination with LBH589 synergistically reduced cell viability in Huh-7, Hep3B, A549 and PANC-1 cells (Fig. 1B and Fig. S1B).
We noticed that, combined treatment with ARS and SAHA or LBH589 displayed extensive cell death, characterized by swollen cells with membrane blowing large bubbles, favoring necrotic features (Fig. 1C and D). Annexin V/PI assay was employed to quantify cell death in Hep3B cells treated with ARS, HDACi or combination of the two compounds for 48 h (Fig. 1E and F). As shown in Fig. 1E, 64.9% of the cells exhibited dying following combination treatment, while monotherapy with either compound caused fewer dying cells (19.2% of the ARS-treated cells, 17.6% of the SAHA-treated cells). Most of the dying cells in combination treatment group were annexin V-positive/PI-positive, whereas, the annexin V-positive/PI-negative cells were rarely detected (Fig. 1E and F). Furthermore, caspase-3 activation and PARP cleavage as well as gasdermin E (GSDME) cleavage42, an indicator of pyroptosis that triggered by activated caspase-3, were not obviously increased in the combination treated cells (data not shown). These data indicated that apoptosis as well as pyroptosis was unlikely the major form of cell death caused by the combination therapy. We also observed that combination therapy induced much more ROS generation compared to monotherapy (Fig. S1C). This data indicated that increased ROS generation might at least partially account for enhanced efficacy of combination therapy.
In humans, 18 isoforms of HDACs have been recognized and are classified into four groups, including class I (HDAC-1, -2, -3, and -8), class II (HDAC-4, -5, -6, -7, and -9), class III (sirtuins 1–7), and class IV (HDAC-11)43. To verify which HDAC isoforms might be critical for the synergistic effect between ARS and HDACi, we tested several isoform-selective inhibitors, including romidepsin (inhibitor of HDAC-1 and HDAC-2), CI994 (inhibitor of HDAC-1–HDAC-3) and tubastatin A (inhibitor of HDAC-6). We found that romidepsin, to a lesser extent, CI994, potentially enhanced Hep3B cells sensitivity to ARS (Fig. 2A and Fig. S1D), while tubastatin A addition did not show additional efficacy. The synergistic manner of ARS and romidepsin was also observed in two non-small cell lung cancer cell lines, A549 and H1975 (Fig. 2B). In agreement with pan-HDACi, romidepsin also induced more cell death upon ARS treatment (Fig. 2C and D; Fig. S1E). The findings suggested that HDAC-1and HDAC-2 inhibition might be were effective to sensitize tumor cells to ARS.
Figure 2.

Effect of HDAC isoform specific inhibitors on the cytotoxicity of ARS. (A) Cell viability of Hep3B cells treated with romidepsin (left), CI994 (middle) or tubastatin A (right) in combination with ARS. MTT assay were performed after 72 h treatment. Error bars represent SD of triplicate experiments. CI values are shown below the corresponding concentrations. (B) Romidepsin synergizes with ARS to inhibit A549 (left) and H1975 (right) cells. CI values were calculated as shown. (C) Representative microphotographs of Hep3B cells treated with ARS (20 μmol/L), romidepsin (5 nmol/L) and combination for 48 h (scale bars 40 μm). (D) Flow cytometric analysis of cell death of Hep3B cells treated with vehicle control, ARS (20 μmol/L), romidepsin (5 nmol/L) or the combination for 48 h by annexin V/PI assay.
3.2. Heme synthesis is required for the synergistic antitumor activity of ARS and HDACi
To test our hypothesis that HDACi increase tumor cell sensitivity to ARS by inducing heme biosynthesis, we firstly need to confirm the relationship between heme synthesis and ARS. Though a couple of previous studies indicated that heme mediated the cytotoxicity of ARTs, by using chemical modulators involved in heme biosynthesis pathway. While some other investigations pointed the intracellular none-heme Fe2+ that mediated the antitumor activity of ARTs. Therefore, re-evaluation of the role of heme synthesis in antitumor action of ARS was required.
We took hepatocellular cancer cell Huh-7 and Hep3B cells as examples to assess the cytotoxicity of ARS in the presence or absence of exogenous molecules that promote or block the heme synthesis pathway (Fig. 3A and B). Notably, treatment of SA, an ALAD inhibitor, markedly rescued the viability of the cancer cells after ARS treatment (Fig. 3A). Meanwhile, increasing heme synthesis by addition of ALA or PpIX increased ARS-induced cytotoxicity (Fig. 3A). To further confirm this, SA was co-treated with ALA in the presence of ARS in both Huh-7 and Hep3B cells, and the results showed that SA completely countered the cytotoxicity-enhancing effect of ALA (Fig. 3A).
Figure 3.

Cytotoxicity of ARS depends on heme synthesis. (A) Huh-7 cells (left) and Hep3B cell (right) were exposed to various concentrations of ARS (0, 5, 10, 20, 40 and 80 μmol/L) alone or in combination with ALA (1 mmol/L), PpIX (5 μmol/L) or succinyl acetone (SA, 0.5 mmol/L) for 48 h followed by CCK-8 assay. Error bars represent SD of quadruplicate experiments. (B) Schematic representation of heme biosynthetic pathway and its exogenous modulators. (C) Hep3B cells were transfected with lentiCRISPR-sgRNA-ALAD or mock and ALAD knockdown was verified by Western blot assay. (D) ALAD knockdown cells and mock cells in the presence or absence of 0.5 mmol/L SA were treated with 0.5 mmol/L ALA or vehicle for 4 h for whole cell heme measurement (n = 4). (E) Cell viability was determined by CCK-8 assay after 72 h treatment with ARS as indicated. Error bars represent SD of triplicate experiments. Asterisks, ALAD KD vs Mock; *P < 0.05, **P < 0.01, ***P < 0.001 (Student's t-test). (F) Western blot analysis of ALAD in Hep3B parental cells, WT and KO clones. Western blot shows ALAD detection from one of three independent experiments. (G) ALAD KO cells, WT cells and parental cells were administered with 0.5 mmol/L ALA for 4 h for whole cell heme measurement (n = 3). (H) Cell viability was measured by MTT assay after 72 h treatment with ARS as indicated. Error bars represent SD of triplicate experiments. Asterisks, KO vs WT; *P < 0.05, **P < 0.01, ***P < 0.001 (Student's t-test). (I) ALAD KO cells and WT cell were treated with various concentrations of ARS alone or in combination with ALA (1 mmol/L) for 48 h. Cell viability was performed by CCK-8 assay. Error bars represent SD of triplicate experiments.
Considering that SA might have some nonspecific effects, we disrupted ALAD using CRISPR/Cas9 technology in Hep3B cells (Supporting Information Fig. S2A). We firstly analyzed the CRISPR-mediated ALAD knockdown (KD) cells. Results from Western blot assay showed that ALAD was efficiently knocked down (Fig. 3C). As expected, exogenous ALA induced heme synthesis was dramatically attenuated in ALAD KD cells, similar to the effect of SA (Fig. 3D). Importantly, a marked reduction in ARS cytotoxic activity was observed in Hep3B KD cells (Fig. 3E and Fig. S2B). To exclude the influence of growth rate, the effects of ARS were also examined in the absence of serum, though no obvious difference in cell viability was observed within four days (Fig. S2C). In agreement with the results in Fig. 3E, cytotoxic activity of ARS was also significantly blocked in KD cells in the absence of serum (Fig. S2D).
We next isolated two ALAD knockout (ALAD KO) clones from Hep3B cells that showed complete loss of ALAD protein expression as well as two clones that had undergone the selection for CRISPR-sgRNA transfection but lacked deletion of ALAD, and hence they expressed wild-type (WT) ALAD as their parental cells that remained intact (Fig. 3F). ALAD null clones exhibited similar growth rate with control cells during the observation period (Fig. S2E). However, functionally, ALAD null clones lost exogenous ALA induced heme generation (Fig. 3G). Importantly, loss of ALAD completely rescued the viability of Hep3B cells after ARS treatment alone or in the presence of ALA (Fig. 3H and I). All these data demonstrate that ARS-induced cytotoxicity is dependent on heme synthesis and increasing heme synthesis could enhance the cytotoxicity of ARS.
Then, we decided to identify whether heme synthesis is involved in the synergistic effect of ARS and HDACi, SA was applied in the presence or absence of ARS, HDACi and the combination treatment. We found that SA significantly rescued the viability of tumor cells exposed to the combination of ARS and HDACi (Fig. 4A and B). Consistently, reduced cell death was observed in SA rescued group (Fig. 4C and D). More importantly, we noticed that the viability of tumor cells exposed to the combination of ARS, HDACi and SA was almost equivalent to that of cells exposed to the combination of HDACi and SA in all the tested solid tumor cell lines (Fig. 4A and B; Supporting Information Figs. S3A and B). As the schematic diagram showed (Fig. 4E), these results indicated that the synergistic antitumor effect of ARS and HDACi mainly relied on heme biosynthesis. Consistently, ALAD knocking down or deletion showed the similar effect to SA treatment (Fig. 4F and G; Figs. S3C–E). Annexin V/PI assay further confirmed that blocking heme synthesis in ALAD KO cells completely abrogated the increased cell death induced by combination treatment with ARS and LBH589 (Fig. 4H).
Figure 4.

Synergistic antitumor effect of ARS and HDACi relies on heme synthesis. (A)–(B) Huh-7 cells (left) and Hep3B cell (right) were exposed to ARS, HDACi [SAHA for (A); LBH589 for (B)] or their combination in the presence or absence of SA (0.5 mmol/L) for 72 h. Cell viability was measured by MTT assay. **P < 0.01, ***P < 0.001 (Student's t-test). (C)–(D) Images of Huh-7 cells (up) and Hep3B cells (down) treated with the combination of ARS (20 μmol/L) and SAHA (2 μmol/L) (C) or LBH-589 (25 nmol/L) (D) with or without SA (0.5 mmol/L) (scale bars 40 μm). (E) Schematic representation of anti-tumor effect of combination treatment with ARS and HDACi in the presence or absence of SA. (F)–(G), ALAD knockout clones or WT cells were treated with 20 μmol/L of ARS or HDACi [2 μmol/L of SAHA for (F) and 25 nmol/L LBH589 for (G)] or the combination for 72 h (F) or 48 h (G) and cell viability was measured. (H) ALAD deletion blocked the combination treatment of ARS (20 μmol/L) and LBH589 (25 nmol/L) induced cell death.
3.3. ARS and HDACi synergistically increase ALAS1 expression
In mammalian cells, heme biosynthesis is mediated by a series of enzymes (Fig. 3B). We first assessed the expression levels of the key enzyme ALAS1. Interestingly, we observed that ALAS1 protein was markedly increased after ARS treatment for 24 h, and it increased even more upon combination treatment with ARS and SAHA (Fig. 5A and Supporting Information Fig. S4A). Whereas, SAHA alone treatment only slightly increased it (Fig. 5A and Fig. S4A). Similar results were achieved with ARS and LBH589 combination treatment (Fig. 5B and Fig. S4B). To confirm that the upregulation of ALAS1 was not the transit effect, a time course of ALAS1 protein levels was analyzed. As shown in Fig. 5C, ALAS1 started to increase at 2 h treatment with ARS alone or in combination with LBH589 and further increased afterwards. Notably, the combination treatment with ARS and LBH589 induced more ALAS1 than that induced by ARS alone treatment, which was more pronounced after 6 h treatment. Nevertheless, LBH589 alone treatment appeared minor increase in ALAS1. Real-time PCR analysis revealed that HDACi alone treatment as well as ARS alone treatment seems only slightly increased the mRNA level of ALAS1, while combination treatment significantly enhanced it (Fig. 5D). These results indicated that an increase in ALAS1 protein in combination treatment group probably attributed to combination treatment-induced ALAS1 transcription together with ARS-induced upregulation of ALAS1 protein likely by post-transcriptional modification.
Figure 5.

HDACi synergizes with ARS to induce ALAS1 expression. (A)–(B) Western blot validation of heme biosynthetic enzyme ALAS1 in Huh-7 cell and Hep3B cell in the absence and presence of ARS, HDACi [SAHA (A) or LBH589 (B)], or both treatment for 24 h. β-Actin served as loading control. (C) Time courses of ALAS1 of Hep3B cell received ARS, LBH589, or both treatments. The values under each blot are the ALAS1 expression relative to controls at the same time taken as 1 after normalization by β-actin. (D) Real-time PCR was performed for ALAS1 of Huh-7 cell and Hep3B cell in the absence (normalized as 1) and presence of ARS, HDACi (SAHA or LBH589), or both treatment for 24 h. (E) Hep3B cells were transiently transfected with three ALAS1 siRNA or control siRNA (si NC) or remained untreated (control). Knock down efficiency of ALAS1 was examined by Real-time PCR analysis (upper) and Western blot assay (lower). GAPDH served as loading control. (F) Hep3B cells were treated with siRNA as described for (E) 24 h prior to administrate with ARS, SAHA or the combination as indicated for 48 h. Cell viability was measured by MTT assay. (G)–(H) Huh-7 cells were treated as described for (E) and (F). ALAS1 knockdown significantly blocked synergistic antitumor effect of ARS and SAHA. *P < 0.05, **P < 0.01, ***P < 0.001 (compared as indicated), #P < 0.05, ##P < 0.01 (compared with si NC group treated with ARS), †P < 0.05, ††P < 0.01, †††P < 0.001 (compared with si NC group treated with ARS + SAHA) (Student's t-test).
To identify whether the synergistic upregulation of ALAS1 mediated the synergistic antitumor activity of ARS and HDACi, ALAS1 was knocked down by siRNA in Hep3B cells. Knockdown efficiency was examined by real-time PCR and Western blot assay (Fig. 5E). ALAS1 knockdown significantly rescued the viability of Hep3B cells after ARS treatment (Fig. 5F). More importantly, the synergistic antitumor effect of ARS and SAHA seen in control and si NC groups was significantly attenuated in the ALAS1 knockdown groups (Fig. 5F). Similar results were obtain in Huh-7 cells (Fig. 5G and H). These results demonstrated an important role of ALAS1 upregulation in mediating synergistic antitumor effect of ARS and HDACi.
We then examined other enzymes involved in heme biosynthesis. HMBS and FECH were not apparently influenced by either ARS, or HDACi or combination treatment either at the protein levels or mRNA levels (Supporting Information Fig. S5A–C). Moreover, both the protein and mRNA levels of ALAD were slightly decreased by ARS treatment, but significantly increased upon HDACi treatment as well as combination treatment (Fig. S5A, E and F). Since ALAD is the redundant protein in cells, it was unlikely that its increment would influence heme production and consequently cell sensitivity to ARS. We also assessed the expression status of ALAS2. Compared to ALAS1, mRNA level of ALAS2 is very low in Huh-7 cells and ARS, SAHA and combination treatment did not apparently altered it (Fig. S5G). To confirm this, another two pairs of ALAS2 primers were used to detect ALAS2 mRNA levels in Huh-7, Hep3B and HCT116 cells and similar results were observed (data not shown). Interestingly, ALAS2 protein was apparently detected in Huh-7 (Fig. S5H), Hep3B and HCT116 cells (data not shown). Yet, the protein levels were not altered after ARS, or SAHA or combination treatment (Fig. S5H).
We next sought to understand the following issues: (i) Why did ARS alone treatment largely increase the protein levels of ALAS1? (ii) Why did HDACi alone not apparently enhance ALAS1 transcription but dramatically increase ALAS1 at both mRNA and protein levels when combined with ARS?
3.4. ARS consumed “free heme” and upregulates ALAS1 by negative feedback
Previous investigations have documented that ALAS1 is undergone the negative feedback regulation by heme. On the other hand, ARTs are thought to react with heme to form covalent heme–artemisinin adducts or some other intermediate44, 45. Thus, we presumed that ARS might react with cellular “free heme” to form heme–ARS adduct, resulting in reduction in free heme. Loss of free heme diminished the repression of ALAS1 expression, and thus more ALAS1 would be expressed to compensate consumed free heme. Consistent to our hypothesis, exogenous ALA or PpIX-induced free heme was significantly reduced after ARS treatment (Fig. 6A and Supporting Information Fig. S6). Moreover, ARS substantially overcame exogenous PpIX-induced repression of ALAS1 expression (Fig. 6B). In contrast, ARS could not further increase ALAS1 in the presence of SA (Fig. 6C). Together, ARS treatment decreased the levels of “free heme”, resulting in upregulation of ALAS1 protein by negative feedback regulation.
Figure 6.

ARS consumed cellular heme and facilitated HDACi to stably induce synergistic increase in ALAS1 expression. (A) Huh-7 cell (left) and Hep3B cell (right) were pretreated with or without ARS (20 μmol/L) for 12 h, followed with 100 μmol/L ALA in serum free medium or 1 μmol/L PpIX treatment for another 4 h. Heme was measured with the cell lysate. n = 3–4, *P < 0.05, **P < 0.01, ***P < 0.01 (Student's t-test). (B) Representative Western blot analysis of ALAS1 in Hep3B cells treated with various concentration of PpIX in the absence or presence of ARS. (C) The protein levels of ALAS1 in Hep3B cell treated with ARS or SAHA in the absence or presence of SA (0.5 mmol/L). (D) Hep3B cells receiving LBH589 as indicated, in the absence or presence of SA were subjected to Western blot validation of ALAS1. (E)–(F) Hep3B WT and ALAD KO cells were treated with concentrations of SAHA (E) or LBH589 (F) as indicated for 24 h and the amounts of ALAS1 and ALAD were analyzed by Western blot assay. (G) Time course of mRNA level of ALAS1 in Huh-7 cells and Hep3B cells treated with SAHA (4 μmol/L), SA (0.5 mmol/L) or both. (H) Western blot validation of ALAS1 in Hep3B cells treated with SAHA (5 μmol/L), in the absence or presence of SA (0.5 mmol/L) or PpIX (1 μmol/L) or both. (I) HDACi promoted ALA biosynthesis in Huh-7 cells. After 24 h treatment with SAHA (5 μmol/L) or LBH-589 (0.5 μmol/L), cells were lysed for ALA measurement. (J) HDACi promoted heme biosynthesis. Hep3B cells were administrated with ARS, SAHA or the combination in the presence or absence of SA.
3.5. HDACi sustainably induce ALAS1 transcription only when the negative feedback regulation is weakened or inhibited
Interestingly, despite HDACi alone caused negligible upregulation of ALAS1 protein, in the presence of SA, HDACi dramatically enhanced ALAS1 protein levels (Fig. 6C and D). Consistently, HDACi markedly increased ALAS1 protein levels in Hep3B ALAD KO cells, but not in Hep3B WT cells (Fig. 6E and F). These data indicated that ALAS1 might be regulated by HDAC. To further confirm this, a time course experiment was performed to detect the transcript of ALAS1. We found that, upon SAHA treatment, the mRNA levels of ALAS1 increased within 2 h and plateaued between 6 and 10 h, then went down to the baseline at about 24 h in Huh-7 cell as well as in Hep3B cell (Fig. 6G). These results illustrated that HDACi indeed induced ALAS1 expression initially, but later on this effect seemed to be overcome by negative feedback regulation. However, when SA was present, the negative feedback regulation was weakened or inhibited through SA-mediated blockade of heme synthesis. In this case, SAHA induced and maintained the high expression levels of ALAS1 (Fig. 6G). Conversely, when exogenous PpIX was added, a strong repression of ALAS1 expression by negative feedback regulation occurred, and in this case SAHA was insufficient to upregulate ALAS1 expression no matter whether SA was present or not (Fig. 6H). Together, we propose that HDACi synergizes with ARS to induce ALAS1 expression as heme-consuming ARS weakens heme-induced negative feedback repression of ALAS1.
3.6. HDACi promotes ALA and heme biosynthesis
Given the fact that ALAS1 catalyzes the production of ALA, we further measured the amount of ALA. The endogenous ALA was below the detection range of our assay, while addition of SA caused dramatic accumulation of ALA (data not shown). We found that LBH589 and SAHA markedly enhanced ALA content in the presence of SA (Fig. 6I). To further verify whether this enhancement of ALA would contribute to heme synthesis, we examined the heme production. Notably, we observed a slight but significant increase in heme levels after SAHA treatment (Fig. 6J). Not surprisingly, ARS somewhat countered the effect of SAHA (Fig. 6J). Collectively, these results revealed that HDACi promoted ALA production and consequently heme synthesis. On the other hand, ARS reduced the amount of SAHA-induced heme, consistent with the above data showing that ARS caused consumption of “free heme”.
3.7. Combination treatment with ARS and SAHA reduces tumor growth rate on xenograft tumor in mice
Having demonstrated the synergistic antitumor activity of ARS and SAHA in various tumor cell lines, we further determined this effect in vivo. HCT116 cells and Hep3B cells were subcutaneously injected in nude mice to establish tumor xenograft. Nude mice bearing tumor xenografts were randomized to the respective treatment groups and tumor sizes were measured. The mice were treated daily by intragastrical administration of ARS (50 mg/kg) and/or SAHA (50 mg/kg) for 3 weeks. As noted, the combination therapy inhibited colorectal tumor growth by 57.6% while the single SAHA treatment only inhibited the tumor growth by 22.6% and single ARS treatment showed no apparent antitumor effect (Fig. 7A and B). Mice receiving single agent or combination treatment didn't yield body weight loss (Fig. 7C), indicating that the drugs at the tested dosage were safe. For the liver tumor, similarly, the mean tumor size of combination treatment was significantly reduced compared with vehicle or single-agent treatment groups (Fig. 7D). No apparent toxic effect on body weight was found in all the treated groups (Fig. 7E). These results indicated that the combination treatment exhibited a greater anti-tumor effect in xenografts in vivo than the single-agent treatment group with no obvious toxicity.
Figure 7.

Combination treatment with ARS and SAHA reduces the tumor growth rate in xenograft models. (A)–(B) Mice transplanted with HCT116 human xenografts were treated daily for 20 days with vehicle, ARS (0.5 mg/kg); SAHA (50 mg/kg) or both agents. Tumor volumes are expressed as mean and SD (n = 10–12). A photograph of representative HCT116 tumors excised from the mice with last treatments (B). (C) Body weight of each group is presented as mean and SD (n = 10–12). (D)–(E) Nude mice bearing Hep3B tumor were treated as (A) for 21 days. Tumor size (D) and body weight (E) are expressed as mean and SD (n = 7). Statistical differences of tumor volume were calculated toward the end treatment. *P < 0.05, **P < 0.01 (Student's t-test).
4. Discussion
Heme is an essential molecule to all eukaryotic cells, and its levels in cancer cells are much higher than in normal cells6, 7. The purpose of this study was to explore a combinatory therapeutic approach to solid tumors with non-erythrocyte nature through modulating heme synthesis pathway.
The antimalarial drugs, artemisinin and its derivatives (ARTs) exhibited heme-dependent antitumor activity. Heme is considered as the activator of ARTs as ferrous iron-containing heme can cleave the endoperoxide moiety of ARTs, resulting in tumor-killing radicals such as carbon centered radicals and ROS10, 11, 46. Moreover, the non-heme iron could also trigger the activation of ARTs47, 48. In the present study, by using heme synthesis modulators (e.g., ALA, PpIX, SA) together with ALAD knockdown and knockout cells, we provided convincing evidence that heme biosynthesis was required for the cytotoxicity of ARS, further supporting the important role of heme in activating ARTs.
HDAC inhibition has been reported to increase heme content via a significant increase in ALAS2 gene transcripts in erythroid cells49, 50, 51. Nevertheless, the effect of HDACi on heme synthesis as well as ALAS1 expression in non-erythrocytes remains elusive. We found that, in solid tumor cells, HDACi initially significantly promoted ALAS1 transcription, but later on the mRNA levels of ALAS1 declined to basal levels. This declining was supposed to be mediated by negative feedback regulation by heme. Actually, a slight but significant increase in heme levels was observed in the presence of HDACi, and more importantly, when negative feedback regulation was blocked by SA or ALAD deletion, HDACi stably increased ALAS1 transcription and subsequent increased ALAS1 protein.
Combination treatment with ARS and pan-HDACi (SAHA and LBH589), as well as isoform specific HDACi (romidepsin) exerted synergistic antitumor activity by inducing cell death in vitro. Zhang et al30. observed that HDACi facilitated DHA-induced apoptosis in hepatocellular cancer cells, and proposed a mechanism involving altered ERK phosphorylation and MCL-1 expression. However, our mechanical studies revealed that, HDACi coordinated with ARS to promote ALAS1 expression, and subsequent heme generation, leading to enhanced cytotoxicity of ARS. As mentioned above, HDACi alone treatment could not stably increase ALAS1 transcription because of the negative feedback repression by heme. However, during the combination treatment, ARS consumed the “free” heme by forming heme-ARS adducts and attenuated the feedback repression of ALAS1, which facilitated HDACi to sustainably increase ALAS1 expression. As noticed, ARS alone treatment increased ALAS1 protein contents. This phenomenon was mainly owing to the removal of heme's inhibitory effect on ALAS1 probably through post-transcriptional regulation, as the levels of ALAS1 mRNA were only negligibly increased. Anyway, the increased ALAS1 protein contents by ARS treatment to compensate the consumed heme seemed not sufficient or might not reach the threshold for the cytotoxicity of ARS in most tumor cells, as the antitumor activity of ARS alone was low both in vitro and in vivo. However, HDACi in combination with ARS further dramatically increased ALAS1 protein levels by promoting ALAS1 transcription, leading to significantly enhanced tumor cell sensitivity to ARS (Fig. 8).
Figure 8.

Schematic representation of HDACi synergizing with ARS to combat against tumor cell through elevated heme synthesis via synergistic upregulation of ALAS1 expression.
The results of in vivo study on xenograft models showed that ARS monotherapy did not significantly inhibit tumor growth, which was different from the study results from some of the literature reports46, 52. This might be due to the different drug delivery, as we used intragastric administration, while others mainly used intraperitoneal injection administration46, 52. Studies from Chou group53 also showed no significant tumor growth retardation effect of DHA when oral administrated. It is supposed that increase in dosage of ARS or change in delivery may improve the antitumor efficacy, as low bioavailability and short elimination half-life of ARTs were found after oral administration in animals54. Notably, the combination treatment with ARS and SAHA reduced the tumor size in a higher extent than vehicle or single-agent treatment group, which is in accordance with the previous study30. In the course of our research, Wang et al46. reported that ART and the heme precursor ALA combination enhanced anti-colorectal cancer activity, which is consistent with our findings.
5. Conclusions
In conclusion, the present study demonstrates that the combination treatment with HDACi and ARS exhibits synergistic antitumor effect in various cancer cells in vitro and in xenograft models. Mechanical studies reveal that the synergistic upregulation of ALAS1 by HDACi and ARS modulates the intracellular heme homeostasis which mediates enhanced tumor cell sensitivity to ARS. This finding promotes the conception that drugs that can increase heme synthesis or ALAS1 protein levels may have the potential to enhance anticancer efficacy of ARTs. Moreover, this combination treatment approach may explore the clinical utility of HDACi in treating solid tumors. Collectively, we have proposed and verified a promising therapeutic approach to solid tumors based on modulating heme synthesis by the combination of ARTs with heme synthesis modulators such as HDACi.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (grants 31501182, 81573299 and 81730094); the “111 Project” from the Ministry of Education of China to Hongbin Sun; the State Administration of Foreign Experts Affairs of China, China (No. 111-2-07); Fundamental Research Funds for the Central Universities, China (2632017ZD05).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2019.05.001.
Appendix A. Supporting information
The following are the Supplementary data to this article:
References
- 1.Zhang Y., Zhang J., An W., Wan Y., Ma S., Yin J. Intron 1 GATA site enhances ALAS2 expression indispensably during erythroid differentiation. Nucleic Acids Res. 2017;45:657–671. doi: 10.1093/nar/gkw901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sardh E., Harper P., Balwani M., Stein P., Rees D., Bissell D.M. Phase 1 trial of an RNA interference therapy for acute intermittent porphyria. N Engl J Med. 2019;380:549–558. doi: 10.1056/NEJMoa1807838. [DOI] [PubMed] [Google Scholar]
- 3.Lathrop J.T., Timko M.P. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science. 1993;259:522–525. doi: 10.1126/science.8424176. [DOI] [PubMed] [Google Scholar]
- 4.Kubota Y., Nomura K., Katoh Y., Yamashita R., Kaneko K., Furuyama K. Novel mechanisms for heme-dependent degradation of ALAS1 protein as a component of negative feedback regulation of heme biosynthesis. J Biol Chem. 2016;291:20516–20529. doi: 10.1074/jbc.M116.719161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J., Li Y., Tong J., Gao J., Guo Q., Zhang L. Long non-coding RNA-dependent mechanism to regulate heme biosynthesis and erythrocyte development. Nat Commun. 2018;9:4386. doi: 10.1038/s41467-018-06883-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Navone N.M., Polo C.F., Frisardi A.L., Andrade N.E., Baille A.M. Heme biosynthesis in human breast cancer—mimetic "in vitro" studies and some heme enzymic activity levels. Int J Biochem. 1990;22:1407–1411. doi: 10.1016/0020-711x(90)90230-z. [DOI] [PubMed] [Google Scholar]
- 7.Sohoni S., Ghosh P., Wang T., Kalainayakan S.P., Vidal C., Dey S. Elevated heme synthesis and uptake underpin intensified oxidative metabolism and tumorigenic functions in non-small cell lung cancer cells. Cancer Res. 2019;79:2511–2525. doi: 10.1158/0008-5472.CAN-18-2156. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z., Liu Y., Chen Y., Li L., Lan P., He D. Transdermal delivery of 5-aminolevulinic acid by nanoethosome gels for photodynamic therapy of hypertrophic scars. ACS Appl Mater Interfaces. 2019;11:3704–3714. doi: 10.1021/acsami.8b17498. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J., Jiang C., Figueiró Longo J.P., Azevedo R.B., Zhang H., Muehlmann L.A. An updated overview on the development of new photosensitizers for anticancer photodynamic therapy. Acta Pharm Sin B. 2018;8:137–146. doi: 10.1016/j.apsb.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mercer A.E., Copple I.M., Maggs J.L., O'Neill P.M., Park B.K. The role of heme and the mitochondrion in the chemical and molecular mechanisms of mammalian cell death induced by the artemisinin antimalarials. J Biol Chem. 2011;286:987–996. doi: 10.1074/jbc.M110.144188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S., Gerhard G.S. Heme mediates cytotoxicity from artemisinin and serves as a general anti-proliferation target. PLoS One. 2009;4:e7472. doi: 10.1371/journal.pone.0007472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo Z. Artemisinin anti-malarial drugs in China. Acta Pharm Sin B. 2016;6:115–124. doi: 10.1016/j.apsb.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo Z. The modification of natural products for medical use. Acta Pharm Sin B. 2017;7:119–136. doi: 10.1016/j.apsb.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Efferth T., Dunstan H., Sauerbrey A., Miyachi H., Chitambar C.R. The anti-malarial artesunate is also active against cancer. Int J Oncol. 2001;18:767–773. doi: 10.3892/ijo.18.4.767. [DOI] [PubMed] [Google Scholar]
- 15.Krishna S., Bustamante L., Haynes R.K., Staines H.M. Artemisinins: their growing importance in medicine. Trends Pharmacol Sci. 2008;29:520–527. doi: 10.1016/j.tips.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhaw-Luximon A., Jhurry D. Artemisinin and its derivatives in cancer therapy: status of progress, mechanism of action, and future perspectives. Cancer Chemother Pharmacol. 2017;79:451–466. doi: 10.1007/s00280-017-3251-7. [DOI] [PubMed] [Google Scholar]
- 17.Slezakova S., Ruda-Kucerova J. Anticancer activity of artemisinin and its derivatives. Anticancer Res. 2017;37:5995–6003. doi: 10.21873/anticanres.12046. [DOI] [PubMed] [Google Scholar]
- 18.Zheng L., Fu Y., Zhuang L., Gai R., Ma J., Lou J. Simultaneous NF-κB inhibition and E-cadherin upregulation mediate mutually synergistic anticancer activity of celastrol and SAHA in vitro and in vivo. Int J Cancer. 2014;135:1721–1732. doi: 10.1002/ijc.28810. [DOI] [PubMed] [Google Scholar]
- 19.Pili R., Liu G., Chintala S., Verheul H., Rehman S., Attwood K. Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in patients with clear-cell renal cell carcinoma: a multicentre, single-arm phase I/II clinical trial. Br J Canc. 2017;116:874–883. doi: 10.1038/bjc.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nervi C., De Marinis E., Codacci-Pisanelli G. Epigenetic treatment of solid tumours: a review of clinical trials. Clin Epigenet. 2015;7:127. doi: 10.1186/s13148-015-0157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai X., Stiff A., Duggan M., Wesolowski R., Carson W.E., 3rd, Friedman A. Modeling combination therapy for breast cancer with BET and immune checkpoint inhibitors. Proc Natl Acad Sci U S A. 2018;115:5534–5539. doi: 10.1073/pnas.1721559115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu C., Zhang Y., Rolfe P.A., Hernández V.M., Guzman W., Kradjian G. Combination therapy with NHS-muIL12 and avelumab (anti-PD-L1) enhances antitumor efficacy in preclinical cancer models. Clin Cancer Res. 2017;23:5869–5880. doi: 10.1158/1078-0432.CCR-17-0483. [DOI] [PubMed] [Google Scholar]
- 23.Bryant K.L., Stalnecker C.A., Zeitouni D., Klomp J.E., Peng S., Tikunov A.P. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25:628–640. doi: 10.1038/s41591-019-0368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kinsey C.G., Camolotto S.A., Boespflug A.M., Guillen K.P., Foth M., Truong A. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019;25:620–627. doi: 10.1038/s41591-019-0367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao M., Guo W., Wu Y., Yang C., Zhong L., Deng G. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharm Sin B. 2019;9:304–315. doi: 10.1016/j.apsb.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang X., Tan L., Shi K., Peng J., Xiao Y., Li W. Gold nanorods together with HSP inhibitor-VER-155008 micelles for colon cancer mild-temperature photothermal therapy. Acta Pharm Sin B. 2018;8:587–601. doi: 10.1016/j.apsb.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leary M., Heerboth S., Lapinska K., Sarkar S. Sensitization of drug resistant cancer cells: a matter of combination therapy. Cancers. 2018;10:E483. doi: 10.3390/cancers10120483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gandhi L., Rodríguez-Abreu D., Gadgeel S., Esteban E., Felip E., De Angelis F. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 29.Neoptolemos J.P., Palmer D.H., Ghaneh P., Psarelli E.E., Valle J.W., Halloran C.M. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet. 2017;389:1011–1024. doi: 10.1016/S0140-6736(16)32409-6. [DOI] [PubMed] [Google Scholar]
- 30.Zhang C.Z., Pan Y., Cao Y., Lai P.B., Liu L., Chen G.G. Histone deacetylase inhibitors facilitate dihydroartemisinin-induced apoptosis in liver cancer in vitro and in vivo. PLoS One. 2012;7 doi: 10.1371/journal.pone.0039870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J., Yesilkanal A.E., Wynne J.P., Frankenberger C., Liu J., Yan J. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature. 2019;568:254–258. doi: 10.1038/s41586-019-1005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song Z., Chen C.P., Liu J., Wen X., Sun H., Yuan H. Design, synthesis, and biological evaluation of (2E)-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)acetate derivatives as anti-proliferative agents through ROS-induced cell apoptosis. Eur J Med Chem. 2016;124:809–819. doi: 10.1016/j.ejmech.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Huang Y., Sun G., Wang P., Shi R., Zhang Y., Wen X. Synthesis and biological evaluation of Complex I inhibitor R419 and its derivatives as anticancer agents in HepG2 cells. Bioorg Med Chem Lett. 2018;28:2957–2960. doi: 10.1016/j.bmcl.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 34.Zhang T., Chen Y., Ge Y., Hu Y., Li M., Jin Y. Inhalation treatment of primary lung cancer using liposomal curcumin dry powder inhalers. Acta Pharm Sin B. 2018;8:440–448. doi: 10.1016/j.apsb.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen C.P., Chen X., Qiao Y.N., Wang P., He W.Q., Zhang C.H. In vivo roles for myosin phosphatase targeting subunit-1 phosphorylation sites T694 and T852 in bladder smooth muscle contraction. J Physiol. 2015;593:681–700. doi: 10.1113/jphysiol.2014.283853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turiján-Espinoza E., Salazar-González R.A., Uresti-Rivera E.E., Hernández-Hernández G.E., Ortega-Juárez M., Milán R. A pilot study of the modulation of sirtuins on arylamine N-acetyltransferase 1 and 2 enzymatic activity. Acta Pharm Sin B. 2018;8:188–199. doi: 10.1016/j.apsb.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z., Guo X., To K.K., Chen Z., Fang X., Luo M. Olmutinib (HM61713) reversed multidrug resistance by inhibiting the activity of ATP-binding cassette subfamily G member 2 in vitro and in vivo. Acta Pharm Sin B. 2018;8:563–574. doi: 10.1016/j.apsb.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holopainen T., Huang H., Chen C., Kim K.E., Zhang L., Zhou F. Angiopoietin-1 overexpression modulates vascular endothelium to facilitate tumor cell dissemination and metastasis establishment. Cancer Res. 2009;69:4656–4664. doi: 10.1158/0008-5472.CAN-08-4654. [DOI] [PubMed] [Google Scholar]
- 41.Mauzerall D., Granick S. The occurrence and determination of δ-aminolevulinic acid and porphobilinogen in urine. J Biol Chem. 1956;219:435–446. [PubMed] [Google Scholar]
- 42.Wang Y., Gao W., Shi X., Ding J., Liu W., He H. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 43.Wang X., Wei X., Pang Q., Yi F. Histone deacetylases and their inhibitors: molecular mechanisms and therapeutic implications in diabetes mellitus. Acta Pharm Sin B. 2012;2:387–395. [Google Scholar]
- 44.Zhang S., Gerhard G.S. Heme activates artemisinin more efficiently than hemin, inorganic iron, or hemoglobin. Bioorg Med Chem. 2008;16:7853–7861. doi: 10.1016/j.bmc.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 45.Robert A., Benoit-Vical F., Claparols C., Meunier B. The antimalarial drug artemisinin alkylates heme in infected mice. Proc Natl Acad Sci U S A. 2005;102:13676–13680. doi: 10.1073/pnas.0500972102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J., Zhang J., Shi Y., Xu C., Zhang C., Wong Y.K. Mechanistic investigation of the specific anticancer property of artemisinin and its combination with aminolevulinic acid for enhanced anticolorectal cancer activity. ACS Cent Sci. 2017;3:743–750. doi: 10.1021/acscentsci.7b00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang N.D., Tan S.H., Ng S., Shi Y., Zhou J., Tan K.S. Artesunate induces cell death in human cancer cells via enhancing lysosomal function and lysosomal degradation of ferritin. J Biol Chem. 2014;289:33425–33441. doi: 10.1074/jbc.M114.564567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamacher-Brady A., Stein H.A., Turschner S., Toegel I., Mora R., Jennewein N. Artesunate activates mitochondrial apoptosis in breast cancer cells via iron-catalyzed lysosomal reactive oxygen species production. J Biol Chem. 2011;286:6587–6601. doi: 10.1074/jbc.M110.210047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han L., Lu J., Pan L., Wang X., Shao Y., Han S. Histone acetyltransferase p300 regulates the transcription of human erythroid-specific 5-aminolevulinate synthase gene. Biochem Biophys Res Commun. 2006;348:799–806. doi: 10.1016/j.bbrc.2006.07.147. [DOI] [PubMed] [Google Scholar]
- 50.Han L., Zhong Y., Huang B., Han L., Pan L., Xu X. Sodium butyrate activates erythroid-specific 5-aminolevulinate synthase gene through Sp1 elements at its promoter. Blood Cells Mol Dis. 2008;41:148–153. doi: 10.1016/j.bcmd.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 51.Canh Hiep N., Kinohira S., Furuyama K., Taketani S. Depletion of glutamine enhances sodium butyrate-induced erythroid differentiation of K562 cells. J Biochem. 2012;152:509–519. doi: 10.1093/jb/mvs097. [DOI] [PubMed] [Google Scholar]
- 52.Wang S.J., Gao Y., Chen H., Kong R., Jiang H.C., Pan S.H. Dihydroartemisinin inactivates NF-κB and potentiates the anti-tumor effect of gemcitabine on pancreatic cancer both in vitro and in vivo. Cancer Lett. 2010;293:99–108. doi: 10.1016/j.canlet.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 53.Moore J.C., Lai H., Li J.R., Ren R.L., McDougall J.A., Singh N.P. Oral administration of dihydroartemisinin and ferrous sulfate retarded implanted fibrosarcoma growth in the rat. Cancer Lett. 1995;98:83–87. [PubMed] [Google Scholar]
- 54.Li Q.G., Peggins J.O., Fleckenstein L.L., Masonic K., Heiffer M.H., Brewer T.G. The pharmacokinetics and bioavailability of dihydroartemisinin, arteether, artemether, artesunic acid and artelinic acid in rats. J Pharm Pharmacol. 1998;50:173–182. doi: 10.1111/j.2042-7158.1998.tb06173.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.