

Abstract
Aryl hydrocarbon receptor (AhR), a cellular chemical sensor, controls cellular homeostasis, and sphingosine-1-phosphate (S1P), a bioactive intermediate of sphingolipid metabolism, is believed to have a role in immunity and inflammation, but their potential crosstalk is currently unknown. We aimed to determine whether there is a functional linkage between AhR signaling and sphingolipid metabolism. We showed that AhR ligands, including an environmental polycyclic aromatic hydrocarbon (PAH), induced S1P generation, and inhibited S1P lyase (S1PL) activity in resting cells, antigen/IgE-activated mast cells, and mouse lungs exposed to the AhR ligand alone or in combination with antigen challenge. The reduction of S1PL activity was due to AhR-mediated oxidation of S1PL at residue 317, which was reversible by the addition of an antioxidant or in cells with knockdown of the ORMDL3 gene encoding an ER transmembrane protein, whereas C317A S1PL mutant-transfected cells were resistant to the AhR-mediated effect. Furthermore, analysis of AhR ligand-treated cells showed a time-dependent increase of the ORMDL3–S1PL complex, which was confirmed by FRET analysis. This change increased the S1P levels, which in turn, induced mast cell degranulation via S1PR2 signaling. In addition, elevated levels of plasma S1P were found in children with asthma compared to non-asthmatic subjects. These results suggest a new regulatory pathway whereby the AhR–ligand axis induces ORMDL3-dependent S1P generation by inhibiting S1PL, which may contribute to the expression of allergic diseases.
Keywords: Aryl hydrocarbon receptor, ORMDL sphingolipid biosynthesis regulator 3, Sphingosine-1-phosphate, Sphingosine-1-phosphate lyase
Introduction
Aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor and has been identified as an important regulator controlling cellular and tissue homeostasis.1,2 Many environmental pollutants and endogenous metabolites have been recognized as ligands for AhR, providing a potential mechanistic link in the environmental exposure–disease relationship.3,4 This is supported by our recent findings that the AhR–ligand axis controls cellular Ca2+ signaling, the endoplasmic reticulum (ER), and mitochondrial stress response in mast cells.5,6 However, the exact mechanisms through which AhR regulates cellular responses are still unclear. ORMDL sphingolipid biosynthesis regulator 3 (ORMDL3) is an ER-located transmembrane protein and was originally believed to negatively regulate serine palmitoyltransferase (SPT), which catalyzes the first and rate-limiting step in sphingolipid (SL) de novo synthesis, in response to changes in SL/ceramide levels and cellular activation.7–12 Furthermore, several single nucleotide polymorphisms of the ORMDL3 gene locus have been associated with an increased risk of childhood asthma and many inflammatory diseases.13–17
Recent experimental models utilizing ectopic over-expression, transgenic, and gene-deficient systems have shown that over-expression or downregulation of ORMDL3 can regulate Ca2+ homeostasis, ER stress response and SL metabolism,8,10–12,18–21 suggesting its potential importance in the control of asthma and inflammatory diseases. However, the accumulated evidence has provided a complex picture, due, in part, to the different models used, particularly in regard to the role of ORMDL3 in the control of SPT and the consequent expression of disease phenotypes. For example, while mice carrying human ORMDL3 transgenes show increased airway hyper-responsiveness spontaneously in the absence of airway inflammation,21 lung Clara cell-specific ORMDL3-null mice exhibit increased allergen-induced airway responsiveness independent of inflammation20 perhaps through the induction of smooth muscle contraction by sphingosine-1-phosphate (S1P).20,22 Mice with reduced SPT levels showed decreased lung ceramide levels but increased bronchial reactivity without inflammation,23 whereas others have reported that inhibition of ceramide biosynthesis attenuated antigen-induced airway inflammation and bronchoconstriction.8 Collectively, the exact role of ORMDL3 in controlling SPT activity and SL metabolism remains unclear, and how ORMDL3 is regulated and contributes to the expression of asthma and inflammatory diseases remains to be elucidated. In fact, several recent studies have refuted the role of ORMDL3 in regulating SPT,8,10,11 suggesting that it instead has a potential effect on the downstream pathway.
In this regard, S1P is a bioactive intermediate of SL metabolism that regulates a diverse range of cellular processes involved in immunity, inflammation, and inflammatory disorders through S1P receptor (S1PR)-dependent and independent pathways.24 In particular, S1P has been shown to have a key role in mediating the mast cell response25 and airway smooth muscle contraction.22 S1P is produced by sphingosine phosphorylation, which is catalyzed by sphingosine kinase (SPHK), and cleaved by an ER-located protein, sphingosine-1-phosphate lyase (S1PL); S1PL is encoded by SGPL1, which catalyzes the final and irreversible degradation step in the SL metabolic pathway by cleaving S1P to generate ethanolamine phosphate and an alkyl aldehyde.26
In this study, we discovered that several AhR ligands, including a prominent environmental PAH, were inducers of ORMDL3-dependent S1P generation in mast cells and epithelial cells, resulting in enhanced cellular responses. Notably, this was mediated through a blockade of S1PL activity via enhanced oxidation of S1PL, providing new mechanistic insight into how ORMDL3 is involved in AhR-mediated S1P generation and its potential functional importance in asthma and inflammatory diseases, particularly as it pertains to exposure to the ubiquitous environmental PAHs.
Materials and methods
The materials and detailed methods used in this study, including generation of mouse and human mast cells, treatment of epithelial A549 cells, S1P measurement, S1PL activity analysis, analysis of the in vivo mouse model, transfection, degranulation, ROS measurement, confocal fluorescence resonance energy transfer (FRET) analysis, recruitment of study subjects, plasma sample collection, and various statistical analyses, are provided in the Supplementary Methods.
Results
AhR ligands induce S1P generation by inhibiting S1P lyase (S1PL) activity
To investigate the functional impact of the AhR–ligand axis on S1P generation in mast cells and epithelial cells as a model, we first assessed a mouse mast cell line, MC/9, and found that the intracellular and secreted S1P levels were significantly increased in a time-dependent manner upon stimulation of the cells with 6-formylindolo[3,2-b]carbazole (FICZ), an endogenous AhR agonistic ligand (Fig. 1a). As expected, increased S1P was found in cells treated with a S1PL inhibitor, THI [2-acetyl-4-(tetrahydroxybutyl) imidazole], as a positive control. Increased levels of intracellular and secreted S1P were also observed in mouse bone marrow-derived mast cells (BMMCs) after stimulation with several known AhR agonistic ligands, including FICZ, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), and indeno[1,2,3-cd]pyrene (IP), compared with those observed in vehicle-treated cells (Fig. 1b). Similar enhancement by AhR ligands was also found in AhR ligand-treated human peripheral blood-derived human mast cells, HCMCs, from two individuals (Fig. 1c, subject #1; Figure S1a, subject #2) and a human mast cell line, HMC-1 (Figure S1b). Addition of the AhR antagonist CH223191 reversed the effects of the AhR ligand (Fig. 1c; Figure S1b).
Fig. 1.

AhR ligands induce S1P synthesis by inhibiting S1PL activity in vitro. S1P levels of a 1 nM FICZ-treated MC/9 cells for the indicated time. At time = 0, the cells were treated with an equal amount of vehicle control; b 1 nM FICZ, 0.1 nM TCDD, and 1 nM IP-treated BMMCs and c 0.1 μM Bap, 1 nM FICZ, 0.1 nM TCDD, and 1 nM IP ± 10 μM CH223191-treated HCMCs (subject #1) for 1 h were assessed using a S1P ELISA kit. FICZ induces mast cell degranulation through the S1P–S1PR2 axis. d MC/9 cells were treated with 1 nM FICZ ± 1 μM JTE-013 for 6 h or e sensitized with E-C1 ± 1 nM FICZ for 16 h in the absence or presence of 1 μM JTE-013 and then stimulated with 10 μg/mL OVA for 30 min, and f BMMCs were treated with 1 nM FICZ ± 1 μM JTE-013 for 6 h. Degranulation was monitored by the release of β-hexosaminidase (Hex). S1PL activity of g MC/9 and h HMC-1 cells treated as described in a. At time = 0, the cells were treated with an equal amount of vehicle control. i BMMCs treated as described in b, and j HCMCs of subject #1 treated as described in c, were detected using a fluorogenic S1PL substrate. THI (2-acetyl-4-(tetrahydroxybutyl) imidazole, 10 μM), S1PL inhibitor, as a positive control. *p < 0.05. Data are representative of three independent experiments
The secreted form of S1P has been shown to have a key role in mediating mast cell degranulation through its receptor, S1PR2.25,27,28 To examine the existence of this likely autocrine loop in AhR ligand-stimulated mast cells, we showed that while FICZ alone induced appreciable levels of degranulation, measured as a percentage of β-hexosaminidase (Hex) release, in MC/9 cells (Fig. 1d), the degranulation was significantly enhanced in AhR ligand-treated cells stimulated with ovalbumin (OVA)-specific IgE mAbs and OVA (Fig. 1e). In both cases, the addition of the S1PR2-specific antagonist JTE-013 inhibited these effects (Fig. 1d, e). In addition, a S1PR2 antagonist was shown to inhibit degranulation in FICZ-treated BMMCs without IgE mAbs/OVA stimulation (Fig. 1f) and IL-13 secretion upon stimulation with IgE and OVA (Figure S1c), suggesting the involvement of the S1P–S1PR2 axis in AhR ligand-induced mast cell activation.
The S1P level is regulated by S1PL, which is involved in its degradation. In MC/9 mouse and HMC-1 human mast cells, AhR ligand (FICZ)-induced S1P generation was concomitant with a significant and time-dependent reduction in the enzymatic activity of S1PL (Fig. 1g, h). Furthermore, the inhibitory effect of AhR ligands, such as IP, could be reversed to the baseline level in BMMCs when the AhR antagonist CH223191 was added (Figure S2a). Reduction of S1PL activity was also observed in BMMCs (Fig. 1i) and human cultured mast cells (HCMCs; Fig. 1j, subject #1; Figure S2b, subject #2) after treatment with AhR ligands, including FICZ, TCDD and IP. In addition, a time-dependent and dose-dependent reduction of S1PL activity was found in epithelial A549 cells treated with AhR ligands, including FICZ, TCDD and BaP (Figs. S2c–e), and, again, this effect could be blocked by addition of the AhR antagonist CH223191 (Figure S2f). These results suggest that the AhR–ligand axis-mediated increase in S1P level was due, at least in part, to the reduction in S1PL activity in both cell types.
Fig. 2.

The AhR ligand IP regulates SL metabolism in vivo. a S1P levels in BALFs from mice immunized with 10 μg OVA or a combination with 2 μM IP or an equal amount of methanol as vehicle control, followed by 3% OVA aerosol challenge. N = 5 in each group. b Intracellular S1P levels (N = 12 in each group) and c S1PL activity (N = 6 in each group) of lung tissues from mice with intra-tracheal administration of IP for 1 h. *p < 0.05, relative to the vehicle control
As increased levels of the lipid mediator S1P in vivo were found in a mouse model of allergic asthma,29 we investigated if AhR ligands could regulate S1P production in a murine model. Although a slight but not significant increase of S1P was observed in the bronchoalveolar lavage fluids (BALFs) from Ag-sensitized and challenged mice, significantly increased S1P was observed in mice chronically exposed to IP alone or in combination with antigen sensitization and challenge compared to that in vehicle-treated mice (Fig. 2a). In fact, mice with a single exposure to IP for 1 h showed a significant increase in the S1P level in the lung tissues (Fig. 2b), concomitant with a significant reduction of S1PL activity (Fig. 2c). These results provide in vivo evidence for the role of AhR in regulating SL metabolism and S1P generation.
Identification of the ORMDL3-S1PhL-S1P complex and its involvement in AhR-mediated S1P generation
To investigate the role of ORMDL3 in regulating AhR-mediated S1P production, we evaluated the S1P levels in cells with ORMDL3 knockdown. The results showed that a significant reduction in the S1P level was found in HMC-1, A549, and MC/9 cells with ORMDL3 knockdown (Figs. 3a, b; Figure S3a, respectively; sh-ORMDL3) compared to that in cells transduced with control sh-RNAs (sh-NC). Similarly, in the presence of FICZ, no apparent induction of S1P was observed in cells with ORMDL3 knockdown (Fig. 3a, b; Figure S3a, respectively). In contrast, in untreated cells, a significant increase in the enzymatic activity of S1PL was observed in cells with ORMDL3 knockdown compared to that in the cells with control shRNA (sh-NC), and the increased S1PL activity was not further enhanced in FICZ-treated cells with ORMDL3 knockdown (Figs. 3c, d; Figure S3b). Conversely, without FICZ stimulation, the S1P level was significantly increased when HMC-1 cells were transfected with Flag-ORMDL3 construct, concomitant with a reduction of S1PL activity, compared to that observed in the vehicle controls. However, no further increase in S1P level and reduction in S1PL activity were observed after FICZ treatment (Fig. 3e, f, respectively). These results suggest that an optimal level of ORMDL3 appeared to be required for the tight regulation of S1P synthesis. A similar functional effect of ORMDL3 knockdown was found in antigen-activated mast cells. As shown in Figure S3c, cells with ORMDL3 knockdown showed a significant reduction in degranulation after the cross-linkage of surface-bound IgE with OVA, and the FICZ-mediated enhancement of mast cell degranulation was lost (Figure S3c; sh-ORMDL3). In addition, although increased IL-13 secretion was observed in FICZ-treated mast cells stimulated with IgE and OVA (Figure S3d; sh-NC), no significant increase of IL-13 was found in cells with ORMDL3 knockdown (Figure S3d; sh-ORMDL3).
Fig. 3.

ORMDL3 affects S1P levels by regulating S1PL activity. Analysis of S1P levels in a HMC-1 and b A549 cells and of S1PL activity in c HMC-1 and d A549 cells with (sh-ORMDL3) or without ORMDL3 (sh-NC) knockdown. Analysis of e S1P levels and f S1PL activity in HMC-1 cells over-expressing Flag-ORMDL3 (insert, the expression of ORMDL3 in cells under the respective conditions). FRET analysis of the CFP–S1PL and YFP–ORMDL3 interaction in g HMC-1 and h A549 cells. *p < 0.05; N = three independent experiments
Interestingly, pulldown analysis of cell lysates with S1P-conjugated beads showed that S1PL and ORMDL3 bound to S1P (Figure S4a; lane 4, upper and lower panels), but the interaction was not detected by using non-lipid-conjugated beads (Figure S4a; lane 3, upper and lower panels). We also observed that pre-incubating the cell extracts with recombinant GST-fused S1PL proteins enhanced the binding of S1PL to S1P (Figure S4a; lane 5, upper panel) but reduced the binding of ORMDL3 to S1P (Figure S4a; lane 5, lower panel). Furthermore, a reduction in the binding of ORMDL3 and S1PL to S1P was also noted in cells after FICZ treatment for 30 min (Figure S4a; lanes 6 and 7 vs. lanes 4 and 5). To further confirm a direct ORMDL3–S1PL interaction, we performed FRET analyses of HMC-1 and A549 cells co-transfected with YFP–ORMDL3 and CFP–S1PL expression constructs. We found that in untreated cells, a direct interaction between ORMDL3 and S1PL was visualized in cells, and enhanced binding was observed 30 min after FICZ stimulation and then decreased at the 1 h time point (Fig. 3g, h). These results suggest that ORMDL3 may negatively regulate the enzymatic activity of S1PL through a direct interaction with S1PL, which led to increased S1P levels.
AhR ligands enhance oxidation of S1PL and inhibit its activity
Consistent with our previous studies,5,6 exposure of mast cells to AhR ligands, including FICZ, increased the intracellular ROS levels. For example, FICZ-treated HMC-1 cells showed an immediate increase in the ROS level, which was reversed following addition of the antioxidant NAC (Fig. 4a). Surprisingly, in HMC-1 cells with ORMDL3 knockdown (sh-ORMDL3), an AhR ligand, FICZ, was unable to induce ROS (Fig. 4a). As reversible oxidation of S1PL has been shown to influence its activity,30 we investigated whether AhR negatively regulates the enzymatic activity of S1PL through increased ROS and hence enhanced oxidation of S1PL. We found that, indeed, in the presence of the antioxidant NAC, FICZ-mediated inhibition of S1PL activity (Fig. 4b) and the resulting increase in S1P level were reversed (Fig. 4c). To examine the oxidation state of S1PL, we first used recombinant S1PL protein as a model and assessed the level of H2O2-induced oxidation in vitro by analyzing the derivation of the carbonyl groups in the S1PL protein side chains to 2,4-dinitrophenylhydrazone (DNP-hydrazone) with 2,4-dinitrophenylhydrazine (DNPH). The results showed that two protein bands representing the oxidized S1PL proteins were detected by Western blotting analysis using anti-DNP Abs (Figure S4b; lane 2). Importantly, S1PL protein derived from HMC-1 cells with forced S1PL expression showed an increase in the levels of oxidized S1PL after FICZ treatment compared to those of the vehicle control (Fig. 4d; lane 3) or those without FICZ treatment (Fig. 4d; lane 2). Furthermore, the enhanced S1PL oxidation was ROS-dependent as it could be blocked with NAC addition (Fig. 4d; lane 4), suggesting that AhR signaling may enhance S1PL oxidation in a ROS-dependent manner, resulting in an increase in S1P.
Fig. 4.

AhR ligand-mediated oxidation of S1PL in HMC-1 cells with or without ORMDL3 knockdown. a ROS generation, b S1PL activity, and c intracellular S1P level. d Western blotting analysis of oxidized S1PL in HMC-1 cells with Flag-S1PL expression. Intracellular S1P levels and S1PL activities in e HMC-1 and f A549 cells expressing S1PL wild-type or the C317A mutant. *p < 0.05. Data are representative of three independent experiments
Two cysteine residues, Cys218 and Cys317, of human S1PL were previously shown to be critical for its enzymatic activity.31–33 We found that a C to A mutation at position 317, but not 218, abolished its physical association with ORMDL3 and S1PL oxidation in cells without FICZ treatment (data not shown). Thus, we focused on the contribution of the Cys317 residue to the direct S1PL–ORMDL3 interaction and the potential modification of S1PL by oxidation in response to AhR signaling. As expected, the C317A mutant lost its ability to directly bind to ORMDL3 in HMC-1 and A549 cells in the presence or absence of FICZ treatment (Figure S5a, left and right panels, respectively), which was confirmed by FRET analysis. In contrast to wild-type S1PL, the C317A mutant was resistant to modification by oxidation in FICZ-treated cells (Figures S5b and S5c). Consequently, the HMC-1 and A549 cells with the C317A mutant showed decreased levels of intracellular S1P but increased enzymatic activities compared to cells with wild-type S1PL (Fig. 4e, f).
Increased levels of plasma S1P in children with asthma
As the generation of S1P, an important regulator in inflammatory diseases, was shown to indicate the enhanced mast cell response upon exposure to AhR ligands, including an environmental PAH, we compared the plasma S1P levels between children with current asthma and healthy subjects. The results showed that the plasma S1P levels were significantly higher in subjects with asthma than normal control subjects (Fig. 5).
Fig. 5.
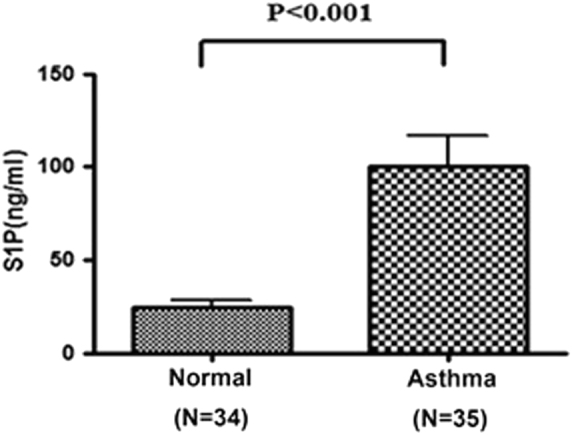
Analysis of plasma S1P levels in subjects with asthma and in healthy controls. Each circle represents each individual result. The line within the vertical points indicates the mean for each group. *p < 0.0001 by Mann–Whitney U tests
Discussion
In this study, we showed that AhR agonistic ligands, including an environmental PAH, inhibited the enzymatic activity of S1PL, which involved a direct ORMDL3–S1PL interaction and oxidation of S1PL. These changes resulted in increased synthesis of S1P, which, in turn, enhanced mast cell response via S1PR2 in an autocrine manner. Reduction of S1PL activity and increased levels of S1P were also noted in mice exposed to AhR ligands alone or in combination with antigen challenge. In addition, elevated levels of plasma S1P were found in children with asthma compared to those in non-asthmatic subjects. These results provide new mechanistic insight into how the AhR–ligand axis regulates ORMDL3-dependent S1P generation in response to environmental insults or endogenous metabolites, which may add a new dimension to the existing regulatory network of asthma and inflammatory diseases.
Accumulated data from studies of ORMDL3’s role in asthma have presented a complex picture thus far. Several mechanisms have been proposed to explain how ORMDL3 and SL intermediates contribute to asthma.8,10,20,21,23 However, there has been no consensus in regard to the exact function of ORMDL3, as it pertains to the proposed inhibitory role of ORMDL3 on SPT activity and SL metabolism. This is also complicated by the fact that the expression of ORMDL3 appears to be constitutive and can be enhanced upon cellular activation by, for example, Th2 cytokines.21 At present, it is uncertain as whether the constitutive level of ORMDL3 and the expression inducible upon cellular activation are functionally different in controlling ER stress and SL metabolism. In this regard, our study demonstrated for the first time a novel function of ORMDL3, seemingly at its constitutive level, in regulating AhR ligand-induced S1P production by negatively regulating S1PL activity.
We noted first that ORMDL3 was required for FICZ-induced ROS generation, and after exposure to AhR ligands, no apparent change in ORMDL3 expression was observed (data not shown). Furthermore, in resting cells, ORMDL3 interacted loosely with S1PL, and upon stimulation with AhR ligands, increased S1PL oxidation and ORMDL3–S1PL complex formation were observed. In addition, reduced ORMDL3 expression significantly inhibited S1P level in resting and AhR ligand-treated cells, concomitant with decreased levels of intracellular ROS. Reversible oxidation of the Cys317 of S1PL has emerged as an important regulatory mechanism,30,33 which may represent a primary regulatory target of the AhR–ligand axis. These results therefore suggest that the basal level of ORMDL3 may serve as “chaperone-like” protein involved in controlling ER stress response and redox balance and that any alteration in the homeostatic state would trigger ER-associated SL metabolism. The constitutive level of ORMDL3 may affect SL metabolism, as shown by the increase in oxidized S1PL and S1P, independent of the newly induced level of ORMDL3. We also noted that over-expression of ORMDL3 resulted in an enhanced increase in ROS generation (data not shown), which may explain the significant inhibition of S1PL activity observed in HMC-1 cells with ORMDL3 over-expression (Fig. 3f) and further support a direct role of ROS in regulating the enzymatic activity of S1PL. In addition, ORMDL3 knockdown delayed the FICZ-induced increase in cytosolic Ca2+, whereas ORMDL3 over-expression enhanced the effect of FICZ on cytosolic Ca2+ (data not shown). These findings are consistent with the function of ORMDL3 in controlling ER Ca2+ dynamics and ER stress response,7,9,11,18,19 and this non-transcriptional event might not be observed in cellular activation by antigens or cytokines. In addition, increased ORMDL3 upon antigen or cytokine stimulation may serve as a feedback mechanism in maintaining homeostasis following the oxidative stress response. Further detailed mechanistic studies are needed to test this hypothesis.
We observed that a chronic and low-dose exposure of IP was sufficient to increase secreted S1P without antigen sensitization and challenge, suggesting a potential contribution of the AhR–ligand axis in priming immune responses and potentiating the development of asthma.5,34 In our recent study, intranasal IP exposure alone without Ag sensitization and challenge resulted in oxidative stress in the lungs and no local inflammation but significantly enhanced the severity of OVA-induced lung inflammation (Wong et al., manuscript submitted). Moreover, we found that the S1PR2 antagonist JTE-013, but not the S1PR1 antagonist W146 (data not shown), abolished the AhR–ligand axis-mediated degranulation and IL-13 secretion, which is consistent with previous reports showing distinct roles of these two receptors in mast cell functions.25,27,28 IgE-mediated mast cell activation was shown to induce SPHK activation and S1P secretion.35 In addition to the inhibition of S1PL activity, FICZ-induced S1P generation was significantly inhibited by the SPHK1 inhibitor PF543 in BMMCs and HMC-1 as well as A549 cells (Figures S6a-c). In addition, increased activation of SPHK1 in FICZ-treated HMC-1 and A549 cells (Figures S6d and 6e, respectively) and of SPHK1 as well as SPHK2 in lung tissues derived from mice treated with IP were observed (Figures S6f and 6g, respectively), suggesting that AhR ligands could not only mediate inhibition of S1PL activity but also activation of SPHK1 and SPHK2. Notably, the AhR–ligand axis did not appear to induce the generation of ceramide-1-phosphate (C1P) in mast cells, and no difference was found in the level of plasma C1P between subjects with asthma and healthy controls (data not shown). Other potential targets of AhR signaling, including S1P phosphohydrolase-1 (SPP1), an enzyme that specifically dephosphorylates S1P, are currently unclear and remain an important subject for future investigation. In addition, although significantly increased levels of plasma S1P were observed in children with asthma in our study population, the S1P level was not correlated with the levels of FEV1, PC20, FeNO, and total IgE (data not shown). Further analysis with a large sample size is needed to determine whether plasma S1P level is correlated with lung function parameters.
Recent awareness of, and concern about, the exposure to ubiquitous environmental pollutants, including PAHs and chemicals with oxidant-generating capacities, has highlighted their impact on human health and disease, particularly respiratory diseases.36,37 Exposure to PAHs associated with air pollution and tobacco smoke is considered an important environmental risk for asthma38 and increases the prenatal risk for allergic sensitization in children.39 In addition, exposure to tobacco smoke is suggested to increase the risk of Chr. 17q12-21 gene loci for asthma.40 Our findings that AhR ligands, including environmental PAHs, initiate a functional AhR-ORMDL3-S1PL-S1P linkage in regulating cellular responses provide new mechanistic insight in the environmental exposure–disease relationship and offer a promising target for investigating the gene–environment interaction in asthma and inflammatory diseases. Furthermore, as S1P has been shown to be critical in regulating mast cell response,25 allergic asthma24,25,41, and other inflammatory diseases,24 strategies targeting S1P generation, degradation and/or receptor signaling may thus have promise in the development of new therapies for asthma and inflammatory diseases.
Electronic supplementary material
Acknowledgements
This work was supported, in part, by grants from the National Health Research Institutes, Taiwan (EOPP10-014 and EOSP07-014 to S.-K.H.), Kaohsiung Medical University “The Talent Plan” (105KMUOR04 to S.-K.H.), the Ministry of Science and Technology, Taiwan (MOST 105-2320-B-039-004 and MOST 106-2320-B-039-037, to H.-C.W.), China Medical University Hospital, Taiwan (DMR-106-154 and DMR-107-117, to H.-C.W.), the Community Medicine Research Center, Chang Gung Memorial Hospital at Keelung (CMRPG3E1183 to L.-C.C.), the 1000 Young Talents Plan Program, China (to Y.Z.), the Initial Funding for New PI, Fudan Children’s Hospital and Fudan University (to Y.Z.), the National Natural Science Foundation of China (81671561, to Y.Z.), and the National Key Research and Development Program of China (2016YFC1305102, to Y.Z.). We thank the Center for Research Resources and Development of Kaohsiung Medical University for providing the Olympus FV1000 confocal microscope.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
5/16/2018
In this article, published online 23 March 2018, the affiliation 10 of Zhou Y was incorrect. The affiliation should be “Children’s Hospital and Institute of Biomedical Sciences, Fudan University. Key Laboratory of Neonatal Disease, Ministry of Health, 201102, Shanghai, China.” The authors regret the errors.
Supplementary Information
Supplementary Information accompanies this paper at (10.1038/s41423-018-0022-2).
References
- 1.Nguyen NT, Hanieh H, Nakahama T, Kishimoto T. The roles of aryl hydrocarbon receptor in immune responses. Int. Immunol. 2013;25:335–43. doi: 10.1093/intimm/dxt011. [DOI] [PubMed] [Google Scholar]
- 2.Sibilano R, Pucillo CE, Gri G. Allergic responses and aryl hydrocarbon receptor novel pathway of mast cell activation. Mol. Immunol. 2015;63:69–73. doi: 10.1016/j.molimm.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Quintana FJ. The aryl hydrocarbon receptor: a molecular pathway for the environmental control of the immune response. Immunology. 2013;138:183–9. doi: 10.1111/imm.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–9. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 5.Zhou Y, Tung HY, Tsai YM, Hsu SC, Chang HW, Kawasaki H, et al. Aryl hydrocarbon receptor controls murine mast cell homeostasis. Blood. 2013;121:3195–204. doi: 10.1182/blood-2012-08-453597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang HC, Zhou Y, Huang SK. SHP-2 phosphatase controls aryl hydrocarbon receptor-mediated ER stress response in mast cells. Arch. Toxicol. 2017;91:1739–48. doi: 10.1007/s00204-016-1861-1. [DOI] [PubMed] [Google Scholar]
- 7.Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, et al. Orm family proteins mediate sphingolipid homeostasis. Nature. 2010;463:1048–53. doi: 10.1038/nature08787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oyeniran C, Sturgill JL, Hait NC, Huang WC, Avni D, Maceyka M, et al. Aberrant ORM (yeast)-like protein isoform 3 (ORMDL3) expression dysregulates ceramide homeostasis in cells and ceramide exacerbates allergic asthma in mice. J. Allergy Clin. Immunol. 2015;136:1035–46. doi: 10.1016/j.jaci.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiefer K, Carreras-Sureda A, Garcia-Lopez R, Rubio-Moscardo F, Casas J, Fabrias G, et al. Coordinated regulation of the orosomucoid-like gene family expression controls de novo ceramide synthesis in mammalian cells. J. Biol. Chem. 2015;290:2822–30. doi: 10.1074/jbc.M114.595116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhakupova A, Debeuf N, Krols M, Toussaint W, Vanhoutte L, Alecu I, et al. ORMDL3 expression levels have no influence on the activity of serine palmitoyltransferase. FASEB J. 2016;30:4289–300. doi: 10.1096/fj.201600639R. [DOI] [PubMed] [Google Scholar]
- 11.Siow D, Sunkara M, Dunn TM, Morris AJ, Wattenberg B. ORMDL/serine palmitoyltransferase stoichiometry determines effects of ORMDL3 expression on sphingolipid biosynthesis. J. Lipid Res. 2015;56:898–908. doi: 10.1194/jlr.M057539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller M, Rosenthal P, Beppu A, Gordillo R, Broide DH. Oroscomucoid such as protein 3 (ORMDL3) transgenic mice have reduced levels of sphingolipids including sphingosine-1-phosphate and ceramide. J. Allergy Clin. Immunol. 2017;139:1373–6. doi: 10.1016/j.jaci.2016.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–3. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 14.Balantic M, Rijavec M, Flezar M, Camlek T, Hudoklin I, Kosnik M, et al. A polymorphism in ORMDL3 is associated not only with asthma without rhinitis but also with chronic obstructive pulmonary disease. J. Investig. Allergol. Clin. Immunol. 2013;23:256–61. [PubMed] [Google Scholar]
- 15.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008;40:955–62. doi: 10.1038/ng.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGovern DP, Gardet A, Torkvist L, Goyette P, Essers J, Taylor KD, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010;42:332–7. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurreeman FA, Stahl EA, Okada Y, Liao K, Diogo D, Raychaudhuri S, et al. Use of a multiethnic approach to identify rheumatoid- arthritis-susceptibility loci, 1p36 and 17q12. Am. J. Hum. Genet. 2012;90:524–32. doi: 10.1016/j.ajhg.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cantero-Recasens G, Fandos C, Rubio-Moscardo F, Valverde MA, Vicente R. The asthma-associated ORMDL3 gene product regulates endoplasmic reticulum-mediated calcium signaling and cellular stress. Hum. Mol. Genet. 2010;19:111–21. doi: 10.1093/hmg/ddp471. [DOI] [PubMed] [Google Scholar]
- 19.Miller M, Tam AB, Cho JY, Doherty TA, Pham A, Khorram N, et al. ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proc. Natl Acad. Sci. USA. 2012;109:16648–53. doi: 10.1073/pnas.1204151109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller M, Tam AB, Mueller JL, Rosenthal P, Beppu A, Gordillo R, et al. Cutting edge: targeting epithelial ORMDL3 increases, rather than reduces, airway responsiveness and is associated with increased sphingosine-1-phosphate. J. Immunol. 2017;198:3017–22. doi: 10.4049/jimmunol.1601848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller M, Rosenthal P, Beppu A, Mueller JL, Hoffman HM, Tam AB, et al. ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J. Immunol. 2014;192:3475–87. doi: 10.4049/jimmunol.1303047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenfeldt HM, Amrani Y, Watterson KR, Murthy KS, Panettieri RA, Jr., Spiegel S. Sphingosine-1-phosphate stimulates contraction of human airway smooth muscle cells. FASEB J. 2003;17:1789–99. doi: 10.1096/fj.02-0836com. [DOI] [PubMed] [Google Scholar]
- 23.Worgall TS, Veerappan A, Sung B, Kim BI, Weiner E, Bholah R, et al. Impaired sphingolipid synthesis in the respiratory tract induces airway hyperreactivity. Sci. Transl. Med. 2013;5:186ra167. doi: 10.1126/scitranslmed.3005765. [DOI] [PubMed] [Google Scholar]
- 24.Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. 2014;510:58–67. doi: 10.1038/nature13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olivera A, Rivera J. An emerging role for the lipid mediator sphingosine-1-phosphate in mast cell effector function and allergic disease. Adv. Exp. Med. Biol. 2011;716:123–42. doi: 10.1007/978-1-4419-9533-9_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serra M, Saba JD. Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv. Enzyme Regul. 2010;50:349–62. doi: 10.1016/j.advenzreg.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oskeritzian CA, Price MM, Hait NC, Kapitonov D, Falanga YT, Morales JK, et al. Essential roles of sphingosine-1-phosphate receptor 2 in human mast cell activation, anaphylaxis, and pulmonary edema. J. Exp. Med. 2010;207:465–74. doi: 10.1084/jem.20091513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jolly PS, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J, et al. Transactivation of sphingosine-1-phosphate receptors by Fc epsilon RI triggering is required for normal mast cell degranulation and chemotaxis. J. Exp. Med. 2004;199:959–70. doi: 10.1084/jem.20030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oskeritzian CA, Milstien S, Spiegel S. Sphingosine-1-phosphate in allergic responses, asthma and anaphylaxis. Pharmacol. Ther. 2007;115:390–9. doi: 10.1016/j.pharmthera.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhan X, Desiderio DM. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Anal. Biochem. 2006;354:279–89. doi: 10.1016/j.ab.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 31.Van Veldhoven PP, Gijsbers S, Mannaerts GP, Vermeesch JR, Brys V. Human sphingosine-1-phosphate lyase: cDNA cloning, functional expression studies and mapping to chromosome 10q22(1) Biochim. Biophys. Acta. 2000;1487:128–34. doi: 10.1016/S1388-1981(00)00079-2. [DOI] [PubMed] [Google Scholar]
- 32.Bourquin F, Riezman H, Capitani G, Grutter MG. Structure and function of sphingosine-1-phosphate lyase, a key enzyme of sphingolipid metabolism. Structure. 2010;18:1054–65. doi: 10.1016/j.str.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 33.Suh JH, Saba JD. Sphingosine-1-phosphate in inflammatory bowel disease and colitis-associated colon cancer: the fat’s in the fire. Transl. Cancer Res. 2015;4:469–83. doi: 10.3978/j.issn.2218-676X.2015.10.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller RL, Garfinkel R, Lendor C, Hoepner L, Li Z, Romanoff L, et al. Polycyclic aromatic hydrocarbon metabolite levels and pediatric allergy and asthma in an inner-city cohort. Pediatr. Allergy Immunol. 2010;21:260–7. doi: 10.1111/j.1399-3038.2009.00980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olivera A, Urtz N, Mizugishi K, Yamashita Y, Gilfillan AM, Furumoto Y, et al. IgE-dependent activation of sphingosine kinases 1 and 2 and secretion of sphingosine 1-phosphate requires Fyn kinase and contributes to mast cell responses. J. Biol. Chem. 2006;281:2515–25. doi: 10.1074/jbc.M508931200. [DOI] [PubMed] [Google Scholar]
- 36.Huang SK, Zhang Q, Qiu Z, Chung KF. Mechanistic impact of outdoor air pollution on asthma and allergic diseases. J. Thorac. Dis. 2015;7:23–33. doi: 10.3978/j.issn.2072-1439.2014.12.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stanek LW, Brown JS, Stanek J, Gift J, Costa DL. Air pollution toxicology—a brief review of the role of the science in shaping the current understanding of air pollution health risks. Toxicol. Sci. 2011;120:S8–27. doi: 10.1093/toxsci/kfq367. [DOI] [PubMed] [Google Scholar]
- 38.Klingbeil EC, Hew KM, Nygaard UC, Nadeau KC. Polycyclic aromatic hydrocarbons, tobacco smoke, and epigenetic remodeling in asthma. Immunol. Res. 2014;58:369–73. doi: 10.1007/s12026-014-8508-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosa MJ, Jung KH, Perzanowski MS, Kelvin EA, Darling KW, Camann DE, et al. Prenatal exposure to polycyclic aromatic hydrocarbons, environmental tobacco smoke and asthma. Respir. Med. 2011;105:869–76. doi: 10.1016/j.rmed.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flory JH, Sleiman PM, Christie JD, Annaiah K, Bradfield J, Kim CE, et al. 17q12-21 variants interact with smoke exposure as a risk factor for pediatric asthma but are equally associated with early-onset versus late-onset asthma in North Americans of European ancestry. J. Allergy Clin. Immunol. 2009;124:605–7. doi: 10.1016/j.jaci.2009.05.047. [DOI] [PubMed] [Google Scholar]
- 41.Jolly PS, Rosenfeldt HM, Milstien S, Spiegel S. The roles of sphingosine-1-phosphate in asthma. Mol. Immunol. 2002;38:1239–45. doi: 10.1016/S0161-5890(02)00070-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.