

Abstract
The chemokine receptor CCR7 and its ligands CCL19 and CCL21 guide the homing and positioning of dendritic and T cells in lymphoid organs, thereby contributing to several aspects of adaptive immunity and immune tolerance. In the present study, we investigated the role of CCR7 in the pathogenesis of collagen-induced arthritis (CIA). By using a novel anti-human CCR7 antibody and humanized CCR7 mice, we evaluated CCR7 as a target in this autoimmune model of rheumatoid arthritis (RA). Ccr7-deficient mice were completely resistant to CIA and presented severely impaired antibody responses to collagen II (CII). Selective CCR7 expression on dendritic cells restored arthritis severity and anti-CII antibody titers. Prophylactic and therapeutic treatment of humanized CCR7 mice with anti-human CCR7 mAb 8H3-16A12 led to complete resistance to CIA and halted CIA progression, respectively. Our data demonstrate that CCR7 signaling is essential for the induction of CIA and identify CCR7 as a potential therapeutic target in RA.
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by the formation of persistent inflammatory cellular infiltrates in the synovial tissues of diarthrodial joints, leading to cartilage and bone erosion. Leukocytes forming these infiltrates extravasate from synovial blood vessels in response to chemokines and adhesion signals.1 Traditionally, chemokines have been classified into homeostatic and inflammatory chemokines. Although homeostatic chemokines and their receptors mainly regulate the steady-state trafficking of immune cells to and within lymphoid organs, inflammatory chemokines guide the recruitment of effector leukocytes into sites of inflammation.2 The homeostatic chemokines CCL19 and CCL21 are constitutively expressed in lymphoid organs and lymphatic vessels, and their receptor, CCR7, is highly expressed on naïve and central memory T cells, as well as activated DCs. The interactions of CCR7 with its ligands orchestrate the structural organization of lymphoid organs, the initiation of immune responses and the induction of immune tolerance.3,4 Several studies have provided evidence for strong expression of homeostatic chemokines at sites of inflammation, especially during chronic inflammatory conditions, such as RA.5,6 CCL19 expression was demonstrated on stromal and endothelial cells in RA synovial tissue,7 while CCL21 was detected on synovial endothelial cells and near perivascular synovial infiltrates in the joints of RA patients.6,8 Furthermore, it was suggested that these chemokines attract naïve T cells to sites of ongoing autoimmune inflammation.6 Synovial CD4+ T cells exhibit an effector memory phenotype but have been shown to express CCR7.7 CCR7 was also detected on fibroblast-like synoviocytes (FLS) of RA patients and was suggested to contribute to angiogenesis through vascular endothelial growth factor (VEGF) and angiotensin I (Ang I) secretion by RA FLS upon CCL19 stimulation.9,10 Synovial CCL21 was shown to recruit endothelial cells and promote angiogenesis.11 Ccr7−/− mice showed reduced early clinical scores in a modified antigen-induced arthritis (AIA) model; however, late joint inflammation and destruction were even more severe than in WT mice.12
These effects prompted us to dissect the role of CCR7 in experimental arthritis and address its potential as a therapeutic target. We show that Ccr7 deficiency confers complete resistance to collagen-induced arthritis (CIA) and that susceptibility to CIA can be restored by selective expression of CCR7 on dendritic cells. Furthermore, we applied a novel anti-human CCR7 mAb in humanized CCR7 mice that demonstrated both prophylactic and therapeutic efficacy in treating CIA. Thus, this study identifies the CCR7/CCR7-ligand axis as an essential mediator of CIA and CCR7 as a potential target for the treatment of RA.
Results
Ccr7-deficient mice are resistant to CIA
To evaluate the contribution of CCR7 in RA, we employed the collagen-induced arthritis (CIA) mouse model. Ccr7−/− mice in a C57BL/6 genetic background were completely resistant to the development of CIA (Fig. 1a, b). Although the joints of arthritic WT mice exhibited severe inflammation, cartilage, and bone resorption, no lesions could be observed in the joints of Ccr7−/− mice, and their joint spaces were well preserved (Fig. 1c). To exclude the possible influence of the BL6 genetic background on the CIA-resistant phenotype in Ccr7−/− mice, we also used DBA/1 mice that are highly sensitive to CIA. Of interest, DBA/1-Ccr7−/− mice were also completely resistant to the development of CIA (Fig. 1d, e), and their paws were free of inflammatory infiltrates (Fig. 1f). Due to a study reporting delayed but exaggerated immune responses in Ccr7−/− mice,13 we monitored BL6-Ccr7−/− mice for 120 days upon CIA induction, boosting twice with CII in CFA. In this prolonged CIA protocol, B6-Ccr7−/− mice did not develop any arthritic symptoms throughout the monitoring period (Fig. 1g). Furthermore, we addressed the role of CCR7 ligands in CIA using plt/plt mice, a naturally occurring mutant strain that is deficient for CCL19 and one of the two CCL21 isoforms.14 Plt/plt mice showed a markedly reduced CIA incidence and score (Fig. 1h, i). Since the anti-CII Ab response is an essential driver of CIA,15 we evaluated serum anti-CII Ab levels. The Ab response to murine CII (mCII) was severely reduced in BL6-Ccr7−/− mice for all isotypes analyzed and was almost absent for IgG1 Abs (Fig. 1j). In plt/plt mice, the anti-CII Ab response was mildly reduced overall (Fig. 1k). Taken together, these data show that CCR7 is an essential factor for the development of CIA and that the presence of CCL21-Leu in plt/plt mice can partially revert the complete resistance to CIA observed in Ccr7−/− mice.
Fig. 1.

Ccr7−/− mice are resistant to CIA. Incidence (a) and clinical score (b) of CIA in Ccr7-deficient mice on a C57BL/6 background. The results are shown from ≥12 mice per group. The data in (b) are the mean ± SEM. c Representative pictures of CIA manifestation (left) and joint histopathology (right) in B6 WT and B6-Ccr7−/− mice. d Incidence and (e) clinical score of CIA in Ccr7-deficient mice on a DBA/1J background. The results are shown from ≥10 mice per group. f Representative pictures of CIA manifestation (left) and joint histopathology (right) in DBA/1 J WT and DBA/1J-Ccr7−/− mice. g Incidence of prolonged CIA in B6-Ccr7−/− mice. Mice were boosted on day 21 and on day 80 post-initial immunization and monitored until day 120. Results are shown from 12 mice. h Incidence and clinical score (i) of CIA in plt/plt mice. The results are shown from 9–11 mice per group. Areas under the curve (AUC) calculated from the CIA scores were compared for statistical significance (Student’s t-test) *P < 0.05; Levels of anti-murine-CII Ab levels were measured by ELISA in serially diluted serum samples from B6-Ccr7−/− mice (j) and plt/plt mice (k) upon CIA induction (3-fold dilution steps, 1:100–1:72900). The results are shown from n = 6–8 mice per group. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001
CCR7-mediated DC migration is essential for the development of CIA
To identify the cell type that specifically prevents the development of CIA in Ccr7-deficient mice, we employed mice with conditional expression of human CCR7 in the absence of murine CCR7. In these mice, the human CCR7 gene is knocked in in the mouse Ccr7 locus and is transcribed only upon cre-mediated excision of a floxed neo stop cassette.16,17 Mice with T cell- or DC-specific human CCR7 expression are designated T-CCR7+/+ and DC-CCR7+/+, respectively. Mice expressing human CCR7 on all CCR7 proficient cells due to neo deletion at the germline level are designated GL-CCR7+/+, and mice carrying neo in both alleles, thus expressing neither murine nor human CCR7, are named CCR7stop/stop. Upon CIA induction, GL-CCR7+/+ mice developed robust arthritis, confirming our previous finding that human CCR7 is fully functional in mice.17 Similar to Ccr7−/− mice, CCR7stop/stop mice were completely resistant to CIA induction. Restoration of human CCR7 expression in DCs in DC-CCR7+/+ mice was sufficient to enable robust development of arthritis, whereas both arthritis incidence and score were significantly reduced in T-CCR7+/+ mice (Fig. 2a, b). Evaluation of the anti-CII Ab levels in sera of human CCR7-expressing mice revealed significantly higher Ab production in DC-CCR7+/+ mice compared to CCR7stop/stop mice (Fig. 2d). These data show that restoration of CCR7 expression in DCs is sufficient for robust arthritis induction.
Fig. 2.
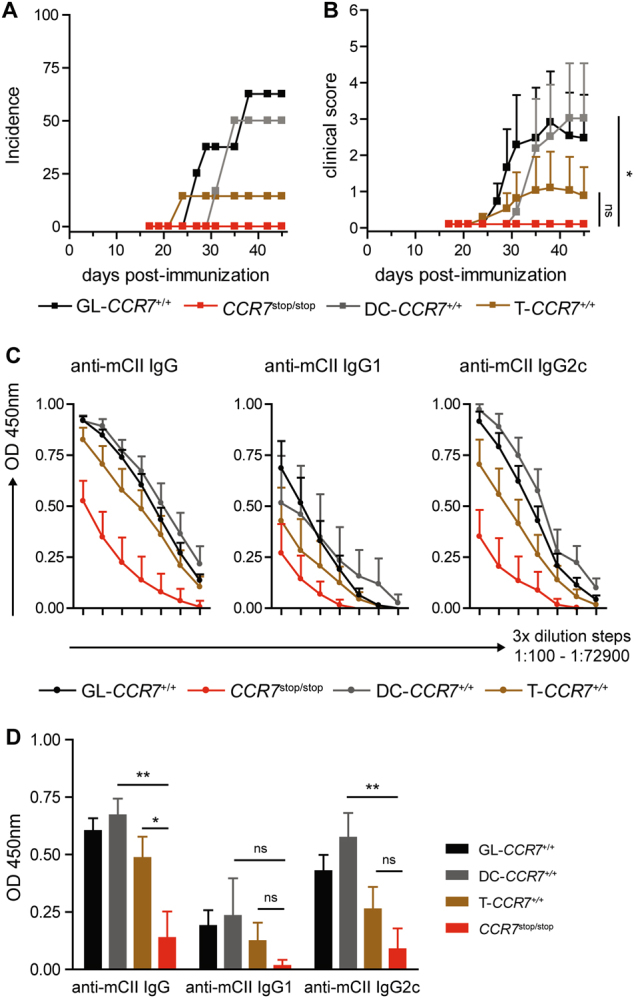
CCR7 expression on DCs is critical for the development of CIA. a Incidence and score (b) of CIA in GL-CCR7+/+, T-CCR7+/+, DC-CCR7+/+, and CCR7stop/stop mice. The data are shown from 6–8 mice per group. The data in (b) are the mean ± SEM. Areas under the curve (AUC) calculated from the CIA scores were compared for statistical significance (Student’s t-test) *P < 0.05; c Levels of anti-murine-CII IgG, IgG1c and IgG2c were measured in serially diluted serum samples (3-fold dilution steps, 1:100–1:72900) from GL-CCR7+/+, T-CCR7+/+, DC-CCR7+/+, and CCR7stop/stop mice upon CIA induction. d Levels of anti-murine-CII IgG, -IgG1, and -IgG2c shown in (c) at a serum dilution of 1:2700 were compared for statistical significance. The data in (c, d) are presented as the mean ± SEM from 6–10 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001
Generation and characterization of anti-human CCR7 mAb
Due to the complete resistance to CIA in Ccr7−/− and CCR7stop/stop mice, we hypothesized that antibody targeting of CCR7 would interfere with arthritis development. We used a murine IgG1 anti-human CCR7 monoclonal antibody (clone 8H3-16A12). The specificity of mAb 8H3-16A12 was confirmed by flow cytometry using lymphocytes isolated from T-CCR7+/+ mice and WT mice. Although mAb 8H3-16A12 demonstrated binding to CCR7+/+ T cells even at very low concentrations, no binding to WT T cells was detectable (Fig. 3a). The specificity was further confirmed by immunohistology (Fig. 3b). On sections of peripheral LNs from WT mice, no staining with mAb 8H3-16A12 could be demonstrated, while strong staining was detectable in T cell areas of the inguinal lymph nodes of DC-T-CCR7+/+ mice that expressed human CCR7 on both T cells and DCs.17 To assess modulation of human CCR7-mediated chemotaxis by 8H3-16A12, transwell assays were performed using T cells from DC-T-CCR7+/+ mice. We observed a concentration-dependent inhibition of T cell migration towards murine CCL19 by mAb 8H3-16A12 (Fig. 3c). However, even at very high mAb concentrations, only partial inhibition could be observed. Subsequently, we performed an in vivo assay to evaluate the effects of mAb 8H3-16A12 on human CCR7-expressing cells. A 1:1 mixture of lymphocytes isolated from DC-T-CCR7+/+ or WT mice was adoptively transferred into WT recipients that were injected with mAb 8H3-16A12 or a mouse IgG1 isotype control Ab. Twenty-four hours after cell transfer, the frequency of each cell group in the pool of transferred cells was determined by flow cytometry. Since the frequency of human CCR7-expressing T cells was significantly reduced in skin-draining lymph nodes, the spleen and, in particular, the blood of mice that were treated with mAb 8H3-16A12, we concluded that mAb 8H3-16A12 likely leads to depletion of human CCR7-expressing cells in vivo rather than inhibiting their homing into lymph nodes.
Fig. 3.

Characterization of anti-human CCR7 antibody 8H3-16A12. a T cells from T-CCR7+/+ LNs were stained with anti-human CCR7 Ab 8H3-16A12 at a concentration of 25 µg/ml (red histogram), 1 µg/ml (orange histogram), and 0.04 µg/ml (green histogram). The black histogram depicts staining of T cells from WT mice with 25 µg/ml 8H3-16A12. Control staining with mIgG1 isotype at a concentration of 25 µg/ml on T cells from T-CCR7+/+ mice is shown in the filled gray histogram. Representative data are shown from more than 3 experiments. b LN sections from DC-T-CCR7+/+ and WT mice were stained with mAb 8H3-16A12 (bar 20 µm). Representative data are shown from more than 12 lymph nodes analyzed per group. c Modulation of CCL19-induced migration of DC-T-CCR7+/+ lymphocytes by 8H3-16A12 was assessed using a transwell assay as described in the material and methods. The percentage of migration inhibition was calculated as (migration index mIgG1 − migration index 8H3-16A12) × 100 / migration index mIgG1. The data are presented as the mean ± SD from triplicate wells and are representative of two independent experiments. d Schematic representation of adoptive cell transfer of lymphocytes from DC-T-CCR7+/+ mice (CD45.2+) mixed at a 1:1 ratio with lymphocytes from WT mice (CD45.1+) in WT recipient mice (CD45.1+CD45.2+) treated with 8H3-16A12 or mIgG1 isotype control Ab. e As depicted in (d), the relative frequency of T cells from WT and DC-T-CCR7+/+ donor mice was assessed in the indicated organs of the recipient mice by flow cytometry 24 h after cell transfer. The data are presented as the mean ± SD from pooled 7–8 mice per group from two experiments. *P < 0.05, **P < 0.01, ***P < 0.001
Treatment with anti-human CCR7 mAb 8H3-16A12 significantly affects the composition of immune cells in the blood and lymphoid organs of humanized CCR7 mice
The in vivo effects of mAb 8H3-16A12 were further assessed in naïve GL-CCR7+/+ mice. GL-CCR7+/+ mice were treated with a single i.p. injection of 2.5 mg/kg 8H3-16A12, 25 mg/kg 8H3-16A12, or mIgG1 isotype control Ab and sacrificed at day 2, 7, 15, and 30 post-treatment. Treatment with 8H3-16A12 led to significant reductions of both CD4+ and CD8+ T cells in the blood and skin-draining lymph nodes (Fig. 4a). Maximal reduction was observed at day 15 post-treatment, while T cell counts recovered at day 30. Mainly naïve CD62L+ CD44− T cells were reduced, whereas there were no significant alterations in the counts of effector memory T cells (CD62L− CD44+). B cells, conventional DCs (cDCs), plasmacytoid DCs (pDCs) and neutrophils were not affected by 8H3−16A12 treatment (Fig. 4a, b). Coating of peripheral blood T cells with anti-human CCR7 Ab 8H3-16A12 was still evident at day 15 upon application, especially in mice that received 25 mg/kg Ab (Fig. 4c). Several studies have demonstrated cytokine release syndrome following the administration of T cell targeting Abs.18–20 Therefore, we measured expression of several pro-inflammatory cytokines in the sera of 8H3-16A12-treated mice throughout the treatment period. For all time points investigated, we were unable to detect increased levels of pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, even in mice receiving 25 mg/kg 8H3-16A12 (Fig. 4d).
Fig. 4.

Single dose toxicity study of anti-human CCR7 mAb 8H3-16A12. GL-CCR7+/+ mice received a single i.p. injection of 2.5 mg/kg 8H3-16A12, 25 mg/kg 8H3-16A12 or 25 mg/kg mIgG1 control Ab and were sacrificed 2, 7, 15, and 30 days post-injection. The cell counts of the indicated immune cell subsets (a, b) were assessed in the blood and pLN by flow cytometry. The data are presented as the mean ± SD from 5–8 mice per group. c Coating of human CCR7 by 8H3-16A12 on T cells in blood. Cells were stained with anti-mIgG1 Ab, and the MFI of the staining is presented as fold change over mIgG1-treated mice. The data are presented as the mean ± SD from 3–7 mice per group. d Levels of TNF-α, IL-6, and IL-1β were measured in sera in a Luminex-based assay. The data are presented as the mean ± SD from 6–9 per group
Treatment with anti-human CCR7 mAb 8H3-16A12 prevents the induction of CIA in humanized CCR7 mice
To assess the effects of anti-human CCR7 mAb 8H3-16A12 in arthritis, CIA was induced in GL-CCR7+/+ mice that were subjected to prophylactic treatment with 8H3-16A12 consisting of i.p. injections of 20 mg/kg mAb every 12 days starting 4 days before primary immunization with CII in CFA. This regimen led to complete resistance to CIA development (Fig. 5a) and significantly diminished the levels of anti-CII antibodies in the sera of treated mice (Fig. 5b). The efficacy of prophylactic treatment with 8H3-16A12 prompted us to evaluate its therapeutic potential in GL-CCR7+/+ mice that started upon the onset of arthritic symptoms and consisted of i.p. injections of 20 mg/kg mAb every 10–11 days. Interestingly, treatment with 8H3-16A12 halted the exacerbation of CIA and led to significantly reduced arthritic scores compared to the control group receiving isotype control Ab (Fig. 5c). Surprisingly, anti-CII Ab levels were not reduced in the sera of 8H3-16A12-treated mice (Fig. 5d). Assessment of the immune cell composition in the joint-draining lymph nodes (JDLN) of 8H3-16A12-treated mice upon CIA manifestation revealed significantly reduced overall counts of T cells, especially naïve T cells, whereas effector memory T cells were not affected (Fig. 5e). Of note, treatment with 8H3-16A12 significantly raised the relative frequency of Foxp3+ cells in the T cell compartment in JDLN (Fig. 5f), whereas the B cell compartment, including GC B cells, was not affected.
Fig. 5.

Treatment with 8H3-16A12 inhibits CIA development. a CIA score of GL-CCR7+/+ mice that were injected i.p. with 20 mg/kg 8H3-16A12 or 20 mg/kg mIgG1 control Ab every 12 days starting at day 4 before CIA induction. The results are shown from ≥18 mice per group. b Sera were collected from mice shown in (a), and levels of anti-murine-CII IgG, IgG1, and IgG2c were measured in serially diluted serum samples (3-fold dilution steps, 1:100–1:72900). The data are presented as the mean ± SEM. c CIA score of GL-CCR7+/+ mice that were treated upon manifestation of arthritis every 10 days with i.p. injections of 20 mg/kg 8H3-16A12 or 20 mg/kg mIgG1 control Ab. The data are shown as the mean ± SEM from ≥11 mice per group. d Anti-murine-CII IgG, IgG1, and -IgG2c levels were measured in serially diluted serum samples from mice shown in (c) (3-fold dilution steps, 1:100–1:72900). The data are presented as the mean ± SEM. e Cell counts of the indicated immune cell subsets and the ratio of FoxP3+/FoxP3- T cells f were assessed upon termination of the observation period in mice shown in (c) in JDLN by flow cytometry. Naïve CD4+ T cells were gated as CD62L+ CD44−, effector memory CD4+ T cells as CD62L− CD44+ and GC B cells as B220+ Fas+ cells. The data are presented as the mean ± SD from ≥11 mice per group. Ns not significant; *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
The present study describes an essential role of the CCR7/CCR7-ligand axis in the induction and maintenance of CIA and identifies CCR7 as a potential target for the treatment of RA. The use of Ccr7-deficient mice, both in a BL6 and in the highly CIA-susceptible DBA/1 genetic background, reveals an absolute requirement for CCR7 signaling in CIA induction, even using a prolonged CIA protocol based on repeated boosting with the disease-inducing antigen. This finding was not expected since different groups have reported delayed but exacerbated immune responses in Ccr7−/− and plt/plt mice using cutaneous contact hypersensitivity (CHS) models and spontaneous formation of ectopic lymphoid tissue in Ccr7−/− mice.13,14,21,22 These aberrant responses were attributed to impaired LN migration and function of regulatory T cells (Tregs). In the CIA model of the present study, we did not observe such reactions in Ccr7−/− mice, even at late time points, and anti-CII Ab titers were strongly reduced compared to WT mice. Likewise, several studies reported that Ccr7-deficient mice are susceptible to mild generalized autoimmunity characterized by the spontaneous development of lymphocytic infiltrates in several organs along with increased titers of autoantibodies.23,24 However, this susceptibility rarely led to the development of overt autoimmune disease, such as Sjögren’s syndrome or diabetes. Possible explanations for the increased susceptibility of Ccr7-deficient mice towards autoimmunity include defective egress of activated lymphocytes from peripheral sites, severe impairment of Treg function due to impaired Treg migration and a break of central and/or peripheral tolerance due to disturbed thymic compartmentalization or impaired steady-state DC migration to LNs. Of interest, we did not observe any signs of spontaneous or induced arthritis in Ccr7−/− mice, and the paws of Ccr7−/− mice were free of lymphocytic infiltrates at day 45 post-CIA induction. These observations suggest a dominant role for the CCR7/CCR7-ligand axis in the induction of the autoreactive response in CIA over the preservation of immune tolerance towards CII.
Although Ccr7-deficient mice were completely resistant to the induction of CIA, few plt/plt mice developed arthritic paws. In plt/plt mice, expression of CCL21-leu is preserved on the lymphatic endothelium, which allows CCR7-expressing cells, such as activated dendritic cells, to migrate to some degree via afferent lymphatic vessels towards draining LNs where they can prime T cells.14 The present study provides further evidence for an essential role for CCR7-mediated DC migration in CIA, since the selective expression of human CCR7 on DCs was sufficient to overcome the resistance of Ccr7-deficient mice to CIA. DC-CCR7+/+ mice showed a robust CIA incidence, albeit lower than that of mice expressing human CCR7 ubiquitously, suggesting that the CCR7-mediated migration of cells, other than DCs, also contributes to the development of CIA. The role of DCs in autoimmunity is complex and versatile. The presentation of self-antigens by DCs can elicit or suppress autoimmune responses depending on the inflammatory signals DCs acquire and the expression of pro-inflammatory or regulatory molecules. The concurrent presentation of self Ag to T cells and the secretion of pro-inflammatory cytokines, such as IL-6, IL-12, and IL-23, promote the differentiation of autoreactive Tfh, Th1, and Th17 cells. In contrast, self Ag presentation coupled to PDL1-PD-1 signaling and/or TGFβ secretion leads to Treg induction or T cell anergy, thereby promoting peripheral tolerance.25 Our results suggest that, in the CIA model, CCR7-expressing DCs are needed to initiate autoimmune responses and not to sustain tolerance to CII.
Anti-CII antibodies are a main disease driver in CIA, and their titers, especially of IgG1 Abs, were severely reduced in the Ccr7-deficient mice. Similar to the Ccr7-deficient mice, anti-CII Ab titers were significantly reduced in the GL-CCR7+/+ mice treated with anti-huCCR7 mAb 8H3-16A12 throughout the course of CIA. B cell numbers in the SLOs of Ccr7-deficient mice are not significantly reduced, and they present an aberrantly activated phenotype with significantly elevated expression of MHC-II and co-stimulatory molecules.26,27 Furthermore, treatment with anti-human CCR7 Ab 8H3-16A12 does not significantly modulate the number of follicular B cells or GC B cells in SLOs. The defective anti-CII Ab response in Ccr7−/− mice and GL-CCR7+/+ mice prophylactically treated with 8H3-16A12 likely reflects the effects of CCR7 deficiency or 8H3-16A12 treatment on the T cell compartment. The LN homing of naïve T cells in Ccr7-deficient mice is severely impaired, and they are efficiently depleted upon treatment with 8H3-16A12, whereas the numbers of effector T cells in both the blood and LNs are not significantly altered. These effects argue for a requirement of steady naïve T cell priming and subsequent B cell help during the induction phase of arthritis for sufficient autoantibody titers needed for disease onset. However, when tolerance is breached due to sufficient anti-CII titers and arthritis develops in humanized CCR7 mice, treatment with 8H3-16A12 is unable to lead to a significant reduction in anti-CII titers, although disease progression ceases. The therapeutic efficacy of 8H3-16A12 treatment in CIA is likely attributed to the selective modulation of the T cell compartment consisting of a selective depletion of naïve CCR7hi T cells and a significant alteration in the ratio of regulatory to non-regulatory T cells in favor of the former. These effects may shift the balance towards the suppression of ongoing autoimmune responses. Treg targeting in CIA was demonstrated to exacerbate disease severity,28 and Treg expansion ex vivo and reinfusion led to significant amelioration of several murine autoreactive disease models, including CIA.28–31 Although adoptive transfer of Tregs led to a significant reduction in CIA severity, T and B cell responses to CII were not affected, and the authors suggested local control of the effector response in the arthritic joints as a mode of action of the transferred Tregs.
Thus far, clinical trials employing T cell targeting therapies in RA have led to largely disappointing results with the exception of blocking co-stimulation with a CTLA4−Ig fusion protein (Abatacept).32 Approaches using mAbs directed against CD4, CD5, and CD52 have shown low efficacy, and side effects are often severe.33,34 More recent developments of non FcR-binding CD3-specific mAbs, used in combination with anti-TNFα mAbs, have shown efficacy in preclinical RA models, such as CIA.35 The suggested effector mechanisms of non FcR-binding CD3-targeting mAbs Abs are effector T cell depletion and Treg preservation.36 Due to the considerable roles of T cells in RA pathogenesis and chronicity, novel and safe T cell targeting therapeutics are needed. In the present study, we report the functional characterization of a novel anti-human CCR7 mAb, 8H3-16A12, in humanized CCR7 mice. Treatment of humanized CCR7 mice with 8H3-16A12 led to complete resistance to CIA in a prophylactic setup and inhibited CIA progression in a therapeutic setup. Of note, injection of 8H3-16A12 did not provoke excessive pro-inflammatory cytokine release, probably predicting a favorable safety profile for human application. Taken together, our data suggest that the humanized 8H3-16A12 mAb has potential for the treatment of RA.
Materials and methods
Mice
Mice were bred under SPF conditions at the Central Animal Facility of Hannover Medical School. Ccr7−/− mice (B6.129P2(C)-Ccr7tm1Rfor/J) backcrossed for at least 15 generations on a BL6 genetic background, plt/plt mice (B6N.DDD-plt/NknoJ), B6 CD45.1 (B6.SJL−Ptprca Pepcb/BoyJ)- and B6 CD45.1/CD45.2 heterozygous mice were described previously.14,37,38 DBA 1/J-Ccr7−/− mice were backcrossed for at least 15 generations on a DBA/1 genetic background. Humanized T-CCR7+/+, DC-CCR7+/+, DC-T-CCR7+/+, GL-CCR7+/+ and CCR7stop/stop mice were described previously.16,17 All animal experiments were performed in accordance with institutional guidelines and approved by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit.
Collagen-induced arthritis
CIA was induced as previously described.39,40 Briefly, 10- to 14-week-old male mice were immunized intradermally at the tail base with 100 µg chicken collagen II in dilute acetic acid (MD Bioproducts, Switzerland) mixed with an equal volume of CFA (Difco), with a 100 µl total injection volume per mouse. All mice received an identical boost immunization 21 days after the primary immunization. Mice were scored several times a week for clinical signs of arthritis as follows: 0 = normal, 1 = mild swelling and erythema affecting single paw joints, 2 = pronounced swelling and erythema affecting one or more paw joints, and 3 = severe joint swelling and erythema and ankylosis/joint deformity. The maximum clinical score per mouse was 12. Arthritis scoring was performed in all experiments in a blinded manner.
Flow cytometry
Cell suspensions isolated from lymph nodes, thymus, spleen, and peripheral blood were stained with the following mAbs: anti-CD44-eFluor 450 (IM7), anti-CD4 PerCp (RM4-5), anti-PD-1 PE-Cy7 (J43), anti-CD45.2 APC-eFluor 780 (104), anti-CD11b PE-Cy7 (M1/70), anti-Ly-6C PerCp-Cy5.5 (HK1.4), anti-Ly-6G PE (1A8), anti-CD11c PE-Cy7 (N418), anti-CD45.1 APC (A20), anti-CD3e PE-Cy7 (145-2C11), anti-CD62L APC-Cy7 (Mel-14), anti-CD45R (B220) eFluor 450 (RA3-6B2), anti-Fas PE (15A7) (all from eBioscience), anti-CD19 Alexa 488 (6D5) (Biolegend), anti-CD19 APC-eFluor 780 (6D5), anti-B220 Pacific Orange (RA3-6B2) (ThermoFisher Scientific), anti-I-Ab FITC (AF6-120.1) (BD Biosciences), anti-CD8β Pacific Orange (Rm CD8-2), and anti-CD11b biotinylated (MAC-1) grown in our laboratories. Staining of biotinylated Abs was followed by incubation with Cy5 or APC-eFluor 780 labeled streptavidin (eBioscience). Human CCR7 was stained using anti-human CCR7 mAb (8H3-16A12) followed by biotinylated anti-murine IgG1 (A85-1) and Cy5 or Alexa-488 labeled streptavidin. Fluorescence-minus-one (FMO) or isotype control samples were used to assess background fluorescence and nonspecific Ab binding. Data were acquired on an LSR II (BD Biosciences) and analyzed with FACSDiva (BD Biosciences) and FlowJo Software (Treestar).
Histological analysis
Paws were decalcified in EDTA upon overnight fixation in 10% (vol/vol) neutral buffered formalin and embedded in paraffin blocks. Five-micron paw sections were stained with H&E and assessed for inflammatory infiltrates, synovial hyperplasia, and cartilage/bone erosion in a blinded fashion. Spleen and JDLNs were embedded in OCT immediately after extraction and frozen on dry ice. Using a cryostat (CM3050; Leica), 8-µm-thick sections were produced and subjected to fixation in ice-cold acetone for 10 min. Cryosections were rehydrated in Tris-buffered saline with 0.05% Tween 20, subsequently blocked in 5% mouse or rat serum and stained at room temperature with the following antibodies: anti-B220 Cy5 (RA3-3A1), anti-huCCR7 Cy3 (all grown and labeled in our laboratories), and DAPI staining (Sigma-Aldrich). A motorized epifluorescence microscope (BX61; UPlanSApo lenses: 10 × /0.4, 20 × /0.75 and 40 × /0.9) with a fluorescence camera (F-View II) and cellSens software (Olympus) were used to acquire images.
Cytokine measurement
Cytokines and chemokines in mouse sera were measured using a Luminex-based cytokine array according to the manufacturer’s instructions (Bio-Rad Laboratories, Hercules, USA).
ELISA
Levels of anti-chicken collagen II and anti-murine collagen II antibodies were measured in serial dilutions of mouse sera by ELISA as described previously.40
Generation of anti-human CCR7 mAb
Female BALB/c mice were immunized with 200 µg of peptide immunogen (derived from the N terminus of human CCR7) in Pepscan adjuvant (Pepscan Presto BV; The Netherlands) divided over four immunizations. Standard hybridoma cloning techniques were applied, and supernatants of the clones were screened for binding to the immunogen and to cells overexpressing human CCR7. The corresponding stretch of murine CCR7 was not bound by the antibody in the ELISA.
Transwell assays
The lower chamber of a Transwell plate with a 5-µm pore-size polycarbonate membrane (Corning Costar) was filled with RPMI1640 medium containing 1 µg/ml murine CCL19 (Peprotech) and 0–50 µg/ml anti-human CCR7 Ab 8H3-16A12 or mIgG1 isotype control Ab. Lymphocytes isolated from LNs of DC-T-CCR7+/+ mice (3 × 106/ml) were preincubated at 37 °C for 30 min with 0–50 µg/ml anti-human CCR7 Ab 8H3-16A12 or mIgG1 isotype control Ab and then transferred to the upper chamber. After 3.5 h of incubation at 37 °C, cells in the lower chamber were resuspended and counted on an LSR II (BD Biosciences) using beads (Fluoresbrite YG Microspheres 6.00 µm, Polysciences, Inc). The migration index (MI) was calculated as specific migration towards CCL19/spontaneous migration. The percent of migration inhibition was calculated as (MI Isotype control − MI 8H3-16A12) × 100/MI Isotype control.
Adoptive cell transfer
WT recipient mice were injected i.v. with a mix of 40 × 106 lymphocytes containing lymphocytes from DC-T-CCR7+/+ and WT mice mixed at a ratio of 1:1. Two hours before cell transfer, recipient mice were injected with 20 mg/ml anti-human CCR7 Ab 8H3-16A12 or mIgG1 isotype control Ab. Twenty-four hours after cell transfer, the recipient mice were sacrificed, and the frequency of the transferred T cells identified by congenic markers in peripheral lymph nodes, spleen, and blood was assessed by flow cytometry.
Statistics
Prism 4 (GraphPad Software, Inc.) was used for all statistical analyses. Unpaired two-tailed Student’s t-tests were performed in order to determine statistical significance between groups. *P < 0.05, **P < 0.01, ***P < 0.001. Where indicated, statistical analysis of CIA scores was performed on area under the curve (AUC) values calculated from the CIA score curves, as previously described41 and according to GraphPad software specifications.
Acknowledgements
We thank Linda Oberdörfer, Kerstin Daemen, and Jana Keil for excellent technical assistance and Svetlana Piter for excellent animal care. This work was supported by Deutsche Forschungsgemeinschaft (DFG) grant KFO 250-FO 334/2-1 to R. Förster.
Author Contributions
G.L.M., A.B., M.F., and J.R. performed experiments; G.L.M. and C.S.F. analyzed experiments; J.W.B. and E.K. provided essential reagents for the study; G.L.M. and R.F. designed experiments and wrote the paper. The manuscript was approved by all authors.
Competing interests
G.L.M., A.B., M.F., J.R., C.S.F., E.K., and R.F. declare no conflicts of interest. J.W.B. is an employee of Pepscan and was named inventor on a patent disclosing anti-CCR7 antibodies.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Georgios L. Moschovakis, Email: Moschovakis.Leandros@mh-hannover.de
Reinhold Förster, Email: Foerster.Reinhold@mh-hannover.de.
References
- 1.Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388:2023–2038. doi: 10.1016/S0140-6736(16)30173-8. [DOI] [PubMed] [Google Scholar]
- 2.Schulz O, Hammerschmidt SI, Moschovakis GL, Forster R. Chemokines and chemokine receptors in lymphoid tissue dynamics. Annu Rev. Immunol. 2016;34:203–242. doi: 10.1146/annurev-immunol-041015-055649. [DOI] [PubMed] [Google Scholar]
- 3.Comerford I, et al. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. 2013;24:269–283. doi: 10.1016/j.cytogfr.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat. Rev. Immunol. 2008;8:362–371. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- 5.Hjelmstrom P, Fjell J, Nakagawa T, Sacca R, Cuff CA, Ruddle NH. Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am. J. Pathol. 2000;156:1133–1138. doi: 10.1016/S0002-9440(10)64981-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weninger W, et al. Naive T cell recruitment to nonlymphoid tissues: a role for endothelium-expressed CC chemokine ligand 21 in autoimmune disease and lymphoid neogenesis. J. Immunol. 2003;170:4638–4648. doi: 10.4049/jimmunol.170.9.4638. [DOI] [PubMed] [Google Scholar]
- 7.Burman A, et al. A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J. Immunol. 2005;174:1693–1700. doi: 10.4049/jimmunol.174.3.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Page G, Lebecque S, Miossec P. Anatomic localization of immature and mature dendritic cells in an ectopic lymphoid organ: correlation with selective chemokine expression in rheumatoid synovium. J. Immunol. 2002;168:5333–5341. doi: 10.4049/jimmunol.168.10.5333. [DOI] [PubMed] [Google Scholar]
- 9.Bruhl H, et al. Functional expression of the chemokine receptor CCR7 on fibroblast-like synoviocytes. Rheumatol. (Oxf.) 2008;47:1771–1774. doi: 10.1093/rheumatology/ken383. [DOI] [PubMed] [Google Scholar]
- 10.Pickens SR, et al. Characterization of CCL19 and CCL21 in rheumatoid arthritis. Arthritis Rheum. 2011;63:914–922. doi: 10.1002/art.30232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pickens SR, et al. Role of the CCL21 and CCR7 pathways in rheumatoid arthritis angiogenesis. Arthritis Rheum. 2012;64:2471–2481. doi: 10.1002/art.34452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wengner AM, et al. CXCR5- and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum. 2007;56:3271–3283. doi: 10.1002/art.22939. [DOI] [PubMed] [Google Scholar]
- 13.Schneider MA, Meingassner JG, Lipp M, Moore HD, Rot A. CCR7 is required for the in vivo function of CD4 + CD25 + regulatory T cells. J. Exp. Med. 2007;204:735–745. doi: 10.1084/jem.20061405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mori S, et al. Mice lacking expression of the chemokines CCL21-ser and CCL19 (plt mice) demonstrate delayed but enhanced T cell immune responses. J. Exp. Med. 2001;193:207–218. doi: 10.1084/jem.193.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stuart JM, Dixon FJ. Serum transfer of collagen-induced arthritis in mice. J. Exp. Med. 1983;158:378–392. doi: 10.1084/jem.158.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ivanov S, et al. CCR7 and IRF4-dependent dendritic cells regulate lymphatic collecting vessel permeability. J. Clin. Invest. 2016;126:1581–1591. doi: 10.1172/JCI84518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wendland M, et al. Lymph node T cell homeostasis relies on steady state homing of dendritic cells. Immunity. 2011;35:945–957. doi: 10.1016/j.immuni.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 18.Chatenoud L, Ferran C, Bach JF. The anti-CD3-induced syndrome: a consequence of massive in vivo cell activation. Curr. Top. Microbiol Immunol. 1991;174:121–134. doi: 10.1007/978-3-642-50998-8_9. [DOI] [PubMed] [Google Scholar]
- 19.Ferran C, et al. Cytokine-related syndrome following injection of anti-CD3 monoclonal antibody: further evidence for transient in vivo T cell activation. Eur. J. Immunol. 1990;20:509–515. doi: 10.1002/eji.1830200308. [DOI] [PubMed] [Google Scholar]
- 20.Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20:119–122. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kocks JR, Davalos-Misslitz AC, Hintzen G, Ohl L, Forster R. Regulatory T cells interfere with the development of bronchus-associated lymphoid tissue. J. Exp. Med. 2007;204:723–734. doi: 10.1084/jem.20061424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mzinza, D. T., et al. Application of light sheet microscopy for qualitative and quantitative analysis of bronchus-associated lymphoid tissue in mice. Cell. Mol. Immunol. (2018). 10.1038/cmi.2017.150 [DOI] [PMC free article] [PubMed]
- 23.Davalos-Misslitz AC, et al. Generalized multi-organ autoimmunity in CCR7-deficient mice. Eur. J. Immunol. 2007;37:613–622. doi: 10.1002/eji.200636656. [DOI] [PubMed] [Google Scholar]
- 24.Winter S, et al. Manifestation of spontaneous and early autoimmune gastritis in CCR7-deficient mice. Am. J. Pathol. 2011;179:754–765. doi: 10.1016/j.ajpath.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat. Rev. Immunol. 2013;13:566–577. doi: 10.1038/nri3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forster R, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/S0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 27.Moschovakis GL, et al. Deficient CCR7 signaling promotes TH2 polarization and B-cell activation in vivo. Eur. J. Immunol. 2012;42:48–57. doi: 10.1002/eji.201141753. [DOI] [PubMed] [Google Scholar]
- 28.Morgan ME, et al. CD25 + cell depletion hastens the onset of severe disease in collagen-induced arthritis. Arthritis Rheum. 2003;48:1452–1460. doi: 10.1002/art.11063. [DOI] [PubMed] [Google Scholar]
- 29.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4 + CD25 + regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol. 2002;169:4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 30.Morgan ME, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25 + regulatory T cells. Arthritis Rheum. 2005;52:2212–2221. doi: 10.1002/art.21195. [DOI] [PubMed] [Google Scholar]
- 31.Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4 + CD25 + regulatory T cells. J. Immunol. 2003;170:3939–3943. doi: 10.4049/jimmunol.170.8.3939. [DOI] [PubMed] [Google Scholar]
- 32.Isaacs JD. Therapeutic T-cell manipulation in rheumatoid arthritis: past, present and future. Rheumatol. (Oxf.) 2008;47:1461–1468. doi: 10.1093/rheumatology/ken163. [DOI] [PubMed] [Google Scholar]
- 33.Epstein WV. Expectation bias in rheumatoid arthritis clinical trials. The anti-CD4 monoclonal antibody experience. Arthritis Rheum. 1996;39:1773–1780. doi: 10.1002/art.1780391102. [DOI] [PubMed] [Google Scholar]
- 34.Strand V, Kimberly R, Isaacs JD. Biologic therapies in rheumatology: lessons learned, future directions. Nat. Rev. Drug Discov. 2007;6:75–92. doi: 10.1038/nrd2196. [DOI] [PubMed] [Google Scholar]
- 35.Depis F, et al. Long-term amelioration of established collagen-induced arthritis achieved with short-term therapy combining anti-CD3 and anti-tumor necrosis factor treatments. Arthritis Rheum. 2012;64:3189–3198. doi: 10.1002/art.34497. [DOI] [PubMed] [Google Scholar]
- 36.Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J. Immunol. 2011;187:2015–2022. doi: 10.4049/jimmunol.1100713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohl L, et al. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity. 2004;21:279–288. doi: 10.1016/j.immuni.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 38.Zietara N, et al. Multicongenic fate mapping quantification of dynamics of thymus colonization. J. Exp. Med. 2015;212:1589–1601. doi: 10.1084/jem.20142143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inglis JJ, et al. Collagen-induced arthritis in C57BL/6 mice is associated with a robust and sustained T-cell response to type II collagen. Arthritis Res Ther. 2007;9:R113. doi: 10.1186/ar2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moschovakis GL, et al. T cell specific Cxcr5 deficiency prevents rheumatoid arthritis. Sci. Rep. 2017;7:8933. doi: 10.1038/s41598-017-08935-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfeifle R, et al. Regulation of autoantibody activity by the IL-23-TH17 axis determines the onset of autoimmune disease. Nat. Immunol. 2017;18:104–113. doi: 10.1038/ni.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]