

ABSTRACT
Melanoma is the deadliest form of skin cancer. In the early stages, melanoma can be treated successfully with surgery alone and survival rates are high, but after metastasis survival rates drop significantly. Therefore, early and correct diagnosis is key for ensuring patients have the best possible prognosis. Melanoma misdiagnosis accounts for more pathology and dermatology malpractice claims than any cancer other than breast cancer, as an early misdiagnosis can significantly reduce a patient’s chances of survival. As far as treatment for metastatic melanoma goes, there have been several new drugs developed over the last 10 years that have greatly improved the prognosis of patients with metastatic melanoma, however, a majority of patients do not show a lasting response to these treatments. Thus, new biomarkers and drug targets are needed to improve the accuracy of melanoma diagnosis and treatment. This article will discuss the major advancements of melanoma diagnosis and treatment from antiquity to the present day.
KEYWORDS: Melanoma, diagnosis, immunohistochemistry, treatment, immunotherapy, epigenetics
Introduction
At the basal level of the epidermis sit the melanocytes, which produce the UV absorbing pigment melanin. Melanocytes make up a minority cell population in the epidermis, with only 1500 melanocytes per square millimeter, and divide infrequently, less than twice per year. In response to UV radiation keratinocytes produce α-melanocyte stimulating hormone (α-MSH), which binds to the melanocortin 1 receptor (MC1R) on melanocytes signaling to induce melanin synthesis.1 The melanocytes then transfer the melanin to surrounding keratinocytes through finger-like projections that reach out into the surrounding cells.2 Sun-exposed keratinocytes accumulate melanin, which shields their nuclei from the mutagenic effects of UV radiation. As the keratinocytes mature, they undergo keratinization, anucleate and die. Thus, the outer layer of the skin is protected by both melanin pigment in keratinocytes and a layer of dead keratinocytes that act as a barrier to protect the living cells beneath.
There are two forms of melanin produced by melanocytes: the black/brown pigment eumelanin, and a red/yellow pigment pheomelanin. The ratio of eumelanin to pheomelanin in the skin determines skin color rather than the number of melanocytes, which is relatively constant in all skin types. The darker eumelanin is a better UV shield, and consequently people with darker skin have a lower skin cancer risk. Pheomelanin not only offers less protection against UV radiation, but the production of pheomelanin produces carcinogens.3-5 Pheomelanin has been shown to produce more ultraviolet-A-induced reactive oxidative species (ROS) leading to greater DNA damage in response to UV radiation.3,4,6-8 It has long been known that melanoma risk was tied to skin, hair and eye coloration: people who have light skin that does not tan, blond or red hair, and light eyes have a much higher risk of having melanoma compared to the population as a whole.1,7,9,10
Coloration of the skin, hair and eyes is controlled, in part, by MC1R. Polymorphisms in the MC1R gene determine the activity level of MC1R. Variants of the MC1R gene that lead to reduced MC1R function result in the production of predominantly the red/yellow pheomelanin pigment and fair skin that does not tan, and light eyes and hair.1,4,11 Fully functional MC1R stimulates production of the black/brown eumelanin.1 People harboring less functional variants of MC1R accumulate more mutations due to increased exposure of the nuclei to UV damage. If mutations accumulate in sensitive regions of the genome, then skin cancers can arise.
Melanoma, a malignant tumor arising from melanocytes, is a rare disease, affecting only 22.1 out of 100,000 people in the US (Cancer statistics from the Center for Disease Control and Prevention). However, it is also a very deadly disease accounting for 75% of skin cancer deaths though it only accounts for 4% of skin cancer cases. In 2019, it is expected that 96,480 new cases of melanoma will be diagnosed, and 7,230 people will die in the US alone (American Cancer Society). This review will go over a brief history of melanoma, the major molecular defects that lead to melanoma progression, the major methods for diagnosing and determining patient prognosis and melanoma treatment options.
History of melanoma
Melanoma is generally thought of as a modern disease exacerbated by migration of fair skinned people to areas nearer the equator and by modern sun-seeking behavior. However, although melanoma incidence rates have certainly skyrocketed in modern times, melanoma is an ancient disease that has been documented throughout history. The first documented case of melanoma was recorded in the 5th century by Hippocrates of Cos, but earlier physical evidence of melanoma has been found in the bones of pre-Columbian mummies thought to be ~2400 years old.12 The first recorded cases of melanoma in Western medical literature are found in the 1651 writings of Drs. Highmore and Bonet, and in the 1757 writings of Henrici and Nothnagel, which all describe fatal black tumors spread throughout the bodies of their patients.12
The first surgical removal of a melanoma tumor was recorded in 1787 by the Scottish surgeon John Hunter, who believed, as most (if not all) doctors did at the time, that melanoma was a fungal growth; it would not be until much later that the exact nature of the disease would be discovered.12,13 He successfully removed a recurring tumor from the jaw of a 35 year-old man, and from this point surgical resection became routine for the treatment of melanoma.12,13
In the 1820s Dr. William Norris described a patient who had ‘fungoid disease’ and noted that the man’s father had also died of a similar disease and postulated that the disease could, in certain cases, be heritable.12,13 Norris observed that melanomas arose from moles, and that individuals from families with hereditary melanoma often had a large number of moles.12,13 Norris was the first to recommend surgical removal of melanoma lesions with wide excision margins, noting that if the margins were not wide enough, the disease tended to recur, and that surgical excision was ineffective if the disease had already disseminated.12,13 Norris’ observations were remarkable for the time, as melanoma was so little understood, and as they were made some 50 years before Mendel’s work on inheritance.
Later that century, in 1892, Dr. Herbert Snow offered rationale for removal of lymph nodes, describing them as traps that prevented the spread of cancer to the blood.12,13 For nearly 100 years after this, melanoma treatment remained much the same, with surgical excision and lymph node removal as the only treatment for primary melanoma. No treatment options existed for the disease after metastasis. It was not until the discovery of chemotherapies in the 1940s and their utility in treating some cancers, that the next small breakthrough in melanoma treatment came.
Molecular defects
When melanoma was first recognized as a disease in the 1800s it was classified based on where the tumor arose. In the 1960s melanomas began to be classified based on histologic patterns. While histology is still important for melanoma diagnosis, one of the most influential shifts in the understanding of cancer progression was the realization that cancer arises due to the accumulation of genetic mutations, leading to misregulation of cellular pathways.12 Since the 1980s there has been a concerted effort to identify individual mutations that commonly occur in cancers such as melanoma, and to develop targeted therapies to treat each patient in a more personalized way based on the mutations they carry.7,9,12-14
Approximately 5–12% of melanomas are hereditary, and hereditary melanomas tend to have different mutation profiles to non-hereditary melanomas.12 CDKN2A is a mutation commonly found in familial melanoma syndromes, though somatic CDKN2A mutations also occur in sporadic melanomas.15 Families that carry CDKN2A mutations tend to have high numbers of clinically atypical (but benign) nevi and a family history of melanoma.12 Of familial melanomas, up to 40% contain CDKN2A mutations, which lead to defects in the proteins p14ARF and p16INK4A. These proteins are important tumor suppressors that regulate the G1 checkpoint and stabilize p53 expression.11,12 P16 binds to CDK4 leading to cell cycle arrest, while mutations in p16 prevent its binding to CDK4 thus disrupting cell cycle arrest.15 Though p14ARF and p16INK4A are not druggable targets, therapeutic targeting of other related proteins, such as CDK4, is being explored.15
A less common but far more virulent group of germline mutations that increase the risk of developing melanoma, are mutations in nucleotide excision repair (NER) pathways. UV radiation creates bulky DNA lesions that must be removed and repaired by repair mechanisms such as NER; if these pathways are disrupted, mutational events will be propagated. The disease xeroderma pigmentosum (XP) is caused by hereditary mutations in one of eight NER genes; XPA, XPB, XPD, XPE, XPF, XPG, or XP-V.10,16 Though mutations in any of these eight nucleases result in XP, XPA and XPC are the most commonly mutated and account for half the cases of XP.10,16 The lack of functional NER pathways results in the accumulation of UV or chemically induced mutations in the skin and a heightened risk of skin malignancies. Patients with XP show extreme sensitivity to UV radiation; often they develop some form of skin cancer before the age of 10 and have a far greater risk of developing cutaneous melanoma on sun exposed skin than the population as a whole.10,16 This disease highlights the importance of nucleotide excision repair in melanoma prevention.
Not all variants of melanoma have the same mutational frequencies, however, there are recurrent somatic mutations that appear frequently in all types of melanoma. Driving mutations in melanoma tend to be in signaling pathways that regulate proliferation (BRAF, NRAS, and NF1),14 growth and metabolism (PTEN and KIT),14 resistance to apoptosis (TP53),14 replicative lifespan (TERT),17,18 cell identity (ARID2),14 and cell cycle control (CDKN2A)4,12,14,17 (Figure 1).
Figure 1.
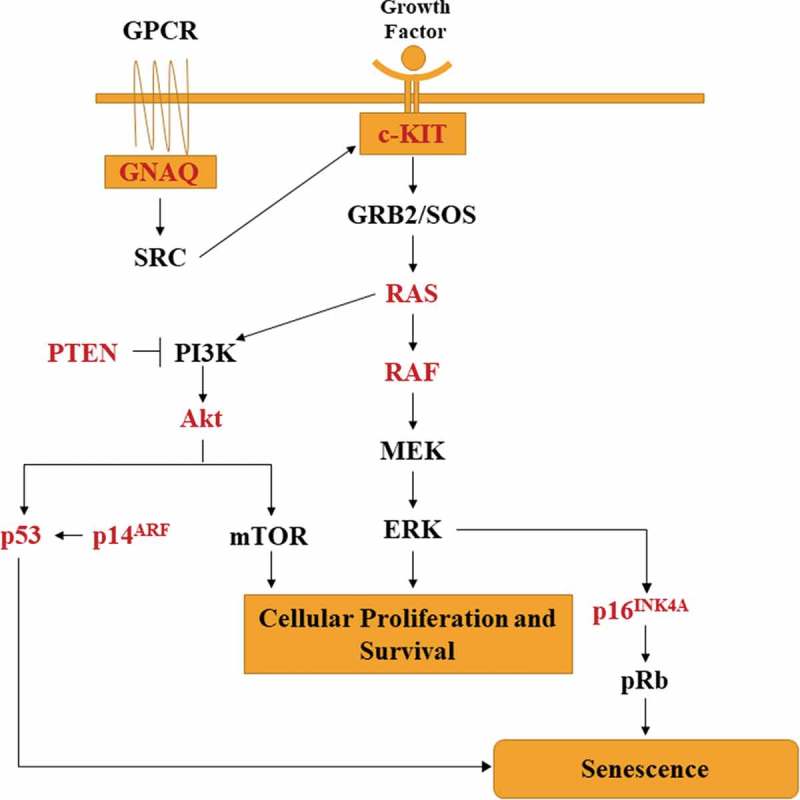
Oncogenic pathways commonly misregulated in melanoma. Mutations in NRAS, BRAF, GNAQ and c-KIT lead to constitutive MAPK signaling leading to unbridled proliferation and survival. The phosphatidylinositol 3ʹ kinase (PI3K) cascade is activated by oncogenic NRAS. Mutations of the CDKN2A gene leads to loss of P16INK4A or P14ARF or both depending on where the mutations occur. CDKN2A mutations are common in hereditary melanoma.
Though a handful of mutations involved in melanoma development may be inherited, the majority of melanomas arise from somatic mutations acquired later in life. One of the most commonly mutated pathways in melanoma, and one that has garnered a lot of interest for targeted therapeutic purposes, is the MAP kinase signaling pathway. Mutations in BRAF and other MAPK mutational events are thought to be early oncogenic events. Nearly 80% of benign nevi have BRAF mutations, indicating that a BRAF mutation alone is insufficient for a malignant phenotype and that more than one mutation is needed to transform normal melanocytes into a malignant tumor.14,15 It is estimated that 70% of melanomas contain mutations in the MAPK signaling pathway;14 ~50% of melanomas contain activating BRAF mutations, the most common of which is the V600E mutation (>85% of BRAF mutations), which lead to constitutive activity of downstream MAPK signaling. This mutation is seen more commonly in melanomas arising from intermittent (rather than chronic) sun exposure and with a superficial spreading melanoma (SSM) phenotype.4,7,9,12,14,19 Another 15–20% of melanomas have NRAS mutations,12,14 2% have CKIT mutations (common in mucosal melanomas),14 and 50% of uveal melanomas have GNAQ mutations.20 The MAPK pathway is involved in controlling proliferation and survival in response to growth factors, so when mutations cause constitutive activity, cells grow unchecked. Therefore, this is a particularly important oncogenic pathway making it a prime target for therapeutic intervention. BRAF is an attractive target in this pathway as it is mutated in approximately half of melanoma patients, it is easy to target and it is not active in healthy cells.14
UV radiation also directly stimulates MAPK signaling and has been shown to be a major pathway involved in UV-induced carcinogenesis.21 ERK, JNK and p38 kinases have all been shown to be activated in response to UV radiation through phosphorylation of threonine and tyrosine in the T-X-Y motif of the activation loop.21 Activation of these proteins results in their translocation into the nucleus where they in turn phosphorylate transcription factors leading to the expression of target genes that control cellular pathways such as proliferation, differentiation, development and cell death.21 UV-induced activation of this pathway is an area of interest for melanoma research, as understanding the mechanism by which UV-induced carcinogenesis occurs may result in the development of drugs to target these pathways, which may lead to new treatment options for skin cancers such as melanoma.
Diagnosis and prognosis of melanoma
The early classification of melanoma was based on where the tumor arose from (existing nevus, acquired melanocytic lesion, blemish free skin), but in the 1960s a prominent dermatologist, Wallace Clark, suggested that melanoma ought to be classified based on histological features instead, thus revolutionizing the way melanoma was diagnosed.12-14 He initially described three histological variants of melanoma, superficial spreading melanoma (SSM), lentigo maligna melanoma (LMM) and nodular melanoma (NM),13,14 and while several new variants such as acral lentiginous melanoma, mucosal melanoma, desmoplastic melanoma and nevoid melanoma have since been described, SSM, LLM and NM are still variants recognized today.14 This was important work as melanoma was once thought of a homogenous disease; Clark was the first to recognize that it is actually a heterogenous disease, to show that the variants behaved differently, had differences in prognosis and that not all melanomas should be treated in the same way.12-14
Furthermore, in 1966 Clark proposed a system for evaluating melanoma based the depth of invasion of melanoma cells into the dermis and subcutaneous fat.12-14 Clark divided the skin into histologically-identifiable anatomic compartments; as melanoma cells traversed each compartment (or Clark “level” as they came to be known), the risk of distant spread theoretically increased:
Level 1: melanoma cells are confined to the epidermis (melanoma in situ)
Level 2: invasion of single cells or very small nests of melanoma into the papillary dermis
Level 3: melanoma cells “fill and expand” the papillary dermis
Level 4: invasion into the reticular dermis
Clark’s observations led to changes in the standard treatment for patients in the early stages of melanoma; he noted that patients with deeper cutaneous invasion (level III-V) were more likely to have lymph node invasion and determined that lymph node dissection should be limited only to patients with melanoma that had moved past the papillary dermis.12,13
The Clark levels still remain a standardized way to convey information related to risk of disease aggressiveness and are reported in melanoma reports by some pathologists, however in 1970 Alexander Breslow independently devised a more accurate method for classifying melanoma based on a measured depth of invasion that captured the thickness of the tumor.12-14,22 Breslow’s classification system, known as Breslow’s depth, was based on the depth of invasion in millimeters rather than the depth by anatomic compartments, which vary in thickness at different anatomic sites.22 Breslow also initially divided melanoma into five stages:
Stage I: less than or equal to 0.75 mm
Stage II: 0.76 − 1.5 mm
Stage III: 1.51–2.25 mm
Stage IV: 2.26–3.0 mm
Stage V: greater than 3.0 mm.22
He showed that patients with thinner melanomas (stages I and II) had a much better chance of survival and a lower risk of regional and distant metastasis.14 This observation allowed for smaller resections than were commonly performed at the time and, like Clark’s method, allowed physicians to assess the risk of the melanoma having spread to the lymph nodes and to determine whether lymph node removal was necessary (patients with a Breslow thickness of 1.5 mm or greater were more likely to benefit from prophylactic lymph node dissection).12-14 Eventually, modification of measurement cut-offs would be made and incorporated into the staging systems that are still used today to guide treatment and predict prognosis; Breslow depth remains one of the best independent predictors of patient outcome.23
In the 1980s it became clear that using the Clark level and Breslow thickness for prognostic purposes resulted in variation in the predicted outcome and therapeutic recommendations between pathologists,24,25 and that new consensus guidelines for reporting melanoma were needed. Over the next 40 years, as data on melanoma patient treatment and survival accumulated and statistical methods improved, new staging systems were created. The American Joint Committee on Cancer (AJCC) has been instrumental in the creation of the TNM (tumor, node, metastasis) staging system.14,23 Most major tumor types have their own TNM staging system and melanoma is no exception; multiple committees and taskforces have worked to analyze the every-growing mountain of data on melanoma patient treatment and prognosis and have put together an internationally recognized melanoma database, which is assessed and updated regularly.12-14,23,26 This database is continually analyzed and used to update the AJCC melanoma TNM staging system. The AJCC staging system provides pathologists and clinicians a guideline for staging patients diagnosed with melanoma. By combining histologic attributes of the primary tumor (T), the presence and extent of regional lymph node disease (N), and the presence and extent of distant metastasis (M), clinicians are able to assign patients a stage grouping that is strongly linked to survival and prognosis. In addition to Breslow depth, attributes of the primary tumor such as ulceration, mitotic rate, tumor-associated inflammation, and regression27 have all been correlated with outcome and therefore incorporated into the AJCC staging system for melanoma.
The most recent revision of the AJCC staging manual (8th edition) was released in 2016 and implemented in 2018.28 Highlights of the evidence-based changes now reflected in current staging criteria include refinements in measurements of tumor thickness, removal of mitotic activity as a reason to upstage a thin melanoma, expansion of the regional lymph node (N) categories based on number of positive regional lymph nodes, expansion of metastasis (M) categories based on location of metastasis, and expanded stage groupings in stage III disease to better stratify long term prognosis.28
Staging is vitally important as it gives clinicians the tools to assess patient prognosis and put together a treatment regimen that will give the patient the best possible chance for recovery or prolonged survival. However, it has long been observed that, though there is an international staging system, the diagnosis of melanoma remains a difficult one to render accurately and consistently.28-30,31,32 Multiple studies show surprisingly high inter- and intra- observer variability between pathologists in the diagnosis of melanocytic neoplasms, particularly those with ambiguous histologic features.14,15,19,21-23 Much of the variability in diagnosis and prognosis for melanoma is due to the somewhat subjective visual observations used for melanoma diagnosis and prognosis, therefore, research into new, (****)) more objective methods to improve the accuracy and reproducibility in the field of melanoma diagnosis is ongoing.33
To improve detection and diagnosis of melanoma (as well as other cancers), non-invasive imaging technology pre-biopsy and more quantitative techniques post-biopsy, such as fluorescence in situ hybridization (FISH), comparative genomic hybridization (CGH), sequencing, mass spectrometry (MS) and IHC are being used more frequently.34,35
Non-invasive imaging for melanoma detection
Recent technological advancements have resulted in the emergence of non-invasive imaging technology to detect melanoma. Several smartphone applications, such as SkinVision, UMSkinCheck, and MoleScope, have been developed with purported goals of wide-spread accessibility, cost-effective screening, and ultimately increased early detection for patients. However, multiple studies have shown that these applications are often inaccurate:36,37 one study found that 3 out of 4 applications incorrectly classified upwards of 30% of melanomas as low-risk lesions.37 These applications could be a very useful tool for melanoma diagnosis if accuracy could be improved and if strict regulatory oversight was ensured,37,38 however, experts caution that reliance on these applications as they are currently available has the potential to harm patients through a false sense of security, potentially resulting in delayed diagnosis. As the technology evolves and becomes more reliable, these applications may become very effective tools for melanoma diagnosis, but at present users should chose applications judiciously to ensure that they are effective.
Other imaging technologies have been developed for use by clinicians as an adjunct to visual screening alone. These devices aim to help clinicians decide whether biopsies are needed for ambiguous lesions. Two devices, MelaFind and SIAscope (Spectrophotometric Intracutaneous Analysis), use visible and near infrared light (~400 nm to ~1000 nm) to visualize lesions and give information to help clinicians decide whether a biopsy is necessary.39-41 MelaFind is a fully automatic diagnostic system that was developed in 2010.39 It uses light to visualize skin lesions up to 2.5 mm deep, and provides information on morphologic disorganization of cells in a lesion that can help clinicians decide whether a lesion needs to be biopsied to rule out melanoma.39 Studies have shown that the use of MelaFind results in more accurate biopsy decisions.39,42 A study was conducted in 2017 in which 160 board certified dermatologists analyzed 25 melanomas and 25 benign nevi with or without multi-spectral digital skin lesion analysis performed with MelaFind: researchers found that evaluation with MelaFind increased biopsy sensitivity from 76% after clinical evaluation alone to 92%, increased specificity from 52% to 79% and increased overall biopsy accuracy from 64% to 86%.39 Though these results are encouraging, many insurance companies will not cover MelaFind use as they consider it to be experimental, and so patients must pay for the procedure out of pocket, which may dissuade patients from agreeing to its use. As multi-spectral imaging technology becomes more refined and if it continues to provide evidence of cost-savings through more directed biopsy practice, it may find its way into routine clinical use.
SIAscope, a device similar to MelaFind, was developed in 2002. It is capable of measuring collagen, blood and melanin.43 The device shows whether melanin is confined to the epidermis and images the vascular network and pigment composition of a lesion.40,41,43 Early versions of the device had some sensitivity issues with a sensitivity 82.7% and specificity 80.1%, which is similar to sensitivity and specificity of dermatoscopy (visual examination of a pigmented lesion with a handheld magnifier) performed by experienced dermatopathologists. As such, some clinicians have questioned whether it provides enough of a benefit to warrant its use for detecting and diagnosing melanoma.43 However, the device may be useful for improving diagnosis of pigmented lesions by primary care physicians, particularly those practicing in more rural environments without easy access to specialized dermatology care.40
Immunohistochemistry use in melanoma diagnosis
Clinical markers
Regardless of the detection method, diagnosis of melanoma requires that a lesion be recognized as clinically atypical and biopsied by a health care provider. Once a lesion has been biopsied, microscopic analysis is performed. Melanoma is generally diagnosed by trained pathologists based on a variety of classic histopathological features. However, melanoma is a heterogenous disease and some histologic variants are not easily recognizable by traditional hematoxylin and eosin (H&E) examination alone.44-46 Likewise, there are numerous histological mimics of melanoma, and distinguishing between melanoma and its mimics can be challenging.47-50 Moreover, as the molecular mechanisms driving melanomagenesis have become better understood, researchers and clinicians have utilized molecular biomarkers to assist in recognition of melanoma, and immunohistochemistry (IHC) has been increasingly utilized in the interpretation of difficult cases.34 IHC is the most common ancillary technique used by pathologists to assist in the diagnosis of melanoma as it is readily available in most laboratories, relatively inexpensive, and reliable and reproducible. Therefore, it is unsurprising that the use of IHC for the diagnosis of melanoma has significantly increased over the last 20 years.34
For melanoma, there are two types of biomarkers most commonly used for diagnostic and prognostic purposes: melanocytic markers and proliferative markers.23,26,33,34,51-56 Melanocytic markers are used to determine if an ambiguous lesion is melanocytic in origin and tend to be proteins involved in melanin synthesis, melanosomes biogenesis, or melanocyte differentiation, while proliferation makers are used to evaluate cell cycle activity in a lesion.54,56 Currently, the determination of tumor proliferation is typically done counting mitotic figures (mitosis/mm2); however, recent studies have shown that IHC detection of proliferative markers can be a robust indicator of proliferative activity with prognostic import.57,58
The use of IHC has even become important in staging systems. Beginning in the 7th ed AJCC, IHC was recommended in the evaluation of sentinel lymph nodes if tumor cells were not evident on H&E, thus improving detection of micrometastasis.23 There are several melanocytic markers that provide robust support for melanocytic origin and, in the appropriate setting, melanoma. Melan-A and MART-1 are two antibodies raised against the same antigen (melanoma antigen recognized by T-cells). MelanA/MART-1 is one of the most widely used biomarkers for melanoma as it is more sensitive than HMB-45 for recognizing melanoma.45,59-61 HMB-45 (which stands for Human Melanoma Black) is an antibody that recognizes the melanocytic antigen gp100 (also known as Pmel 17).35,52,59,61-63 Gp100 plays a role in melanin polymerization, melanosome biogenesis and melanogenesis. Other common melanocytic markers used for melanoma diagnosis include S100 protein, micropthlamia transcription factor (MITF), Tyrosinase and SOX10.33,34,58,59,61,64,65 Melan-A/MART-1, and HMB-45 have some of the highest specificity, their expression being limited to melanocytic tumors and only a few other, rarely encountered entities.26,52,56 The choice of the melanocytic marker used in the evaluation of a melanocytic lesion depends on the goal of the outcome: sensitive markers such as S100 protein and Sox10 are useful when pathologists wish to detect a possible melanocytic neoplasm (although non-melanocytic neoplasms may also be detected with these stains), while specific markers are useful to establish unequivocal melanocytic derivation (although some melanocytic tumors may be missed using only these markers). Ideally, a melanocytic marker is both sensitive and specific.
Melanocytic markers have their shortcomings, however, namely they stain all melanocytes, which means that they have little to no role in distinguishing between melanoma and a benign melanocytic proliferation.26,55,56,61,62 Furthermore, some types of melanoma (specifically desmoplastic melanomas) tend to lack expression of the most specific melanocytic markers, which can potentially result in a false negative diagnosis if more sensitive markers are not emplyed.45,54
Prognostic markers
Proliferation markers are used to determine the number of cells in the cell cycle in a given lesion, with benign melanocytic tumors expected to have low proliferative indices and malignant tumors expected to have high proliferative indices. The most commonly used proliferation marker for melanoma is Ki-67, a protein involved in ribosomal transcription, the expression of which is associated with cell proliferation and is highly elevated in the most aggressive melanomas.45,57,66,67 In 1997 a second mitotic maker, phosphohistone H3 (PHH3), was introduced.57,68 PHH3 is a histone modification that is associated with chromatin condensation in late G2/M phase of the cell cycle, and is therefore, more specific in detecting cells actually undergoing mitosis, and not just cells in cell cycle as detected by Ki-67.45,57 The use of PHH3 for diagnostic and prognostic purposes has been investigated in numerous cancers, and has been shown to be useful in distinguishing melanomas from benign nevi, though it is not a significantly better prognostic indicator than Ki-67.57,58 Both Ki-67 and PHH3 have been shown to provide some limited independent prognostic information for melanoma, although their use is not currently mandated in melanoma reporting nor does their use affect staging. One major limitation of the use of proliferative indicators, however, is their lack of specificity for melanocytes. In tumors with high infiltration of mitotically active immune cells, both of these markers can overestimate proliferation.34
Ultimately, there is no perfect diagnostic or prognostic biomarker for melanoma. It is too diverse a disease to expect to find a single biomarker that can diagnose melanoma, which is why IHC screenings are often done in panels.54,69 Using multiple biomarkers can overcome the shortcomings of the individual biomarkers, and to this end, research into identifying new melanoma biomarkers is ongoing.54,70,71
Serological prognostic biomarkers
While IHC tests for biomarkers expressed in or on the surface of tumor cells, some biomarkers can be detected in patient serum. One for the strongest independent prognostic indicators for malignant melanoma is lactate dehydrogenase (LDH).27,72 LDH catalyzes the conversion of pyruvate into lactate in anoxic conditions, such as in the oxygen depleted tumor microenvironment. In the AJCC 8th edition melanoma staging system elevated LDH is recognized as an adverse prognostic indicator for patients with stage IV melanoma and indicates a lower chance of survival.72,73
Emerging markers: epigenetic misregulation in melanoma
Other promising biomarkers for cancer diagnosis that have garnered a lot of interest in cancer research are chromatin remodeling and epigenetic regulatory proteins.74,75 Every cell in multicellular organisms contains the same genetic material, however, each type of cell has a unique function and set of specialized proteins that carry out that function. Furthermore, as an organism ages the protein make-up of cells changes; the proteins that are necessary for embryonic development can lead to illness if expressed later in life. Therefore, controlling which proteins are expressed in each cell type and when they are expressed is a vitally important process for the health of an organism.
The human genome, composed of three billion base pairs and coding for ~20,000 proteins, is tightly compacted into chromatin, and fits into the nucleus of the cell, which is only 6 μm in diameter. The organization of genetic material is imperative, and to this end the DNA is wrapped around protein complexes composed of proteins called histones, to create nucleosomes. There are 8 core histones in each nucleosome, two each of histones H2A, H2B, H3 and H4, and from each histone the N-terminal tails extends from the complex and is the sites of posttranslational modifications (PTMs). Chemical modifications added to the DNA or histones aid in the regulation of cellular processes such as DNA damage repair,76,77 DNA replication,78,79 cell cycle regulation,79,80 and apoptosis.81 These modifications can directly affect chromatin compaction by affecting how nucleosomes interact with each other, and the level of compaction allows or denies access to cellular machinery. Epigenetic modifications can also act as binding sites for transcription factors and cellular machinery. In this way epigenetic modifications and the chromatin remodeling complexes that add and remove these modifications regulate cellular processes. Epigenetic modifiers have been shown to be dysregulated in several diseases, the most prevalent being cancer. Epigenetic misregulation in cancer is a key area of diagnostic and therapeutic cancer research at present.70,74,82-87
Several epigenetic modifications have been linked to melanoma. Deacetylation of lysine has been shown to be involved in the silencing of key tumor suppressor genes such as CDNK1A, which leads to the down-regulation of important proteins such as p21cip1.53,88,89 Histone hypoacetylation has also been shown to contribute to melanoma progression leading to the down-regulation of proapoptotic proteins such as Bim, Bax and Bak, and PIB52A, which is needed to block PI3K/Akt signaling and for inhibition of proliferation.88 When hypoacetylation leads to down-regulation of PIB52A then melanoma cells can grow unchecked.88
H3k27me3 and melanoma
Proper regulation of histone methyltransferases is also important for keeping cells operating properly, and dysregulation has been shown to contribute to melanoma development. The polycomb repressive complexes (PRC1 and PRC2) are important chromatin remodeling complexes in the cell. The catalytic subunit of the PRC2 is the enhancer of zeste homologue 2 (EZH2) methyltransferase. EZH2 catalyzes the trimethylation of histone 3 lysine 27 (H3K27me3). H3K27me3 is an important gene silencing mark that is involved in X-inactivation and embryogenesis, however, later in life, high expression or activating mutations of EZH2 have been shown to be a factor in the pathogenesis of cancers such as melanoma.74,90-92 PRC2 is an important regulator of key tumor suppressor genes such as the CDKN2A locus and the pRB-E2F pathway.20,75,90,93 The CDKN2A locus plays a role in cell cycle regulation and apoptosis. It is silenced during embryogenesis and in proliferating cells, however, when silenced in tumor cells, inappropriate proliferation results.90-92 EZH2 and H3K27me3 have also been shown to regulate expression of the cell adhesion molecule E-cadherin, and EZH2 overexpression results in downregulation of E-cadherin, leading to a decrease in cellular adhesion and increase in cell mobility and invasiveness.94
Low levels of EZH2 and H3K27me3 have also been reported in a subset of melanoma patients and in other cancers as well. However, this has not been as extensively studied, and the mechanisms by which loss of EZH2 contributes to cancer pathology is poorly understood.48,95,96
Treatment
Surgical resection
Surgical removal of the tumor and surrounding healthy tissue is the primary treatment for localized melanoma, and sentinel lymph node biopsy is performed in patients whose tumors are greater than 0.8 mm thick or are thinner than this but ulcerated (stage pT1b or greater).13 If melanoma cells are found in the sentinel lymph nodes, then the remaining lymph nodes in the area are sometimes removed. In some situations, metastatic tumors can be surgically removed as well, but surgical treatment in the setting of known metastatic disease is not meant to be curative and will require other treatment options as well.
Chemotherapy
For patients with metastatic disease, surgical treatment alone will not be curative and drug therapies are the next line of defense. Until recently, the only treatment options for patients with metastatic melanoma was chemotherapy. The idea of developing chemical treatments for cancer, or chemotherapies, was first explored in the early 20th century by the German chemistry Paul Ehrlich.12,13,97-99 Ehrlich theorized that chemical agents could target defined sub-populations of cells, and to this end he developed the first primitive alkylating agents.12,13,97-99 Ehrlich, incidentally, was also the first to suggest that immune cells were involved in eliminating transformed cells, though immune-based therapies would not be developed until the end of the 20th century.98
In 1910 George Clowes developed the first transplantable tumor system in rodents, which led to the development of model systems leading to a major breakthrough that allowed researchers and clinicians to screen prospective chemotherapeutic agents in a living organism.97 The early chemotherapy agents developed by Ehrlich and others were largely ineffectual, and it was not until the 1940s that progress was made in the development of chemical agents that were effective for treating cancer.
The use of mustard gas during WWI led to the observation that exposure to these compounds resulted in depletion of the bone marrow and lymph nodes.97 In the 1940s two pharmacologists from Yale, Alfred Gilman and Louis Goodman, were asked by the US Office of Scientific Research and Development, to explore therapeutic uses for these chemicals.12,97 Gilman and Goodman found that one mustard compound, nitrogen mustard, effectively treated mice with transplanted lymphoid tumors; nitrogen mustard was then administered to lymphoma patients and a marked regression was observed, however, the regressions did not last and side effects were severe.12,97 Regardless, the development of chemotherapeutics made it possible to treat metastatic cancers for the first time in history.
In 1968, the first chemotherapy clinical trials in metastatic melanoma were conducted using the drug 1-phenylalanine mustard, commonly known as melphalan, however, it was not effective and had extremely high toxicity.12,13 The first and only chemotherapeutic drug to be approved by the FDA for the treatment of melanoma was dacarbazine in 1975.12,13,98 Until more recently, dacarbazine remained the standard of care for metastatic melanoma, though the response is partial at best, with median survival from 5–11 months and a 1 year survival rate of only 27%.12,13,97 Yet no other chemotherapeutic developed for melanoma treatment since has been more effective or less toxic. Thus, early detection of melanoma was vital for improving patient prognosis, and in the 1960s doctors began to focus on public education about the disease and to stress the importance of regular skin cancer screening, especially for at-risk populations.12
More recent drug discoveries have greatly improved the prognosis for patients with metastatic melanoma, and as targeted therapies and immunotherapies are being used more, chemotherapy is being used much less frequently.
Targeted therapies
Multiple targeted therapies have been developed to combat molecular defects present in melanoma. The most promising of these include the BRAF inhibitors, vemurafenib and dabrafenib, that were approved for the treatment of metastatic and unresectable BRAF-mutated melanomas in 2011 and 2013 respectively.12,19 However, though these drugs are highly effective for approximately half of patients with BRAF mutated melanomas, a majority of patients develop secondary resistance within a relatively short amount of time.12,14,19 Some of the mechanisms by which this secondary resistance develops have been discovered,100,101 and researchers have been working to develop new drugs and new drug combinations to try to achieve a more lasting effect.12
Immune response in melanoma and immunotherapies
While early and correct diagnosis gives patients their best chance for survival, some melanoma cases will not be diagnosed until the late stages, and so developing new therapies for melanoma treatment is still a priority, and in the last 30 years there have been major breakthroughs in immunotherapy treatments for metastatic melanoma.
The human immune system has the difficult job of distinguishing between self and non-self. To communicate to the cells of the immune system that a somatic cell is a healthy self-cell, all nucleated cells have major histocompatibility complex (MHC) class I proteins on their surfaces. These complexes display epitopes that come from proteins that were expressed and then degraded within the cell. These proteins can either be self-proteins or non-self-proteins. If the epitopes displayed on the MHC class I proteins are non-self, the immune system is alerted that there is a pathogen present within the cell, and the cell needs to be neutralized. Cancer cells are essentially self-cells, but they are highly mutagenic, and the epitopes displayed by MHC proteins on their surfaces can be recognized by immune cells as aberrant, signaling that the cells are diseased and need to be neutralized.71 Melanoma has the highest rate of mutations of any cancer,102 and as a result, they produce a wide range of tumor antigens that are recognized by the immune system and are highly immunogenic. Tumor antigens are classed into two groups; tumor associated antigens (TAA), which are mostly found in tumor cells though are expressed in normal cells as well, and tumor specific antigens (TSA), which are solely expressed in tumor cells.71 Melanoma tumors also have a group of antigens specific to melanoma cells; these antigens are involved in melanoma differentiation and are called melanoma differentiation antigens (MDA). TAAs, TSAs and MDAs are all proteins that are displayed on MHC class I proteins on melanoma cell surfaces, which can alert the immune system that the cells are diseased.
The antigens displayed on the MHC class I proteins are recognized by T-cell receptors (TCR) found on CD8+ cytotoxic T lymphocytes (CTL), which can kill cells that are non-self or diseased self-cells. MHC class II proteins are present on the surface of antigen presenting cells (APCs) such as macrophages and dendritic cells. APCs take up extracellular proteins from the environment and display them to CD4 + T helper (Th) cells, which then activates an immune response to specific antigens.
Once the immune system has been alerted to the presence of non-self or diseased self-cells, lymphocytes invade the tissue and begin to kill the aberrant cells. These lymphocytes, when histologically visualized in the microscope in the evaluation of a tumor, are called tumor infiltrating lymphocytes (TILs), and the number of TILs present in a tumor is a prognostic indicator in some tumors, including melanomas.71 If there are no TILs present in a tumor, this is a sign that the tumor has successfully evaded the immune system and correlates with a worse prognosis. The battle between the immune system and malignant tumors, therefore, comes down to a tumor’s ability to evolve mechanisms to escape immune detection before the immune system can destroy the tumor. This process is known as immune editing.
There are three phases of immune editing: elimination, equilibrium and evasion.103 During the elimination phase, natural killer (NK), dendritic cells (DC), CTLs and B cells enter the tumor microenvironment or peripheral tissue and begin killing cancer cells faster than they can grow.103 In the equilibrium phase the cancer cells are constantly growing but are suppressed by the immune cells, therefore, the growth and expansion of cancer cells is counterbalanced by the immune system.103 Finally, if tumors enter the evasion phase of immune editing, tumor cells are able to develop strategies to neutralize the immune response enabling them to grow unchecked.71,103
Due to the highly mutagenic nature of melanoma cells, they quickly develop mechanisms to evade the immune response. Particularly virulent melanoma tumors have even been shown to have reduced levels of MHC class I proteins on the cell surfaces, thereby reducing antigen presentation.71,104 It has been shown that some melanomas downregulate TAAs and MDAs, which inhibits recognition and clearance of tumor cells by CTLs.71,104 Melanomas have also been shown to secrete immune inhibitory molecules like transformation growth factor beta (TGF-β), prostaglandin E2 (PGE2) and immune suppressive cytokines.26,51,71,105
Another common way that melanoma cells avoid immune detection is through manipulation of immune checkpoints. On the surface of T-cells there is a protein called the programed cell death protein 1 (PD-1).103 When an immune cell recognizes the PD-1 ligand (PD-L1/2) on the surface of somatic cells, this further communicates to the immune cells that the cell is a self-cell leading to inhibition of the immune system, promoting self-tolerance and preventing autoimmunity.103 The PD-1 immune checkpoint normally regulates the immune system by inducing apoptosis of maturing T-cells that recognize self-antigens in the lymph nodes.103 PD-1 also prevents apoptosis of regulatory T-cells, which are anti-inflammatory cells that repress the immune response to self-cells.103 This mechanism normally protects tissue from damage during anti-microbial immune responses, however, PD-L1/2 is commonly overexpressed in cancers, including melanoma, enabling tumor cells to effectively “turn off” the immune response and evade immune destruction.103,106
As knowledge of the immune system’s response to cancer, and the mechanisms by which cancer cells evade the immune system has grown, new therapies have been developed to try to redirect the immune system and stimulate an anti-tumor response. There are three main types of immune treatments for cancer: cancer vaccines, adoptive cell therapies and immunomodulatory strategies.
One of the first immune therapies developed for metastatic melanoma was an interleukein-2 (IL2) treatment that was approved for use in 1992.12,107 IL-2 is a pro-proliferative cytokine, which promotes expansion of melanoma specific T-cells. IL-2 treatments elicited a response in a small percent of patients, with 6% showing a complete response.12,98,104,107-109 At the time, even such a small response was considered promising, but IL-2 treatment proved to be highly toxic, and more recent treatments have shown to be less toxic and more effective.
In the 1990’s researchers also started developing cancer vaccines. Cancer vaccines, unlike prophylactic vaccines that are administered to healthy people to prevent disease, are therapeutic vaccines administered to patients in the late stages of cancer to try to sensitize the immune system to cancer antigens. Creating effective therapeutic cancer vaccines is challenging, however, due to the adaptations that allow cancer cells to hide from the immune system. Several melanoma vaccines were developed and tested in the 1990s and early 2000s, however, none of the early vaccines developed were effective and no melanoma vaccines have been approved for clinical use.110-113
The most effective treatments for metastatic melanoma to date are the immune checkpoint inhibitors, the first of which was approved for clinical use in 2011.71,107,108 Melanoma manipulation of immune checkpoints can be overcome by treatment with antibodies against PD1, PD-L1/2 and CTLA-4 (Figure 2).71,98,114 Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is another immune checkpoint receptor that acts in a comparable way to PD-1. CTLA-4 is constitutively expressed on T-reg cells and recognizes B7-1/2 receptor on APCs.98,114 CTLA-4 competes with CD28 on T-cells for binding to B7-1/2; when CD28 binds B7-1/2 an immune response is activated while CTLA-4 binding represses the immune response.106,114,115 Treatment with antibodies to PD-1, PD-L1/2 and CTLA-4 effectively block binding to the respective ligands and the corresponding signal that causes tolerance, allowing for stimulation of an immune response.71,98,106,114,115
Figure 2.
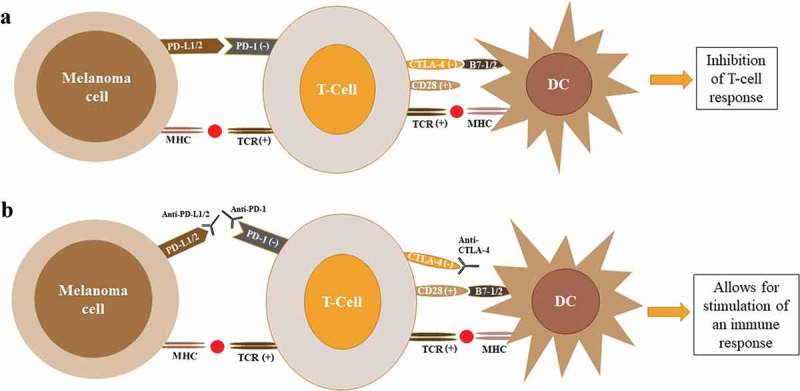
Regulation of T-cell response by CTLA4 and PD1. (A) T-cell activation by dendritic cells requires signaling by both the MHC and CD28 complexes. Binding of the CTLA4 to B7-1/2 (CD80/86) suppresses T-cell activation and acts as a feedback mechanism to prevent ongoing immune response. Binding to the MHC class I complex on melanoma cells leads to T-cell activation. After persistent activation, T-cells upregulate PD-1 expression. When PD-1 binds to the PD-L1/2 expressed by tumor cells, this leads to deactivation of T-cells. (B) Antibodies against CTLA4, PD1 or PD-L1/2 prevent binding to associated ligands leading to activation of T-cells and stimulation of an immune response to tumor cells. Abbreviations: DC, dendritic cell; CTLA4, cytotoxic T lymphocyte antigen 4; PD1, programmed death 1; TCR, T-cell receptor; MHC, major histocompatibility complex; PD-L1, programmed death-ligand 1; PD-L2, programmed death-ligand 2.
There are three immune checkpoint inhibitor drugs that have been approved for use in melanoma treatment: the anti-CTLA-4 antibody ipilimumab and two anti-PD-1 antibodies nivolumab and pembrolizumab.106 There are also several PD-L1/2 antibody drugs currently in clinical trials, and a few that have been approved for clinical use, though not for melanoma.106
Treatment with iplimumab showed durable survival of up to 10 years in 20% of cases; compared to the median survival rate of less than one year in stage IV melanoma patients, this is a great advancement.106,115-118 Pembrolizumab has a response rate of ~37–38% in patients with metastatic melanoma, and an overall survival of 74% at 12 months.118 Treatment with nivolumab showed a ~40% response rate with a 12 month overall survival rate of 73% compared to 43% of patients treated with dacarbazine.109 Ipilimumab combined with nivolumab treatment has resulted in a ~57% response rate and 11.5 months of progression free survival.72,107
While checkpoint inhibiters are promising, there are necessarily complications involved in inhibiting mechanisms that promote tolerance of self-cells. Therefore, the side-effects of checkpoint inhibiters can be severe. The side effects are typically immune-related inflammatory conditions of the skin, GI system, and endocrine organs.118,119 Recognizing and managing side effects of the these treatments is important, and though the toxicity of these drugs can be offset in some cases by treatment with corticosteroids, some patients cannot tolerate the side-effects and treatment must be terminated.120
Though immune checkpoint inhibitors have been a breakthrough for cancer therapeutics and have revolutionized the treatment of metastatic melanoma, a significant subset of patients still does not respond to these drugs, and many patients who do respond develop a secondary resistance. Research into why some patients respond and other do not is ongoing. As treatments are expensive and side effects can be severe, there is great interest in finding biomarkers that can predict whether a patient will respond to treatment or not.71,107-109 Research into other drug targets is also a priority as it is clear that there is no “magic bullet” for melanoma, and a more personalized medicine approach is needed.
Conclusion
Melanoma is an incredibly virulent disease. It is a heterogenous and complex disease, which can make it difficult to diagnose and treat. Understanding the mechanisms that lead to melanomagenesis and allow melanomas to evade the immune system will give us new strategies for diagnosis and treating the disease. Furthermore, new technologies that can provide more objective methods for diagnosing and prognosticating melanoma are being developed, which will improve patient outcomes.
Over the last 20 years the use of IHC as an ancillary test in the diagnosis of melanoma has increased. The development of robust sensitive and specific cancer biomarkers in the field of tissue immunohistochemistry remains a focus of research. Though it can be a useful tool for diagnostic (and possibly prognostic) purposes, this technique has its shortcomings. IHC scoring can be subjective, and thus developing diagnostic systems using multiple biomarkers will require coincident development of strict interpretation criteria and standardization procedures to ensure reproducibility from laboratory to laboratory and between pathologists. Though IHC is a useful method for biomarker recognition, newer, more objective and repeatable methods are currently being developed and could potentially revolutionize the way cancers like melanoma are diagnosed.
Recently there has been a lot of interest in using a high-resolution and high-precision ion monitoring technique known as parallel reaction monitoring (PRM) for detection of biomarkers in diseases such as cancer.121,122 PRM uses mass spectrometry (MS) to detect specific peptides of known masses and is a very powerful technique for the sensitive detection and quantitation of proteins and peptides such as histone PTMs. Using this method, the MS instrument can be directed to only fragment and sequence ions of a determined size. These targeted MS approaches provide for greater sensitivity compared to discovery-based workflows.
The treatment of metastatic melanoma has greatly improved in recent years with the introduction of treatments such as BRAF, CTLA4 and PD1 inhibitors. Some of the mechanism by which secondary resistance develops have been discovered, and researchers have been working to develop new drugs and new drug combinations to try to achieve a more lasting effect.12 Research into how and why these treatments succeed or fail is ongoing. Biomarker discovery for the prediction of patients who will or will not respond is a priority with the hope that eventually clinicians will be able to stratify patients and develop more personalized treatments based on mutational and biomarker profiles. Developing a more personalized approach to treating melanoma patients will not only improve prognosis, it will also cut down on the cost of treatment . Drugs that will not help a patient will not be administered, and it will also reduce patient suffering due to side effects of treatments that will be ineffective for them.
Metastatic melanoma has long been one of the most virulent and deadly diseases, which has, until recently, evaded all attempts at treatment. However, we are beginning to understand the underlying genetic and mechanistic causes of the disease, allowing for the development of more effective therapies and giving hope that one day, a cure may be within reach.
Funding Statement
This work was supported by the National Institutes of Health [P20GM121293].
Acknowledgments
We acknowledge support from NIH grant P20GM121293 (AJT), the Scharlau Family Endowed Chair in Cancer Research to AJT, and the UAMS Experimental Pathology Core.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Williams PF, Olsen CM, Hayward NK, Whiteman DC.. 2011. Melanocortin 1 receptor and risk of cutaneous melanoma: A meta-analysis and estimates of population burden. Int J Cancer. 129(7):1730–1740. doi: 10.1002/ijc.25804. [DOI] [PubMed] [Google Scholar]
- 2.Seiberg M. 2001. Keratinocyte–melanocyte interactions during melanosome transfer. Pigm Cell Res. 14(4):236–242. doi: 10.1034/j.1600-0749.2001.140402.x. [DOI] [PubMed] [Google Scholar]
- 3.Morgan AM, Lo J, Fisher DE. 2013. How does pheomelanin synthesis contribute to melanomagenesis?: two distinct mechanisms could explain the carcinogenicity of pheomelanin synthesis. Bioessays. 35(8):672–676. doi: 10.1002/bies.201300020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Israeli E. 2012. An ultraviolet radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Isr Med Assoc J. 14(12):770. doi: 10.1038/nature11624. [DOI] [Google Scholar]
- 5.Premi S, Wallisch S, Manu CM, Weiner AB, Bacchiocchi A, Wakamatsu K, Bechara EJ, Halaban R, Douki T, Brash DE. 2015. Chemexcitation of melanin derivatives induces DNA photoproducts long after UV exposure.pdf. Science. 347(6224):842–847. doi: 10.1126/science.1256022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Premi S, Wallisch S, Mano CM, Weiner AB, Bacchiocchi A, Wakamatsu K, Bechara EJH, Halaban R, Douki T, Brash DE. 2015. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science. 347(6224):842–847. doi: 10.1126/science.1256022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robles-espinoza CD, Roberts ND, Chen S, Leacy FP, Alexandrov LB, Pornputtapong N, Halaban R, Krauthammer M, Cui R, Bishop DT, et al. 2016. Germline MC1R status influences somatic mutation burden in melanoma. Nat Commun. 7(May):1–7. doi: 10.1038/ncomms12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gray-schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. 2007;445(7130):851–857. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- 9.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. 2015. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 348(6237):880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Orazio J, Marsch A, Veith J. Skin pigmentation and melanoma risk In: Advances in malignant melanoma - clinical research perspective; 2011. 39–68. doi: 10.5772/18681. [DOI] [Google Scholar]
- 11.Shain AH, Bastian BC. 2016. From melanocytes to melanomas. Nat Rev Cancer. 16(6):345–358. doi: 10.1038/nrc.2016.37. [DOI] [PubMed] [Google Scholar]
- 12.Rebecca VW, Sondak VK, KSMS . A brief history of melanoma : from mummies to mutations. Melanoma Res. 2013;22(2):114–122. doi: 10.1097/CMR.0b013e328351fa4d.A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee C, Collichio F, Ollila D, Moschos S. 2013. Historical review of melanoma treatment and outcomes. Clin Dermatol. 31(2):141–147. doi: 10.1016/j.clindermatol.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Scolyer RA, Long GV, Thompson JF. 2011. Evolving concepts in melanoma classification and their relevance to multidisciplinary melanoma patient care. Mol Oncol. 5(2):124–136. doi: 10.1016/j.molonc.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehnert JM, Kluger HM. 2012. Driver mutations in melanoma: lessons learned from bench-to-bedside studies. Curr Oncol Rep. 14(5):449–457. doi: 10.1007/s11912-012-0249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li C, Hu Z, Liu Z, Wang L-E, Strom SS, Gershenwald JE, Lee JE, Ross MI, Mansfield PF, Cormier JN, et al. 2006. Polymorphisms in the DNA repair genes XPC, XPD, and XPG and risk of cutaneous melanoma: a case-control analysis. Cancer Epidemiol Biomarkers Prev. 15(12):2526–2532. doi: 10.1158/1055-9965.EPI-06-0672. [DOI] [PubMed] [Google Scholar]
- 17.Read J, Wadt KAW, Hayward NK. 2015. Melanoma genetics. J Med Genet. 53(1):1–14. doi: 10.1136/jmedgenet-2015-103150. [DOI] [PubMed] [Google Scholar]
- 18.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. 2013. TERT promoter mutations in familial and sporadic melanoma. Science. 339(6122):959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 19.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. 2011. Paul Lorig and B-3 SG. Improved Survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 26(364):2507–2516. doi: 10.1056/NEJMoa1103782.Improved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwämmle V, Sidoli S, Ruminowicz C, Wu X, Lee C-F, Helin K, Dk K, Jensen ON. 2016. Systems level analysis of histone H3 post-translational modifications reveals features of PTM crosstalk in chromatin regulation. Molecular an Cell Proteomics. 15(8):2715–2729. doi: 10.1074/mcp.M115.054460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bode AM, Dong Z. 2003. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci Signal. 2003(167):re2–re2. doi: 10.1126/stke.2003.167.re2. [DOI] [PubMed] [Google Scholar]
- 22.Breslow A. 1970. Thickness, cross-sectional areas and depth of invasion in the prognosis of cutaneous melanoma. Ann Surg. 175(5):902–908. doi: 10.1097/00000658-197011000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.P V D, Gershenwald JE. 2011. Staging and prognosis of cutaneous melanoma. Surg Oncol Clin N Am. 20(1):1–17. doi: 10.1016/j.soc.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prade M, Sancho-Garnier H, Cesarini JP, Cochran A. 1980. Difficulties encountered in the application of clark classification and the Breslow thickness measurement in cutaneous malignant melanoma. Int J Cancer. 26(2):159–163. doi: 10.1002/ijc.2910260206. [DOI] [PubMed] [Google Scholar]
- 25.Colloby PS, West KP, Fletcher A. 1991. Observer variation in the measurement of Breslow depth and Clark’s level in thin cutaneous malignant melanoma. J Pathol. 163(3):245–250. doi: 10.1002/path.1711630310. [DOI] [PubMed] [Google Scholar]
- 26.Pitcovski J, Shahar E, Aizenshtein E, Gorodetsky R. 2017. Melanoma antigens and related immunological markers. Crit Rev Oncol Hematol. 115:(May):36–49. doi: 10.1016/j.critrevonc.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 27.Bartlett EK, Karakousis G. 2015. Current staging and prognostic factors in melanoma. Surg Oncol Clin N Am. 24(2):215–227. doi: 10.1016/j.soc.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Gerami P, Busam K, Cochran A, Cook MG, Duncan LM, Elder DE, Fullen DR, Guitart J, Leboit PE, Mihm MC, et al. 2014. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathology. 38(7):934–940. doi: 10.1097/PAS.0000000000000198. [DOI] [PubMed] [Google Scholar]
- 29.Corona R, Mele A, Amini M, De Rosa G, Coppola G, Piccardi P, Fucci M, Pasquini P, Faraggiana T. 1996. Interobserver variability on the histopathologic diagnosis of cutaneous melanoma and other pigmented skin lesions. J Clin Oncol. 14(4):1218–1223. doi: 10.1200/JCO.1996.14.4.1218. [DOI] [PubMed] [Google Scholar]
- 30.Farmer ER, Gonin R, Hanna MP. 1996. Discordance in the histopathologic diagnosis of melanoma and melanocytic nevi between expert pathologists. Hum Pathol. 27(6):528–531. doi: 10.1016/S0046-8177(96)90157-4. [DOI] [PubMed] [Google Scholar]
- 31.Troxel DB. Pitfalls in the diagnosis of malignant melanoma: findings of a risk management panel study. Am J Surg Pathol. 2003;27(9):1278–1283. doi: 10.1097/00000478-200309000-00012. [DOI] [PubMed] [Google Scholar]
- 32.McGinnis KS, Lessin SR, Elder DE, IV DP G, Schuchter L, Ming M, Elenitsas R. 2002. Pathology review of cases presenting to a multidisciplinary pigmented lesion clinic. Arch Dermatol. 138(5):617–621. doi: 10.1001/archderm.138.5.617. [DOI] [PubMed] [Google Scholar]
- 33.Elmore JG, Barnhill RL, Elder DE, Longton GM, Pepe MS, Reisch LM, Carney PA, Titus LJ, Nelson HD, Onega T, et al. Pathologists’ diagnosis of invasive melanoma and melanocytic proliferations: observer accuracy and reproducibility study. BMJ. 2017;357:j2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim RH, Meehan SA. 2017. Immunostain use in the diagnosis of melanomas referred to a tertiary medical center: a 15-year retrospective review (2001–2015). J Cutan Pathol. 44(3):221–227. doi: 10.1111/cup.12867. [DOI] [PubMed] [Google Scholar]
- 35.Hudson AR, Smoller BR. 1999. Immunohistochemistry in diagnostic dermatopathology. Dermatol Clin. 17(3):667–689. doi: 10.1016/S0733-8635(05)70115-7. [DOI] [PubMed] [Google Scholar]
- 36.Rat C, Hild S, Rault Sérandour J, Gaultier A, Quereux G, Dreno B, Nguyen J-M. 2018. Use of smartphones for early detection of melanoma: systematic review. J Med Internet Res. 20(4):e135–e135. doi: 10.2196/jmir.9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolf JA, Moreau JF, Akilov O, Patton T, English JC 3rd, Ho J, Ferris LK. 2013. Diagnostic inaccuracy of smartphone applications for melanoma detection. JAMA Dermatol. 149(4):422–426. doi: 10.1001/jamadermatol.2013.2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.W JV, C LW, K M. Challenges to smartphone applications for melanoma detection. Dermatol Online J. 2016;23(2):4–6. [PubMed] [Google Scholar]
- 39.Monheit G, Cognetta AB, Ferris L, Rabinovitz H, Gross K, Martini M, Grichnik JM, Mihm M, Prieto VG, Googe P, et al. 2011. The performance of MelaFind: a prospective multicenter studythe performance of MelaFind. Arch Dermatol. 147(2):188–194. doi: 10.1001/archdermatol.2010.302. [DOI] [PubMed] [Google Scholar]
- 40.Emery JD, Hunter J, Hall PN, Watson AJ, Moncrieff M, Walter FM. 2010. Accuracy of SIAscopy for pigmented skin lesions encountered in primary care: development and validation of a new diagnostic algorithm. BMC Dermatol. 10:9. doi: 10.1186/1471-5945-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Claridge E, Cotton S, Moncrieff M, Hall P. Spectrophotometric Intracutaneous Imaging (SiaScopy In: Serup J, Jemec GBCGG, editors. CRC Press; 2013. doi: 10.3109/9781420003307-44. [DOI] [Google Scholar]
- 42.Farberg AS, Winkelmann RR, Tucker N, White R, Rigel DS. The impact of quantitative data provided by a multi-spectral digital skin lesion analysis device on dermatologists’decisions to biopsy pigmented lesions. J Clin Aesthet Dermatol. 2017;10(9):24–26. [PMC free article] [PubMed] [Google Scholar]
- 43.Moncrieff M. Use of a spectrophotometric intracutaneous analysis device in the real-time diagnosis of melanoma. 2008; doi: 10.1111/j.1365-2133.2007.08325.x. [DOI] [PubMed] [Google Scholar]
- 44.Blokhin E, Pulitzer M, Busam KJ. 2013. Immunohistochemical expression of p16 in desmoplastic melanoma. J Cutan Pathol. 40(9):796–800. doi: 10.1111/cup.12186. [DOI] [PubMed] [Google Scholar]
- 45.Kucher C, Zhang PJ, Pasha T, Elenitsas R, Wu H, Ming ME. 2004. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 26(6):452–457. doi: 10.1097/00000372-200412000-00002. [DOI] [PubMed] [Google Scholar]
- 46.Busam KJ, Zhao H, Coit DG, Kucukgol D, Jungbluth AA, Nobrega J, Viale A. 2005. Distinction of desmoplastic melanoma from non-desmoplastic melanoma by gene expression profiling. J Investigative Dermatol. 124(2):412–419. doi: 10.1111/j.0022-202X.2004.23600.x. [DOI] [PubMed] [Google Scholar]
- 47.Prieto-Granada CN, Wiesner T, Messina JL, Jungbluth AA, Chi P, Antonescu CR. 2016. Loss of H3K27me3 expression is a highly sensitive marker for sporadic and radiation-induced MPNST. Am J Surg Pathol. 40(4):479–489. doi: 10.1097/PAS.0000000000000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le Guellec S, Macagno N, Velasco V, Lamant L, Lae M, Filleron T, Malissen N, Cassagnau E, Terrier P, Chevreau C, et al. 2017. Loss of H3K27 trimethylation is not suitable for distinguishing malignant peripheral nerve sheath tumor from melanoma: A study of 387 cases including mimicking lesions. Mod Pathol. 30(12):1677–1687. doi: 10.1038/modpathol.2017.91. [DOI] [PubMed] [Google Scholar]
- 49.Schaefer I-M, Fletcher CD, Hornick JL. 2016. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol. 29(1):4–13. doi: 10.1038/modpathol.2015.134. [DOI] [PubMed] [Google Scholar]
- 50.Cleven AH, Al SGA, Briaire-de Bruijn I, Ingram DR, van de Rijn M, Rubin BP, de Vries MW, Watson KL, Torres KE, Wang W-L, et al. 2016. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol. 29(6):582–590. doi: 10.1038/modpathol.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicolaou A, Estdale SE, Tsatmali M, Herrero DP, Thody AJ. 2004. Prostaglandin production by melanocytic cells and the effect of α-melanocyte stimulating hormone. FEBS Lett. 570(1):223–226. doi: 10.1016/j.febslet.2004.06.041. [DOI] [PubMed] [Google Scholar]
- 52.Skelton HG, Smith KJ, Barrett TL, Lupton GPGJ. HMB-45 staining in benign and melignant melanocytic lesions. A reflection of cellular activation. Am J Dermatopathol. 1991;13(6):543–550. doi: 10.1097/00000372-199113060-00004. [DOI] [PubMed] [Google Scholar]
- 53.Chervona Y, Costa M. Histone modifications and cancer: biomarkers of prognosis? Am J Cancer Res. 2012;2(5):589–597. [PMC free article] [PubMed] [Google Scholar]
- 54.Compton LA, Murphy GF, Lian CG. 2015. Diagnostic Immunohistochemistry in cutaneous neoplasia: an update. Dermatopathology. 2(1):15–42. doi: 10.1159/000377698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ivan D, Prieto VG. Use of immunohistochemistry in the diagnosis of melanocytic lesions: applications and pitfalls. Future Oncol. 2010;6. doi: 10.2217/fon.10.81. [DOI] [PubMed] [Google Scholar]
- 56.Prieto VG, Shea CR. 2008. Use of immunohistochemistry in melanocytic lesions. J Cutan Pathol. 35(2):1–10. doi: 10.1111/j.1600-0560.2008.01130.x. [DOI] [PubMed] [Google Scholar]
- 57.Ladstein RG, Bachmann IM, Straume O, Akslen LA. 2010. Ki-67 expression is superior to mitotic count and novel proliferation markers PHH3, MCM4 and mitosin as a prognostic factor in thick cutaneous melanoma. BMC Cancer. 10(1):140. doi: 10.1186/1471-2407-10-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nasr MR, El-Zammar O. 2008. Comparison of pHH3, Ki-67, and survivin immunoreactivity in benign and malignant melanocytic lesions. Am J Dermatopathol. 30(2):117–122. doi: 10.1097/DAD.0b013e3181624054. [DOI] [PubMed] [Google Scholar]
- 59.Willis B, Johnson G, Wang J, Cohen C. 2014. SOX10: A useful marker for identifying metastatic melanoma in sentinel lymph nodes. Applied Immunohistochem & Mol Morphol. 23(2):109–112. doi: 10.1097/PAI.0000000000000097. [DOI] [PubMed] [Google Scholar]
- 60.Lee JJ, Granter SR, Laga AC, Saavedra A, Zhan Q, Guo W, Xu S, Murphy GF, Lian CG. 2015. 5-hydroxymethylcytosine expression in metastatic melanoma versus nodal nevus in sentine lymph node biopsies. Mod Pathol. 28(2):218–229. doi: 10.1038/modpathol.2014.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nielsen PS, Riber-Hansen R, Steiniche T. 2011. Immunohistochemical double stains against Ki67/MART1 and HMB45/MITF: promising diagnostic tools in melanocytic lesions. Am J Dermatopathol. 33(4):361–370. doi: 10.1097/DAD.0b013e3182120173. [DOI] [PubMed] [Google Scholar]
- 62.Bergman R, Dromi R, Trau H, Cohen I, Lichtig C. 1995. The pattern of HMB-45 antibody staining in compound Spitz nevi. Am J Dermatopathol. 17(6):542–546. doi: 10.1097/00000372-199512000-00002. [DOI] [PubMed] [Google Scholar]
- 63.Uguen A, Talagas M, Costa S, Duigou S, Bouvier S, De Braekeleer M, Marcorelles P. 2015. A p16-Ki-67-HMB45 immunohistochemistry scoring system as an ancillary diagnostic tool in the diagnosis of melanoma. Diagn Pathol. 10(1):195. doi: 10.1186/s13000-015-0431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Biernacka A, Linos KD, DeLong PA, Suriawinata AA, Padmanabhan V, Liu X. 2016. A case of S-100 negative melanoma: A diagnostic pitfall in the workup of a poorly differentiated metastatic tumor of unknown origin. CytoJournal. 13(21):1–16. doi: 10.4103/1742-6413.190914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stefanaki C, Chardalias L, Soura E, Katsarou A, Stratigos A. 2017. Paediatric melanoma. J Eur Acad Dermatol Venereol. 31(10):1604–1615. doi: 10.1111/jdv.14299. [DOI] [PubMed] [Google Scholar]
- 66.Lindboe CF, Torp SH. 2002. Comparison of Ki-67 equivalent antibodies. J Clin Pathol. 55(6):467–471. doi: 10.1136/jcp.55.6.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chorny JA, Barr RJ, Kyshtoobayeva A, Jakowatz J, Reed RJ. 2003. Ki-67 and p53 expression in minimal deviation melanomas as compared with other nevomelanocytic lesions. Mod Pathol. 16(6):525–529. doi: 10.1097/01.MP.0000072747.08404.38. [DOI] [PubMed] [Google Scholar]
- 68.Nielsen PS, Riber-Hansen R, Jensen TO, Schmidt H, Steiniche T. 2013. Proliferation indices of phosphohistone H3 and Ki67: strong prognostic markers in a consecutive cohort with stage I/II melanoma. Mod Pathol. 26(3):404–413. doi: 10.1038/modpathol.2012.188. [DOI] [PubMed] [Google Scholar]
- 69.Ohsie SJ, Sarantopoulos GP, Cochran AJ, Binder SW. 2008. Immunohistochemical characteristics of melanoma. J Cutan Pathol. 35(5):433–444. doi: 10.1111/j.1600-0560.2007.00891.x. [DOI] [PubMed] [Google Scholar]
- 70.Micevic G, Theodosakis N, Bosenberg M. 2017. Aberrant DNA methylation in melanoma: biomarker and therapeutic opportunities. Clin Epigenetics. 9(1):34. doi: 10.1186/s13148-017-0332-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee N, Zakka LR, Mihm MC, Schatton T. 2016. Tumour-infiltrating lymphocytes in melanoma prognosis and cancer immunotherapy. Pathology. 48(2):177–187. doi: 10.1016/j.pathol.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 72.Palmer SR, Erickson LA, Ichetovkin I, Knauer DJ, Markovic SN. 2011. Circulating serologic and molecular biomarkers in malignant melanoma. Mayo Clinic Proc. 86(10):981–990. doi: 10.4065/mcp.2011.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Keung EZ, Balch CM, Gershenwald JE, Halpern AC. Key changes in the AJCC eighty edition melanoma staging system. Melanoma Letter. 2018;36(1):1–10. [Google Scholar]
- 74.Besaratinia A, Tommasi S. 2014. Epigenetics of human melanoma: promises and challenges. J Mol Cell Biol. 6(5):356–367. doi: 10.1093/jmcb/mju027. [DOI] [PubMed] [Google Scholar]
- 75.Zhang T, Cooper S, Brockdorff N. 2015. The interplay of histone modifications - writers that read. EMBO Rep. 16(11):1467–1481. doi: 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dulev S, Tkach J, Lin S, Batada NN. 2014. SET8 methyltransferase activity during the DNA double-strand break response is required for recruitment of 53BP1. EMBO Rep. 15(11):1163–1174. doi: 10.15252/embr.201439434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lukas J, Lukas C, Bartek J. 2011. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 13(10):1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 78.Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. 2010. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol. 12(11):1086–1093. doi: 10.1038/ncb2113. [DOI] [PubMed] [Google Scholar]
- 79.Sims RJ, Nishioka K, Reinberg D. 2003. Histone lysine methylation: A signature for chromatin function. Trends in Genet. 19(11):629–639. doi: 10.1016/j.tig.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 80.van Nuland R, Gozani O. 2016. Histone H4 Lysine 20 (H4K20) methylation, expanding the signaling potential of the proteome one methyl moiety at a time. Mol Cell Proteomics. 15(3):755–764. doi: 10.1074/mcp.R115.054742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jacquet K, Fradet-Turcotte A, Avvakumov N, Lambert J-P, Roques C, Pandita RK, Paquet E, Herst P, Gingras A-C, Pandita TK, et al. 2016. The TIP60 complex regulates bivalent chromatin recognition by 53BP1 through direct H4K20me binding and H2AK15 acetylation. Mol Cell. 62(3):409–421. doi: 10.1016/j.molcel.2016.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW, Dutta S, Appella E, Gozani O, Blum G, et al. 2014. PR-set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 300(5):795069. doi: 10.1371/journal.pgen.1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bartke T, Borgel J, DiMaggio PA. 2013. Proteomics in epigenetics: new perspectives for cancer research. Brief Funct Genomics. 12(3):205–218. doi: 10.1093/bfgp/elt002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chopra M, Bohlander SK. 2015. Disturbing the histone code in leukemia: translocations and mutations affecting histone methyl transferases. Cancer Genet. 208(5):192–205. doi: 10.1016/j.cancergen.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 85.Yamaguchi H, Hung MC. 2014. Regulation and role of EZH2 in cancer. Cancer Res Treat. 46(3):209–222. doi: 10.4143/crt.2014.46.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lyko F, Brown R. 2005. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J Natl Cancer Inst. 97(20):1498–1506. doi: 10.1093/jnci/dji311. [DOI] [PubMed] [Google Scholar]
- 87.Tsai CT, So CWE. 2017. Epigenetic therapies by targeting aberrant histone methylome in AML: molecular mechanisms, current preclinical and clinical development. Oncogene. 36(13):1753–1759. doi: 10.1038/onc.2016.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sarkar D, Leung EY, Baguley BC, Finlay GJ, Askarian-amiri ME. Epigenetic regulation in human melanoma : past and future. Epigenetics. 2015;10(2):103–121. doi: 10.1080/15592294.2014.1003746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Minucci S, Pelicci PG. 2006. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 6(1):38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 90.Albert M, Helin K. 2010. Histone methyltransferases in cancer. Sem Cell Dev Biol. 21(2):209–220. doi: 10.1016/j.semcdb.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 91.Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, Arenas-Ramirez N, Haeusel J, Zhang Y, Bonalli M, et al. 2015. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 6:6051. doi: 10.1038/ncomms7051. [DOI] [PubMed] [Google Scholar]
- 92.Sato T, Kaneda A, Tsuji S, Isagawa T, Yamamoto S, Fujita T, Yamanaka R, Tanaka Y, Nukiwa T, Marquez VE, et al. 2013. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci Rep. 3(1911):1–9. doi: 10.1038/srep01911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ezponda T, Licht JD. 2014. molecular pathways: deregulation of histone H3 lysine 27 methylation in cancer — different paths, same destination. Clin Cancer Res. 20(19):5001–5008. doi: 10.1158/1078-0432.CCR-13-2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sengupta D, Byrum SD, Avaritt NL, Davis L, Shields B, Mahmoud F, Reynolds M, Orr LM, Mackintosh SG, Shalin SC, et al. 2016. Quantitative histone mass spectrometry identifies elevated histone H3 Lysine 27 (Lys 27) trimethylation in melanoma. Mol Cell Proteomics. 15(3):765–775. doi: 10.1074/mcp.M115.053363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pfister SX, Ashworth A. 2017. Marked for death: targeting epigenetic changes in cancer. Nat Rev Drug Discovery. 16(4):241–263. doi: 10.1038/nrd.2016.256. [DOI] [PubMed] [Google Scholar]
- 96.Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DTW, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. 2013. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 24(5):660–672. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 97.DeVita VT, Chu E. 2008. A history of cancer chemotherapy. Cancer Res. 68(21):8643LP– 8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 98.Koller KM, Wang W, Schell TD, Cozza EM, Kokolus KM, Neves RI, Mackley HB, Pameijer C, Leung A, Anderson B, et al. 2016. Malignant melanoma—the cradle of anti-neoplastic immunotherapy. Crit Rev Oncol Hematol. 106(2016):25–54. doi: 10.1016/j.critrevonc.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 99.Kaufmann SHE. Paul Ehrlich: founder of chemotherapy. Nat Rev Drug Discovery. 2008;7:373. doi: 10.1038/nrd2582. [DOI] [PubMed] [Google Scholar]
- 100.Martínez-Cardús A, Bugés C, Vila L, Manzano JL, Martínez-Balibrea E, Layos L, de Los Llanos Gil M. 2016. Resistant mechanisms to BRAF inhibitors in melanoma. Ann Trans Med. 4(12):237. doi: 10.21037/atm.2016.06.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Arozarena I, Wellbrock C. 2017. Overcoming resistance to BRAF inhibitors. Ann Trans Med. 5(19):387. doi: 10.21037/atm.2017.06.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Passarelli A, Mannavola F, Stucci LS, Tucci M, Silvestris F. 2017. Immune system and melanoma biology: a balance between immunosurveillance and immune escape. Oncotarget. 8(62):106132–106142. doi: 10.18632/oncotarget.22190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schatton T, Schütte U, Frank NY, Zhan Q, Hoerning A, Robles SC, Zhou J, Hodi FS, Spagnoli GC, Murphy GF, et al. 2010. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 70(2):697–708. doi: 10.1158/0008-5472.CAN-09-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gorelik L, Flavell RA. 2001. Immune-mediated eradication of tumors through the blockade of transforming growth factor-β signaling in T cells. Nat Med. 7:1118–1122. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 106.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. doi: 10.1016/j.ccell.2015.03.00110.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Byrne EH, Fisher DE. 2017. Immune and molecular correlates in melanoma treated with immune checkpoint blockade. Cancer. 123(11):2143–2153. doi: 10.1002/cncr.30444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weber J. 2011. Immunotherapy for melanoma. Curr Opin Oncol. 23(2):163–169. doi: 10.1097/CCO.0b013e3283436e79. [DOI] [PubMed] [Google Scholar]
- 109.George DD, Armenio VA, Katz SC. 2017. Combinatorial immunotherapy for melanoma. Cancer Gene Ther. 24(3):141–147. doi: 10.1038/cgt.2016.56. [DOI] [PubMed] [Google Scholar]
- 110.Berd D, Maguire HC Jr, McCue PMM. 1990. Treatment of metastatic melanoma with an autologous tumor-cell vaccine: clinical and immunologic results in 64 patients. J Clin Oncol. 8(11):1858–1867. doi: 10.1200/JCO.1990.8.11.1858. [DOI] [PubMed] [Google Scholar]
- 111.Morton DL, Foshag LJ, Hoon DS, Nizze JA, Famatiga E, Wanek LA, Chang C, Davtyan DG, Gupta RK, Elashoff R. Prolongation of survival in metastatic melanoma after active specific immunotherapy with a new polyvalent melanoma vaccine. Ann Surg. 1992;216(4):463–482. doi: 10.1097/00000658-199210000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang X-Y. 2013. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res. 119:421–475. doi: 10.1016/B978-0-12-407190-2.00007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Baars A, van Riel JM, Cuesta MA, Jaspars EH, Pinedo HMVDEA. Metastasectomy and active specific immunotherapy for a large single melanoma metastasis. Hepatogastroenterology. 2002;49(45):691–693. [PubMed] [Google Scholar]
- 114.Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, White DE, Levy CL, Rosenberg SA, Phan GQ CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012;18(7):2039–2047. doi: 10.1158/1078-0432.CCR-11-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, Waterfield W, Schadendorf D, Smylie M, Guthrie T, et al. 2010. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 11(2):155–164. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- 116.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen -T-T, Berman DM, Wolchok JD. 2015. Pooled analysis of long-term survival data from phase ii and phase iii trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 33(17):1889–1894. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Méndez R, Ruiz-Cabello F, Rodríguez T, Del Campo A, Paschen A, Schadendorf DGF. 2007. Identification of different tumor escape mechanisms in several metastases from a melanoma patient undergoing immunotherapy. Cancer Immunol Immunother. 56(1):88–94. doi: 10.1007/s00262-006-0166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sharma P, Allison JP. 2015. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 161(2):205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fellner C. Ipilimumab (yervoy) prolongs survival in advanced melanoma: serious side effects and a hefty price tag may limit its use. P & T. 2012;37(9):503–530. [PMC free article] [PubMed] [Google Scholar]
- 120.Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, Chau I, Ernstoff MS, Gardner JM, Ginex P, et al. 2018. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: american society of clinical oncology clinical practice guideline. J Clin Oncol. 36(17):1714–1768. doi: 10.1200/JCO.2017.77.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. 2012. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics. 11(11):1475–1488. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Creech AL, Taylor JE, Maier VK, Wu X, Feeney CM, Udeshi ND, Peach SE, Boehm JS, Lee JT, Carr SA, et al. 2015. Building the connectivity map of epigenetics: chromatin profiling by quantitative targeted mass spectrometry. Methods. 72(C):57–64. doi: 10.1016/j.ymeth.2014.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]