

ABSTRACT
Hydroxyurea (HU) has been widely used in sickle cell disease. Its potential long-term risk for carcinogenesis or leukemogenic risk remains undefined. Here, we report a 26 y old African-American female with Sickle Cell Disease (SCD) who developed refractory/relapsed acute myeloid leukemia (AML) 6 months after 26 months of HU use. That patient’s cytogenetics and molecular genetics analyses demonstrated a complex mutation profile with 5q deletion, trisomy 8, and P53 deletion (deletion of 17p13.1). P53 gene sequence studies revealed a multitude of somatic mutations that most suggest a treatment-related etiology. The above-mentioned data indicates that the patient may have developed acute myeloid leukemia with myelodysplasia-related changes (AML-MRC) as a direct result of HU exposure.
Keywords: Hydroxyurea, acute myeloid leukemia with myelodysplasia-related changes, sickle cell disease, P53 gene
Introduction
Sickle cell disease (SCD) is a congenital hemoglobin disorder that reduces both life expectancy and quality. It affects approximately 100,000 Americans.1 Hydroxyurea (HU) has been increasingly used in SCD patients since the FDA approved it for SCD in 1998. Currently HU is the only standard disease-modifying therapy for SCD.2 On December 21, 2017, the FDA further approved HU as the first drug for treating sickle cell anemia. Several case reports of HU-related leukemia have raised the concern of its leukemogenic risk. However, the incidence of HU-related leukemia remains undetermined and leukemogenic risk of HU remains controversial partially due to lack of long-term follow-up. In this case report, we report an HU-treated SCD patient developing acute myeloid leukemia with myelodysplasia-related changes (AML-MRC).
Case representation
A 26 y old African-American female with sickle cell anemia diagnosed as a child presented with severe abdominal pain, hematuria, leukocytosis (35.4 x 109/L), anemia (Hgb 69 g/L), and thrombocytosis (405 x 109/L). Past medical history was significant for frequent sickle cell crises, pulmonary fibrosis, asthma, recurrent pneumonia, and avascular necrosis of the hips bilaterally. The patient received chronic red blood cell transfusion/exchange therapy since childhood. She received HU for approximately 26 months but discontinued it 6 months prior to this presentation due to lack of clinical response.
After admission, the patient was diagnosed with peritonitis of unknown etiology with a negative CT abdomen/pelvis, negative blood cultures, and negative pelvic inflammatory disease workup. She received a course of piperacillin-tazobactam with subsequent clinical improvement. The patient went on to experience a sickle cell crisis, managed via hydration and a patient-controlled analgesia pump.
The patient’s leukocytosis persisted despite antibiotic therapy, and subsequent peripheral blood smear showed blasts concerning for leukemia. A bone marrow biopsy confirmed acute myeloid leukemia (AML). The patient received induction 7 + 3 chemotherapy with cytarabine and idarubicin. She initially tolerated treatment well, with mild nausea and vomiting. The patient developed neutropenic fever then sepsis at the end of induction chemotherapy. She received meropenem, voriconazole, and vancomycin. Her antibiotic course was complicated by hypotension, responsive to aggressive hydration. The patient also developed superficial thrombophlebitis of the left upper extremity, managed with warm compresses, vancomycin, and ketorolac. The patient afterward experienced acute kidney injury, likely secondary to NSAID administration and vancomycin in the setting of poor PO intake. Her AKI was managed with aggressive fluid resuscitation.
On Day 15, a repeat bone marrow biopsy showed 40% blasts with remnant leukemia. That night, the patient’s course was complicated by seizures and a new occipital lobe lesion on a CT scan concerning for an infarction secondary to sickle cell disease. The patient was managed with plasma exchange (8 units). MRI confirmed an ischemic etiology and, in addition, demonstrated leptomeningeal enhancement concerning for leukemic involvement. A lumbar puncture 2 d later demonstrated no infectious etiology with cytology negative for malignancy. That day the patient also experienced a seizure refractory to lorazepam and phenytoin. Her seizures were ultimately controlled with levetiracetam and phenytoin, and the patient was maintained on valproic acid. The patient subsequently received sirolimus alongside valproic acid for salvage therapy. Another bone marrow biopsy showed normocellular marrow with 12% blasts residual acute myeloid leukemia. The patient was discharged on sirolimus 1 mg PO daily and valproic acid 250 mg PO TID for salvage therapy. Outpatient follow up was scheduled with a long-term goal of curative bone marrow transplant. The patient’s AML quickly deteriorated, however, and she died 4 months after initial diagnosis.
Methods
This study was approved by the Institutional Review Board (IRB). Informed consent was obtained from this patient in this study in accordance with the declaration of Helsinki and IRB requirements.
Flow cytometry and leukemic blasts cell sorting
Peripheral blood cells were obtained from venous blood and bone marrow cells were obtained from bone marrow aspirates taken from the posterior iliac crest. Cell sorting for the leukemic blasts was conducted under BSL-2 conditions with a 16-color BD FACSAria SORP high-speed cell sorter (Becton Dickinson) in the Penn State Hershey Flow Cytometry Core Facility. CD34+ cells that expressed all five (i.e. HLA-DR, CD34, CD13, CD117, and CD45) surface markers were sorted as leukemic cells otherwise they were sorted as non-leukemic cells.3
Immunofluorescence staining of bone marrow biopsy samples
Immunofluorescence staining of bone marrow biopsies was conducted using antibodies against H2AX, BAX, p53, and p21. The primary antibody-treated slides were then incubated with fluorescent conjugated secondary antibodies and then nuclear stained with DAPI. Images were acquired using a Leica SP8 Inverted confocal microscope. Please read reference by Teye et al. for detailed experimental procedures.4
P53 sequencing analysis
One (1) microgram of DNA isolated from non-leukemic cells and leukemic cells (AML) was amplified using primers covering exon 2–11 of the P53 gene including intron/exon boundaries according to instructions in the International Agency for Research on Cancer (IARC) database (version R19) for p53.5 Seven PCR reactions per sample (non-leukemic and leukemic) were spin column purified and sequenced using a 3130XL capillary sequencer (ABI systems) with the same primers in both reversed and forward directions. The obtained sequence was analyzed using the software FinchTV version 1.4.0 and nucleotide BLAST.6,7 Sequencing products were aligned to the P53 GenBank sequence NC_000017.9 and sequence alterations were identified partially through manual inspection. Sequence alterations were correlated with the coding sequence of the p53 protein and the impact on the protein Sorting Intolerant From Tolerant (SIFT) was determined using the International Agency for Research on Cancer (IARC) database.5
Results
Flow cytometry, bone marrow biopsy, and FISH analysis
This patient was immediately admitted and workup was started. Review of peripheral blood smear demonstrated 25% cells were myeloblasts, marked myelodysplasia, and numerous nucleated red blood cells (Figure 1). Neutrophils had nucleus-to-cytoplasmic dysynchrony, hypolobated nuclei, and hypogranular cytoplasm. Red blood cells had marked anisopoikilocytosis including rare sickle cells, microspherocytes, target cells, acanthocytes, hypochromic cells, and Pappenheimer bodies. Platelets were medium to large with normal granularity. The flow cytometry of peripheral blood showed 16% of myeloblasts with CD45 (dim), CD13, CD33 (dim), MPO (dim), CD117, CD34, HLA-DR, CD11c (dim) with aberrant expression of CD56 (heterogeneous), CD5 (partial, dim) and CD22 (partial, dim).
Figure 1.
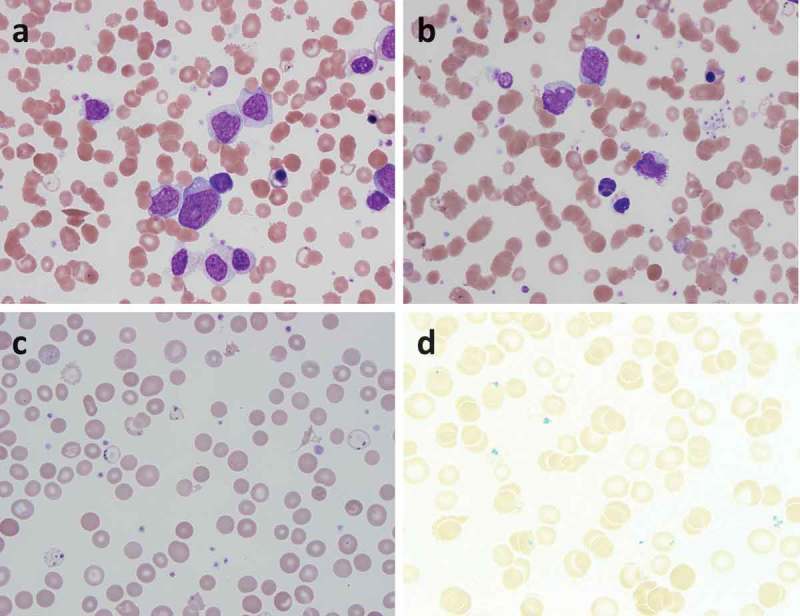
Images from peripheral blood smear. (a) myeloblasts, immature granulocytes, sickle cell, and nucleated red blood cells. (b) Dysplastic nucleated red blood cells with multilobated nucleus. (c) Pappenheimer bodies. (d) Prussian blue stain highlights the pappenheimer bodies.
The findings of the bone marrow biopsy and aspirate were consistent with acute myeloid leukemia with myelodysplasia-related changes: hypercellular marrow (100% cellularity) with 32% blasts and marked left shift (Figure 2). Megakaryocytes were increased with dysmegakaryopoiesis including hypolobated nuclei. Myeloid lineage was increased and markedly left-shifted with dysplastic features including nucleus-to-cytoplasmic dysynchrony, hypogranular cytoplasm, and hypolobated nuclei. Erythroid lineage was decreased with dyserythropoiesis including megaloblastic change, cytoplasmic vacuoles, nuclear budding, nuclear irregularities, and Pappenheimer bodies in mature forms.
Figure 2.

Images from bone marrow biopsy and aspirate. (a) Low power magnification showing a hypercellular marrow (100%). (b) Bone marrow aspirate showing myeloblasts with dysmyelopoiesis.
Interphase FISH analysis revealed 5q deletion, trisomy 8, P53 deletion (deletion of 17p13.1) and hyperdiploidy of chromosomes 5, 7, 11, 15, 16, 17 and 20. The allele-specific PCR analysis showed negative JAK2 V617F mutation. Cytogenetic analysis showed significant karyotypic heterogeneity. The composite karyotype had 45–46 chromosome with del(5)(q13q35), der(7)t(7;12)(q32;q13), i(8)(q10)[16], +i(8)(q10)[2], −12, add(12)(p11.2), del(17)(p11.2), −18, der(19)t(11;19)(q13;p13.3), −20, add(21)(p11.2), add(21)(p11.2), add(22)(p11.2)x2[2], +add(22)(p11.2)[16], +0-1r, +0-2mar[cp18]/90–92, idemx2[2]/46,XX[1].
P53 sequencing and analysis in non-leukemic and leukemic cells
To further explore the etiology of her leukemia and the cause of massive myelodysplastic changes, we analyzed this patient’s P53 gene. Approximately 2,300 bp of DNA from non-leukemic and leukemic cells encoding for intron/exon and exons 2–11 of the P53 gene was analyzed by Sanger sequencing. In total twenty-eight (28) sequence alterations were identified in the non-leukemic and leukemic cells. However, only three (3) sequence alterations could be verified – one conserved deletion and two missense mutations between the different cell types. These included missense mutations at codons 9 and 80, and a frameshift deletion at codon 131. Please read Teye et al. for a detailed description of those mutations.4
We previously reported no significant difference in sequence identity (percent) between non-leukemic and leukemic DNA when analyzing both introns and exons of the P53 gene. Neither did we find any significant differences with respect to the qualitative nature of the sequence alterations between non-leukemic and leukemic P53-DNA. Normalizing the number of sequence alterations found in non-leukemic and leukemic P53 gene DNA to kbp analyzed suggested that the overall mutation rate was similar between non-leukemic and leukemic cells (11.4 and 12.5/kbp, respectively). However, non-leukemic cells displayed in total 14 sequence alterations in coding sequences whereas leukemic cells displayed 22 alterations.4
PCR analyses of the P53 exons from the patient’s non-leukemic and leukemic cells suggest that the leukemic cells lack exons 2–4 and part of 5, which are present in the non-leukemic cells (Figure 3). These exons code for the trans-activation (TA), proline-rich (PRD), and part of the DNA binding domain (DBD) of p53 (Figure 4). Deletion of these exons renders p53 inactive.
Figure 3.
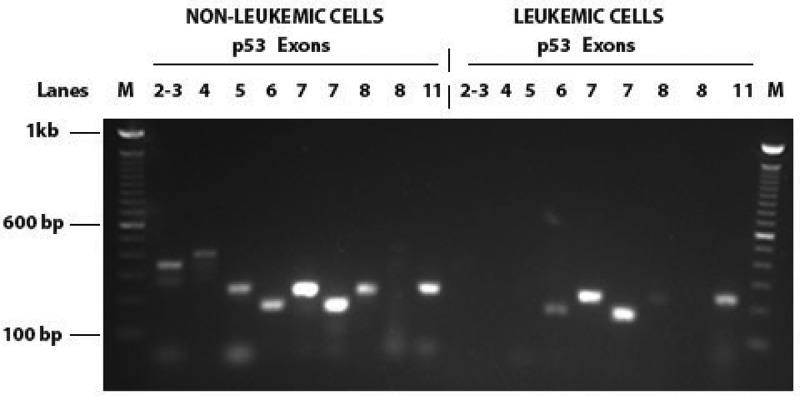
PCR for TP53 exons from DNA isolated from non-leukemic and leukemic cells from a patient treated with Hydroxyurea.
Figure 4.
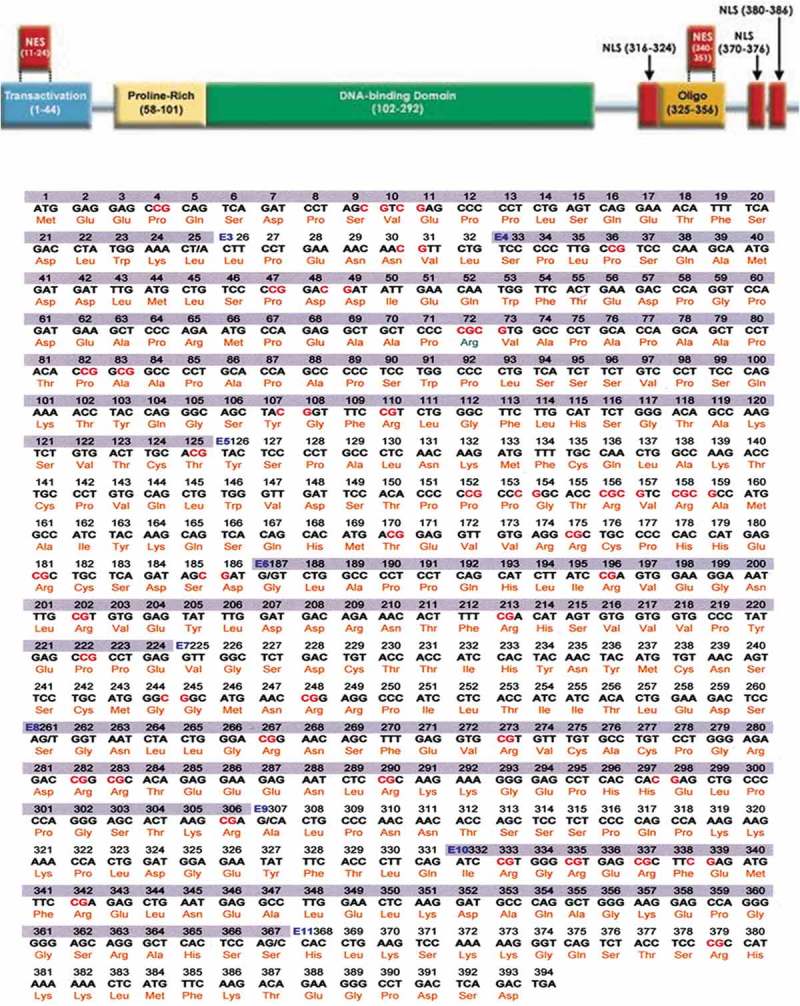
TP53 domains and sequencing data.
Discussion
Patients with SCD have a reduced life quality and expectancy due to a variety of complications including pain crisis, acute chest syndrome, stroke, and leg ulcers.8 HU is the only approved medication for sickle cell disease mainly due to its ability to increase the production of fetal hemoglobin. Since the FDA first approved it in 1967 for the treatment of neoplastic diseases, HU has been widely used for melanoma, hematological malignant diseases, and solid carcinomas. Interestingly, studies have shown that HU is beneficial for sickle cell disease patients by significantly reducing the frequency of pain crises, transfusion, and mortality.2 Long-term and large-scale studies have demonstrated that HU had an excellent safety profile with minor complications.9,10 The adverse effects of HU that have been reported include moderate drowsiness, anorexia, nausea, vomiting, diarrhea, constipation, rash, skin ulceration, dermatomyositis-like skin, skin cancer, peripheral erythema, squamous dysplasia, nail changes, facial erythema, neutropenia, interstitial pneumonia, hepatitis and rarely renal toxicity.2,11
In the 2016 revision to the World Health Organization’s classifications of hematologic malignancies, therapy-related neoplasms are defined as those that develop after a patient receives a cytotoxic therapy.12 However, HU is not on the list of cytotoxic agents from the IARC and instead is listed as “Not classifiable as to its carcinogenicity to humans”.13 The IARC states there is “inadequate evidence” in both human and animal models to determine the carcinogenicity of HU.14 Additionally, a wide variety of malignancies have been reported in both children and adults with SCD. SCD patients may have some additional cancer risks including transfusion-associated viral infection and cellular damage due to chronic inflammation. The incidence of malignancy in SCD patients has not been accurately determined due to a lack of long-term follow-up.11
Although HU has been proposed to be safe for the treatment of children with SCD and improves outcomes,15–17 concerns have been raised with respect to long-term safety following exposure to HU at an early age. Exposure of developing and proliferating tissues to genotoxicity may result in an increased rate of (oncogenic) mutagenesis that may manifest later in life as increased susceptibility to tumorigenesis. Multiple mechanisms have been proposed to explain HU’s genotoxicity, mainly as a cell-cycle-specific agent to interfere with both DNA synthesis and repair mechanisms.2 HU may trigger deoxyribonucleotide pool imbalance and DNA damage through its function as a ribonucleotide reductase inhibitor. Such imbalances decrease the availability of nucleotides for DNA polymerases needed for DNA repair and have been shown to result in genotoxic damage such as DNA strand breaks and micronuclei formation.11,18,19 On the molecular level, HU triggers replicative stress and stalled replication forks that may inhibit nucleotide excision repair (NER).20 In vitro data has shown that HU has mutagenic activity in the Salmonella strains TA98 and TA100 in doses above 117 μmol/plate and 234 μmol/plate, respectively. In vivo experiments have shown significantly increased the formation of micronuclei in reticulocytes when HU was administered to mice at doses of 50, 75, or 100 mg/kg, compared to control- suggesting chromosomal damage.21 In a cross-sectional and longitudinal study of pediatric sickle cell disease patients treated with HU, genotoxicity increased with HU administration, as indicated by micronuclei formation in reticulocytes. However, they found there were substantial variability in micronuclei between patients, suggesting that individuals may have different susceptibilities to hydroxyurea.22 In a study of patients with SCD treated with HU, patients had significantly increased DNA damage in blood leukocytes compared to matched controls, and damage was positively correlated with the mean HU dose.19 A cross-sectional study of SCD patients who were either receiving either HU or no therapy, and a healthy control group, found at baseline SCD patients have significantly greater Damage Index (indicating chromosomal damage) and micronuclei formation. HU treatment increased these damage markers, though the difference from untreated SCD patients was non-significant.16 This suggests SCD patients may already have an elevated risk for malignancy that could be augmented by HU. A similar study of pediatric SCD patients treated with HU for ≥2 y, never treated with HU, or healthy controls found a mild but significant increase in DNA damage in lymphocytes in SCD patients compared to controls. There was no significant difference in DNA damage between the HU-treated SCD patients and those that did not receive treatment.23 Ultimately, the potential for an increased malignancy risk for SCD patients on HU treatment cannot yet be excluded, and further research is needed to determine if HU may induce malignancy.
HU has been reported to be associated with some hematologic malignancies in different patient populations, although HU is not currently classifiable as to its carcinogenicity in humans (currently IARC group 3).13 Attempts have been made to characterize unique genetic signatures of treatment-induced changes.24 A study on 16 cases of t(3:21)(q26:q22) translocation in chronic myeloproliferative disorders found that 15 patients had been previously treated with HU, suggesting hydroxyurea was strongly implicated in the pathogenesis of t(3:21) transformation.25 Another study of essential thrombocythemia (ET) patients who progressed to MDS or AML found that a high proportion of hydroxyurea-treated patients had 17p deletions, while patients who did not receive hydroxyurea did not develop 17p deletions.26 Another study of treated-related MDS/AML in ET found that 17p deletions were present and associated with 5q deletions. That report also demonstrated that none of the untreated patients developed MDS/AML nor had a 17p deletion on FISH analysis.27 P53 gene mutations have also been shown to correlate with 17p deletions, and are significantly associated with 5q deletions in MDS and AML induced by alkylating agents or platinum-based chemotherapies, though this study did not include HU.28,29 The rate of p53 gene mutations in treatment-induced MDS or AML was 27%, compared to a rate of 7% in de novo MDS/AML, and these patients frequently demonstrated loss of heterogeneity of the normal p53 gene.29 Complex karyotypes have also been associated with treatment-related MDS/AML.30 Complex karyotypes also have been shown to be associated with 17p deletions in MDS/AML, and it has been suggested that p53 gene inactivation is the cause of these cytogenetic aberrancies.28 In a study of alkylating agent-induced MDS/AML, a complex karyotype was significantly associated with p53 gene mutations and either 5q or 17 p deletions, as well as a poor prognosis.29 A similar association has been described in AML developing after pipobroman or hydroxyurea administration in essential thrombocythemia patients.27
In vitro, HU can result in the accumulation of somatic mutations and chromosomal damages due to its interference with DNA repair. However, the number of acquired DNA mutations did not increase in patients with long-term exposure to HU.31 Further, no significant increase of illegitimate VDJ recombination was found in the patients after 5 y on HU in the Duke Series.23 Analysis of the pediatric SCD patients treated with HU on the BABY-HUG phase III trial32 found no evidence of increased genotoxicity with HU administration. There was an increase in chromosomal breaks in both the HU-treated and placebo groups at the end of the trial, no change in the number of illegitimate VDJ gene rearrangements, and no difference in the number of micronucleated reticulocytes.32 In the cross-sectional and prospective studies by Flanagan et al.,22 despite increased micronuclei production with hydroxyurea administration in pediatric SCD patients, no evidence of malignancy or myelodysplasia was found despite a maximum of 12 y of HU exposure. In a prospective study of long-term efficacy and safety of HU in SCD patients, no malignancy was found in 131 patients with an average 8 y follow-up and 1161 patient years.33 Steinberg et al.9 found that SCD patients on HU had a similar incidence of malignancy to SCD patients without HU in a large multicenter study of a 17.5-year follow-up. It was suggested that the risk of SCD patients developing leukemias was lower than that seen in myeloproliferative disorders, such as essential thrombocythemia. Further, Lanzkron et al. concluded that hydroxyurea does not increase the risk of leukemia in patients with sickle cell disease, although admitted that evidence may be less than sufficient.2 In an analysis of 20 different randomized controlled trials of patients receiving hydroxyurea for indications other than SCD, the hydroxyurea groups did not have a higher incidence of leukemias compared to the control groups.2 The patient populations studied included HIV, chronic myeloid leukemia, polycythemia vera, and essential thrombocythemia. As the aims and designs of these studies varied greatly, ranges in sample size (40 to 809 patients) and follow-up times (24 weeks to over 10 y) were notably different.24,34–36 These differences make it difficult to draw conclusions about the long-term effects of HU.
Based on the literature review, we identified eight cases of hydroxyurea-treated patients with sickle cell disease who developed leukemia and two reports of developing Hodgkin’s Lymphoma. A 27-year-old SCD female presented with myelodysplasia after HU treatment for 8 y.37 A 42-year-old female had AML after 6 y of HU treatment.38 A 10-year-old girl developed acute lymphoblastic leukemia with the Philadelphia chromosome, however this occurred only 7 weeks after HU treatment was initiated and as such the authors felt there was no causal relationship.39,40 An 8-year-old male, heterozygous for HbS and β0-thalassemia, developed Hodgkin’s lymphoma 6 months after receiving HU treatment. Complete remission was achieved after treatment with adriamycin-bleomycin-vinblastine-dacarbazine and a bone marrow transplant.41 A 25-year-old female developed AML after 2 y of HU treatment.42 A 14-year-old female developed ALL after 3 months of HU treatment.11 A 21-year-old female was diagnosed with acute promyelocytic leukemia after 8 y of HU treatment; she went into remission after treatment with all-trans-retinoic acid and chemotherapy.43,44 An 18-year-old male presented with stage IIa Hodgkin lymphoma after 4 y of HU therapy.45 A 33-year-old man developed AML of erythroid origin after 5 y of HU treatment.46 A 41-year-old male developed myelodysplastic syndrome and acute myeloid leukemia after 15 y of HU treatment. This individual was found to have a complex karyotype involving chromosomes 5, 7, and 17.47
Here, we reported a 26-year-old African-American SCD female developing acute myeloid leukemia with myelodysplasia-related changes (AML-MRC) 6 months after 26 months of HU use. We suspect that this was a direct result of HU exposure resulting in a secondary AML, rather than a de novo occurrence. The patient was a young female who only received hydroxyurea and no other disease-modifying therapies, nor chemotherapy or radiation therapy. She was a college student at the time of diagnosis, with no occupational exposures, yet developed a particularly aggressive AML-MRC. The cytogenetic studies revealed a multitude of somatic mutations that most suggest a treatment-related etiology. The patient’s FISH analysis showed 5q deletion, trisomy 8, P53 (17p) deletion, and a complex karyotype. In line with the aforementioned reports,26–30 it is plausible that the 5q and 17p deletions are not random gene sequence changes, but rather are post-insult damages. Furthermore, we previously found numerous DNA changes associated with the P53 gene, rather than a single isolated mutation. P53 exon analysis via Sanger sequence revealed 28 sequence alterations in non-leukemic and leukemic cells, three of which were verified as either deletion or missense mutations.4 The first was a missense mutation to Serine-9. This residue is phosphorylated following UV- and ionizing radiation, Casein kinase I and DNA-PK interacting with structured ssDNA, and by homeodomain-interacting protein kinase-like 4 (HIPK4) in vitro and in vivo.48–51 Overexpression of HIPK4 in p53-functional A549 cells repress the activity of the human survivin gene promoter indicating that phosphorylation of serine-9 might be important for p53’s ability to repress the expression of the oncogene survivin.52 However, mutation of this residue does not alter P53’s transactivation potential.48,49 Thus, neither of the missense mutations present in codons 9 and 80 is believed to be oncogenic since they do not negatively impact p53’s ability to execute downstream activation of its target genes that are believed to be essential for p53’s ability to function as a tumor suppressor.53 Notably, the frameshift deletion at codon 131 may be deleterious to the expression of the affected P53 allele. We propose that this mutation may represent an initiating oncogenic event that inactivates one P53 allele and predisposes to the deletion of chromosome 17.
A large number of sequence alterations found in the P53 gene were present in both non-leukemic and leukemic cells.4 In the vast majority of cases, the presence of conserved mutations between the cell-types could not be verified, which might indicate that many of the sequence alterations could result from sequencing artifacts near product ends. Another possible explanation is that this patient received extensive hydroxyurea (HU) treatment for her sickle cell disease. The presence of p53 mutations in non-leukemic cells may represent pre-malignant changes. The deletions of exons 2–4 and part of 5 found within the leukemic cells are extensive and lead to inactivation of the p53 gene, and may have contributed to leukemogenesis in this patient. Consistent with the clinical diagnosis of P53-deletion, we found it increasingly difficult to PCR-amplify exons of the P53 gene when using DNA isolated from leukemic cells. Difficult to amplify reactions were typically low-yield reactions expected to be within the linear range of PCR-amplification (data not shown). Thus, this is likely a reflection of the lower P53 gene dosage in the leukemic cells isolated from this patient. Aligning with the previously mentioned reports,28–30 the loss of p53 in this patient may have resulted in the complex karyotype observed. The complex karyotype may also be indicative of a treatment-related etiology. In conclusion, the multiple varied cytogenetic alternations seen here are most characteristic of chemotherapy-induced changes, rather than randomly acquired somatic mutations or germline mutations. In this case, these signature changes are best explained by the patient’s hydroxyurea exposure.
To our knowledge, this is the first case of sickle cell disease associated with AML-MRC. The leukemogenicity of HU has been suggested but not been clarified, and long-term administration may play a role in the etiology of treatment-induced AML. The incidence of HU-related leukemia also remains undetermined. This hinders the expansion of hydroxyurea treatment, which is the only disease-modifying medication for sickle cell disease. Further laboratory research and clinical data analysis are necessary to determine the genotoxic risk of hydroxyurea. Multiple centers participating in large-scaled, long-term continuous follow-up and prospective registrations of SCD patients would be the ways to accurately evaluate the role that HU plays in the etiology of hematologic malignancies.
Acknowledgments
This study is supported by: NIDA/FDA research grant to JJP (P50 DA036107), AA & MDS International Foundation Research Grant to JJP (146818), American Cancer Society research grant to JJP (124171-IRG-13-043-02), Paige’s Cancer Researcher Fund to JJP (Pu33860), JTTAI Foundation Research Grant to JJP (RF83919) and a SUNY Upstate Medical University research grant to JJP.
Authorship
Contributions: JJP designed this study. JJP and NKF conducted experiments. JJP, SR, and XY collected data. JJP provided patient care. JJP, SR, XY, NKF, and WSE participated in manuscript formation.
Disclosure of Potential Conflicts of Interest
The authors declare no competing financial interests.
References
- 1.Centers for Disease Control and Protection Data & statistics on Sickle Cell Disease. [Internet]. 2017. [accessed 2018 November15]. https://www.cdc.gov/ncbddd/sicklecell/data.html.
- 2.Lanzkron S, Strouse JJ, Wilson R, Beach MC, Haywood C, Park H, Witkop C, Bass EB, Segal JB.. Systematic review: hydroxyurea for the treatment of adults with sickle cell disease. Ann Intern Med. 2008;148:939–955. doi: 10.7326/0003-4819-148-12-200806170-00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandes AF, Kerbauy DMB, Matarraz S, Chauffaille MDLLF, Lopez A, Orfao A, Yamamoto M.. Combined flow cytometric assessment of CD45, HLA-DR, CD34, and CD117 expression is a useful approach for reliable quantification of blast cells in myelodysplastic syndromes. Cytometry B Clin Cytom. 2013;84:157–166. doi: 10.1002/cyto.b.21087. [DOI] [PubMed] [Google Scholar]
- 4.Teye EK, Sido A, Xin P, Finnberg NK, Gokare P, Kawasawa YI, Salzberg AC, Shimko S, Bayerl M, Ehmann WC, et al. PIGN gene expression aberration is associated with genomic instability and leukemic progression in acute myeloid leukemia with myelodysplastic features. Oncotarget. 2017;8:29887–29905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37:865–876. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- 6.FinchTV Version 1.4.0. [software]. Seattle (WA): Geospiza; 2006. January 18. [Google Scholar]
- 7.National Center for Biotechnology Information Basic local alignment search tool [Internet]. 2019. https://blast.ncbi.nlm.nih.gov/Blast.cgi.
- 8.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. [DOI] [PubMed] [Google Scholar]
- 9.Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, Ataga K, Swerdlow P, Kutlar A, DeCastro L, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol. 2010;85:403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonioli E, Guglielmelli P, Pieri L, Finazzi M, Rumi E, Martinelli V, Vianelli N, Luigia Randi M, Bertozzi I, De Stefano V, et al. Hydroxyurea-related toxicity in 3,411 patients with Ph’-negative MPN. Am J Hematol. 2012;87:552–554. doi: 10.1002/ajh.23160. [DOI] [PubMed] [Google Scholar]
- 11.Schultz WH, Ware RE. Malignancy in patients with sickle cell disease. Am J Hematol. 2003;74:249–253. doi: 10.1002/(ISSN)1096-8652. [DOI] [PubMed] [Google Scholar]
- 12.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 13.International Agency for Research on Cancer IARC monographs on the identification of carcinogenic hazards to humans [Internet]. 2019. [accessed 2019 March30]. monographs.iarc.fr.
- 14.International Agency for Research on Cancer Some antiviral and antineoplastic Drugs, and other pharmaceutical agents. Lyon (France): IARC Working Group on the Evaluation of Carcinogenic Risk to Humans; 2000. [Google Scholar]
- 15.Wang WC, Ware RE, Miller ST, R V I, Casella JF, Minniti CP, Rana S, Thornburg CD, Rogers ZR, Kalpatthi RV, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377:1663–1672. doi: 10.1016/S0140-6736(11)60355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maia Filho PA, Pereira JF, Almeida Filho TPD, Cavalcanti BC, Sousa JCD, Lemes RPG. Is chronic use of hydroxyurea safe for patients with sickle cell anemia? An account of genotoxicity and mutagenicity. Environ Mol Mutagen. 2019;60:302–304. doi: 10.1002/em.22260. [DOI] [PubMed] [Google Scholar]
- 17.Zimmerman SA, Schultz WH, Davis JS, C V P, Mortier NA, Howard TA, Ware RE. Sustained long-term hematologic efficacy of hydroxyurea at maximum tolerated dose in children with sickle cell disease. Blood. 2004;103:2039–2045. doi: 10.1182/blood-2003-07-2475. [DOI] [PubMed] [Google Scholar]
- 18.Osterman Golkar S, Czene S, Gokarakonda A, Haghdoost S. Intracellular deoxyribonucleotide pool imbalance and DNA damage in cells treated with hydroxyurea, an inhibitor of ribonucleotide reductase. Mutagenesis. 2013;28:653–660. doi: 10.1093/mutage/ges056. [DOI] [PubMed] [Google Scholar]
- 19.Friedrisch JR, Prá D, Maluf SW, Bittar CM, Mergener M, Pollo T, Kayser M, Da Silva MAL, Henriques JAP, Da Rocha Silla LM. DNA damage in blood leukocytes of individuals with sickle cell disease treated with hydroxyurea. Mutat Res Genet Toxicol Environ Mutagen. 2008;649:213–220. doi: 10.1016/j.mrgentox.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 20.Tsaalbi-Shtylik A, Moser J, Mullenders LHF, Jansen JG, de Wind N. Persistently stalled replication forks inhibit nucleotide excision repair in trans by sequestering replication protein A. Nucleic Acids Res. 2014;42:4406–4413. doi: 10.1093/nar/gkt1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santos JL, Bosquesi PL, Almeida AE, Chin CM, Varanda EA. Mutagenic and genotoxic effect of hydroxyurea. Int J Biomed Sci. 2011;7:263–267. [PMC free article] [PubMed] [Google Scholar]
- 22.Flanagan JM, Howard TA, Mortier N, Avlasevich SL, Smeltzer M, Wu S, Dertinger S, Ware RE. Assessment of genotoxicity associated with hydroxyurea therapy in children with sickle cell anemia. Mutat Res Genet Toxicol Environ Mutagen. 2010;698:38–42. doi: 10.1016/j.mrgentox.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez A, Duez P, Dedeken L, Cotton F, Ferster A. Hydroxyurea (hydroxycarbamide) genotoxicity in pediatric patients with sickle cell disease. Pediatr Blood Cancer. 2018;65:e27022. doi: 10.1002/pbc.v65.7. [DOI] [PubMed] [Google Scholar]
- 24.Kiladjian -J-J, Rain J-D, Bernard J-F, Briere J, Chomienne C, Fenaux P. Long-term incidence of hematological evolution in three French prospective studies of hydroxyurea and pipobroman in polycythemia vera and essential thrombocythemia. Semin Thromb Hemost. 2006;32:417–421. doi: 10.1055/s-2006-942762. [DOI] [PubMed] [Google Scholar]
- 25.Yin CC, Cortes J, Barkoh B, Hayes K, Kantarjian H, Jones D. t(3;21)(q26;q22) in myeloid leukemia: an aggressive syndrome of blast transformation associated with hydroxyurea or antimetabolite therapy. Cancer. 2006;106:1730–1738. doi: 10.1002/cncr.21730. [DOI] [PubMed] [Google Scholar]
- 26.Sterkers Y, Preudhomme C, Lai J-L, Demory J-L, Caulier M-T, Wattel E, Bordessoule D, Bauters F, Fenaux P. Acute myeloid leukemia and myelodysplastic syndromes following essential thrombocythemia treated with hydroxyurea: high proportion of cases with 17p deletion. Blood. 1998;91:616–622. [PubMed] [Google Scholar]
- 27.Bernasconi P, Boni M, Caviglianio PM, Calatroni S, Brusamolino E, Passamonti F, Volpe G, Pistorio A, Giardini I, Rocca B, et al. Acute myeloid leukemia (AML) having evolved from essential thrombocythemia (ET): distinctive chromosome abnormalities in patients treated with pipobroman or hydroxyurea. Leukemia. 2002;16:2078–2083. doi: 10.1038/sj.leu.2402641. [DOI] [PubMed] [Google Scholar]
- 28.Soenen V, Preudhomme C, Roumier C, Daudignon A, Laï JL, Fenaux P. 17p Deletion in acute myeloid leukemia and myelodysplastic syndrome. Analysis of breakpoints and deleted segments by fluorescence in situ. Blood. 1998;91:1008–1015. [PubMed] [Google Scholar]
- 29.Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol. 2001;19:1405–1413. doi: 10.1200/JCO.2001.19.10.2674. [DOI] [PubMed] [Google Scholar]
- 30.Merlat A, Lai JL, Sterkers Y, Demory JL, Bauters F, Preudhomme C, Fenaux P. Therapy-related myelodysplastic syndrome and acute myeloid leukemia with 17p deletion. A report on 25 cases. Leukemia. 1999;13:250–257. doi: 10.1038/sj.leu.2401298. [DOI] [PubMed] [Google Scholar]
- 31.Hanft V, Fruchtman S, Pickens C, Rosse W, Howard T. Acquired DNA mutations associated with in vivo hydroxyurea exposure. Blood. 2000;95:3589–3593. [PubMed] [Google Scholar]
- 32.McGann PT, Flanagan JM, Howard TA, Dertinger SD, He J, Kulharya AS, Thompson BW, Ware RE. Genotoxicity associated with hydroxyurea exposure in infants with sickle cell anemia: results from the BABY-HUG Phase III Clinical Trial. Pediatr Blood Cancer. 2012;59:254–257. doi: 10.1002/pbc.v59.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voskaridou E, Christoulas D, Bilalis A, Plata E, Varvagiannis K, Stamatopoulos G, Sinopoulou K, Balassopoulou A, Loukopoulos D, Terpos E. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS). Blood. 2010;115:2354–2363. doi: 10.1182/blood-2009-12-255992. [DOI] [PubMed] [Google Scholar]
- 34.Harrison CN, Campbell PJ, Buck G, Wheatley K, East CL, Bareford D, Wilkins BS, van der Walt JD, Reilly JT, Grigg AP, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med. 2005;353:33–45. doi: 10.1056/NEJMoa043800. [DOI] [PubMed] [Google Scholar]
- 35.Seminari E, Lisziewicz J, Tinelli C, Foli A, Lori F, Maserati R. Hydroxyurea toxicity combined with didanosine (ddl) in HIV-1-seropositive asymptomatic individuals. Int J Clin Pharmacol Ther. 1999;37:514–518. [PubMed] [Google Scholar]
- 36.Frank I, Bosch RJ, Fiscus S, Valentine F, Flexner C, Segal Y, Ruan P, Gulick R, Wood K, Estep S, et al. Activity, safety, and immunological effects of hydroxyurea added to didanosine in antiretroviral-naive and experienced HIV type 1-infected subjects: a randomized, placebo-controlled trial, ACTG 307. AIDS Res Hum Retroviruses. 2004;20:916–926. doi: 10.1089/aid.2004.20.916. [DOI] [PubMed] [Google Scholar]
- 37.Rauch A, Borromeo M, Ghafoor A, Khoyratty B, Maheshwari J. Leukemogenesis of hydroxyurea in the treatment of sickle cell anemia. Blood. 1999;Abstract:415a. [Google Scholar]
- 38.Wilson S. Acute leukemia in a patient with sickle-cell anemia treated with hydroxyurea. Ann Intern Med. 2000;133:925–926. doi: 10.7326/0003-4819-133-11-200012050-00029. [DOI] [PubMed] [Google Scholar]
- 39.de Montalembert M, Begue P, Bernaudin F, Thuret I, Bachir D, Micheau M. Preliminary report of a toxicity study of hydroxyurea in sickle cell disease. French study group on sickle cell disease. Arch Dis Child. 1999;81:437–439. doi: 10.1136/adc.81.5.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Montalembert M, Davies SC. Is hydroxyurea leukemogenic in children with sickle cell disease? Blood. 2001;98:2878–2879. doi: 10.1182/blood.v98.2.351. [DOI] [PubMed] [Google Scholar]
- 41.Moschovi M, Psychou F, Menegas D, Tsangaris G, Tzortzatou-Stathopoulou F, Nikolaidou P. Hodgkin’s disease in a child with sickle cell disease treated with hydroxyurea. Pediatr Hematol Oncol. 2001;18:371–376. doi: 10.1080/088800101316921985. [DOI] [PubMed] [Google Scholar]
- 42.Al-Jam’a AH, Al-Dabbous IA, Al-Khatti AA, Esan FG. Are we underestimating the leukemogenic risk of hydroxyurea. Saudi Med J. 2002;23:1411–1413. [PubMed] [Google Scholar]
- 43.Ferster A, Sariban E, Meuleman N. Malignancies in sickle cell disease patients treated with hydroxyurea. Br J Haematol. 2003;123:368–369. doi: 10.1046/j.1365-2141.2003.04614.x. [DOI] [PubMed] [Google Scholar]
- 44.Gulbis B, Haberman D, Dufour D, Christophe C, Vermylen C, Kagambega F, Corazza F, Devalck C, Dresse M-F, Hunninck K, et al. Hydroxyurea for sickle cell disease in children and for prevention of cerebrovascular events: the Belgian experience. Blood. 2005;105:2685–2690. doi: 10.1182/blood-2004-07-2704. [DOI] [PubMed] [Google Scholar]
- 45.Couronne L, Schneider P, de Montalembert M, Dumesnil C, Lahary A, Vannier JP. Hodgkin lymphoma in a sickle cell anaemia child treated with hydroxyurea. Ann Hematol. 2009;88:597–598. doi: 10.1007/s00277-008-0632-3. [DOI] [PubMed] [Google Scholar]
- 46.Taylor JG, Darbari DS, Maric I, McIver Z, Arthur DC. Therapy-related acute myelogenous leukemia in a hydroxyurea-treated patient with sickle cell anemia. Ann Intern Med. 2011;155:722–724. doi: 10.7326/0003-4819-155-10-201111150-00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baz W, Najfeld V, Yotsuya M, Talwar J, Terjanian T, Forte F. Development of myelodysplastic syndrome and acute Myeloid Leukemia 15 years after hydroxyurea use in a patient with sickle cell anemia. Clin Med Insights Oncol. 2012;6:149–152. doi: 10.4137/CMO.S8810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milne DM, Palmer RH, Campbell DG, Meek DW. Phosphorylation of the p53 tumour-suppressor protein at three N-terminal sites by a novel casein kinase I-like enzyme. Oncogene. 1992;7:1361–1369. [PubMed] [Google Scholar]
- 49.Qin J-Z, Chaturvedi V, Denning MF, Bacon P, Panella J, Choubey D, Nickoloff BJ. Regulation of apoptosis by p53 in UV-irradiated human epidermis, psoriatic plaques and senescent keratinocytes. Oncogene. 2002;21:2991–3002. doi: 10.1038/sj.onc.1205404. [DOI] [PubMed] [Google Scholar]
- 50.Soubeyrand S, Schild-Poulter C, Hache RJG. Structured DNA promotes phosphorylation of p53 by DNA-dependent protein kinase at serine 9 and threonine 18. Eur J Biochem. 2004;271:3776–3784. doi: 10.1111/j.1432-1033.2004.04319.x. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Eckhart W. Phosphorylation sites in the amino-terminal region of mouse p53. Proc Natl Acad Sci U S A. 1992;89:4231–4235. doi: 10.1073/pnas.89.10.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arai S, Matsushita A, Du K, Yagi K, Okazaki Y, Kurokawa R. Novel homeodomain-interacting protein kinase family member, HIPK4, phosphorylates human p53 at serine 9. FEBS Lett. 2007;581:5649–5657. doi: 10.1016/j.febslet.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 53.International Agency for Research on Cancer IARC TP53 database [Internet]. 2019. [accessed 2018 November11]. http://p53.iarc.fr/TP53GeneVariations.aspx.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Centers for Disease Control and Protection Data & statistics on Sickle Cell Disease. [Internet]. 2017. [accessed 2018 November15]. https://www.cdc.gov/ncbddd/sicklecell/data.html.
- National Center for Biotechnology Information Basic local alignment search tool [Internet]. 2019. https://blast.ncbi.nlm.nih.gov/Blast.cgi.
- International Agency for Research on Cancer IARC monographs on the identification of carcinogenic hazards to humans [Internet]. 2019. [accessed 2019 March30]. monographs.iarc.fr.