

Abstract
Autoreactive CD8+ T cells, which play an indispensable role in β cell destruction, represent an emerging target for the prevention of type 1 diabetes (T1D). Altered peptide ligands (APLs) can efficiently induce antigen-specific T cells anergy, apoptosis or shifts in the immune response. Here, we found that HLA-A*0201-restricted CD8+ T cell responses against a primary β-cell autoantigen insulin epitope InsB15–14 were present in both NOD.β2mnull.HHD NOD mice and T1D patients. We generated several APL candidates for InsB15–14 by residue substitution at the p6 position. Only H6F exhibited an inhibitory effect on mInsB15–14-specific CD8+ T cell responses in vitro. H6F treatment significantly reduced the T1D incidence, which was accompanied by diminished autoreactive CD8+ T cell responses to mInsB15-14, inhibited infiltration of CD8+ and CD4+ T cells in the pancreas and reduced pro-inflammatory cytokine production in pancreatic and splenic T cells in NOD.β2mnull.HHD mice. Mechanistically, H6F treatment significantly augmented a tiny portion of CD8+CD25+Foxp3+ T cells in the spleen and especially in the pancreas. This subset exhibited typical Treg phenotypes and required peptide-specific restimulation to exert immunosuppressive activity. Therefore, this APL H6F may be a promising candidate with potential clinical application value for antigen-specific prevention of T1D.
Keywords: Type 1 diabetes, Altered peptide ligand, CD8+CD25+Foxp3+ regulatory T cells, InsB15–14, NOD.β2mnull.HHD mice
Introduction
Type 1 diabetes (T1D) in both humans and nonobese diabetic (NOD) mice is a spontaneous organ-specific autoimmune disease resulting from autoreactive CD4+ and CD8+ T-cell-mediated elimination of insulin-producing pancreatic islet β-cells.1 Emerging data have shown that the major histocompatibility complex (MHC) class I-restricted CD8+ T-cells play an indispensable role in the initiation and progression of T1D.2–4 Antigen-specific immunotherapies aimed at silencing autoreactive CD8+ T-cell responses may be promising approaches for the prevention of T1D development.5 Altered peptide ligands (APLs) with subtle changes at one or a few amino acid residues may provide considerable benefits in antigen-specific immunotherapy for autoimmune disease as they can modulate antigen-specific T-cell responses ranging from induction of T-cell anergy to apoptosis and shifts in T-cell responses.9 Autoreactive CD8+ T-cell tolerance has been successfully induced to prevent T1D in NOD mice by systemic administration of soluble APLs derived from a known immunodominant CD8+ T-cell epitope10,11 or nanoparticles coated with APL-MHCs complexes.12 However, no APLs targeting human histocompatibility leukocyte antigen (HLA)-restricted autoreactive CD8+ T-cell responses have been generated for potential clinical applications.
HLA-A*0201 is the most commonly expressed HLA class I allele in Caucasians and Asians (50%) and contributes to the susceptibility to T1D.6 HLA-A*0201-transgenic NOD.β2mnull.HHD mice, which express a monochain chimeric HLA-A*0201 molecule consisting of human β2-microglobulin covalently linked to the α1 and α2 domains of human HLA-A*0201, followed by the α3 transmembrane and cytoplasmic domains of murine H-2Db, show an increased incidence of diabetes as well as significant acceleration of T1D onset and progression despite considerably reduced CD8+ T-cell numbers compared with NOD mice.6,7 HLA-A*0201-restricted autoreactive CD8+ T-cell responses against peptides derived from insulin (mIns1L3–11, mIns1B5–14, and mIns1/2A2–10) and islet-specific glucose-6-phosphatase catalytic subunit-related proteins (IGRP228–236, IGRP265–273, and IGRP337–345) have been demonstrated in NOD.β2mnull.HHD mice.6,7 Among these peptides, the IGRP228−236 and IGRP265−273 epitopes have also been found to be targets of HLA-A*0201-restricted autoreactive CD8+ T-cells in T1D patients.13,14 We recently found that HLA-A*0201-restricted CD8+ T-cells against two peptides derived from chromogranin A were present in NOD.β2mnull.HHD mice and T1D patients.15 Therefore, NOD.β2mnull.HHD mice represent an ideal humanized model for developing potential clinically translatable interventions targeting diabetogenic HLA-A*0201-restricted CD8+ T-cell responses.16
Insulin is a pivotal autoantigen that initiates the immune response leading to T1D;8 therefore, inducing insulin-reactive T-cell tolerance is particularly important for the prevention of T1D. We found that HLA-A*0201-restricted CD8+ T-cell responses against Ins1B5−14 were present in both NOD.β2mnull.HHD mice and T1D patients. However, administration of mIns1B5−14 could not prevent T1D in NOD.β2mnull.HHD mice. Here, a series of APL candidates of mInB15−14 with substitution at TCR contact sites (p6) were generated. One APL, H6F, was identified as a therapeutic candidate for in vivo studies. Systemic treatment with H6F significantly reduced the T1D incidence in NOD.β2mnull.HHD mice. Most surprisingly, a tiny portion of CD8+CD25+Foxp3+ regulatory T cells (Tregs) was increased in the spleen and especially in the pancreas with H6F treatment. Notably, the suppressive ability of the CD8+CD25+ Tregs was markedly stronger than that of conventional CD4+CD25+ Tregs. Moreover, these CD8+CD25+ Tregs required peptide-specific restimulation to exert their immunosuppressive activity. The results of this study represent the first report of the protective activity of an APL derived from an islet β-cell antigen targeting diabetogenic HLA-A*0201-restricted CD8+ T-cell responses in NOD.β2mnull.HHD mice with potential clinical application value.
Materials and methods
Mice and T1D subjects
NOD.β2mnull.HHD mice were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). The mice were bred and maintained in specific pathogen-free facilities and handled according to “Principles of Laboratory Animal Care and Use in Research” (Ministry of Health, Beijing, China). Fresh blood samples were obtained from T1D subjects as previously described.15 All experimental protocols were approved by the Ethics Committee of the Third Military Medical University, and informed consent was obtained from all participating subjects in this study.
Blood glucose monitoring
Blood glucose was monitored using a glucometer (OneTouch Ultral; LifeScan, Milpitas, CA, USA) at weekly intervals, beginning at 10 weeks of age. According to several previous studies,17–19 diabetes was defined as two consecutive blood glucose values above 11.1 mmol/l.
Modeling of HLA-A*0201-insulin peptide complexes
The 3D structure of the HLA-A*0201 molecule in complex with the mIns1B5−14 decamer peptide was established based on PDB 1I4F, a crystal structure of the complex of HLA-A*0201 with a MAGE-A4 peptide (GVYDGREHTV), as it has the highest resolution of all known crystal structures of the complexes of HLA-A*0201 with decamer peptides.20 The conformations of the p1, p2, and pc (C-terminate) positions of mIns1B5−14 were determined by mutating the corresponding positions in 1I4F, while the conformations of the middle positions (p3–p8) were generated using the loop refinement protocol in the Discovery Studio 2.55 program (Accelrys Inc., San Diego, USA). Energy minimization and molecular dynamics simulation were performed using the standard dynamic cascade protocol in the DS2.55 package as described in Mou et al.21 In brief, the generated conformation was solvated in 0.145 mol/l NaCl using the solvation protocol, and then 5-ns molecular dynamics simulation was performed at 300 K after minimization of the explicit periodic boundary models using the steepest descent and conjugate gradient method. The structures of the HLA-A*0201 molecule in complex with the APLs of the mIns1B5−14 peptide were generated based the optimized model of HLA-A*0201-mIns1B5−14, and another 1-ns molecular dynamic simulation was performed to generate the final models. Finally, the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method22 was used to calculate the binding free energy between peptides and the MHC molecule in the generated conformation.
Peptide treatment
Insulin-derived peptides mIns1B5−14 (HLCGPHLVEA), hInsB5−14 (HLCGSHLVEA), H6E (HLCGPELVEA), H6F (HLCGPFLVEA), H6Q (HLCGPQLVEA), and H6Y (HLCGPYLVEA) and control peptides OVA257–264 (SIINFEKL) and HIVpol476–484 (ILKEPVHGV) were synthesized with purities >95% at the Chinese Peptide Company (Hangzhou, China). We injected cohorts of 4-week-old female NOD.β2mnull.HHD mice with 100 μg (1 μg/μl) of peptide in PBS intraperitoneally. We repeated this procedure every week until the sixth injection.
Histology
Pancreatic tissues from different peptide-treated nondiabetic female NOD.β2mnull.HHD mice at 12 weeks old (ten mice per group) were fixed in 10% neutral-buffered formalin and stained with hematoxylin and eosin (H&E). A minimum of 10 islets from each mouse were microscopically observed by two different observers, and insulitis scoring was performed according to the following criteria: 0, no infiltration; 1, peri-insulitis; 2, insulitis with <50% islet area infiltration; 3, insulitis with 50% islet area infiltration.
IFN-γ ELISPOT assays
Briefly, 2 × 105 CD8+ T cells were purified from the spleens of NOD.β2mnull.HHD mice treated with each peptide by magnetic separation and were then incubated with T2 cells loaded with 50 μg/ml mInsB5−14 or OVA257–264 (negative control) at a rate of 10:1 in anti-mIFN-γ mAb-precoated ELISPOT plates (MabTech) for 36 h at 37 °C. After incubation, the cells were removed and plates were processed according to the manufacturer’s instructions. Spots were counted using a spot reader system (Saizhi, Beijing, China).
Real-time RT-PCR
The mRNA levels of the pro-inflammatory cytokines of pancreatic biopsy samples were detected as previously described.23
Isolation of pancreas-infiltrating cells
Pancreas-infiltrating cells from immunized NOD.β2mnull.HHD mice were isolated as previously described.24
Flow cytometry
Surface markers and intracellular cytokines were detected as previously described.15 Anti-CD3-FITC (145−2C11), anti-CD4-PE (GK1.5) or FITC (RM4-4), anti-CD8-PerCP-Cy5.5 (53-6.7), anti-CD25−PE or APC (PC61.5), anti-CD103-APC (2E7), anti-CD62L-APC (MEL-14), anti-CTLA-4-APC (UC10-4B9), anti-Foxp3-PE (FJK-16s), anti-IFN-γ-APC (XMG1.2), anti-IL-17A-PE (eBio17B7), and isotype control antibodies were obtained from eBiosciences, San Diego, CA, USA.
Suppression assays
CD4+CD25+, CD4+CD25−, and CD8+CD25+ T cells were purified from the spleens of 12-week-old nondiabetic, H6F-treated female NOD.β2mnull.HDD mice (all >90% purity) using the magnetic cell isolation kit (StemCell Technologies) according to the manufacturer’s instructions. Varied numbers of CD8+CD25+ or CD4+CD25+ T cells were co-cultured in triplicate with CD4+CD25− effector T cells (100,000 cells/well) stimulated with anti-CD3/CD28-coupled beads (Life Technologies) for 3 days in 96-well plates at different suppressor/responder ratios, and then [3H] thymidine (1 μCi/well) was added for an additional 16 h of culture. Then, the uptake of [3H] thymidine was determined using a liquid scintillation counter (Beckman Coulter).
To further determine the presence of antigen-specific suppressive CD8+CD25+ T cells, freshly isolated autologous CD4+CD25− T cells (50,000 cells/well) were pre-stimulated with anti-CD3/CD28-coupled beads (Life Technologies, Grand Island, NY) for 14 h and then co-cultured with an equal number of CD8+CD25+ T cells purified from the spleens of H6F-treated NOD.β2mnull.HDD mice and mitomycin C-treated syngeneic spleen cells (250,000 cells/well) loaded with 10 μg/ml H6F, 10 μg/ml OVA257-264 (negative control), or no peptide in triplicate. In some cultures, conditioned medium was removed after 72 h of incubation at 37 °C in 5% CO2 and analyzed for cytokine levels of IFN-γ and IL-17 by enzyme-linked immunosorbent assay (ELISA) kits (Dakewe Biotech., Shenzhen, China) according to the manufacturer’s instructions. In other cultures, [3H] thymidine (1 μCi/well) was added for an additional 16 h of culture. Then, the uptake of [3H] thymidine was determined using a liquid scintillation counter (Beckman Coulter).
Statistics
An unpaired t-test or the Mann–Whitney test was used for comparisons between different treatment groups depending on the distribution of the data. A paired t-test was used to compare autoreactive CD8+ T cell responses to the control peptide or mIns1B5–14 of an individual within a certain treatment group. P values < 0.05 were considered statistically significant.
Results
Design and selection of a potential antagonist peptide from Ins1B5-14
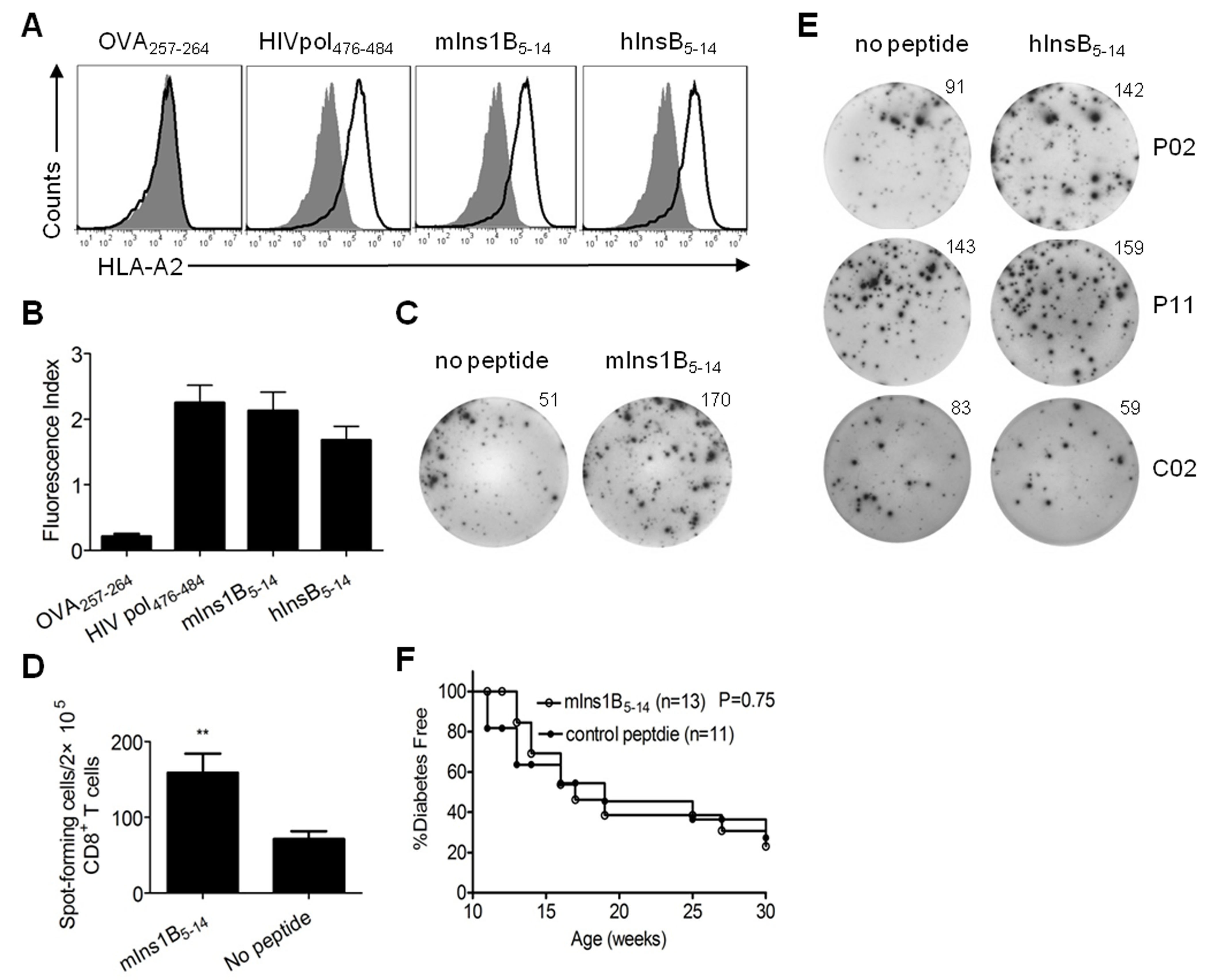
Previous studies have shown that autoreactive CD8+ T cells can be tolerized by administering soluble antigenic peptides to prevent the onset of T1D.10,25 We and others have demonstrated that Ins1B5–14 is an immunodominant target of autoreactive CD8+ T cells in both NOD.β2mnull.HHD mice7 and HLA-A*0201-positive T1D patients (Supplementary Table 1 and Supplementary Figure 1). Administration of soluble mIns1B5–14 could not prevent the development of T1D in NOD.β2mnull.HHD mice (Supplementary Figure 1f). Several studies have demonstrated that administration of soluble APLs rather than native peptides efficiently prevented the development of T1D in NOD mice.10,17,26 Therefore, we sought to identify APLs derived from mIns1B5–14 capable of inducing tolerance of mIns1B5–14-specific pathogenic CD8+ T cells.
The modeled structure of peptide mIns1B5–14 in complex with the HLA-A*0201 molecule was generated and optimized using Discovery Studio 2.55 software. As shown in Fig. 1a, the p5 and p6 positions bulged out of the binding groove and were more accessible for TCR inspection. Moreover, the residue Pro at p5 was a key residue in the turn conformations of peptides, and the substitution of p5 with other residues induced a large change in the peptide conformation. Therefore, a series of the APL candidates of mInB15-14 was generated by residue substitution at the p6 position with F, G, L, R, E, Q, T, and Y. Here, F, G, and L represent residues lacking donors and acceptors of H-bonds in the side chains; R represents the conservative substitution of H; E and Q represent the acidic residues and their amide residues; and T and Y represented the neutral residues sharing donors and acceptors of H-bonds in the side chains. After energy minimization and molecular dynamics simulation of all modeled structures, the eight structures of APLs in complex with the HLA-A*0201 molecule were aligned to the structure of HLA-A*0201-mInsB5–14. The root mean square deviations (RMSDs) of APLs relative to mInsB5-14 were calculated based on the Cα atoms, and the binding free energies between peptide ligands and the HLA-A*0201 molecule were calculated using MM-PBSA analysis. Four APLs (H6F, H6E, H6Q, and H6Y) that were predicted to be good HLA-A*0201 binders with minimal changes in binding conformation (Table 1) were synthesized. The actual binding affinity of these APL candidates for the HLA-A*0201 molecule was evaluated in vitro using a T2-cell-peptide binding test as previously described.15 As shown in Fig. 1b, c, H6F, H6E, H6Q, and H6Y showed similar binding affinities to HLA-A*0201 compared with the native peptide mIns1B5–14. We further tested their ability to competitively inhibit the proliferation of splenocytes from 12-week-old nondiabetic female NOD.β2mnull.HHD mice upon stimulation with mIns1B5–14 in vitro.15 Only H6F significantly inhibited mIns1B5–14-derived splenocyte proliferation in a dose-dependent manner (Fig. 1d, e), indicating that H6F may have an antagonistic effect on the TCR-mIns1B5–14-MHC interaction. Therefore, H6F was selected as a potential antagonist APL for the in vivo treatment study.
Fig. 1.

Design and selection of a potential antagonist peptide for mIns1B5–14. a The modeled structure of the mIns1B5–14-HLA-A*0201 complex showing that the p5 and p6 positions of mIns1B5–14 bulged out of the binding groove, which were more accessible for TCR inspection. b T2 cell-based peptide binding assays for HLA-A*0201 were performed by FACS analysis. T2 cells were incubated with or without the indicated peptides (10 μg/ml) and the level of surface HLA-A2 molecules was detected by flow cytometry. The H-2Kb-binding peptide OVA257-264 was used as a negative control. Filled histograms, no peptide; open histograms, plus peptide. c The fluorescence index (FI) was calculated as follows: FI = (mean fluorescence intensity with the given peptide―mean fluorescence intensity without peptide)/(mean fluorescence intensity without peptide). Bars represent the mean ± SEM of three independent experiments. d, e The proliferation of splenocytes stimulated with mIns1B5–14 (10 μg/ml) plus itself (10 μg/ml) or the indicated APLs (10 μg/ml) (d) and H6F at four different concentrations (e) was measured by [3H] thymidine incorporation. Bars represent the mean ± SEM of seven independent experiments. Significance was determined by an unpaired t-test
Table 1.
ΔΔ G and RMSDC-α calculation results
| APL candidates | ΔΔ G (kcal/mol) | RMSDC-α (Å) | APL candidates | ΔΔ G (kcal/mol) | RMSDC-α (Å) |
|---|---|---|---|---|---|
| H6F | −7.747 | 0.38 | H6E | −1.243 | 0.467 |
| H6G | 2.988 | 0.567 | H6Q | −0.445 | 0.512 |
| H6L | 11.087 | 0.523 | H6T | 23.057 | 0.934 |
| H6R | 2.545 | 1.432 | H6Y | −3.372 | 0.603 |
ΔΔ G: binding free energies of APLs relative to mIns1B5–14; RMSDC-α: RMSDs of APLs relative to mInsB5–14 calculated based on the Cα atoms
Intraperitoneal immunization with the APL H6F prevented the onset of T1D in NOD.β2mnull.HDD mice
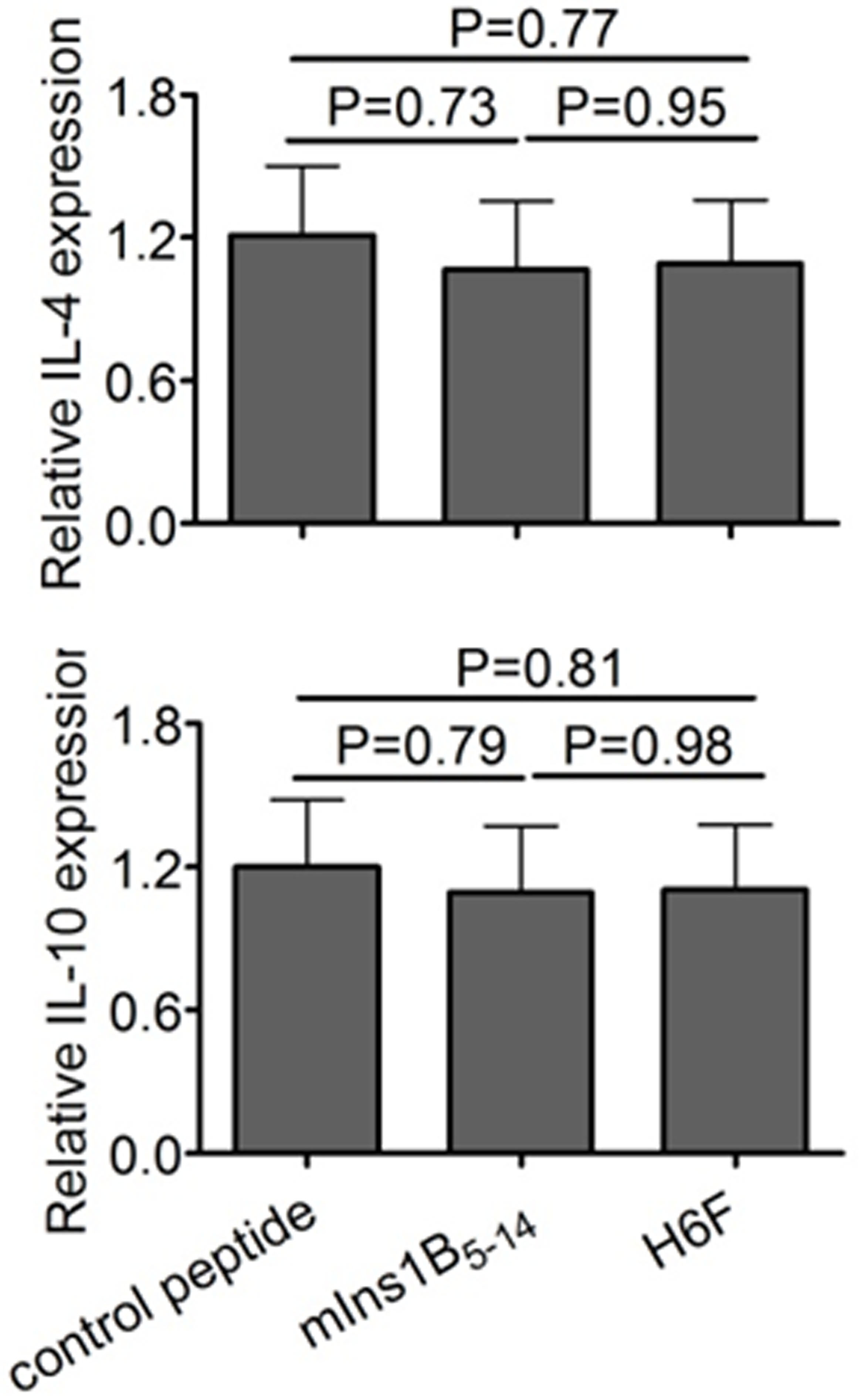
To investigate whether the APL H6F had antidiabetogenic activity, we treated female NOD.β2mnull.HHD mice with repeated i.p. injections of H6F, mIns1B5–14 and OVA257–264 (negative control). As shown in Fig. 2a, administration of H6F significantly prevented the onset of T1D compared with treatment with native peptide or control peptide. Histopathological analysis revealed that H6F treatment significantly inhibited the development of insulitis in NOD.β2mnull.HHD mice at 12 weeks of age compared to control peptide or native peptide treatment (Fig. 2b, c). Notably, the absolute numbers and percentages of infiltrating CD4+ and CD8+ T cells in the pancreata of H6F-treated NOD.β2mnull.HHD mice were much lower than those detected in mIns1B5–14- and control peptide-treated mice (Fig. 2d). Furthermore, H6F-treated NOD.β2mnull.HHD mice exhibited significant decreases in the mRNA levels of the pro-inflammatory cytokines IFN-γ, IL-17, IL-1β, IL-6, and TNF-α but showed no changes in the mRNA levels of anti-inflammatory cytokines IL-4 and IL-10 in the pancreas compared with those in mIns1B5–14 or control peptide-treated NOD.β2mnull.HHD mice (Fig. 2e and Supplementary Figure 2).
Fig. 2.

Intraperitoneal immunization with the APL H6F prevented the onset of T1D in NOD.β2mnull.HDD mice. Female NOD.β2mnull.HHD mice received weekly intraperitoneal injections of 100 μg of peptide H6F, mIns1B5-14, or OVA257-264 from 4 to 9 weeks of age (n = 12 or 13). a The mice were monitored for diabetes development. Data were obtained from three independent experiments. *P < 0.05 and ** P < 0.01. b, c Histopathological evaluation of pancreatic sections from indicated peptide-treated NOD.β2mnull.HHD mice at the age of 12 weeks. Pancreatic sections were stained with H&E and scored for insulitis. Representative micrographs (200× magnification) (b) and histologic scores (c) from each group (n = 10) are shown. Insulitis scoring was performed according to the following criteria: 0, no infiltration; 1, peri-insulitis; 2, insulitis with <50% islet area infiltration; and 3, insulitis with >50% islet area infiltration. d Frequencies and absolute numbers of CD4+ and CD8+ T cells (pre-gated on a live CD3+ population) infiltrated in pancreata from the indicated peptide-treated NOD.β2mnull.HHD mice at the age of 12 weeks. The results for absolute numbers of these CD4+ and CD8+ T cells are expressed as the mean ± SD (each symbol represents a sample of pooled pancreatic infiltrating cells from four mice). e The mRNA expression levels of indicated cytokines in the pancreas in each peptide-treated group of nondiabetic mice (n = 6) were quantified by real-time RT-PCR. The data are presented as fold-change compared to the mRNA levels expressed in pancreata from control peptide-treated mice. Significance was determined by an unpaired t-test
Moreover, compared with NOD.β2mnull.HHD mice that received mIns1B5–14 or control peptide injection, H6F-treated NOD.β2mnull.HHD mice displayed significant decreases in the frequencies of both IFN-γ- and IL-17A-producing CD4+ T cells as well as IFN-γ- and IL-17A-producing CD8+ T cells in the spleen (Fig. 3). These results indicated that H6F had strong protective activity, which was associated with inhibition of insulitis development, T-cell infiltration and pro-inflammatory cytokine production in the pancreas, as well as extensive suppression of peripheral pro-inflammatory T cell responses.
Fig. 3.

Treatment with H6F notably reduced IFN-γ and IL-17 secretion by splenic T cells in the NOD.β2mnull.HDD mice. The percentages of CD4+IFN-γ+ (a), CD8+IFN-γ+ (b), CD4+IL-17A+ (c), and CD8+IL-17A+ (d) T cells in spleens from the indicated peptide-treated nondiabetic NOD.β2mnull.HHD mice at 12 weeks of age (n = 8) were determined by flow cytometry. Representative FACS density plots and statistical analyses are shown. The data are expressed as the mean ± SD. Significance was determined by an unpaired t-test
H6F treatment resulted in loss of mIns1B5-14 autoreactive CD8+ T-cell responses
To determine whether H6F treatment can induce tolerance of autoreactive CD8+ T-cell responses towards the native peptide mIns1B5–14, we analyzed the peptide mIns1B5–14-stimulated IFN-γ spots formed by CD8+ T cells in spleens from NOD.β2mnull.HHD mice that received different treatments as previously described.15 Compared with the control peptide, mIns1B5–14 stimulated a much higher level of IFN-γ production in purified splenic CD8+ T cells from control peptide- and mIns1B5-14-treated NOD.β2mnull.HHD mice. In contrast, no significant specific IFN-γ-producing cell reactivity was detected in splenic CD8+ T cells stimulated with mIns1B5-14 or control peptide in H6F-treated NOD.β2mnull.HHD mice (Fig. 4a, b). These results indicated that treatment with H6F efficiently induced the loss of mIns1B5-14 autoreactive CD8+ T cell responses in NOD.β2mnull.HHD mice.
Fig. 4.

Treatment with H6F resulted in loss of mIns1B5–14 autoreactive CD8+ T cell responses. a Representative statistical analysis of the average number of IFN-γ-positive spots derived from splenic CD8+ T cell responses to control peptide or mIns1B5-14 within a treatment group (n = 5) are shown. The data are expressed as the mean ± SEM. Significance was determined by a paired t-test. b A representative image of IFN-γ ELISPOT assay for monitoring mIns1B5-14-specific splenic CD8+ T cell responses in each peptide-treated group of nondiabetic NOD.β2mnull.HHD mice at the age of 12 weeks. The average number of IFN-γ-positive spots per 2 × 105 splenic CD8+ T cells in triplicate cultures was calculated. T2 cells pulsed with mIns1B5-14 or control peptide (50 µg/ml) were used as stimulators
Treatment with H6F augmented a tiny portion of CD8+CD25+Foxp3+ T cells in the spleen and pancreas
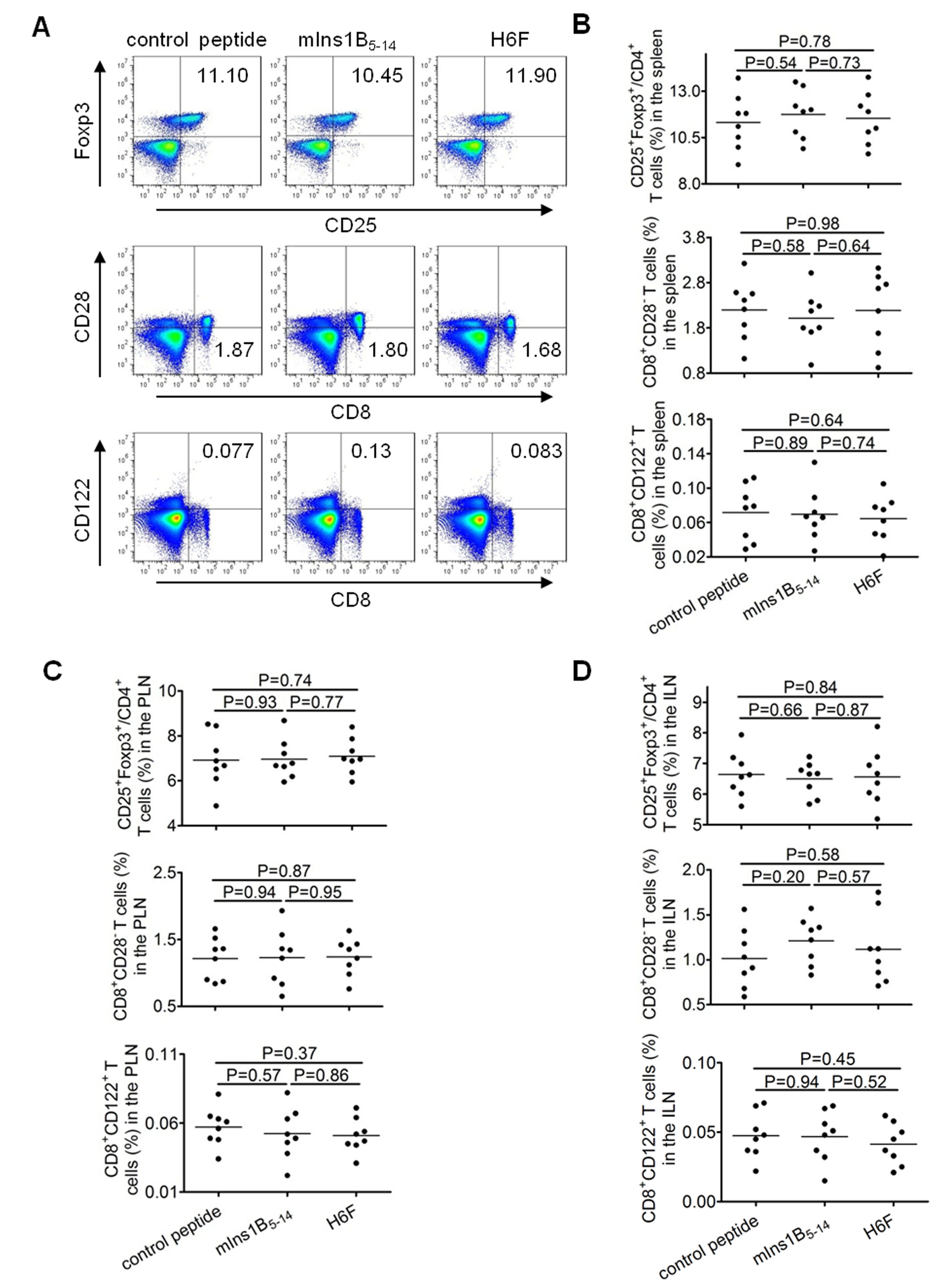
We further investigated the cellular mechanism underlying the protective effect of H6F administration. Induction or expansion of antigen-specific Tregs has been reported to be involved in antigen-specific immunotherapies for autoimmune diseases.12,27,28 Therefore, we examined the frequencies of classic CD4+ Tregs, which highly express CD25 and Foxp3, and CD8+ Treg subsets described previously, such as CD8+CD28- Tregs, CD8+CD122+ Tregs, and CD8+CD25+Foxp3+ Tregs, in different peptide treated-NOD.β2mnull.HHD mice at 12 weeks of age. Very interestingly, a tiny subset of CD8+CD25+Foxp3+ T cells showed an obvious increase in frequency in the spleen and pancreatic lymph nodes (PLNs), but not inguinal lymph nodes (ILNs), from H6F-treated NOD.β2mnull.HHD mice compared with those detected in mIns1B5-14- or control peptide-treated mice (Fig. 5a and data not shown). However, no significant difference was identified among the frequencies of CD4+CD25+Foxp3+, CD8+CD28-, CD8+CD122+ T cells in the spleen, PLNs and ILNs (P > 0.05) from each group of peptide-treated NOD.β2mnull.HHD mice (Supplementary Figure 3 and data not shown). Moreover, the frequency of CD8+CD25+Foxp3+ T cells in PLNs was the highest compared with those detected in ILNs and spleens from H6F-treated NOD.β2mnull.HHD mice (Fig. 5b). Notably, H6F treatment also induced an increase in the frequency of CD8+CD25+Foxp3+ T cells among the pancreas-infiltrating (PIL) T cells, which was much higher than that assayed in PLNs from NOD.β2mnull.HHD mice treated with H6F but not the native peptide (Fig. 5c, d). Taken together, our data indicated that treatment with H6F augmented a tiny portion of CD8+CD25+Foxp3+ T cells in spleens and especially in pancreata from NOD.β2mnull.HHD mice.
Fig. 5.

Treatment with H6F notably increased the frequency of CD8+CD25+Foxp3+ T cells in NOD.β2mnull.HHD mice. Spleen-, pancreatic lymph node (PLN)-, inguinal lymph node (ILN)- and pancreas-infiltrating (PIL) cells were freshly isolated from the indicated peptide-treated nondiabetic NOD.β2mnull.HHD mice at the age of 12 weeks (n = 8–16). a The percentages of CD8+CD25+ T cells and CD8+CD25+Foxp3+ T cells in the spleen and PLNs were determined by flow cytometry. The data are expressed as the mean ± SD. b Comparison of the frequencies of CD8+CD25+ and CD8+CD25+Foxp3+ T cells in ILNs, PLNs, and spleens from H6F-treated nondiabetic NOD.β2mnull.HHD mice (n = 8). The data are expressed as the mean ± SD. c Representative FACS plots showing the frequencies of CD8+CD25+ and CD8+CD25+Foxp3+ T cells among PIL cells pooled from four nondiabetic NOD.β2mnull.HHD mice with the indicated peptide treatments. d Comparison of the frequencies of CD8+CD25+Foxp3+ T cells among PIL cells and PLNs from the same group of NOD.β2mnull.HHD mice treated with mIns1B5-14 or H6F (n = 16). The results of the PIL cells are expressed as the mean ± SD (n = 4, each sample derived from pooled pancreatic infiltrating cells from four mice), and the PLN results are expressed as the mean ± SD (n = 16). Significance was determined by the Mann–Whitney test
CD8+CD25+ T cells harbored a typical regulatory T cell phenotype
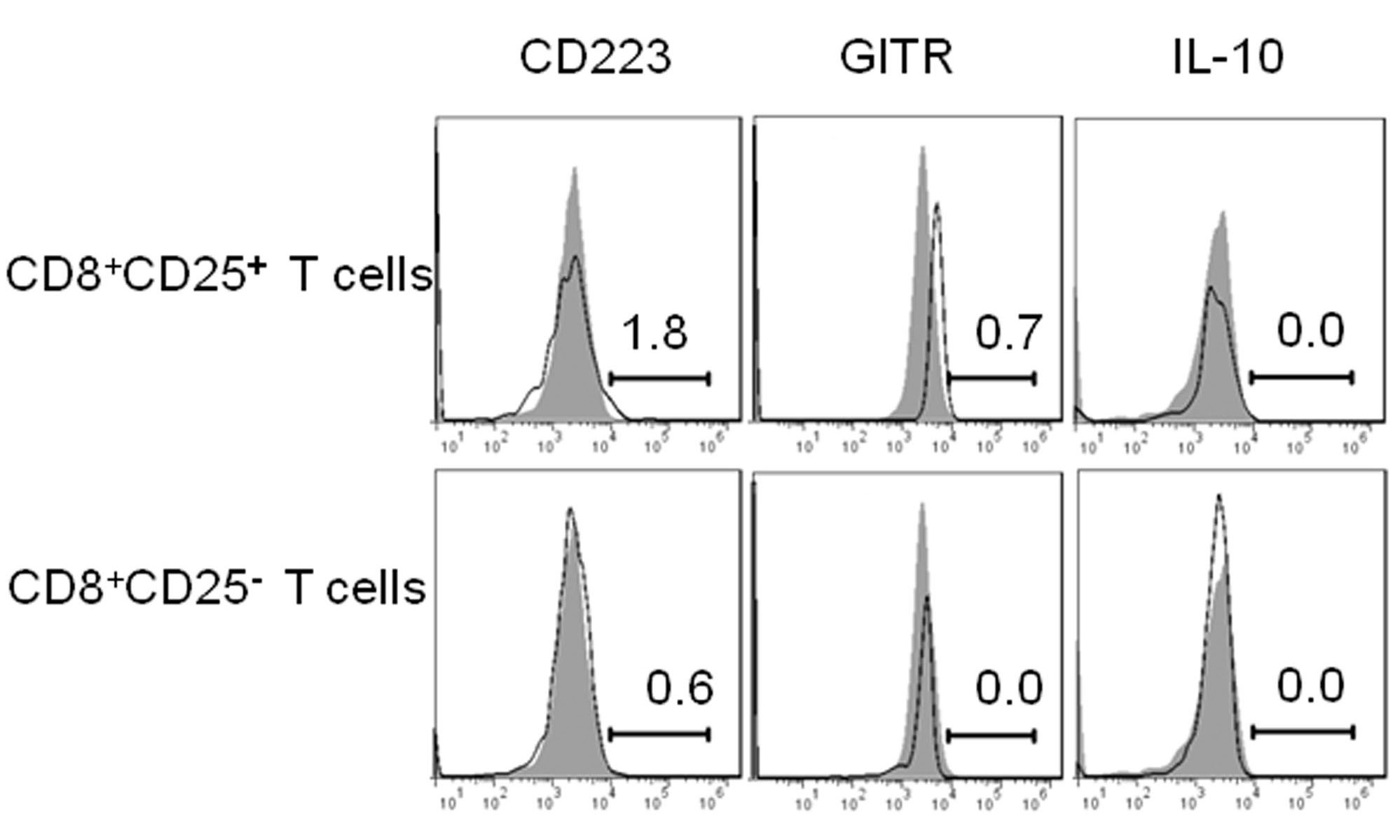
We found the splenic CD8+CD25+ T cells isolated from H6F-treated NOD.β2mnull.HDD mice highly expressed Foxp3, and this subset of cells expressed high levels of CTLA-4, CD62L, and CD103, moderate levels of CD223 and GITR, a low level of IFN-γ, and minimal IL-17A. Splenic CD8+CD25- T cells expressed minimal Foxp3, much lower levels of CTLA-4, CD62L and CD103, a much higher level of IFN-γ and minimal IL-17A. IL-10 was not detected in either cell population (Fig. 6a, b and Supplementary Figure 4). Notably, much higher levels of CD103, CD62L, Foxp3, and CTLA-4 secreted by splenic CD8+CD25+ T cells, and much lower levels of IFN-γ and IL-17A secreted by splenic CD8+CD25- T cells were found in H6F-treated NOD.β2mnull.HDD mice compared with those in mice treated with mIns1B5–14 or control peptide (Fig. 6c, d). Collectively, these results demonstrated that CD8+CD25+ T cells in NOD.β2mnull.HDD mice may have competent regulatory activity because of their high expression levels of similar phenotypic regulatory markers compared to conventional CD4+CD25+ Tregs.
Fig. 6.

Phenotypic characterization of CD8+CD25+ T cells in the NOD.β2mnull.HHD mice. CD8+CD25+ and CD8+CD25− T cells were freshly isolated from the spleens of H6F-treated nondiabetic NOD.β2mnull.HHD mice at the age of 12 weeks (n = 5) and these cells were phenotyped by flow cytometry analysis. a Representative flow cytometry phenotypic profiles of CD8+CD25+ and CD8+CD25- T cells in H6F-treated NOD.β2mnull.HHD mice. The numbers above the line indicate the event numbers of cells with a positive phenotype in the indicated cell subsets, and the numbers below the line indicate the total numbers of the indicated CD8+CD25+ T cells or CD8+CD25- T cells. b The mean percentages of Foxp3+, CTLA-4+, CD62L+, CD103+, IFN-γ+ and IL-17A+ in CD8+CD25+ (open bars) or CD8+CD25- (gray bars) T cells from H6F-treated NOD.β2mnull.HHD mice (n = 5). ***Compared to the same phenotype or cytokine production of CD8+CD25+ T cells, P < 0.001. The data are expressed as the mean ± SD. c The mean percentages of Foxp3+, CTLA-4+, CD103+, and CD62L+ in splenic CD8+CD25+ T cells or d the mean percentages of IFN-γ+ and IL-17A+ in splenic CD8+CD25- T cells isolated from different peptide-treated NOD.β2mnull.HHD mice (n = 5). The data are expressed as the mean ± SD. Significance was determined by an unpaired t-test
Splenic CD8+CD25+ T cells in H6F-treated mice exerted ligand-specific suppressive activity
To further confirm whether CD8+CD25+ T cells in H6F-treated NOD.β2mnull.HDD mice have suppressive activity, syngeneic CD4+CD25- T cells were co-cultured with CD8+CD25+ or CD4+CD25+ T cells purified from the spleens of these mice at different ratios in the presence of anti-CD3/CD28-coupled beads, and then the proliferation of these cells was detected. Unexpectedly, we found that CD8+CD25+ T cells inhibited the proliferation of CD4+CD25- T cells more efficiently than CD4+CD25+ T cells at equal numbers (Fig. 7a). Furthermore, we examined whether splenic CD8+CD25+ T cells isolated from H6F-treated mice require ligand-specific reactivation to exhibit inhibitory effects. As shown in Fig. 7b, c, the proliferation and IFN-γ and IL-17A production of pre-activated CD4+CD25- responder T cells in the same cultures were significantly inhibited only when CD8+CD25+ T cells derived from H6F-treated mice were co-cultured with mitomycin C-treated syngeneic splenocytes pulsed with H6F peptide, but not with control peptide or no peptide. These results indicated that splenic CD8+CD25+ T cells in H6F-treated mice possessed a strong immunosuppressive function and required antigenic peptide ligand-specific reactivation to exert their immunosuppressive activity.
Fig. 7.

Splenic CD8+CD25+ T cells in the H6F-treated NOD.β2mnull.HHD mice exerted ligand-specific suppressive activity. a Suppression of CD4+CD25- T cell proliferation by splenic CD8+CD25+ T cells or CD4+CD25+ T cells. CD4+CD25- T cells (100,000 cells/well) were co-cultured in triplicate with varied numbers of CD8+CD25+ (open bars) or CD4+CD25+ (gray bars) T cells isolated from the same H6F-treated mice at 1∶0, 10∶1, 5∶1, 2∶1, 1∶1 and 0∶1 ratios in the presence of anti-CD3/CD28-coupled beads for 3 days. *P < 0.05 and **P < 0.01 compared with the [3H] thymidine uptake of CD4+CD25- T cells alone. b The proliferation of pre-activated CD4+CD25- T cells (50,000 cells/well) cocultured with mitomycin C-treated syngeneic splenocytes (250,000 cells/well) loaded with 10 μg/ml OVA257-264 (gray bars), 10 μg /ml H6F (open bars), or no peptide (black bar) in the presence of an equal number of CD8+CD25+ T cells purified from the spleens of H6F-treated NOD.β2mnull.HHD mice. **P < 0.01 compared with the [3H] thymidine uptake of CD4+CD25- T cells cocultured with mitomycin C-treated syngeneic splenocytes alone. c IFN-γ and IL-17A in the cell culture supernatants from (b) were assayed by ELISA. ***P < 0.001 and **P < 0.01 compared with IFN-γ and IL-17A in the cell culture supernatants of CD4+CD25- T cells cocultured with mitomycin C-treated syngeneic splenocytes alone. The data are presented as the mean of triplicate cultures and are representative of three independent experiments. Significance was determined by an unpaired t-test
Discussion
Here, we reported for the first time that the peptide H6F, a novel APL derived from an HLA-A*0201-restricted immunodominant peptide (InsB15–14), displayed strong protective activity in NOD.β2mnull.HDD mice, which was associated with inhibition of insulitis and T1D development, suppression of T-cell infiltration and pro-inflammatory cytokine production in the pancreas, and induction of insulin-specific CD8+ T cell tolerance. Very intriguingly, H6F treatment selectively led to expansion of a tiny population of CD8+CD25+Foxp3+ T cells in vivo, which exhibited a typical Treg phenotype and required peptide-specific stimulation to exert their immunosuppressive activity.
TCR engagement by APLs with amino acid substitutions at TCR contact residues or at residues in the binding pocket of the native peptide usually alters the strength or type of T cell responses induced by the native peptide. Recently, numerous APLs have been designed based on the autoreactive CD4+ T cell epitopes for the treatment of T1D.17,26 Currently, few APLs designed to modulate diabetogenic autoreactive CD8+ T cells have been studied. A superagonist APL with a single amino acid substitution at TCR contact residue p6 has been reported to effectively protect against autoimmune diabetes by promoting elimination of pathogenic CD8+ T cells.11 Another study suggested that APLs with “low-avidity” rather than APLs with “very high-avidity” or wild-type self-peptide had superior antidiabetogenic activities in NOD mice.10 One APL generated via substitution at p6 of the dominant epitope InsB15–23 has been identified to antagonize the TCR of the highly pathogenic InsB15–23-reactive CD8+ T cell clone G9C8 in vitro, but its protective effect on diabetes has not yet been studied in NOD mice.29 A previous study based on the crystal structure demonstrated that one amino acid substitution at p6, which led to only a minor conformational change in the contact with the TCR and not with the MHC, had a profound influence on the TCR-peptide-MHC interaction.30 Therefore, in this study, a set of APLs was designed by residue substitution at p6 using in silico analysis. Multiple T cell clones with different avidity levels towards the same peptide are always present in organisms. We speculated that these APLs mentioned above, which were all screened using a single specific CD8+ T cell clone, may have an unknown or even opposite effect on other clones with the same antigen specificity. Therefore, we screened out an APL, H6F, as a candidate for in vivo treatment by detecting the inhibitory effect on native peptide-induced polyclonal splenocyte responses. Furthermore, repeated treatment with H6F in vivo resulted in a protective effect against T1D in NOD.β2mnull.HDD mice. Therefore, the present study provides a convenient approach for in silico design and in vitro screening of APLs for modulation of multiple CD8+ T cell responses to an antigenic peptide.
Previous studies have demonstrated that administration of APLs derived from CD8+ T cell epitopes can efficiently induce elimination of pathogenic CD8+ T cells in the islets of transgenic mouse models of T1D.11 Consistently, we observed that H6F treatment diminished peripheral CD8+ T cell responses against the native peptide. In addition, H6F treatment also decreased the infiltration of both CD8+ and CD4+ T cells in the pancreas and the production of pro-inflammatory cytokines in pancreatic and splenic T cells. We suppose that H6F treatment may induce extensive bystander suppressive effects, including inhibition of native peptide-specific CD8+ T cell responses. The protective effects of antigen-specific immunotherapies for autoimmune diabetes have frequently been associated with induction of antigen-specific Tregs.10,27,31 However, compared with CD4+ Tregs,32 few studies on the induction of CD8+ Tregs in autoimmune diabetes are available in the literature. One study showed that treatment of NOD mice with nanoparticles coated with disease-relevant peptide-major histocompatibility complexes expanded cognate low-avidity CD8+CD122+CD44hi Tregs, which suppressed local presentation of autoantigens and resulted in disease prevention in prediabetic mice.12 Another study reported that CD8+Foxp3+ Tregs induced by glutamate decarboxylate 65-immunoglobulin G-transduced splenocytes were responsible for the induction of GAD-specific immune tolerance in NOD mice.33 A recent study demonstrated that the Chinese medicine Ginseng and Astragalus granule ameliorated T1D in NOD mice by increasing the numbers of both CD4+Foxp3+ and CD8+CD122+PD-1+ Tregs in spleens and lymph nodes of NOD mice.34 Here, we showed that repeated treatment with H6F significantly augmented a rare population of CD8+CD25+Foxp3+ T cells in the spleen, PLNs and especially in the pancreas, which possessed a typical and enhanced functional Treg phenotype. Especially compared with CD8+CD25+ Tregs in the native or control peptide-treated NOD.β2mnull.HDD mice, CD8+CD25+ Tregs in H6F-treated NOD.β2mnull.HDD mice expressed much higher levels of Foxp3, CTLA-4, CD62L, and CD103, reflecting strong immunosuppressive properties. CD62L(high) Tregs have been reported to express increased amounts of CTLA-4 and were more potent than CD62L(low) Tregs in suppressing proliferation and inducing apoptosis in effector T cells.35 CD8+CD25+Foxp3+ Tregs expressing high levels of CTLA-4 and CD62L were reduced in the cerebrospinal fluid of multiple sclerosis patients during acute exacerbations.36 Fleissner D et al. reported that intestinal hemagglutinin expression led to peripheral induction of hemagglutinin-specific CD8+Foxp3+ Tregs expressing a high level of CD103.37 CD8+CD25+ Tregs have been shown to share similar phenotypic features with CD4+CD25+ Tregs, such as high expression levels of CTLA-4 and Foxp3, and both Treg subsets inhibited effector T cell responses with similar efficiency.27,38 We found that the immunosuppressive capacity of CD8+CD25+ Tregs isolated from H6F-treated NOD.β2mnull.HDD mice was markedly higher than that of CD4+CD25+ Tregs isolated from the same mice at an equal suppressor to responder ratio. This finding was also supported by another study showing that CD8+CD25+Foxp3+ Tregs suppress effector T cell proliferation more effectively than CD4+CD25+Foxp3+ Tregs in vitro.39 We also showed that this population of CD8+CD25+ Tregs isolated from H6F-treated mice required H6F-specific reactivation to exert their inhibitory effect on the proliferation and IFN-γ and IL-17 secretion of polyclonally activated responder T cells, indicating that antigen-specific CD8+ Tregs were induced by H6F treatment. Our study together with these prior studies indicates that antigen-specific CD8+CD25+ Tregs represent an important subset of regulatory T cells and may play an important role in the maintenance of self-tolerance even at a very low frequency in the target organ and the peripheral lymphoid tissue; therefore, this may be a plausible mechanism to explain why H6F treatment led to decreased infiltration of both autoreactive CD8+ and CD4+ T cells and reduced pro-inflammatory cytokine production in both the pancreas and spleen in NOD.β2mnull.HDD mice. However, we did not explore the mechanism for the expansion of CD8+CD25+Foxp3+ Tregs by H6F treatment. Some studies have suggested that weak, prematurely terminated, or suboptimal TCR signaling favored the expansion of Foxp3+ Tregs.40,41 Therefore, we speculate that the expansion of CD8+Foxp3+ Tregs induced by H6F may result from a weakened TCR signaling cascade induced by MHC/H6F/TCR interactions, which may also explain why H6F exhibited a competitive inhibitory effect on the proliferation of murine splenocytes induced by the native peptide in vitro.
In conclusion, our current findings indicate that a novel APL, H6F, designed from an insulin-derived HLA-A*0201-restricted immunodominant CD8+ T cell epitope has a protective effect against T1D associated with the expansion of a rare population of immunosuppressive antigen-specific CD8+CD25+Foxp3+ Tregs in humanized NOD mice. Given that hInsB5-14 was an immunodominant target of autoreactive CD8+ T cells in HLA-A*0201-positive T1D patients42 and that more importantly, mIns1B5-14, on which the APL was based, differs from its human equivalent at position 5 but displayed an appreciable cross-reaction with hInsB5-14,7 we speculated that the APL H6F may be a promising candidate with potential clinical application value for antigen-specific prevention of T1D. However, whether the human equivalent of H6F would have similar properties to those of the mouse peptide used in our study remains an intriguing question that we will explore in our future studies.
Electronic supplementary material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31570931 and No. 31771002) and the National Key Project for Research & Development of China (Grant no. 2016YFA0502204).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
The online version of this article (10.1038/s41423-018-0058-3) contains supplementary material.
References
- 1.Zheng Y, Wang Z, Zhou Z. miRNAs: novel regulators of autoimmunity-mediated pancreatic β-cell destruction in type 1 diabetes. Cell Mol. Immunol. 2017;14:488–496. doi: 10.1038/cmi.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serreze DV, Leiter EH, Christianson GJ, Greiner D, Roopenian DC. Major histocompatibility complex class I-deficient NOD-β2mnull mice are diabetes and insulitis resistant. Diabetes. 1994;43:505–509. doi: 10.2337/diab.43.3.505. [DOI] [PubMed] [Google Scholar]
- 3.Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes. 1993;42:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- 4.Standifer NE, et al. Identification of Novel HLA-A*0201-restricted epitopes in recent-onset type 1 diabetic subjects and antibody-positive relatives. Diabetes. 2006;55:3061–3067. doi: 10.2337/db06-0066. [DOI] [PubMed] [Google Scholar]
- 5.Pinkse GG, et al. Autoreactive CD8 T cells associated with beta cell destruction in type 1 diabetes. Proc. Natl Acad. Sci. USA. 2005;102:18425–18430. doi: 10.1073/pnas.0508621102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takaki T, et al. HLA-A*0201-restricted T cells from humanized NOD mice recognize autoantigens of potential clinical relevance to type 1 diabetes. J. Immunol. 2006;176:3257–3265. doi: 10.4049/jimmunol.176.5.3257. [DOI] [PubMed] [Google Scholar]
- 7.Jarchum I, et al. In vivo cytotoxicity of insulin-specific CD8+ T-cells in HLA-A*0201 transgenic NOD mice. Diabetes. 2007;56:2551–2560. doi: 10.2337/db07-0332. [DOI] [PubMed] [Google Scholar]
- 8.von Herrath M. Immunology: insulin trigger for diabetes. Nature. 2005;435:151–152. doi: 10.1038/435151a. [DOI] [PubMed] [Google Scholar]
- 9.Degano M, et al. A functional hot spot for antigen recognition in a superagonist TCR/MHC complex. Immuntiy. 2000;12:251–261. doi: 10.1016/S1074-7613(00)80178-8. [DOI] [PubMed] [Google Scholar]
- 10.Han B, et al. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: high efficiency of a low-affinity peptide. Nat. Med. 2005;11:645–652. doi: 10.1038/nm1250. [DOI] [PubMed] [Google Scholar]
- 11.Hartemann-Heurtier A, et al. An altered self-peptide with superagonist activity blocks a CD8-mediated mouse model of type 1 diabetes. J. Immunol. 2004;172:915–922. doi: 10.4049/jimmunol.172.2.915. [DOI] [PubMed] [Google Scholar]
- 12.Tsai S, et al. Reversal of autoimmunity by boosting memory-like autoregulatory Tcells. Immuntiy. 2010;32:568–580. doi: 10.1016/j.immuni.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 13.Mallone R, et al. CD8+ T-cell responses identify beta-cell autoimmunity in human type 1 diabetes. Diabetes. 2007;56:613–621. doi: 10.2337/db06-1419. [DOI] [PubMed] [Google Scholar]
- 14.Jarchum I, Nichol L, Trucco M, Santamaria P, DiLorenzo TP. Identification of novel IGRP epitopes targeted in type 1 diabetes patients. Clin. Immunol. 2008;127:359–365. doi: 10.1016/j.clim.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, et al. Identification of autoreactive CD8+T cell responses targeting chromogranin A in humanized NOD mice and type 1 diabetes patients. Clin. Immunol. 2015;159:63–71. doi: 10.1016/j.clim.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 16.Niens M, et al. Prevention of “Humanized” diabetogenic CD8 T-cell responses in HLA-transgenic NOD mice by a multipeptide coupled-cell approach. Diabetes. 2011;60:1229–1236. doi: 10.2337/db10-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alleva DG, et al. Immunological characterization and therapeutic activity of an altered-peptide ligand, NBI-6024, based on the immunodominant type 1 diabetes autoantigen insulin B-chain (9-23) peptide. Diabetes. 2002;51:2126–2134. doi: 10.2337/diabetes.51.7.2126. [DOI] [PubMed] [Google Scholar]
- 18.Martinez NR, et al. Disabling an integral CTL epitope allows suppression of autoimmune diabetes by intranasal proinsulin peptide. J. Clin. Investig. 2003;111:1365–1371. doi: 10.1172/JCI200317166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ablamunits V, Elias D, Reshef T, Cohen IR. Islet T cells secreting IFN-gamma in NOD mouse diabetes: arrest by p277 peptide treatment. J. Autoimmun. 1998;11:73–81. doi: 10.1006/jaut.1997.0177. [DOI] [PubMed] [Google Scholar]
- 20.Hillig RC, et al. High-resolution structure of HLA-A*0201 in complex with a tumour-specific antigenic peptide encoded by the MAGE-A4 gene. J. Mol. Biol. 2001;310:1167–1176. doi: 10.1006/jmbi.2001.4816. [DOI] [PubMed] [Google Scholar]
- 21.Mou Z, et al. Identification of broadly conserved cross-species protective Leishmania antigen and its responding CD4+ T cells. Sci. Traasl Med. 2015;7:167r–310r. doi: 10.1126/scitranslmed.aac5477. [DOI] [PubMed] [Google Scholar]
- 22.Kollman PA, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 23.Zhang M, et al. Caffeic acid reduces cutaneous tumor necrosis factor alpha (TNF-alpha), IL-6 and IL-1beta levels and ameliorates skin edema in acute and chronic model of cutaneous inflammation in mice. Biol. Pharm. Bull. 2014;37:347–354. doi: 10.1248/bpb.b13-00459. [DOI] [PubMed] [Google Scholar]
- 24.Trembleau S, et al. Pancreas-infiltrating Th1 cells and diabetes develop in IL-12-deficient nonobese diabetic mice. J. Immunol. 1999;163:2960–2968. [PubMed] [Google Scholar]
- 25.Bercovici N, et al. Systemic administration of agonist peptide blocks the progression of spontaneous CD8-mediated autoimmune diabetes in transgenic mice without bystander damage. J. Immunol. 2000;165:202–210. doi: 10.4049/jimmunol.165.1.202. [DOI] [PubMed] [Google Scholar]
- 26.Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J. Exp. Med. 2011;208:1501–1510. doi: 10.1084/jem.20110574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fousteri G, et al. Subcutaneous insulin B:9-23/IFA immunisation induces Tregs that control late-stage prediabetes in NOD mice through IL-10 and IFNgamma. Diabetologia. 2010;53:1958–1970. doi: 10.1007/s00125-010-1777-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hjorth M, et al. GAD-alum treatment induces GAD65-specific CD4+CD25highFOXP3+ cells in type 1 diabetic patients. Clin. Immunol. 2011;138:117–126. doi: 10.1016/j.clim.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Petrich DML, et al. Functional inhibition related to structure of a highly potent insulin-specific CD8 T cell clone using altered peptide ligands. Eur. J. Immunol. 2008;38:240–249. doi: 10.1002/eji.200737762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomson CT, Kalergis AM, Sacchettini JC, Nathenson SG. A structural difference limited to one residue of the antigenic peptide can profoundly alter the biological outcome of the TCR-peptide/MHC class I interaction. J. Immunol. 2001;166:3994–3997. doi: 10.4049/jimmunol.166.6.3994. [DOI] [PubMed] [Google Scholar]
- 31.Bisikirska B, Colgan J, Luban J, Bluestone JA, Herold KC. TCR stimulation with modified anti-CD3 mAb expands CD8+ T cell population and induces CD8+CD25+ Tregs. J. Clin. Invest. 2005;115:2904–2913. doi: 10.1172/JCI23961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang P., Lu Q. Genetic and epigenetic influences on the loss of tolerance in autoimmunity. Cell. Mol. Immunol. 2018; in press. 10.1038/cmi.2017.137. [DOI] [PMC free article] [PubMed]
- 33.Wang R, et al. CD8+ regulatory T cells are responsible for GAD-IgG gene-transferred tolerance induction in NOD mice. Immunology. 2009;126:123–131. doi: 10.1111/j.1365-2567.2008.02884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Xie Q, Liang CL, Zeng Q, Dai Z. Chinese medicine Ginseng and Astragalus granules ameliorate autoimmune diabetes by upregulating both CD4+FoxP3+ and CD8+CD122+PD1+ regulatory T cells. Oncotarget. 2017;8:60201–60209. doi: 10.18632/oncotarget.18732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lange C, Scholl M, Melms A, Bischof F. CD62L(high) Treg cells with superior immunosuppressive properties accumulate within the CNS during remissions of EAE. Brain Behav. Immun. 2011;25:120–126. doi: 10.1016/j.bbi.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 36.Correale J, Villa A. Role of CD8+CD25+Foxp3+ regulatory T cells in multiple sclerosis. Ann. Neurol. 2010;67:625–638. doi: 10.1002/ana.21944. [DOI] [PubMed] [Google Scholar]
- 37.Fleissner D, Hansen W, Geffers R, Buer J, Westendorf AM. Local induction of immunosuppressive CD8+T cells in the gut-associated lymphoid tissues. PLos One. 2010;5:e15373. doi: 10.1371/journal.pone.0015373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bienvenu B, et al. Peripheral CD8+CD25+ T lymphocytes from MHC class II-deficient mice exhibit regulatory activity. J. Immunol. 2005;175:246–253. doi: 10.4049/jimmunol.175.1.246. [DOI] [PubMed] [Google Scholar]
- 39.Churlaud G, et al. Human and mouse CD8+CD25+FOXP3+ regulatory T cells at steady state and during interleukin-2 therapy. Front. Immunol. 2015;6:171. doi: 10.3389/fimmu.2015.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J. Immunol. 2009;183:4895–4903. doi: 10.4049/jimmunol.0901459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sauer S, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl Acad. Sci. USA. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Enée E, et al. Equivalent specificity of peripheral blood and islet-infiltrating CD8+ T lymphocytes in spontaneously diabetic HLA-A2 transgenic NOD mice. J. Immunol. 2008;180:5430–5438. doi: 10.4049/jimmunol.180.8.5430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.