

Abstract
The CBF signal pathway is responsible for a significant portion of plant responses to low temperature and freezing. Overexpression of CBF genes in model organisms such as Arabidopsis thaliana enhances abiotic stress tolerance but also reduces growth. In addition to these effects, overexpression of the peach (Prunus persica [L.] Batsch) CBF1 gene in transgenic apple (Malus x domestica Borkh.) line T166 also results in early entry into and late exit from dormancy. Although the regulation of dormancy-induction and dormancy-release occur while the CBF regulon is operative in perennial, woody plants, how overexpression of CBF1 affects these dormancy-related changes in gene expression is incompletely understood. The objective of the present study was to characterize global changes in gene expression in peach CBF1-overexpressing and non-transformed apple bark tissues at different states of dormancy via RNA-seq. RNA-seq bioinformatics data was confirmed by RT-qPCR on a number of genes. Results indicate that the greatest number of significantly differentially expressed genes (DEGs) occurred in April when dormancy release and bud break normally occur but are delayed in Line T166. Genes involved in storage and inactivation of auxin, GA, and cytokinin were generally upregulated in T166 in April, while those for biosynthesis, uptake or signal transduction were generally downregulated in T166. Genes for cell division and cambial growth were also downregulated in T166 relative to the non-transformed line. These data suggest that overexpression of the peach CBF1 gene impacts growth hormone homeostasis and as a result the activation of growth in the spring, and most likely growth cessation in the fall as well.
Subject terms: Plant molecular biology, Abiotic
Introduction
The domesticated apple, Malus × domestica Borkh., is a member of the family Rosaceae, tribe Pyreae, and is grown world-wide. Over 100 varieties are grown commercially in the United States, with an annual average of 240 million bushels of apples worth close to $4 billion (wholesale) (http://usapple.org/). Global production of apples for 2016/2017 exceeded 77 million metric tons (https://www.statista.com/statistics/279555/global-top-apple-producing-countries/).
Erratic weather patterns, as a symptom of climate change, pose an increasing threat to industry profitability. Data from the USDA Risk Management Agency for the years 2007–2017 indicates that apple growers claimed $157,177,390 in insured losses from freeze damage (https://www.rma.usda.gov/informationtools/). The majority of this damage occurred in the spring, when abnormally warm temperatures were followed by freezing temperatures1. For example, spring frost events in 2007 and 2017 each resulted in $1 billion in losses from all crops2 (https://www.ncdc.noaa.gov/billions/events/US/1980-2017). During these events, unseasonably warm temperatures in early spring (late February–March) resulted in the deacclimation of ecodormant buds and early bud break in many temperate, perennial fruit crops. The occurrence of subsequent freezing temperatures several weeks later (April) when flowers and early vegetative growth with little freezing tolerance were present resulted in high levels of frost injury3.
While dormancy and cold hardiness in perennial, woody plants are closely related, they each exhibit distinct regulatory aspects and phenology. Dormancy progresses seasonally through several phases regulated by both intrinsic (paradormancy and endodormancy) and extrinsic (ecodormancy) factors4. While growth during paradormancy and endodormancy is arrested by specific endogenous signals from within the plant, regulation of growth during ecodormancy is regulated by temperature and/or day length. Collectively, entry into and exit from dormancy represents a dynamic process involving both hereditary and epigenetic regulated changes in gene expression5–8.
Cold acclimation, the ability to tolerate freezing temperatures, is a complex and dynamic process. Low temperature survival depends on a combination of biophysical and biochemical factors, often driven by the upregulation or downregulation of specific sets of genes9–12. Among several other factors, research on cold acclimation has identified several COld-Responsive (COR) proteins, such as dehydrins, and transcription factors that control the expression of suites of COR genes, often referred to as a cold regulon. CBF/DREB transcription factors are integral to responses to low temperature and drought and are responsible for approximately 12–20% of cold-induced transcriptional changes in Arabidopsis13,14.
CBFs/DREBs are members of the AP2/ERF family of transcription factors that bind to a promoter motif consisting of A/GCCGAC, commonly referred to as the C-repeat or Drought Response Element. CBF genes have been identified in all investigated higher angiosperm species, including woody perennials. All CBF amino acid sequences feature common motifs in addition to the characteristic AP2 DNA binding domain13. The number of CBF genes in a genome varies from species to species, with some examples displaying the conserved motifs but not actually participating in low temperature responses. Apple and peach (Prunus persica [L.] Batsch) have five and six CBF genes, respectively11. The importance of CBF genes to cold hardiness has been demonstrated repeatedly in different plant systems1. Regulation of CBF genes is complex in Arabidopsis, with a variety of kinases, transcription factors, light- and photoperiod-related proteins, hormones, and degradation factors such as ubiquitin potentially playing roles15. Similar genes and proteins are found in apple, peach and other woody, perennial species suggesting that their CBF genes are regulated in a similar fashion1,11.
Wisniewski et al.16 reported that over-expression of a peach CBF gene (PpCBF1) in apple results in enhanced freezing tolerance, growth reduction, early onset of dormancy triggered by short days, and delayed bud break in the spring. The response to short days was especially novel, since growth cessation in apple is typically not impacted by short days17. These phenotypes were confirmed in a field planting maintained for three years18. Wisniewski et al.19 provided a model that suggested that CBF regulated cold acclimation, dormancy, and growth through COR, DAM, and RGL genes, respectively. It was also suggested that overexpression of PpCBF1 regulated the expression of an apple Early Bud Break gene (EBB) that had been shown to regulate bud break in poplar20.
The growth-related and dormancy-related genes examined by Wisniewski et al.19, however, represent only a small number of the potential genes whose expression may be altered directly or indirectly by over-expression of the peach CBF gene in apple. Understanding complex pleiotropic effects requires studies of global gene transcription over time. In the current study, field samples of apple bark tissues were collected over several months from non-transgenic “M.26” apple trees and transgenic “M.26” apple trees overexpressing a peach CBF (PpCBF1) gene to provide a comprehensive overview of the differentially expressed genes (DEGs) associated with the phenotypes of these two genotypes from Ecodormancy (February, March, and April) to active growth (July), with an emphasis during bud break (April).
Results
High-throughput sequencing resulted in 191.78 million high-quality, single-end reads (Table 1). An average of 89% of the clean reads were successfully aligned to the apple reference genome21, and an average of 31,483 genes or protein coding transcripts were identified between both genotypes and over all timepoints. This represents approximately 54.9% of the total predicted transcripts in the apple reference genome v1.0. Log2-transformation of the counts display similar distributions between the samples (Supplemental Fig. 1).
Table 1.
Total RNA-seq reads from the T166 and “M.26” bark tissue samples
| Sample | Genotype | Month | Replicate | Reads | Overall alignment (%) | Genes identified |
|---|---|---|---|---|---|---|
| 1 | M26 | Feb | 1 | 10,788,833 | 89.74 | 31,982 |
| 2 | M26 | Feb | 2 | 8,194,111 | 90.65 | 30,761 |
| 3 | M26 | Feb | 3 | 6,576,203 | 88.92 | 29,834 |
| 4 | M26 | Mar | 1 | 7,113,401 | 89.29 | 29,874 |
| 5 | M26 | Mar | 2 | 8,381,946 | 89.52 | 30,608 |
| 6 | M26 | Mar | 3 | 8,751,294 | 90.12 | 30,712 |
| 7 | M26 | Apr | 1 | 7,564,861 | 89.00 | 33,168 |
| 8 | M26 | Apr | 2 | 8,398,890 | 89.18 | 33,147 |
| 9 | M26 | Apr | 3 | 7,597,188 | 89.04 | 32,173 |
| 10 | M26 | Jul | 1 | 8,013,981 | 88.03 | 33,558 |
| 11 | M26 | Jul | 2 | 8,391,855 | 90.87 | 34,167 |
| 12 | M26 | Jul | 3 | 8,111,513 | 90.75 | 33,732 |
| 13 | T166 | Feb | 1 | 7,497,785 | 88.67 | 29,546 |
| 14 | T166 | Feb | 2 | 8,400,121 | 89.25 | 30,119 |
| 15 | T166 | Feb | 3 | 7,955,107 | 88.86 | 29,948 |
| 16 | T166 | Mar | 1 | 8,862,755 | 88.44 | 30,172 |
| 17 | T166 | Mar | 2 | 7,300,094 | 88.24 | 29,207 |
| 18 | T166 | Mar | 3 | 7,088,792 | 88.60 | 28,988 |
| 19 | T166 | Apr | 1 | 8,302,823 | 88.03 | 31,814 |
| 20 | T166 | Apr | 2 | 6,878,138 | 86.27 | 30,711 |
| 21 | T166 | Apr | 3 | 7,411,775 | 86.34 | 31,294 |
| 22 | T166 | Jul | 1 | 8,358,582 | 89.16 | 33,605 |
| 23 | T166 | Jul | 2 | 8,225,370 | 89.40 | 33,327 |
| 24 | T166 | Jul | 3 | 7,617,277 | 88.91 | 33,149 |
| Total | NA | NA | NA | 191,782,695 | 89.02 | 31,483 |
Each genotype had three biological replicates per timepoint
Overall analysis of differentially upregulated and downregulated genes based on the experimental variables: (1) comparisons between the genotypes (transgenic vs. non-transgenic) at each of the sampled months; (2) genes that were differentially expressed over time within a genotype; (3) genes that were differentially expressed in one sampling point (month) to the next sampling point, and; (4) genes that were differentially expressed based on an interaction between genotype and time. Upregulation and downregulation was based on log2 fold-change >1 and log2 fold-change <−1, respectively, for genes with adjusted p-value < 0.05.
The four comparisons between the experimental variables provide different types of information. Comparison (1), “M.26” vs. T166 by month, gives a direct view of the differences in gene expression between the two genotypes at specific timepoints. The number of DEGs in the transgenic genotype (T166) during the latter months of winter (February and March) were similar, averaging about 1400 genes. Notably, there were a much greater number of downregulated genes in the February samples than in the March samples. The greatest number of DEGs between the two genotypes occurred in the April sampling with over 4000 DEGs. This result indicates that significant differences in gene expression between the transgenic (T166) and non-transgenic (“M.26”) exist at the time bud break was beginning in the “M.26” trees but not in the T166 trees (Fig. 1).
Fig. 1. Bud break data for field-grown M.26 and T166 trees in 2013.
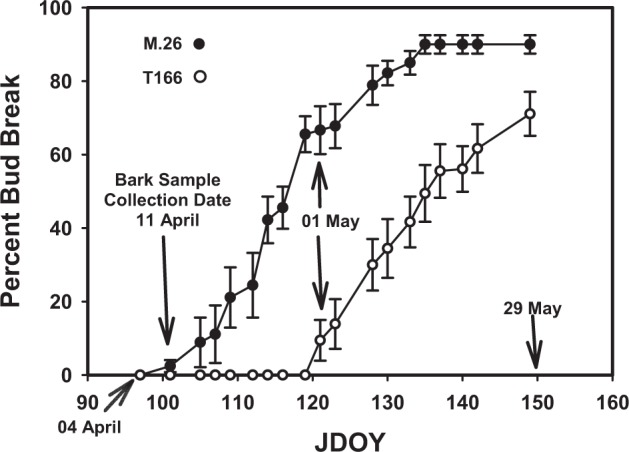
JDOY is Julian Day of Year. Filled symbols, M.26. Open symbols, T166. Three shoots on each of three trees of M.26 and T166 were tagged and bud break from 20 individual lateral buds from the terminal bud were tracked. Percent bud break is mean ± s.d., n = 60
Comparison (2) indicates the effect of time on the expression level of genes within each genotype. Genes being identified as DEGs would be any that have expression levels that significantly changed (up or down) over time. Overall, the number of differentially expressed genes were similar in “M.26” (11,469) and T166 (11,097). This does not indicate similar expression patterns over time, only that a similar number of genes exhibited significant changes in expression over the sampled period (Feb–July).
Comparison (3) provides information on changes in gene expression from one sampling timepoint to the next within each genotype. One of the notable differences in this set of comparisons is in the number of DEGs in the Feb–Mar comparison for each genotype. The non-transgenic “M.26” genotype exhibited a much greater number of differentially expressed genes (2813) in this monthly comparison than the transgenic T166 genotype (530), indicating that “M.26” was undergoing a greater shift in gene expression than T166 in the late winter to early spring.
Comparison (4) indicates the number of genes who expression was significantly affected by the interaction between genotype and time. Genes identified as being DEGs in this comparison are those whose expression was significantly different between the two genotypes over the time course of sampling (Feb–Jul). These genes would be the most appropriate for further investigation, as their expression differs between the “M.26” and T166 genotypes over time.
The data depicted in comparison 1 (Table 2) can also be depicted by Principal Coordinate Analysis (PCoA, Fig. 2a), a correlation matrix (Fig. 2b), and a sample distance matrix (Fig. 2c). A PCoA analysis of DEGs in “M.26” vs. T166 was conducted using Qlucore software (v. 3.2). As illustrated in Fig. 2a, gene expression in T166 (yellow dots) clustered together in February and March and more distantly with the “M.26” samples (blue dots) from February and March. In fact, a finer grouping of the clusters indicates that both the “M.26” and T166 samples in February and March cluster separately from each other. The separation of “M.26” and T166 is most evident in the April samples where the two genotypes clustered independent of each other while the individual biological replicates within each genotype clustered together. Both genotypes clustered together with each other in the July samples, indicating that there were only minor differences in gene expression between the two groups.
Table 2.
Upregulated and downregulated gene count comparisons of T66 vs. “M.26”
| Comparison | Upregulated | Downregulated | Total | ||
|---|---|---|---|---|---|
| (1) M26 vs. T166 | February | 498 | 1189 | 1687 | |
| March | 734 | 384 | 1118 | ||
| April | 1834 | 2177 | 4011 | ||
| July | 146 | 56 | 202 | ||
| (2) Time main effect | M26 | 7075 | 4394 | 11,469 | |
| T166 | 7090 | 4007 | 11,097 | ||
| (3) Month-to-Month | M26 | Feb–Mar | 1004 | 1809 | 2813 |
| Mar–Apr | 3399 | 2336 | 5735 | ||
| Apr–Jul | 3906 | 2043 | 5949 | ||
| T166 | Feb–Mar | 255 | 275 | 530 | |
| Mar–Apr | 3096 | 2419 | 5515 | ||
| Apr–Jul | 5918 | 4225 | 10,143 | ||
| (4) Time–strain interaction | 1888 | 895 | 2783 | ||
Comparisons include genotype–genotype, time main effect, month-to-month by genotype, and time–genotype interaction comparisons. Upregulated and downregulated gene counts are provided based on log2 fold-change >1 and log2 fold-change p-value < 0.05
Fig. 2. Graphical representations of Comparison 1 (genotype by month) data.
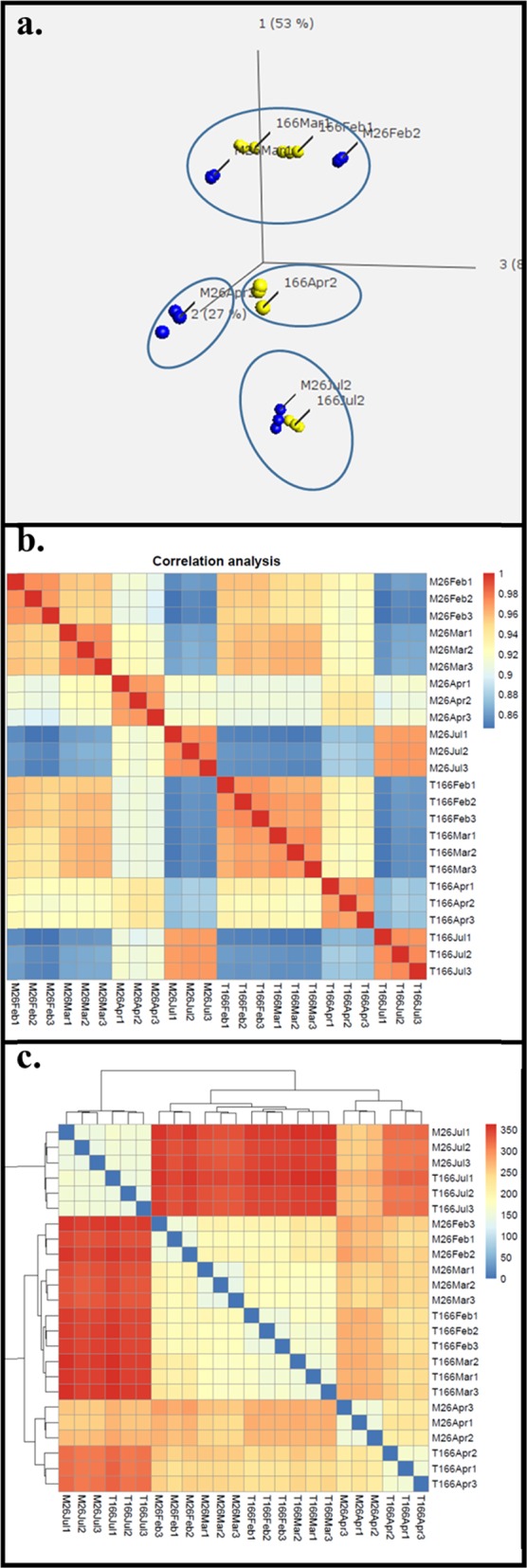
a PCoA analysis. b Correlation-matrix: results of the correlation analysis displayed in matrix format. Each row and each column represents a single sample. Colored cells indicate the correlation value between the row and column sample based on the read count for each gene. Blue colors indicate lower correlation, and red colors indicate higher correlation. c Sample-distances: a sample distance matrix with accompanying clustering for each sample. Distances are calculated using a Euclidean distance, with larger distances (red) indicating more dissimilarity between the two samples in terms of genetic expression. Lower distances (blue) indicate more similar gene expression patterns, with identical expression patterns being represented by darker blue
The correlation matrix (Fig. 2b) provides a picture of the similarity of the samples, with the color spectrum indicating how similar/dissimilar each sample is to another. The darker the red, the more directly similar the samples are, with the darkest red indicating a Pearson correlation of 1. Conversely, the blue cells indicate samples that are very dissimilar. In general, the results confirm the results obtained in the PCoA. For example, July “M.26” samples are similar to July T166 samples and both are highly dissimilar to February and March samples of both genotypes. Furthermore, April T166 and “M.26” samples are only slightly similar to each other, as was evident in the PCoA. The sample distance matrix figure (Fig. 2c) is comparable to the correlation matrix in that it shows how similar any two samples are. The distance metric used here is a basic Euclidean distance. Red indicates a more dissimilar pair of samples and blue indicates a more similar value. The main difference between the analyses in Fig. 2a–c is the metric used to determine similarity. Similar trends emerge, however, in each figure. Gene expression in “M.26” and T166 is most similar in July, less so in February and March, and most dissimilar in April. These data suggest that overexpression of PpCBF1 has many pleiotropic effects during winter and early spring, and these downstream effects are minimized during summer.
How “M.26” and T166 compare in terms of upregulation and downregulation of gene expression is important to understanding the differences in dormancy and freezing tolerance in T166 relative to “M.26”. A complex pattern of differential gene expression was evident, with numerous genes being upregulated or downregulated in T166 trees relative to “M.26” trees (Fig. 3). The greatest disparity in the number of significantly upregulated and downregulated DEGs between T166 and M.26 occurred in February, followed by April. The number of significantly upregulated DEGs greatly exceeded the number of downregulated DEGs in those months. Conversely, the number of significantly downregulated DEGs exceeded the number of upregulated DEGs in March and July. The low number of DEGs in either direction in July suggests that few differences exists between T166 and “M.26” trees at that time of year.
Fig. 3. DGE-overview.
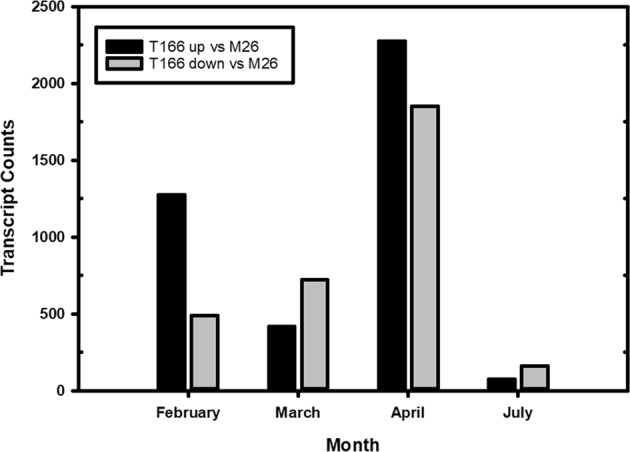
Bar plots showing the number of upregulated and downregulated differentially expressed genes for the monthly comparison between T166 and “M.26” genotypes. The black bars represent the upregulated genes, i.e., genes with an adjusted p-value ≤ 0.05 and log fold-change ≥ 1. The grey bars represent the number of downregulated genes, i.e., genes with an adjusted p-value ≤ 0.05 and log fold-change ≤ −1. Upregulation and downregulation of genes is based on the change from T166 to “M.26”. Upregulated would indicate T166 has a higher average expression than “M.26” and lower for downregulated
The Venn diagrams (Fig. 4) of month to month differences in the number of unique DEGs within each genotype indicate important differences in the level of gene expression that may reflect differences in the timing at which growth was activated in the two genotypes. There were only a relatively small number of DEGs (179) in the Feb–March comparison in T166, while the “M.26” genotype exhibited 1789 unique DEGs in the same comparison. Such a high number of unique DEGs was not observed in the T166 trees until the comparison between Mar and April samples, suggesting that the activation of growth, as reflected in levels of DEGs, occurred later in T166 than it did in “M.26” trees.
Fig. 4.

Venn diagrams of the changes in differentially expressed genes in T166 and “M.26” over time. T166 lags behind “M.26” gene expression changes in late winter—spring
Eight genes were chosen to validate the model by RT-qPCR (Fig. 5). The relative expression levels determined by RT-qPCR over time are comparable with the raw RNA-seq reads for the same samples. These trends can be compared to Base Mean data in Table 3. The base mean represents the average expression of that gene over both genotypes at a particular timepoint. The log2 fold change is then calculated to assess difference of individual genotype means from the base mean. The Wald test approach, which performs a parametric significance test of the selected factor level uses a negative binomial distribution. Significant p-values result from the factor being determined as significant in the Wald test. A positive log2 fold change indicates highly differential expression of the gene in T166 over that seen in “M.26”, while a negative log2 fold change indicates the converse—highly differential expression of the gene in T166 lower than that seen in “M.26”. The trends seen in Table 3 correspond to that seen in Fig. 4, thus validating the overall model for calling highly differentially expressed genes.
Fig. 5. Model validation of the bioinformatics pipeline, with RT-qPCR and raw reads.

RT-qPCR (top panels of each lettered pair) was conducted on selected genes to validate the results obtained by the RNA-Seq (bottom panels of each lettered pair). Solid circles, “M.26”. Open circles, T166. a RuBisCO large subunit-binding protein subunit alpha. b Dehydrin8 (MdDHN8). c Dehydrin4 (MdDHN4). d Dormancy-associated MADS-box1 (MdDAM1). e Universal stress protein A. f Early light-induced protein (ELIP). g α-1,4 glucan phosphorylase L isozyme. h Probable phosphatase phosphor 2. Means ± sd. n = 9 for RT-qPCR (3 biological replicates x 3 technical replicates; n = 3 for raw reads (3 biological replicates). Relative abundance for RT-qPCR graphs is abundance relative to the LTL1 endogenous reference gene, deemed as most stable across time by the NormFinder software71
Table 3.
Base mean and Log2 fold change data of genes used in RT-qPCR bioinformatics pipeline validation
| Gene description | MDP model | Feb BM | Feb l2FC | Mar BM | Mar l2FC | Apr BM | Apr l2FC | Jul BM | Jul l2FC |
|---|---|---|---|---|---|---|---|---|---|
| Universal stress protein A-like protein | MDP0000688187 | 3319.822 | 0.432 | 2575.369 | 0.817 | 3196.586 | 1.965 | ||
| MdDHN4 | MDP0000360414 | 11,386.714 | 0.489 | 12,444.817 | 1.591 | 1064.200 | 2.117 | 5807.932 | 1.405 |
| MADS-box protein AGL24 | MDP0000322567 | 2980.377 | 0.318 | 2356.479 | 0.427 | 659.739 | 0.561 | 780.211 | −0.714 |
| Alpha-1,4 glucan phosphorylase L isozyme | MDP0000129346 | 1487.964 | 0.298 | 3354.163 | −0.880 | 1768.988 | −1.603 | ||
| MdDHN8/ COR47 | MDP0000529003 | 814.447 | 0.445 | 604.875 | −0.761 | 2615.074 | 0.837 | ||
| RuBisCO large subunit-binding protein subunit alpha | MDP0000631455 | 839.777 | 0.540 | 1271.574 | 0.271 | 1294.958 | 0.320 | 1136.768 | −0.320 |
| Early light-induced protein, chloroplastic | MDP0000182592 | 35,671.891 | 0.523 | 54,225.772 | 0.470 | 3232.784 | 2.359 | ||
| Probable phosphatase phospho2 | MDP0000404331 | 178.894 | 1.257 | 143.378 | 2.306 | 301.816 | 3.230 |
The data can be compared to those presented in Fig. 5. BM, base mean, which represents the average expression of that gene over both genotypes. L2FC, log2 fold change of T166 relative to “M.26”. Missing data indicates that the gene was not significantly differentially expressed between T166 and “M.26”
Constitutive overexpression of the peach CBF1 gene in T166 would be expected to result in the upregulation of downstream, stress-associated DEGs. In fact, a large number Dehydrin genes were highly upregulated in T166 vs. “M.26” in all months (Supplemental Table 2, Supplemental Figs. 2–4). Additionally, several LEA homologs, salt tolerance genes, two superoxide dismutase (SOD) genes, a cold acclimation WCOR413-like gene, and eleven out of twelve genes encoding a Universal Stress Protein domain were also upregulated in T166 compared to “M.26” in April. Two Defender Against Cell Death genes, involved in programmed cell death (PCD), were slightly upregulated in T166 relative to “M.26” in April. Sixty heat shock proteins (HSPs), HSP cognates, or heat stress transcription factors were identified among the expressed genes. Most of the small HSP genes were slightly to strongly upregulated in T166, while the majority of large HSP genes were downregulated in April. The heat-stress transcription factors were also largely upregulated in April. Numerous genes associated with redox reactions or responses to reactive oxygen species (ROS) were upregulated in T166, except for ascorbate oxidases, which were all downregulated in T166 relative to “M.26”. A majority of genes associated with biotic stress were downregulated in T166 relative to “M.26” in April. Eight of nine wound-induced or wound-responsive genes were upregulated, along with two defense-related genes in T166 compared to “M.26” in April. In like manner, seven probable WRKY transcription factors were also upregulated in T166 relative to “M.26” in April. In contrast, pathogenesis-related (PR) group 5 genes encoding thaumatin-like and osmotin-like proteins were largely downregulated. Thirteen Senescence Associated Genes (SAGs) were found to be highly upregulated in T166 relative to “M.26” in April. Twelve disease resistance genes were also identified that exhibited patterns of both upregulation and downregulation. Native CBF genes were not significantly differentially expressed in the April samples of T166 compared to “M.26”.
Even though the greatest disparity in the number significantly upregulated and downregulated DEGs between T166 and M.26 was observed in February, April samples are of particular importance due to the fact that “M.26” begins to exhibit bud break at that time, while buds inT166 trees are still ecodormant (Fig. 1)18. Most of the genes coding for cell division cycle proteins were downregulated in T166 compared to “M.26” in the April samples, including cyclins, CDKs, and all G2/ mitotic-specific cyclins (Supplemental Table 3; Supplemental Figs. 2 and 3). Nearly all the identified kinesin genes were downregulated as was an LFR (LEAF AND FLOWER RELATED) gene involved in leaf and flower development. Eleven genes encoding Expansins were all found to be downregulated in T166 compared to “M.26”.
Only a limited number of signal transduction pathway genes for growth-associated hormones (auxins, cytokinins, and gibberellins) were differentially expressed in April, mainly auxin-related and GA-related (Supplemental Table 4), and these were largely downregulated in T166 relative to “M.26”, including auxin influx and efflux carrier genes. In contrast, genes for inactivation by glycosylation or storage pathways were upregulated. These data are consistent with ecodormancy and the lack of bud break or growth observed in T166 trees compared to “M.26” trees16,18,19.
Abscisic acid (ABA) and ethylene, two inhibitory or senescence-related hormones, would be expected to perhaps exhibit some degree of upregulation in the T166 genotype relative to the “M.26” genotype given that bud break is not readily evident in T166 trees in April. In particular, biosynthetic or signal transduction pathways for these hormones might be upregulated in T166 relative to “M.26”. Instead, the pattern of gene expression for these pathways was inconsistent for both hormones (Supplemental Table 5). This includes genes encoding Phospholipase D alpha 1 and Phospholipase D delta 1, which are involved in signal transduction pathways for both ABA and ethylene.
Numerous genes, other than those associated with hormone biosynthetic or signal transduction pathways, are associated with dormancy and growth. Therefore, the expression of genes reported in models for bud break, vernalization, and floral initiation were examined, recognizing that these processes may differ in herbaceous and woody perennials. The upregulation and downregulation of different light signal transduction pathway genes varied in T166 relative to “M.26” (Supplemental Table 6; Supplemental Figs. 2 and 3). Two PHYB genes exhibited contrasting patterns of expression, while both PHYC genes were upregulated in T166 relative to “M.26”. In contrast, a PIF1 homolog and two PIF3 homologs were all downregulated in T166 compared to “M.26”. Circadian clock genes (REVILLE-like, TIC, XAP5 CIRCADIAN TIMEKEEPER, PFT1, and LHY) had no consistent upregulated or downregulated expression trends in T166 relative to “M.26”. Similarly, genes identified with the autonomous and vernalization pathways were not uniformly differentially upregulated or downregulated in T166 relative to “M.26”. Partial homologs of the target of these pathways, FLC, exist in apple. Differentially expressed Agamous-like (AGL) MADS-box genes were identified, one of which was identified as FLC-like. The FLC-like gene was differentially downregulated in April in T166 relative to “M.26”. Two of these MADS-box/ AGL DEGs were previously identified as Dormancy Associated MADS-box (DAM) homologs, MdDAM1 and MdDAM219. MdDAM1 was highly upregulated in T166 compared to “M.26” in early spring, but down-regulated in summer. In contrast, MdDAM2 was differentially expressed only during bud break, again in T166 relative to “M.26”.
Vegetative or vascular growth initiation genes, such as SOC, and ANT/ AIL1, were found to be highly differentially expressed, with SOC upregulated and ANT/AIL1 downregulated between T166 and “M.26”, respectively (Supplemental Table 6; Supplemental Figs. 2 and 3), but in opposite directions. Four CONSTANS (CO)—like homologs were upregulated in T166 compared to “M.26”, but all the gene sequences were poor matches to either the Arabidopsis or Populus versions. Nine Squamosa promoter-binding-like (SPL) protein genes were highly differentially expressed, with only one being upregulated in T166 relative to “M.26”. Five CLAVATA1 genes, associated with meristem and floral initiation in Arabidopsis, were downregulated in T166 vs. “M.26”. No evidence of differential expression patterns was observed for FT and TFL. These genes are typically expressed in buds22,23, so it is not surprising that expression, differential or otherwise, was not detected since bark tissues were examined in the current study. The gene encoding the PXY receptor, associated with cambial cell division24, was strongly downregulated in T166 relative to “M.26” in both Feb and April. In contrast, several knotted-1-like (KNAP) genes, also involved with cambial cell division, were upregulated in T166 relative to “M.26”.
The expression of genes associated with the plasmodesmata (PD) hypothesis of dormancy regulation25 were also examined. This hypothesis suggests that PD sphincters and their degradation regulate the onset and release of dormancy in buds (Supplemental Table 7). All callose synthase genes and most Glucan endo-1,3-beta-glucosidase (glucan hydrolase group 17) genes were downregulated in T166 relative to M.26. Five remorin family genes were detected, with a heterogeneous expression pattern.
Discussion
The role of CBF transcription factors in low temperature/freezing and water deficit responses has been well documented in a variety of plant families and genera, including woody perennials1,13,26,27. Numerous abiotic-stress-responsive genes possessing the canonical A/GCCAG core motif in their promoters have been shown to be downstream targets of CBF binding28,29. Studies in plants overexpressing CBF genes have demonstrated increased survivability under artificial and natural conditions of freezing or water limitation11,13,26,27. These studies reported an improvement of −2 to −5 °C in freezing tolerance depending on the plant system. Wisniewski et al.16 reported increases of −3 to −4 °C in both non-cold-acclimated and cold-acclimated greenhouse-grown apple trees overexpressing the peach CBF1 gene (PpCBF1, line T166). This effect was later confirmed in field studies, where non-acclimated trees in mid-summer had similar increases in freezing tolerance18. Artlip et al.30 reported that the effect on cold hardiness was not graft transmissible through a transgenic T166 rootstock, but juvenility (time to flowering) was affected in non-transgenic scions grafted to transgenic T166 rootstock.
Plant processes other than stress tolerance have been reported to be improved by CBF overexpression. Constitutive overexpression of CBF genes has been reported to result in diminished growth in both herbaceous and woody plant systems, including apple16,18,19. Overexpression of peach CBF1 in apple also affects entry into and exit from dormancy. Wisniewski et al.16, Artlip et al.18, and Wisniewski et al.19 demonstrated that apple gained a novel sensitivity to short day (SD) photoperiods, manifested by early leaf senescence and bud set. Several genes known to participate in dormancy processes were examined by Wisniewski et al.19. They reported a correlation between gene expression patterns in spring and the bud break patterns exhibited by M.26 and T166. While Wisniewski et al.19 provided information on the expression of a few genes related to growth and dormancy, a more complete analysis is provided in the present study. This information provides a more comprehensive picture of how the peach CBF1 gene affects so many physiological processes and pathways, especially those related to dormancy and growth. Samples taken from trees grown in the field provides an excellent measure of the impact of CBF proteins in a natural setting.
Based on the DEGs identified in the present study it appears that the overexpression of CBF in apple impacts gene expression in a complex manner that potentially impacts numerous different processes and pathways. The RNA-seq analysis conducted in the present study detected approximately 46,500 genes constituting ≈73% of the predicted apple genome (Table 1). While there is considerable overlap of the genes being expressed between T166 and “M.26” during any given month (Supplemental Figs. 5–12), a significant level of difference was also detected (Figs. 3, 4, 5). PCoA indicated that gene expression in the July samples of the two genotypes was much more similar to each other than it was to gene expression in other months. It is likely that the effect of PpCBF1 overexpression on gene expression is at a minimum during the summer months when tree growth and general metabolism are extremely active. Similarly, clustering or the relatedness of Feb and Mar samples for both genotypes, when growth processes are at a near minimum, were somewhat similar to each other and are distinctly separated from the Apr samples of both genotypes.
The Feb–Mar–Apr months, encompassing late winter and early spring, are a time of transition for perennial, woody plants in the northern hemisphere, as during that time they overcome ecodormancy and undergo bud break. A combination of photoperiod and temperature typically fosters extensive changes in gene expression in most perennial woody plants1,6,31,32. In contrast, apple (and pear) depend solely on temperature to induce entry and release from ecodormancy17. The novel sensitivity to short days reported by Wisniewski et al.16, Artlip et al.18, Wisniewski et al.19, and Artlip et al.30 are reflected in the DEGs that were identified in the current study.
Several types of stress-related protein genes were examined to determine if they were upregulated by overexpression of PpCBF1. Dehydrin and Late Embyrogenesis Abundant (LEA) genes were upregulated in T166, relative to the non-transgenic parent genotype “M.26”, as has been previously documented in other species. These genes typically contain a C-repeat/DRE binding site for CBF/DREBs and are recognized as targets of CBF/DREB proteins. The results of the RNA-seq analysis agree with Wisniewski et al.16,19. DREB2A genes were also upregulated in T166, along with the CBF/ DREB1 Tiny gene. It is possible that the latter genes may respond to multiple cues or have C-repeat/DREB sites in their promoters. Notably, no native CBF genes were differentially expressed, despite the presence of C-repeats in the promoters of MdCBF1, 2, 3, and 519. Other factors, however, also play a role in CBF expression and regulation1,13,15, and it is possible that regulation of apple CBF genes may differ in regard to the Arabidopsis model. Indeed, few of the MdCBFx promoters contain regulatory motifs consistent with Shi et al.15 or Wisniewski et al.1. Additional research is clearly required on how native MdCBFx expression is regulated.
Many additional stress proteins were identified as being up-regulated in T166. Both small (<30 kDa) and large (>70 kDa) Heat Shock Protein (HSP) genes were consistently up-regulated in T166 in Feb and Mar. HSPs are known to be induced by low temperatures33, and so PpCBF1 overexpression may have contributed to the observed upregulation. In April samples of T166 the upregulation of small HSPs was maintained while large HSPs were largely downregulated. Concurrently, several HSP transcription factors were also upregulated in April samples, suggesting that these may primarily regulate small HSP gene expression. Numerous genes encoding proteins with a bacterial Universal Stress Protein motif were also differentially expressed, generally upregulated in T166 compared to “M.26”. As noted by Kerk et al.34, the functionality of these domains in Arabidopsis thaliana is uncertain. Thus, additional research is needed to discern what the functions of the identified proteins are in apple bark. Genes associated with redox or ROS homeostasis were inconsistent in overall trends, perhaps reflecting the relatively dormant state of T166. Transcripts for osmotin-like and thaumatin-like genes were decidedly downregulated in T166 relative to ‘M.26’ in the monthly samples. These ABA-responsive and wound-responsive proteins are members of the pathogenesis-related 5 (PR5) group of pathogen-response proteins33. Notably, many of other wound-induced and pathogen-induced genes were upregulated in T166, suggesting a complex interplay between CBF and signal transduction pathways responsible for the expression of defense-related genes.
Comparisons between T166 and “M.26” made in the current study indicate that large differences in gene expression exist at the time bud break begins to occur in “M.26” but not T166. Differences in the timing of bud break were also noted in Artlip et al.18, who reported that bud break in “M.26” was evident in about 5% trees at the time (April) the samples were taken for the RNA-seq study. In contrast bud break in T166 trees was not readily apparent until after an additional two weeks (see also Fig. 1, this study).
Wisniewski et al.19 examined a limited suite of genes associated with growth and dormancy (RGL/ DELLA genes, MdDAMs 1-3 and MdEBB1) in T166 and “M.26” trees and determined that some temporal differences in the level of expression of these genes were evident in these two genotypes. In the present study, a more comprehensive analysis of gene expression was conducted that could serve as a basis for understanding the effect of CBF overexpression on freezing tolerance and dormancy, as well as several other non-target parameters, such as growth and flowering.
Plant growth regulator (PGR) levels impact every aspect of plant growth and development, including dormancy. Growth-promoting PGRs such as auxins, gibberellins, and cytokinins have been reported to be affected by CBF overexpression35–37. Genes responsible for the storage or catabolism of auxins were largely upregulated in April samples of T166 compared to “M.26”, suggesting that auxin levels were low and thus may have inhibited the onset of growth in T166. Indeed, cambial transcriptomic and cambial cell dynamic studies in aspen strongly suggest that auxin transport and perception are vital to transitions from growth to dormancy and the converse5,7,29,38.
GA biosynthetic-enzyme encoded genes such as ent-kaurene oxidases are noticeably upregulated in T166 relative to “M.26”, however, several GA2 oxidase (catabolic) genes were also upregulated in T166 in Apr. Achard et al.35, Suo et al.37, and Niu et al.36 reported on changes in the expression levels of GA-biosynthetic and deactivating genes in plants over-expressing CBF genes in Arabidopsis thaliana, soybean (Glycine max), and tobacco (Nicotiana tabacum), respectively. Suo et al.37 and Niu et al.36 reported decreased GA levels in CBF-overexpressing plants. Low levels of GA stabilize growth-inhibiting RGL/DELLA proteins39, which can be exacerbated by CBF overexpression35. Transcripts from four RGL/ DELLA genes were detected, primarily in April, but were slightly downregulated in T166 relative to “M.26”. The expression of two of these genes, RGL1a and 1b, was previously characterized by Wisniewski et al.19, who also found that expression level of these genes in April was not substantially different between T166 and “M.26”. Wisniewski et al.19, however, did find differences during other months, suggesting that those months may be critical to the impact of CBF overexpression on growth inhibition. Important GA-responsive genes may also be downregulated in T166, and thus contribute to reduced growth. For example, transcripts encoding numerous putative GID1 GA-receptors were identified in the current transcriptome analysis and found to be significantly downregulated in T166. GID1 proteins bind to GA and foster the degradation of RGL/DELLA proteins, thus relieving RGL/DELLA growth inhibition40. Data from the current study, however, are equivocal in terms of evident expression patterns and the subject needs to be examined further.
Bhalerao and Fischer7 noted that reduced response to cytokinins can lead to reduced cambial cell division rates, and it is well known that cytokinin levels can also impact growth. Therefore, genes encoding cytokinin storage and inactivation enzymes were examined. In general, these genes were upregulated in T166 relative to “M.26”, suggesting another potential mechanism by which PpCBF1 overexpression may negatively affect growth and development. Indeed, genes encoding ANT/AIL (ANTINTEGUMENTA/ AINTEGUMENTA-LIKE) and CYCD (D-type cyclins) are all downregulated in T166 relative to “M.26” in April. ANT/ AIL positively regulate CYCD, increasing cell division7.
Inhibitory PGR-related genes associated with ABA and ethylene were examined. Genes encoding multiple members of the ABA signal transduction pathway (PYL/ PYR receptors, SnRK2 and 3 homologs, mitogen protein kinase cascade, Phospholipase D) were found to be differentially expressed over the entire sampling period, particularly in April. There was no consistent trend in expression, however, suggesting that PpCBF1 overexpression does not impact ABA signal transduction directly. The expression of ethylene-related genes was similarly inconsistent with no evident pattern within or between the two genotypes. While terminal biosynthetic enzyme ACC oxidase homologs were generally upregulated in April T166 samples, as well as in a few other months, ethylene receptor genes varied in their level of expression.
Numerous genes and models have been reported that couple PGRs with entry into and exit from dormancy. Models of the regulation of dormancy in woody perennials have been reported that incorporate aspects of herbaceous models6,7,41–43. The general pathway includes photoperiod perception by phytochromes A and B which interact with the circadian rhythm proteins (LHY, TIC, and TOC). CO/FT modules are then stimulated, leading to AP1 homolog activation, followed by ANT/AIL1, and resulting in the induction of vegetative growth. Rinne et al.25 proposed the plasmodesmata (PD) hypothesis, wherein callose sphincters close off the PD during autumn, preventing stimulatory PGRs from reaching the apical meristem. As daylength increases, GA-inducible 1,3-β-glucanases degrade the callose plug, allowing stimulatory PGRs to enter meristematic cells and induce bud break. Tylewicz et al.44 further refined this this hypothesis by reporting that dormancy onset is ABA-dependent, with photoperiod playing a role. ABA has also been implicated in pear flower bud endodormancy, as reported by Li et al.45. Apple and pear are closely related, so it is possible that they share this mechanism as well. Indeed, apple and pear have closely-related DAM genes and expression patterns in common as assessed in seasonal tissue collections and growth chamber experiments19,28,46,47.
DEGs that are included in data or models presented by Schrader et al.5, Ding and Nilsson40, Bhalerao and Fischer7, and Xing et al.48 were observed in the current study. In April, PhyA and B expression was slightly downregulated in T166 compared to “M.26”, as was CRYPTOCHROME1. PhyC genes were upregulated also but have not been implicated in current models of dormancy regulation. Other prominent members of the circadian regulatory module were either not differentially expressed (e.g., TIC, CCA1) or exhibit mixed expression (upregulated or downregulated differential expression in T166 relative to “M.26”). Mixed expression was also observed for GI, CO, the FT homolog MdFT2, and ANT/ AIL1. It is perhaps unsurprising that GI, CO, and ANT/AIL display expression in cambial tissues, as light perception and transmission has been observed in bud and vascular tissues7,30,49. However, the presence of MdFT2 in cambial tissues is apparently novel. Previous reports in apple indicate that MdFT2 is highly expressed in developing fruit23. Given the known role of FT in floral induction, Mimida et al.50 suggested that MdFT2 could have roles in floral organs and fruit development. The present study suggests that MdFT2 could have roles in early-season cambial growth dynamics as well. Autonomous and Vernalization pathway genes were inconsistently differentially expressed. These pathways are typically considered part of the dormancy/flowering cycle of herbaceous plants, but also function in perennial, woody flowering plants as well48. The primary target of the autonomous and vernalization pathways is FLC. An FLC paralog, termed MdMAF2 and previously reported in buds51, was slightly down regulated in T166 relative to “M.26”. Kumar et al.52 disagree with the assignment of MdMAF2 as an FLC paralog, however. Such disagreement, coupled with the low transcript number observed, renders the relevance of MdMAF2 expression in bark, differential or not, murky at best.
Other genes associated with either vegetative or vascular growth such as SOC, SPL, and CLAVATA5,7,23,53 were varied in their expression as well. PXY, associated with cambial cell fate decisions7, was negatively DE in T166 relative to “M.26”. The thinner stem diameters reported for T166 compared to “M.26” by Artlip et al.18 may reflect this downregulated expression. General genes related to growth, such as cyclins, cell division control proteins, and expansins5,7 were generally found to be downregulated in T166 relative to “M.26”, over the course of the study, especially in April. This may reflect the overall downregulation of growth-stimulating PGRs, resulting in the late bud break and reduced growth observed in T166. Expression of the knotted1-like (MdKNAP) genes were generally upregulated in T166 relative to “M.26”. The MdKNAP1 and 2 genes are known to be expressed during growth and development54 in apple stem internode tissue, but only MdKNAP1 was differentially expressed, with upregulation in T166 in the present study. It is also known that Populus knotted1 homologs can have overlapping, but discrete functions in these processes55,56. In depth examination of the apple KNAP genes in T166 and “M.26” is clearly warranted to clarify what roles they may play in dormancy and growth.
Genes related to the PD hypothesis were generally downregulated in T166 (Callose Synthases, remorins, glucan endo-1,3-beta-glucosidase (Group 17 glucanases). Downregulated glucan endo-1,3-beta-glucosidases could contribute to delayed dormancy in T166, as Rinne et al.25 reported a positive correlation between their expression and dormancy release.
Dormancy associated MADS-box (DAM) proteins have been reported to regulate dormancy in peach57–59 and implicated in other species. The expression of apple DAM genes has been reported on by several groups, including Wisniewski et al.19 in bark tissues as MdDAMs1-3, and by Wu et al.47 in apical buds as MdDAMa, MdDAMc, and MdSVPb, respectively. In the present study, MdDAM1/ MdDAMa was detected as being slightly, but statistically significantly, upregulated in T166 in Feb, Mar, and April. MdDAM2/ MdDAMc was upregulated in T166 relative to “M.26” only in April, with no statistically significant differential expression data from Feb, Mar, and July. A lack of statistically significant differential expression data does not imply a lack of expression in either T166 or “M.26”. Rather, the expression levels in both genotypes were so similar that they simply were filtered out of the bioinformatics pipeline used in this study. As such, MdDAM2/ MdDAMc, along with MdDAM3/ MdSVPb, were not differentially expressed during the other months examined in this study. The expression data observed in the present study generally agrees with data presented by Wisniewski et al.19 with little difference in the expression of these genes between T166 and “M.26” bark tissues. Wu et al.47 focused primarily on MdDAM and MdSVP expression in apical buds. Similarly, Falavigna et al.60 compiled data from these studies and others, creating a model of DAM expression in apical buds as a function of dormancy. Their model implies elevated expression of MdDAM2/ MdDAMc coincident with the end of the growth cycle, followed by elevated expression of MdDAM1/ MdDAMa as a component of endodormancy. Elevated expression of a third DAM, MdDAMb (not reported in this study), fosters ecodormancy; its decreased expression then heralds budbreak60. It is likely that MdDAMb is not a DEG in bark tissues, or was not a DEG at the sampling timepoint. Overall, the data and interpretations of Wu et al.47 and Falavigna et al.60 largely agree with those presented by Wisniewski et al.19 and the present study, which both focused on bark (cambial) tissues rather than buds. In addition, many of the findings of the current study reflect those presented by Schrader et al.5 in poplar, indicating that conserved cambial growth dynamics exist between two species from different taxonomic families.
Collectively, data in this study extend the findings reported by Wisniewski et al.19 and Artlip et al.30 A hypothetical framework of potential direct and indirect effects of PpCBF1 overexpression is presented in Fig. 6. The current study indicates that the effect of PpCBF1 overexpression in Line T166 is complex as evidenced by the significant differences in gene expression observed in the two genotypes in any given month (Supplemental Tables 2–7; Supplemental Figs. 5–12). PpCBF1 overexpression appears to affect the biosynthesis, signal transduction, and inactivation of PGRs. The effect on PGR-related gene expression may then impact the complex process associated with bud break. In general, the changes in gene expression that were observed in “M.26” during the time of bud break and the re-activation of growth were somewhat delayed in T166. Closer examination of the genes identified in the present study is warranted to better understand the pleiotropic effects of PpCBF1 overexpression in transgenic T166 apple.
Fig. 6. Potential interaction network of CBF overexpression effects in bark tissues primarily in April.

Genes that are downregulated in T166 compared to M.26 are in red, while upregulated genes in T166 compared to M.26 are in green. Gene names in black indicate no difference between T166 and M.26 or non-uniform expression patterns of several gene family members. Solid lines indicate evidence in the literature or deduced in this study, dotted lines denote speculative interactions, arrowheads denote stimulatory actions, and T ends denote inhibitory actions. TFs, transcription factors; SAUR, Small auxin-up RNA; KNAP, Knotted1-likeAPple; CRY, cryptochrome; PHY, phytochrome; GI, GIgantea; CO, COnstans; SOC, suppressor of overexpression of constans 1; LFY, LeaFY; SPL, squamosa promoter binding like; PIF, phytochrome interacting factor; GNC/GNL, GATA Nitrate-inducible, Carbon-metabolism involved/GNC-like; CKIIα, Casein Kinase II alpha subunit; TOC, timing of CAB expression 1; LHY, late elongated HYpocotyl, CCA1, circadian and clock associated 1; ELF3, EarLy Flowering3; ZTL/ADO; Zeitlupe/Adagio; RVE, Reveille; PFT1, phytochrome and flowering integrator 1; FKF1, flavin-binding kelch repeat F box protein; FCA, FT, flowering time; FLC, flowering locus C; LD, LuminiDependens; FLK, flowering late KH motif; FRI, FRIgida; FCA, flowering control locus A; VRN2, vernalization2; VIN3, vernalization insensitive3; FIE, fertilization-independent endosperm; VIP3, Vernalization IndePendence 3; ANT/AIL, AiNTegumenta/AIntegumenta-Like; CYCD, Cyclin D Type; ARR-A/B, type-A/B/C Arabidopsis response regulator; CLV, Clavata; WUS, Wuschel; SnRK2, Sucrose Non-fermenting kinase 1 Related protein Kinase 2; PP2C, Mg2+-dependent and Mn2+-dependent serine-threonine phosphatases type 2C; NCED, 9-cisepoxycarotenoid Dioxygenase; EBB, Early Bud Break. Pathways adapted from information presented in the refs. 19,20,25,30,39,40,44,48,55,60,72,73 and at KEGG (https://www.genome.jp/dbget-bin/www_bget?pathway:mdm04016 and https://www.genome.jp/dbget-bin/www_bget?pathway:mdm04075). Data regarding EBB in buds taken from the ref. 19
Materials and methods
Plant material
Non-transgenic “M.26” and transgenic “M.26” (line T166) trees that were approximately three-years-old were used in this study. The T166 (PpCBF1-OX) line was initially described by Wisniewski et al.16. Briefly, M.26 leaves were subjected to Agrobacterium-mediated transformation with a vector consisting of a pBINPLUSARS backbone and the peach PpCBF1 gene driven by a dual 35S enhancer segment derived from pRTL2. Plants were maintained in tissue culture, roots initiated, and plantlets established in growth chambers and greenhouse prior to being planted in October 2010 at the Appalachian Fruit Research Station, USDA-ARS, Kearneysville, WV per Artlip et al.18 Three trees each of “M.26” and T166 were used in this study as biological replicates.
Young, lateral branches were collected from “M.26” and T166 trees monthly in February, March, April, and July 2013. Bark tissues (cambium, phloem, and epidermis and/or phellem) were scraped from current year and one-year-old shoots and immediately placed in liquid nitrogen, lyophilized, and stored at −20 °C until use. Tissues from three trees of each genotype were sampled and maintained as independent biological replicates. Temperature and precipitation data were recorded daily during 2013. Trees had not been pruned during their initial three years of growth.
Dates of bud break for each tree were recorded during spring 2013. Percent bud break was determined thusly: three shoots on each of three trees of M.26 and T166 were tagged and bud break from 20 individual lateral buds from the terminal bud were tracked.
RNA extraction, library construction, and sequencing
RNA was isolated from bark samples as described by Bai et al.61, with purification and library construction as previously described by Ballester et al.62. In brief, 1 ml Sarkosyl of 20% (w/v) was added to 10 ml of extraction buffer (2% CTAB, 2% polyvinylpyrrolidone (PVP) K-30 (soluble), 100 mM Tris HCl (pH 8.0), 25 mM EDTA, 2.0 M NaCl, 0.5 g/l spermidine (free acid) (HRS), 2% β-mercaptoethanol (added just before use). The extracted total RNA was dissolved in EB buffer (Qiagen, Germantown, MD) supplemented by 1× Ambion RNA secure (Invitrogen/Life Technologies, Carlsbad, CA). To activate RNA secure, the samples were incubated at 60 °C (in a water bath) for 10 min and then immediately put on ice. RNA quantity and quality were evaluated using a Nanodrop 1000 (Thermo Scientific, Waltham, MA). Immediately prior to mRNA isolation, RNA samples were treated with DNase I (amplification grade, Invitrogen) at 37 °C for 30 min followed by heat inactivation at 65 °C for 15 min. Each RNA sample was adjusted to contain 5 μg of total RNA. Library construction was performed using the protocol outlined in Zhong et al.63 and run in two lanes using the Illumina HiSeq2000 platform to obtain 51-bp single-end reads. Libraries from three independent biological replicates of each genotype at each timepoint were sequenced and analyzed.
Bioinformatic analyses
The analyzed data comprised 24 datasets, 12 for the “M.26” wild-type genotype and 12 for the T166 transgenic genotype of Malus x domestica. Each genotype was sampled once during February, March, April, and July and each sampling timepoint contained three independent bioreplicates (trees) at each of the timepoints. After the 24 datasets were sequenced, each was run through an optimized RNA-seq pipeline to determine statistically significant differences in gene expression between specific comparisons. The v1.0 apple reference genome and annotation used for the assembly and annotation was downloaded from Phytozome64.
A computational pipeline consisting of optimized tools was developed to identify differences in gene expression between the T166 and “M.26” genotypes. The pipeline consisted of: (1) read quality check using FastQC65; (2) data trimming using Btrim66; (3) reference genome indexing using HISAT267; (4) alignment of trimmed reads to indexed reference genome using HISAT267; (5) read count quantification using HTSeq68; and (6) differential expression analysis using DESeq269 in R.
Four distinct comparisons of differential gene expression were considered: (1) pairwise comparisons of “M.26” vs. T166 at each timepoint; (2) separate time main effect for each genotype (“M.26” and T166; (3) pairwise comparisons of each consecutive timepoint for “M.26” and T166; and (4) interaction effect of genotype and time. The four types of comparisons provided a total of 13 comparisons, with comparison 1 being responsible for four, comparison 2 being responsible for two, comparison three being responsible for six, and comparison 4 being responsible for one. The four types of comparisons provided a comprehensive overview of the changes in gene expression corresponding to genotype (comparison 1) and time differences (comparisons 2 and 3) and which genes exhibited expression patterns that differ due to genotype over the course of the entire study (comparison 4).
The specific results for differential gene expression were determined using DESeq2, which implements a Wald test or Likelihood Ratio test to determine which genes exhibit different transcript levels within a respective comparison. The pairwise comparisons (1 and 3) utilize the Wald Test approach, which performs a parametric significance test of the selected factor level using a negative binomial distribution. Significant p-values result from the factor being determined as significant in the Wald test. The more complex comparisons (2 and 4) utilize a Likelihood Ratio test, which compares a full linear model considering appropriate additive and interactive effects and compares the fit against a reduced linear model with the selected factor(s) removed. Significant p-values result from a significant fitted improvement in the full model over the reduced model.
DESeq2 compiles a results file containing the gene ID, mean expression value, log2 fold-change and standard error, statistical test value, p-value, and adjusted p-value. DESeq2 adjusts the p-values to account for multiple testing using an FDR method. For this study, genes were considered differentially expressed if their adjusted p-value was below 0.05. Principal Coordinates Analysis (PCoA) was performed using Qlucore v3.2 (Qlucore, Lund, Sweden) bioinformatic software, with statistical significance set at 0.05.
Reverse transcription-quantitative PCR (RT-qPCR)
RT-qPCR was conducted on selected genes to validate the results obtained by the RNA-Seq analysis as previously described19. Total RNA was diluted to 12.5 ng/μl. RT-qPCR analysis was performed using the Invitrogen SuperScript III Platinum SYBR Green One-Step RT-qPCR Kit with ROX (ThermoFisher Scientific, Waltham, MA, USA), with each reaction containing 25 ng of input RNA and 2 pmol of each primer; no-RT control reactions were included to ensure that there was no residual DNA contamination. The Applied Biosystems ViiA 7 (ThermoFisher Scientific,Waltham, MA, USA) was set to cycle as follows: cDNA synthesis at 48.0 °C for 30 min; 95.0 °C denaturation for 5 min; 40 cycles of 95.0 °C for 15 s followed by 55 °C annealing for 1 min; followed by the default ViiA 7 hold and melt curve stages. Gene-specific primers were designed using CLC Genomics Workbench (Qiagen, Valencia, CA, USA) (Supplementary Table 1). Primers were verified for specificity by using genomic DNA templates and assessing the resulting amplicon by agarose gel electrophoresis and by RT-qPCR with a subset of the sample RNA on the ViiA7. All primers produced a single band and single peak. Primer efficiency was also verified for all primer sets by RT-qPCR analysis of a standard curve constructed by serially diluting RNAs from the sample set starting at some concentration above what was used in unknown samples and ending at a concentration well below it. Three technical replicates were used for each of three biological replicates. Several endogenous reference genes (FYPP3, LTL1, translation elongation factor 2, and CKB4) were assessed as to the stability of their expression within the two genotypes and across timepoints70. LTL1 was deemed the best overall reference gene using NormFinder software71. Expression levels of each of the analyzed genes were calculated using the comparative ΔΔCt (threshold cycle) method. Data from biological replicates were used to calculate mean ± standard error (SE) expression values.
Supplementary information
Acknowledgements
The authors thank Phil Welser and Erik Burchard for their technical assistance performing the RT-qPCR validation. This research was done under CRIS 8080-21000-026. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Authors' contributions
M.W. conceived of the study, Q.M. and A.M. conducted the bioinformatics analysis, and T.A. conducted the study, provided biological context to the bioinformatics analysis, and wrote the first draft.
Data availability
The raw reads and comparisons may be obtained from the corresponding authors upon request.
Conflict of interest
The authors declare that they have no conflict of interest.
Contributor Information
Timothy Artlip, Phone: +304-725-3451, Email: tim.artlip@usda.gov.
Michael Wisniewski, Phone: +304-725-3451 x210, Email: Michael.Wisniewski@usda.gov.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41438-019-0168-9).
References
- 1.Wisniewski M, Nassuth A, Arora R. Cold hardiness in trees: a mini-review. Front. Plant Sci. 2018;9:1394. doi: 10.3389/fpls.2018.01394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gu L, et al. The 2007 Eastern US Spring Freeze: increased cold damage in a warming wWorld? BioSci. 2008;58:253–262. doi: 10.1641/B580311. [DOI] [Google Scholar]
- 3.Wisniewski M, Artlip T, Norelli J. Dealing with frost damage and climate change in tree fruit crops. New Y. Fruit. Q. 2016;24:25–28. [Google Scholar]
- 4.Lang GA, Early JD, Martin GC, Darnell RL. Endo-, para-, and ecodormancy: physiological terminology and classification for dormancy research. HortSci. 1987;22:371–377. [Google Scholar]
- 5.Schrader J, et al. Cambial meristem dormancy in trees involves extensive remodeling of the transcriptome. Plant J. 2004;40:173–187. doi: 10.1111/j.1365-313X.2004.02199.x. [DOI] [PubMed] [Google Scholar]
- 6.Cooke JEK, Eriksson ME, Junttila O. The dynamic nature of bud dormancy in trees: environmental control and molecular mechanisms. Plant Cell Environ. 2012;35:1707–1728. doi: 10.1111/j.1365-3040.2012.02552.x. [DOI] [PubMed] [Google Scholar]
- 7.Bhalerao RP, Fischer U. Environmental and hormonal control of cambial stem cell dynamics. J. Exp. Bot. 2016;68:79–87. doi: 10.1093/jxb/erw466. [DOI] [PubMed] [Google Scholar]
- 8.Tarancon C, Gonzalez-Grandio E, Oliveros JC, Nicolas M, Cubas P. A conserved carbon starvation response underlies bud dormancy in woody and herbaceous species. Front. Plant Sci. 2017;8:788. doi: 10.3389/fpls.2017.00788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gusta LV, Wisniewski M. Understanding plant cold hardiness: an opinion. Physiol. Plant. 2013;147:4–14. doi: 10.1111/j.1399-3054.2012.01611.x. [DOI] [PubMed] [Google Scholar]
- 10.Welling A, Palva ET. Molecular control of cold acclimation in trees. Physiol. Plant. 2006;127:167–181. doi: 10.1111/j.1399-3054.2006.00672.x. [DOI] [Google Scholar]
- 11.Wisniewski M, et al. Genomics of cold hardiness in woody plants. Crit. Rev. Plant Sci. 2014;33:92–124. doi: 10.1080/07352689.2014.870408. [DOI] [Google Scholar]
- 12.Kaplan F, et al. Transcript and metabolite profiling during cold acclimation of Arabidopsis reveals an intricate relationship of cold-regulated gene expression with modifications in metabolite content. Plant J. 2007;50:967–981. doi: 10.1111/j.1365-313X.2007.03100.x. [DOI] [PubMed] [Google Scholar]
- 13.Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta. 2012;1819:86–96. doi: 10.1016/j.bbagrm.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Vogel JT, Zarka DG, Van Buskirk HA, Fowler SG, Thomashow MF. Roles of the CBF2 and ZAT12 transcription factors in configuring the low temperature transcriptome of Arabidopsis. Plant J. 2005;41:195–211. doi: 10.1111/j.1365-313X.2004.02288.x. [DOI] [PubMed] [Google Scholar]
- 15.Shi Y, Ding Y, Yang S. Molecular regulation of CBF signaling in cold acclimation. Trends Plant Sci. 2018;23:623–637. doi: 10.1016/j.tplants.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 16.Wisniewski M, Norelli J, Bassett C, Artlip T, Macarisin D. Ectopic expression of a novel peach (Prunus persica) CBF transcription factor in apple (Malus x domestica) results in short-day induced dormancy and increased cold hardiness. Planta. 2011;233:971–983. doi: 10.1007/s00425-011-1358-3. [DOI] [PubMed] [Google Scholar]
- 17.Heide OM, Prestrud AK. Low temperature, but not photoperiod, controls growth cessation and dormancy induction and release in apple and pear. Tree Physiol. 2005;25:109–114. doi: 10.1093/treephys/25.1.109. [DOI] [PubMed] [Google Scholar]
- 18.Artlip TS, Wisniewski ME, Norelli JL. Field evaluation of apple overexpressing a peach CBF gene confirms its effect on cold hardiness, dormancy, and growth. Environ. Exp. Bot. 2014;106:79–86. doi: 10.1016/j.envexpbot.2013.12.008. [DOI] [Google Scholar]
- 19.Wisniewski M, Norelli J, Artlip T. Overexpression of a peach CBF in apple: a model for understanding the integration of growth, dormancy, and cold hardiness in woody plants. Front. Plant Sci. 2015;6:85. doi: 10.3389/fpls.2015.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yordanov YS, Ma C, Strauss SH, Buscov VB. EARLY BUD-BREAK 1 (EBB1) is a regulator of release from seasonal dormancy in poplar trees. Proc. Natl Acad. Sci. 2014;111:10001–10006. doi: 10.1073/pnas.1405621111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Velasco R, et al. The genome of the domesticated apple (Malus x domestica Borkh.) Nat. Genet. 2010;42:833–841. doi: 10.1038/ng.654. [DOI] [PubMed] [Google Scholar]
- 22.Kotoda N, Wada M. MdTFL1, a TFL1-like gene of apple, retards the transition from vegetative to reproductive phase in transgenic. Arabidopsis. Plant Sci. 2004;168:95–104. doi: 10.1016/j.plantsci.2004.07.024. [DOI] [Google Scholar]
- 23.Kotoda N, et al. Molecular characterization of FLOWERING LOCUS T-LIKE genes of apple (Malus x domestica Borkh.) Plant Cell Physiol. 2010;51:561–575. doi: 10.1093/pcp/pcq021. [DOI] [PubMed] [Google Scholar]
- 24.Etchells JP, Mishra LS, Kumar M, Campbell L, Turner SR. Wood formation in trees is increased by manipulating PXY-regulated cell division. Curr. Biol. 2015;25:1050–1055. doi: 10.1016/j.cub.2015.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rinne PLH, et al. Chilling of dormant buds hyperinduces FLOWERING LOCUS T and recruits GA-inducible 1,3-β-glucansases to reopen signal conduits and release dormancy in Populus. Plant Cell. 2011;23:130–146. doi: 10.1105/tpc.110.081307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medina J, Catala R, Salinas J. The CBFs: three Arabidopsis transcription factors to cold acclimate. Plant Sci. 2011;180:3–11. doi: 10.1016/j.plantsci.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 27.Qin F, Shinozaki K, Yamaguchi-Shinozaki K. Achievements and challenges in understanding plant abiotic stress responses and tolerance. Plant Cell Physiol. 2011;52:1569–1582. doi: 10.1093/pcp/pcr106. [DOI] [PubMed] [Google Scholar]
- 28.Stockinger EJ, Gilmour SJ, Thomashow MF. Arabidopsis thaliana CBF1 encodes an AP2 domain transcriptional activator that binds to the C-repeat cis-acting DNA regulatory element that stimulates in response to low temperature and water deficit. Proc. Natl Acad. Sci. 1997;94:1035–1040. doi: 10.1073/pnas.94.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maruyama K, et al. Identification of cold-inducible downstream genes of the Arabidopsis DREB1A/CBF3 transcriptional factor using two microarray systems. Plant J. 2004;38:982–993. doi: 10.1111/j.1365-313X.2004.02100.x. [DOI] [PubMed] [Google Scholar]
- 30.Artlip TS, Wisniewski ME, Arora R, Norelli JL. An Apple rootstock overexpressing a peach CBF gene alters growth and flowering in the scion but does not impact cold hardiness or dormancy. Hortic. Res. 2016;3:16006. doi: 10.1038/hortres.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shim D, et al. A molecular framework for seasonal growth-dormancy regulation in perennial plants. Hortic. Res. 2014;1:14059. doi: 10.1038/hortres.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zohner CM, Renner SS. Perception of photoperiod in individual buds of mature trees regulates leaf-out. New Phytol. 2015;208:1023–1030. doi: 10.1111/nph.13510. [DOI] [PubMed] [Google Scholar]
- 33.Artlip, T. S. & Wisniewski, M. E. in Handbook of Plant and Crop Physiology, 2nd edn. (ed. Pessarakli, M.) Ch. 33 (Marcel Dekker, Inc., New York, 2001).
- 34.Kerk D, Bulgrien J, Smith DW, Gribskov M. Arabidopsis proteins containing similarity to the universal stress protein domain of bacteria. Plant Phys. 2003;131:1209–1219. doi: 10.1104/pp.102.016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Achard P, et al. The cold-inducible CBF1 factor-dependent signaling pathway modulates the accumulation of the growth-repressing DELLA proteins via its effect on gibberellin metabolism. Plant Cell. 2008;20:2117–2129. doi: 10.1105/tpc.108.058941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niu S, Gao Q, Li Z, Chen X, Li W. The role of gibberellin in the CBF1-mediated stress-response pathway. Plant Mol. Biol. Rep. 2014;32:852–863. doi: 10.1007/s11105-013-0693-x. [DOI] [Google Scholar]
- 37.Suo H, et al. Overexpression of AtDREB1A causes a severe dwarf phenotype by decreasing endogenous gibberellin levels in soybean [Glycine max (L.) Merr.] PLoS One. 2012;7:e45568. doi: 10.1371/journal.pone.0045568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baba K, et al. Activity-dormancy transition in the cambial meristem involves stage-specific modulation of auxin response in hybrid aspen. Proc. Natl Acad. Sci. USA. 2011;108:3418–3423. doi: 10.1073/pnas.1011506108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Achard P, Genschik P. Releasing the brakes of plant growth: how GAs shutdown DELLA proteins. J. Exp. Bot. 2009;60:1085–1092. doi: 10.1093/jxb/ern301. [DOI] [PubMed] [Google Scholar]
- 40.Xu H, Liu Q, Yao T, Fu X. Shedding light on integrative GA signaling. Curr. Opin. Plant Biol. 2014;21:89–95. doi: 10.1016/j.pbi.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 41.Ding J, Nilsson O. Molecular regulation of phenology in trees—because the seasons they are a-changin’. Curr. Opin. Plant Biol. 2016;29:73–79. doi: 10.1016/j.pbi.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 42.Petterle A, Karlberg A, Bhalerao RP. Daylength mediated control of seasonal growth patterns in perennial trees. Curr. Opin. Plant Biol. 2016;16:301–306. doi: 10.1016/j.pbi.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 43.Preston JC, Sandve SR. Adaptation to seasonality and the winter freeze. Front. Plant Sci. 2013;4:167. doi: 10.3389/fpls.2013.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tylewicz S, et al. Photoperiodic control of seasonal growth is mediated by ABA acting on cell-cell communication. Science. 2018;360:212–215. doi: 10.1126/science.aan8576. [DOI] [PubMed] [Google Scholar]
- 45.Li J, Niu Q, He L, Teng Y, Bai S. Abscisic acid (ABA) promotes the induction and maintenance of pear (Pyrus pyrifolia White pear group) flower bud endodormancy. Int. J. Mol. Sci. 2018;19:310. doi: 10.3390/ijms19010310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saito T, et al. Expression and genomic structure of the dormancy-associated MADS box genes MADS13 in Japanese pears (Pyrus pyrifolia Nakia) that differ in their chilling requirement for endodormancy release. Tree Physiol. 2013;33:654–667. doi: 10.1093/treephys/tpt037. [DOI] [PubMed] [Google Scholar]
- 47.Wu R, et al. SVP-like MADS box genes control dormancy and budbreak in apple. Front. Plant Sci. 2017;8:477. doi: 10.3389/fpls.2017.00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xing L-B, et al. Transcription profiles reveal sugar and hormone signaling pathways mediating flower induction in apple (Malus x domestica Borkh.) Plant Cell Physiol. 2015;56:2052–2068. doi: 10.1093/pcp/pcv124. [DOI] [PubMed] [Google Scholar]
- 49.Sun Q, Yoda K, Suzuki M, Suzuki H. Vascular tissue in the stem and roots of woody plants can conduct light. J. Exp. Bot. 2003;54:1627–1635. doi: 10.1093/jxb/erg167. [DOI] [PubMed] [Google Scholar]
- 50.Mimida N, et al. Apple FLOWERING LOCUS T proteins interact with transcription factors implicated in cell growth and organ development. Tree Physiol. 2011;31:555–566. doi: 10.1093/treephys/tpr028. [DOI] [PubMed] [Google Scholar]
- 51.Porto DD, et al. Transcription profiling of the chilling requirement for bud break in apples: a putative role for FLC-like genes. J. Exp. Bot. 2015;66:2659–2672. doi: 10.1093/jxb/erv061. [DOI] [PubMed] [Google Scholar]
- 52.Kumar G, et al. Comparative phylogenetic analysis and transcriptional profiling of MADS-box gene family identified DAM and FLC-like genes in apple (Malus x domestica) Sci. Rep. 2016;6:20695. doi: 10.1038/srep20695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cseke LJ, Xheng J, Podila GK. Characterization of PTM5 in aspen trees: a MADS-box gene expressed during woody vascular development. Gene. 2003;318:55–67. doi: 10.1016/S0378-1119(03)00765-0. [DOI] [PubMed] [Google Scholar]
- 54.Watillon B, Kettmann R, Boxus P, Burny A. Knotted1-like homeobox genes are expressed during apple (Malus domestica [L.] Borkh) growth and development. Plant Mol. Biol. 1997;33:757–763. doi: 10.1023/A:1005708700636. [DOI] [PubMed] [Google Scholar]
- 55.Groover AT, et al. The Populus homeobox gene ARBORKNOX1 reveals overlapping mechanisms regulating the shoot apical meristem and the vascular cambium. Plant Mol. Biol. 2006;61:917–932. doi: 10.1007/s11103-006-0059-y. [DOI] [PubMed] [Google Scholar]
- 56.Du J, Mansfield SD, Groover AT. The Populus homeobox gene ARBORKNOX2 regulates cell differentiation during secondary growth. Plant J. 2009;60:1000–1014. doi: 10.1111/j.1365-313X.2009.04017.x. [DOI] [PubMed] [Google Scholar]
- 57.Bielenberg DG, et al. Sequencing and annotation of the evergrowing locus in peach (Prunus persica [L.] Batsch) reveals a cluster of six MADS-box transcription factors as candidate genes for regulation of terminal bud formation. Tree Genet. Genomes. 2008;4:495–507. doi: 10.1007/s11295-007-0126-9. [DOI] [Google Scholar]
- 58.Li Z, Reighard GL, Abbott AG, Bielenberg DG. Dormancy-associated MADS genes from the EVG locus of peach (Prunus persica [L.] Batsch) have distinct seasonal and photoperiodic expression patterns. J. Exp. Bot. 2009;60:3521–3530. doi: 10.1093/jxb/erp195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jimenez S, Reighard GL, Bielenberg DG. Gene expression of DAM5 and DAM6 is suppressed by chilling temperatures and inversely correlated with bud break rate. Plant Mol. Biol. 2010;73:157–167. doi: 10.1007/s11103-010-9608-5. [DOI] [PubMed] [Google Scholar]
- 60.Falavigna VS, Guitton B, Costes E, Andres F. I want to (bud) break free: the potential role of DAM and SVP-like genes in regulating dormancy cycle in temperate fruit trees. Front. Plant Sci. 2019;9:1990. doi: 10.3389/fpls.2018.01990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bai Y, Dougherty L, Xu K. Towards an improved apple reference transcriptome using RNA‑seq. Mol. Genet. Genom. 2014;289:427–438. doi: 10.1007/s00438-014-0819-3. [DOI] [PubMed] [Google Scholar]
- 62.Ballester AR, et al. Transcriptomic response of resistant (PI61983 – Malus sieversii) and susceptible (‘Royal Gala’) parents of the GMAL4593 mapping population of apple to blue mold infection (Penicillium expansum). Front. Plant Sci. 2017;8:1981. doi: 10.3389/fpls.2017.01981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhong S, Joung J-G, Zheng Y. High-throughput Illumina strand specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011 doi: 10.1101/pdb.prot5652. [DOI] [PubMed] [Google Scholar]
- 64.Goodstein DM, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2011;40(D1):D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Andrews, S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
- 66.Kong Y. Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics. 2011;98:152–153. doi: 10.1016/j.ygeno.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 67.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anders S, Huber W. Differential Expression of RNA-Seq Data at the Gene Level—the DESeq Package. Heidelberg: European Molecular Biology Laboratory (EMBL); 2012. [Google Scholar]
- 70.Bowen J, Ireland HS, Crowhurst R. Selection of low-variance expressed Malus x domestica (apple) genes for use as quantitative PCR reference genes (housekeepers) Tree Genet. Genomes. 2014;10:751–759. doi: 10.1007/s11295-014-0720-6. [DOI] [Google Scholar]
- 71.Anderson CL, Jensen JL, Ømtoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer datasets. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 72.Hwang I, Sheen J, Müller B. Cytokinin signaling networks. Ann. Rev. Plant Biol. 2012;63:353–380. doi: 10.1146/annurev-arplant-042811-105503. [DOI] [PubMed] [Google Scholar]
- 73.Korasick DA, Enders TA, Strader LC. Auxin biosynthesis and storage forms. J. Exp. Bot. 2013;64:2541–2555. doi: 10.1093/jxb/ert080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw reads and comparisons may be obtained from the corresponding authors upon request.