

There is need for rapid and accurate methods to detect viable bacterial cells of foodborne pathogens. Conventional culture-based methods are time-consuming and unable to detect bacteria in a viable-but-nonculturable state. The high sensitivity and specificity of the quantitative PCR (qPCR) are negated by its inability to differentiate the DNAs from viable and dead cells. The combination of propidium monoazide (PMA), a DNA-intercalating dye, with qPCR assays is promising for detection of viable cells. Despite encouraging results, these assays still encounter various challenges, such as false-positive signals by dead cells and the lack of an internal control identifying these signals per sample. The significance of our research lies in enhancing the selective entrance of PMA into dead Campylobacter coli cells via spheroplasting and in developing an internal sample process control, thus delivering reliable results in pure cultures and meat samples, approaches that can be applicable to other Gram-negative pathogens.
KEYWORDS: Campylobacter, PMA-qPCR, spheroplast, lysozyme, EDTA
ABSTRACT
A viability quantitative PCR (qPCR) utilizing propidium monoazide (PMA) is presented for rapid quantification of viable cells using the foodborne pathogen Campylobacter coli as a bacterial model. It includes optimized spheroplast formation via lysozyme and EDTA, induction of a mild osmotic shock for enhancing the selective penetration of PMA into dead cells, and exploitation of an internal sample process control (ISPC) involving cell inactivation to assess residual false-positive signals within each sample. Spheroplasting of bacteria in exponential phase did not permit PMA entrance into viable cells since a strong linear relationship was detected between simple qPCR and PMA-qPCR quantification, and no differences were observed regardless of whether spheroplasting was utilized. The PMA-qPCR signal suppression of dead cells was elevated using spheroplast formation. With regard to the ISPC, cell inactivation by hydrogen peroxide resulted in higher signal suppression during qPCR than heat inactivation did. Viability quantification of C. coli cells by optimized spheroplasting-PMA-qPCR with ISPC was successfully applied in an aging pure culture under aerobic conditions and artificially inoculated meat. The same method exhibited a high linear range of quantification (1.5 to 8.5 log10 viable cells ml−1), and results were highly correlated with culture-based enumeration. PMA-qPCR quantification of viable cells can be affected by their rigidity, age, culture media, and niches, but spheroplast formation along with osmotic shock and the use of a proper ISPC can address such variations. The developed methodology could detect cells in a viable-but-nonculturable state and might be utilized for the quantification of other Gram-negative bacteria.
IMPORTANCE There is need for rapid and accurate methods to detect viable bacterial cells of foodborne pathogens. Conventional culture-based methods are time-consuming and unable to detect bacteria in a viable-but-nonculturable state. The high sensitivity and specificity of the quantitative PCR (qPCR) are negated by its inability to differentiate the DNAs from viable and dead cells. The combination of propidium monoazide (PMA), a DNA-intercalating dye, with qPCR assays is promising for detection of viable cells. Despite encouraging results, these assays still encounter various challenges, such as false-positive signals by dead cells and the lack of an internal control identifying these signals per sample. The significance of our research lies in enhancing the selective entrance of PMA into dead Campylobacter coli cells via spheroplasting and in developing an internal sample process control, thus delivering reliable results in pure cultures and meat samples, approaches that can be applicable to other Gram-negative pathogens.
INTRODUCTION
Rapid and accurate quantification of viable bacterial cells is essential for monitoring foodborne pathogens along the food chain. Classical culture-based methods are time-consuming and unable to detect viable-but-nonculturable (VBNC) cells that can maintain metabolic and virulent activity (1–7). Equally, molecular methods, such as real-time quantitative PCR (qPCR), detect DNA originating not only from culturable and VBNC bacteria but also from dead cells (1–2). In order to address these shortcomings, viability PCR (v-PCR) using nucleic acid-intercalating dyes has been launched as a promising technique to rapidly quantify cells and concurrently provide viability information (8). Membrane integrity is a widely used and accepted viability criterion during v-PCR that characterizes viable bacteria (culturable and VBNC cells) and is regarded as a natural barrier for DNA-intercalating dyes, of which propidium monoazide (PMA) displays superior selectivity (8, 9). Ideally, a viability dye exclusively penetrates compromised-membrane dead cells, induces DNA cleavage upon photoactivation, and, therefore, inhibits DNA amplification during PCR (8).
Despite implementations with encouraging results, recently reviewed data suggest that v-PCR still encounters various practical challenges (1, 8). In particular, suppression of dead-cell qPCR signals by PMA is not absolute but varies among different matrices and experimental conditions, leading to overestimated counts of viable cells with intact membranes (8, 10–13). A variety of conditions have been used to minimize these false-positive qPCR signals from dead cells, such as the chosen dye concentration, incubation and photoactivation conditions, lengths of the amplified target genes, and membrane-destabilizing factors (14–17). Nonetheless, an internal sample process control (ISPC) for PMA-based quantifications that would identify variable process factors, such as dye concentration, matrix influence, and cross-linking efficiency, remains a crucial research target that has not been addressed as yet (18).
Campylobacter remains the most commonly reported gastrointestinal bacterial pathogen in humans in the European Union (EU) since 2005, related to the consumption of food of animal origin and especially poultry (19). Campylobacter species information is provided for approximately half of the confirmed human infections in the EU, among which Campylobacter jejuni followed by Campylobacter coli account for over 95% of clinical cases (respectively responsible for 80 to 85% and 10 to 15% of cases) (19). In percentage terms, the importance of C. coli might only initially seem smaller than that of C. jejuni, since its health burden is substantial and much greater than previously thought (20). In particular, official reports and recent scientific evidence indicate alarming levels of multidrug resistance being observed predominantly in C. coli (up to 99%) (21–26). Moreover, an increased risk of C. coli infection in older people and in habitants of rural areas, particularly during summer months, has been reported (27–30). Interestingly, C. coli has been indicated as the prevailing Campylobacter species isolated from various animal origin sources, including poultry and products thereof, in Greece (31–34) and other southern EU member states (23, 26, 35–37).
The aim of the present study was to develop step by step a reliable PMA-qPCR method for the accurate quantification of viable bacterial cells by introducing (i) a methodology of spheroplast formation as a sample pretreatment for enhancing the selective penetration of PMA into dead cells in order to minimize the false-positive qPCR signals and (ii) an ISPC in order to monitor variations in PMA performance and any residual false-positive qPCR signals from dead cells within each sample. C. coli has been chosen as a bacterial model in this study due to the elevated public health concern about it as a result of the emergence of multidrug-resistant strains, its morphological resemblance to C. jejuni, and its predominance over C. jejuni in food samples of animal origin in southern Europe, as mentioned above. Pure suspensions of C. coli and artificially inoculated meat samples were used for the development and evaluation of the optimized PMA-qPCR method.
RESULTS
qPCR linear range, sensitivity, specificity, and repeatability.
The qPCR assay showed a linear range of 100 to 107 copies per reaction (R2 > 0.99), with a PCR efficiency of 93.4% and a limit of detection (LOD) of 7.046 copies per reaction (95% confidence interval). The standard curve was described by the equation , where y stands for the number of genome copies (one genome copy equals one cell equivalent) per qPCR and CT is cycle threshold. The assay cutoff was set at a CT of 40.3. However, it should be noted that the selection of a cutoff value is an in-house procedure depending on the thermal cycler and inherent to analysis.
The intra-assay CT values of the three runs (a measure of repeatability; means ± standard deviations [SD]) were 36.99 ± 0.50, 37.15 ± 0.33, and 37.47 ± 0.20, the interassay CT value was 37.20 ± 0.25, and the intra-assay and interassay coefficients of variation were 0.90% and 0.67%, respectively. The specificity of the primer-probe set for the detection of the C. coli-specific glyA gene by real-time PCR was confirmed by the positive signals generated only for the tested C. coli strains, in contrast to the negative signals generated for the C. jejuni reference and in-house collection strains as well as the reference strains of the other Gram-positive and -negative bacteria tested. This specificity was not influenced by the presence of 6 log10 CFU of heat-killed Escherichia coli ATCC 8739 cells that were added to serial dilutions with low C. coli populations in order to form a visible pellet during cell harvesting by centrifugation while we investigated the quantification range of the optimized PMA-qPCR.
Evaluation of PMA cross-linking to extracellular DNA.
The results from the amplification of the standard DNA of C. coli ATCC 43478 that was treated with various concentrations of PMA are presented in Table 1. The amplification of the extracellular DNA was not completely inhibited by PMA at all the tested dye concentrations.
TABLE 1.
Amplification of extracellular DNA per assay in the presence of various PMA concentrations
| Sample type | Mean CT value ± SD | Mean no. of C. coli copiesa | DNA amplification (%) |
|---|---|---|---|
| DNA standard | 23.07 ± 0.41 | 2.26E+05 | 100.00 |
| 200 μM PMA + DNA standard | 26.35 ± 0.31 | 2.60E+04 | 11.49 |
| 100 μM PMA + DNA standard | 25.22 ± 0.02 | 5.50E+04 | 24.30 |
| 50 μM PMA + DNA standard | 25.08 ± 0.01 | 6.03E+04 | 26.65 |
There were 2.3 × 106 C. coli ATCC 43478 genome copies per assay, which were quantified by using the standard curve equation and the mean value of quadruplicates (one genome copy equals one cell equivalent).
Spheroplast formation and PMA-qPCR performance.
The destabilization of the C. coli ATCC 43478 cell wall (spheroplast formation), induced by the synergic effect of lysozyme and EDTA (Lys/EDTA), was verified by monitoring every 5 min the value of the optical density at 600 nm (OD600) in three replicates of 6 to 8 log10 CFU of viable and dead bacterial suspensions ml−1 (Fig. 1). A noticeable reduction in OD600 was observed within the first 30 min of incubation with the hyperosmotic buffer S1 (which contains phosphate-buffered saline [PBS], sucrose, EDTA, and lysozyme) at 37°C. The succeeding addition of S2 buffer (which contains PBS and EDTA) at a ratio of 1:1 resulted in a reasonably sudden drop of OD600 (osmotic shock), followed by a smoother decrease with continuance of the incubation at 37°C. No noticeable reduction in OD600 was observed 20 min after the addition of S2 buffer. A similar pattern of reduction in OD600 and final stabilization to a minimum value was observed for all the tested EDTA concentrations (Fig. 1). Based on these results, the incubation time at 37°C with S1 buffer was determined at 30 min and at 20 min under dark conditions after the addition of S2 buffer supplemented with PMA. No differences in CT values were observed for samples illuminated for 2 and 4 min. Therefore, the shorter exposure time to light-emitting diode (LED) light of 2 min was chosen for PMA photoactivation.
FIG 1.

Monitoring of spheroplast formation by measuring the optical density at 600 nm (OD600) during incubation of C. coli cultures at 37°C with S1 and S2 buffers containing various EDTA concentrations (12.5 to 100 mM). S2 buffer was added at a ratio of 1:1 immediately after 30 min of incubation with S1 buffer. L1 to L4, viable C. coli cells in exponential phase; D1 and D2, heat-inactivated (72°C for 10 min) C. coli cells.
The results of qPCR and PMA-qPCR with or without spheroplast formation for triplicate aliquots of viable (exponential-phase) and heat-inactivated (72°C for 10 min) C. coli ATCC 43478 cells are presented in Fig. 2. For cells in exponential phase, no differences were observed between the CT values of qPCR and PMA-qPCR combined with either spheroplasting or simple PMA addition. On the other hand, the observed signal suppression of heat-inactivated cells with respect to simple qPCR was higher for PMA-qPCR with spheroplasts (mean ΔCT = 9.12) than for PMA-qPCR with simple PMA addition (mean ΔCT = 3.24), indicating a superior selective entrance of PMA to dead cells when spheroplasting was applied.
FIG 2.

(A to C) Amplification plots of qPCR (A) and PMA-qPCR with (B) or without (C) spheroplast formation for triplicate aliquots of viable (exponential-phase) and heat-inactivated (72°C for 10 min) C. coli cells (6.45 log10 CFU ml−1). (D) Comparison of total and viable-cell counts (expressed as log10 numbers of copies per milliliter for qPCR and log10 numbers of CFU per milliliter for culture-based enumeration) of the corresponding C. coli cells. Each bar represents the mean ± SD of results from three independent repetitions. RFU, relative fluorescence units.
Spheroplasting of C. coli in exponential phase, induced by the synergic effect of lysozyme and various concentrations of EDTA, did not enhance the entrance of PMA into the viable cells, as indicated by the quantification results of qPCR and PMA-qPCR. More precisely, the cell equivalents attained by the two methods were highly correlated in the independent experiments, since a strong linear relationship (R2 > 0.91) was detected, indicating that the cytoplasmic membrane of the spheroplasts remained intact in all cases (Fig. 3).
FIG 3.

Correlation of C. coli cell equivalent counts (mean value from three aliquots) in bacterial suspensions in exponential phase (CAMHB) obtained by qPCR and PMA-qPCR during independent experiments (each symbol represents the result of an independent experiment). The concentrations of EDTA in S1 and S2 buffers for spheroplast formation (PMA-qPCR) were 12.5 mM (A), 25 mM (B), and 50 mM (C). The corresponding linear-regression equations and correlation coefficient (R2) values are presented in the diagrams.
ISPC.
No residual CFU were detected on Columbia blood agar (CBA) plates, irrespective of the inactivation method. The treatment with H2O2 (oxidative stress) yielded the largest ΔCT during PMA-qPCR with respect to qPCR, compared to the values from the two heat inactivation methods, with regard to both the overnight and aged cultures of C. coli (Fig. 4). In particular, as regards C. coli cells in exponential phase that were inactivated by lethal heat treatments at 95°C for 5 min and 75°C for 30 min or inactivated by treatment with 1.5% H2O2 at 42°C for 50 min, the reductions of the dead-cell qPCR signal (ΔCT) were 9.17 ± 0.51, 10.01 ± 1.09, and 13.01 ± 1.33, respectively. The corresponding results for the 56-day-old C. coli culture were 7.55 ± 0.50, 9.37 ± 0.45, and 11.54 ± 0.22. Therefore, the H2O2 treatment was chosen as the most suitable method for generating the ISPC.
FIG 4.
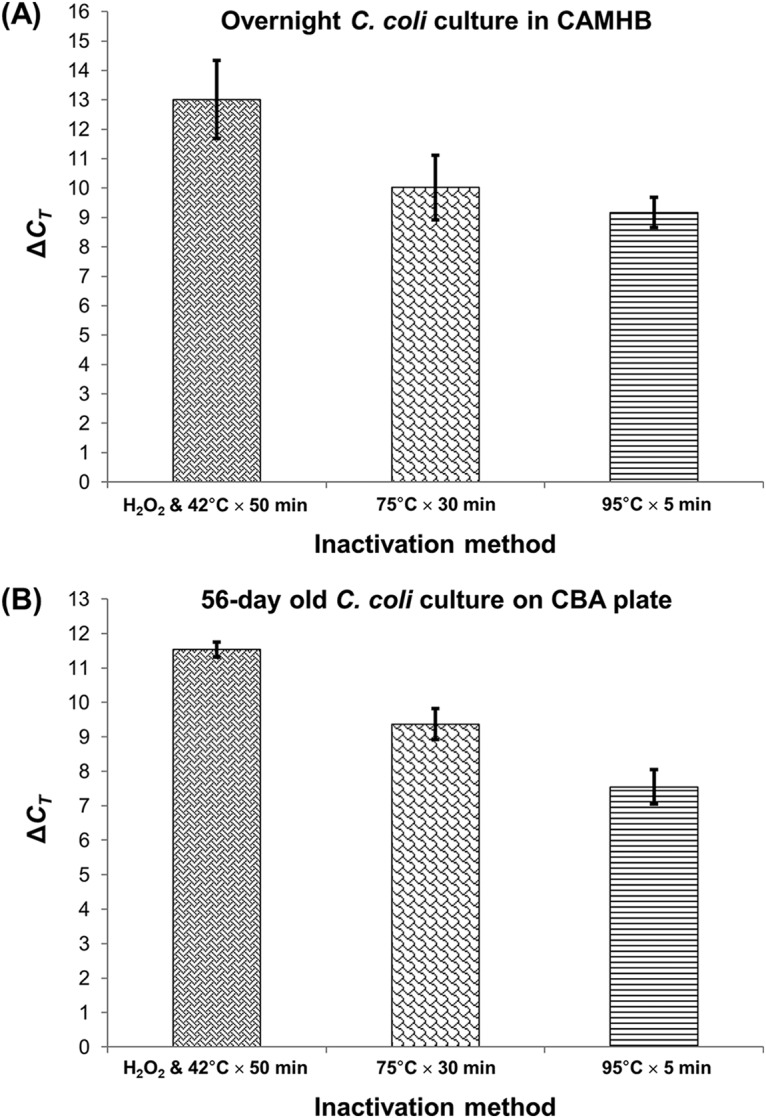
Magnitude of the shift among the observed CT values (ΔCT) of PMA-qPCR (viable-cell-equivalent count) compared to those of qPCR (total cell equivalent count) of an overnight culture in CAMHB (A) and a 56-day-old culture on CBA plate of C. coli that were subjected to three inactivation methods (1.5% H2O2 and incubation at 42°C for 50 min, lethal heat treatment at 75°C for 30 min, lethal heat treatment at 95°C for 5 min) (B). Each bar represents the mean ± SD of results from three independent repetitions (triplicate aliquots).
Application of the final quantification procedure in an aging C. coli population.
Figure 5 illustrates the quantification results of a naturally stressed culture of the reference C. coli strain by the methods presented in Table 2 at three different time points. On each experimental occasion, the steps (Fig. 6) and calculation methods described in the final quantification procedure were applied. The total cell equivalent counts obtained by qPCR at all times were higher than the CFU counts obtained from CBA plates, and this difference was always statistically significant (P < 0.05) (Fig. 5), indicating that a considerable fraction of the C. coli population, which was unable to grow on culture media (VBNC and dead cells), generated signals during qPCR.
FIG 5.

Quantification results of a naturally aging C. coli ATCC 43478 culture in CAMHB stored at room temperature under aerobic conditions at three different time points (42 h, 15 days, and 42 days). (Left) Numbers of genome copies (one genome copy equals one cell equivalent) per milliliter of culture (mean of results from triplicates ± SD) detected by PCR-based (qPCR, PMA-qPCR) and numbers of CFU ± SD obtained by culture-based (plating) methods (see Table 2). (Right) Numbers of genome copies (means of results from triplicates ± SD) detected per qPCR assay after treatment with H2O2 (ISPC). Counts with no statistically significant difference (P > 0.05) from each other are indicated with the same lowercase letter. Counts with a statistically significant difference (P < 0.05) from each other are indicated with a different lowercase letters. *, culture-based enumeration was below the limit of quantification.
TABLE 2.
Methods used for the quantification of C. coli in a pure culture in CAMHB stored at room temperature under aerobic conditions at 42 h, 15 days, and 42 days
| Method | Coded name (concn [mM]) |
S1 buffer composition | Remark |
|---|---|---|---|
| PMA-qPCR | Lys (0), EDTA (0) | 10 mM PBS, 500 mM sucrose | Negative control for lysozyme and EDTA (simple osmotic shock) |
| PMA-qPCR | Lys, EDTA (0) | 10 mM PBS, 500 mM sucrose, 0.5 mg ml−1 lysozyme | Negative control for EDTA |
| PMA-qPCR | Lys, EDTA (12.5) | 10 mM PBS, 500 mM sucrose, 0.5 mg ml−1 lysozyme, 25 mM EDTA | |
| PMA-qPCR | Lys, EDTA (25) | 10 mM PBS, 500 mM sucrose, 0.5 mg ml−1 lysozyme, 50 mM EDTA | |
| PMA-qPCR | Lys, EDTA (50) | 10 mM PBS, 500 mM sucrose, 0.5 mg ml−1 lysozyme, 100 mM EDTA | |
| PMA-qPCR | Lys, EDTA (100) | 10 mM PBS, 500 mM sucrose, 0.5 mg ml−1 lysozyme, 200 mM EDTA | |
| PMA-qPCR | Simple PMA | 0.9% NaCl | Negative control for spheroplasting |
| qPCR | qPCR | Negative control for PMA treatment | |
| Culture based | CBA plates |
FIG 6.

Final procedure for the quantification of culturable, total, and viable C. coli cells using plating, qPCR, and optimized spheroplasting-PMA-qPCR with ISPC, respectively. The dashed red line incorporates the aliquot treatments prior to spheroplasting for the generation of the ISPC. ddH2O, double-distilled water; buf, buffer.
At 42 h, similar C. coli counts were observed between qPCR and the negative controls with regard to spheroplasting (simple PMA addition; P = 0.565) and to Lys/EDTA addition (simple osmotic shock; P = 0.556). The counts obtained by the aforementioned methods were significantly higher (P < 0.05) than those by CBA plating. Contrarily, no statistically different results (P > 0.05) were observed between the CBA plates and PMA-qPCR with spheroplasting, irrespectively of the EDTA concentration used (Fig. 5).
An overall reduction in numbers of C. coli cells was observed on the 15th day of the experiment with respect to the population at 42 h (Fig. 5). The enumeration of the viable fraction of the stressed C. coli cells indicated similar (P > 0.05) results among the applied PMA-qPCR methods with spheroplasting irrespectively of the EDTA concentration used. Moreover, the results of these methods were not significantly different (P > 0.05) from the results with the negative control for Lys/EDTA. On the other hand, the counts obtained by the aforementioned methods were significantly lower (P < 0.05) than those of qPCR and the negative control for spheroplasting (simple PMA addition) and higher (P < 0.05) than those of the culture-based enumeration.
Finally, the quantification of C. coli on the 42nd day revealed a further decline of the total cell count (Fig. 5). No viable and culturable campylobacters were detected on CBA plates, indicating that their population was below the limit of quantification. The total count as quantified by qPCR was significantly higher (P < 0.001) than the counts obtained by all PMA-qPCR methods. Contrarily, all PMA-qPCR methods with spheroplasting, irrespective of the EDTA concentration used, and the negative control for Lys/EDTA yielded similar viable-cell counts (P > 0.05). These counts were found to be significantly (P < 0.008) lower than those of PMA-qPCR with simple PMA addition, with the exception of those of PMA-qPCR with 100 mM EDTA during spheroplasting, which produced similar results (P = 0.060).
With regard to the ISPC for each applied method during the monitoring of the aging C. coli culture, the obtained PMA-qPCR counts are also presented in Fig. 5. The false-positive signals of the cells collectively inactivated by H2O2 varied among the applied methods, though lysozyme-based protocols with up to 25 mM EDTA generated similar results (P > 0.05) at all time points tested. The higher EDTA concentrations (50 and 100 mM) generated the highest qPCR signals compared to those of the rest of the methods at 42 h and 15 days. Simple PMA addition resulted in higher overall false-positive signals than those produced with spheroplasting with 12.5 mM EDTA at all time points and with all spheroplasting methods with up to 100 mM EDTA at day 42.
Application of the final quantification procedure in inoculated meat samples.
The quantification of C. coli cells on inoculated meat surfaces (25 cm2) for each time point are presented in Table 3. The applicability of the final quantification procedure (Fig. 6) in meat samples was verified, since spheroplasting-PMA-qPCR with ISPC and plating on modified charcoal cefoperazone deoxycholate agar (mCCDA) yielded similar counts (P > 0.05) when a two-way analysis of variance (ANOVA) examining the effects of experimental occasion and quantification method was applied (Table 3).
TABLE 3.
Quantification of C. coli cells on three artificially inoculated meat pieces by three methods
| Expt(s) | Cell count obtained by indicated quantification methoda |
||
|---|---|---|---|
| qPCR | Spheroplasting-PMA-qPCR with ISPC |
Plating on mCCDA | |
| 1 | 6.59 ± 0.20 A | 6.31 ± 0.36 A | 5.44 ± 0.15 B |
| 2 | 6.57 ± 0.24 A | 5.71 ± 0.29 B | 5.64 ± 0.24 B |
| 3 | 6.60 ± 0.16 A | 5.36 ± 0.40 B | 5.92 ± 0.32 B |
| 1, 2, 3 | 6.59 ± 0.18 C | 5.79 ± 0.51 D | 5.67 ± 0.30 D |
Testing was carried out in triplicate during three independent experiments by qPCR (log10 cell equivalents per 25 cm2), spheroplasting-PMA-qPCR with ISPC (log10 cell equivalents per 25 cm2), and plating on mCCDA (log10 CFU per 25 cm2). Means with “A” or “B” after them indicate that counts were compared by ANOVA within each experiment. Means with “C” or “D” after them indicate that counts were compared by two-way ANOVA. The factors examined were experimental occasion and quantification method in all three experiments. Counts with the same letter within the same experiment are not statistically significantly different (P > 0.05) from each other, but they differ significantly (P < 0.05) from counts with a different letter.
Quantification range of PMA-PCR using spheroplasts and ISPC.
Figure 7 illustrates the results of the correlation between the CT-based quantification of viable-cell equivalents by spheroplasting-PMA-qPCR with ISPC and the weighted means of the culture-based enumeration on CBA plates with regard to pure C. coli overnight cultures (exponential phase) and overnight cultures mixed with aged cultures. In all cases, the observed high linear relationship between the results of the two methods (R2 > 0.96) indicated the negligible variation between the respective obtained counts over several decimal dilutions (1.5 to 8.5 log10 cell equivalents ml−1) of viable and culturable C. coli bacteria (exponential phase) even in the concurrent presence of dead cells. However, a lower variability of the counts obtained by spheroplasting-PMA-qPCR was observed in triplicate aliquots with 2.5 to 8.5 log10 C. coli cell equivalents ml−1.
FIG 7.
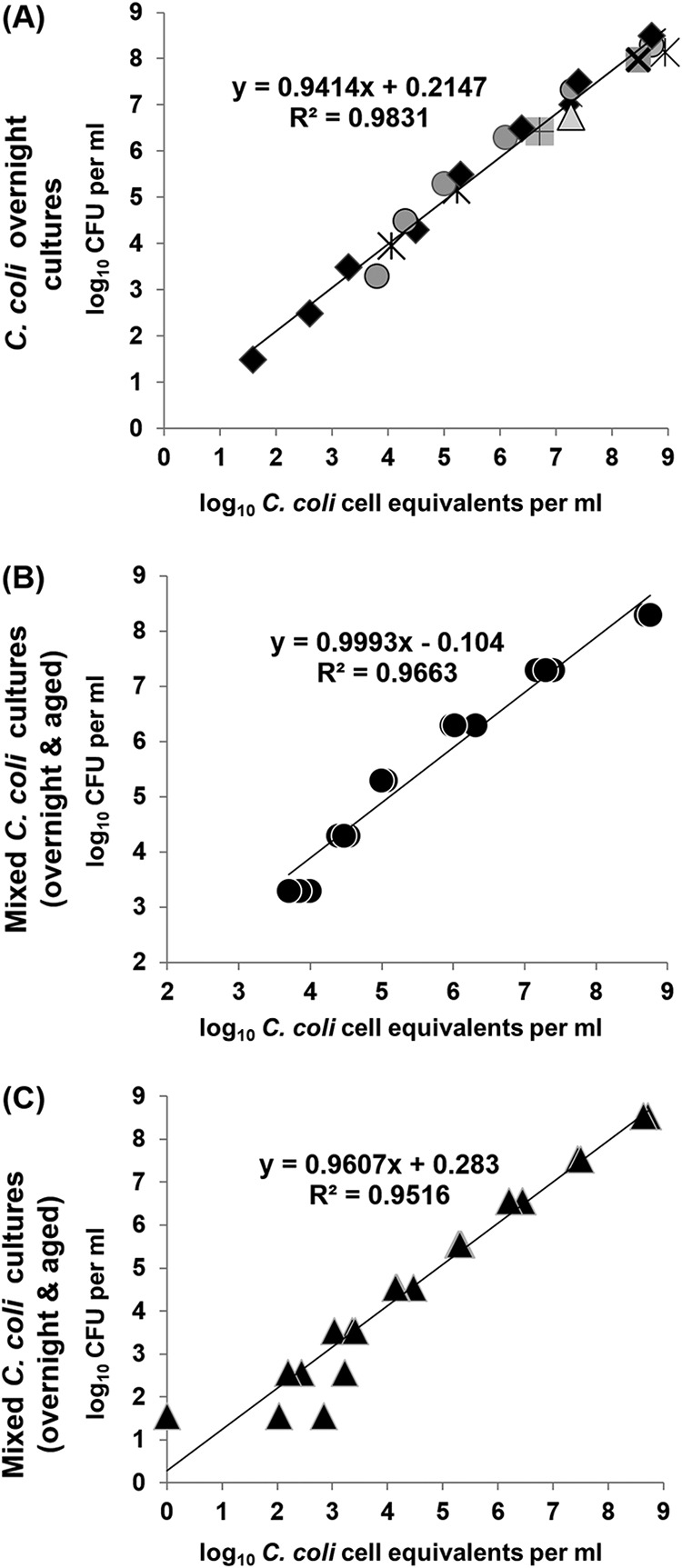
Correlation of viable C. coli counts obtained by the weighted means of numbers of CFU on duplicate CBA plates (y axis) and cell equivalents in triplicate aliquots obtained by spheroplasting-PMA-qPCR with ISPC (x axis). (A) Various overnight cultures during seven independent experiments (four original suspensions and three original suspensions with 10-fold serial dilutions; 1.5 to 8.5 log10 CFU ml−1); (B) original suspension and 10-fold serial dilutions of an overnight culture (3.3 to 8.3 log10 CFU ml−1), all mixed individually with 6 log10 CFU of a 96-day-old culture in CAMHB; (C) original suspension and 10-fold serial dilutions of a single overnight culture (1.5 to 8.5 log10 CFU ml−1), all mixed individually with 5 log10 CFU of a 103-day-old culture in CAMHB. Each symbol represents the mean value of triplicate aliquots (A) and the value of one aliquot (B and C). The corresponding linear-regression equations and correlation coefficient (R2) values are presented in the diagrams.
DISCUSSION
The DNA-binding capacity of PMA is a vital parameter during PMA-qPCR in view of the fact that PMA concentration has been recognized as one of the factors contributing to the efficiency of the dye to reduce the signal from dead bacterial cells (8, 38). In the present study, this capacity was addressed primarily by adding various concentrations of PMA to a pure double-stranded DNA solution of the C. coli reference strain. The DNA-binding capacity of PMA was not absolute even for the highest PMA concentration used, indicating that the amplification of free (extracellular) DNA can lead to erroneous overestimations of viable-bacterium counts during PMA-qPCR. In order to eliminate such bias, an initial centrifugation step and then removal of the supernatant, washing, and harvesting of the pelleted bacterial cells were included in the final quantification procedure.
In this study, the selective entrance of PMA into dead C. coli cells was facilitated by launching a spheroplasting pretreatment prior to the addition of PMA. This approach was based on the fact that during spheroplast formation, outer membranes of Gram-negative bacteria, such as Campylobacter, are fragmented by lysozyme and EDTA, and these changes lead to an increased permeability of the cell wall, though the cytoplasmic membrane remains intact (39–43). However, high EDTA concentrations or prolonged action may induce bacterial cell rupture (41). The present study investigated the effect of different EDTA concentrations in the presence of lysozyme and sucrose (S1 buffer) and the optimum incubation that would create an increased osmotic concentration in the extracellular space of the bacteria while avoiding cell rupture. The succeeding addition of S2 buffer without the presence of the osmotic stressor (sucrose) induces an osmotic shock to spheroplasts accompanied by cell swelling due to the diffusion of water (44). This physiological phenomenon has been exploited in this study for enhancing the selective entrance of the soluble PMA via diffusion into the dead C. coli cells, namely, by adding the dye into the S2 buffer immediately before the latter was added to the spheroplasts. The illumination of the samples was also enhanced by using transparent PCR tubes for sample treatments. Eventually, the superiority of the PMA treatment with spheroplasting compared to the simple addition of PMA for viable-cell quantification by qPCR was verified in the present study. In particular, the observed signal reduction (ΔCT) of heat-inactivated C. coli cells during PMA-qPCR was higher for the treatment with spheroplast formation than for simple PMA addition with respect to qPCR (non-PMA-treated cells). These results are in line with the fact that the magnitude of DNA inactivation during PMA-qPCR in bacterial cells depends principally on the kinetics of the membrane permeability of the dye, which is essentially governed by diffusion without regard to active export conducted by viable cells (18). Moreover, the absence of cell rupture due to swelling and the integrity of the cytoplasmic membranes of spheroplasts have been verified in several independent experiments of this study via the observed high linear relationship between the counts obtained by qPCR and PMA-qPCR for different concentrations of EDTA with regard to viable C. coli cells in exponential phase.
Levels of suppression of dead-cell qPCR signals by PMA have been reported to vary among different matrices and experimental conditions, leading to false-positive signals (8, 10–13). These signals may be addressed by an internal sample process control (ISPC), which would allow for the heterogeneity of samples and variations during treatments within each sample subjected to PMA-qPCR. Preferably, an ISPC for PMA-qPCR should be comprised of inactivated cells subjected to the same treatments and with populations equal to those in the original sample. The necessity of such an ISPC has been explicitly highlighted previously (18). In the current study, an ISPC addressing the aforementioned experimental needs has been developed and incorporated into the final quantification procedure. For the generation of dead Campylobacter cells, lethal heat treatments have been used mainly in previous PMA-qPCR-relevant studies (10, 13, 18, 45). Inactivation of campylobacters by oxidative stress has also been applied previously using H2O2 (18). In this study, two lethal heat treatments and an H2O2 treatment were evaluated on the basis of the relative magnitude of CT shift (ΔCT) compared to that of the non-PMA-treated controls during qPCR with regard to both overnight and aged C. coli cultures, with H2O2-treated cells yielding the highest ΔCT values in all cases. These results may be explained by the inconsistent loss of DNA from bacterial cells upon heat inactivation (18), the amplification of which was not absolutely suppressed by PMA. Therefore, false-positive signals during PMA-qPCR may originate not only from metabolically inactive “ghost” bacteria that have an intact cell wall/membrane (7, 46) but also from extracellular DNA. The abrupt and “nonphysiological” damage of the cytoplasmic membrane due to heat treatment along with the conditional DNA-binding capacity of PMA induce uncontrolled bias during the interpretation of qPCR results. Contrarily, the broad-spectrum biocidal action of H2O2 is well recognized (47). Oxidative stress probably stands for the most common cause of the physiological death of Campylobacter once it is outside the intestine niche (e.g., on foodstuffs), and treatment with H2O2 resembles this natural procedure (18). Consequently, H2O2 treatment was chosen as the most appropriate inactivation method for generating the ISPC in this study.
The simple osmotic shock induced by S1 and S2 buffers without lysozyme and EDTA (Lys/EDTA) resulted in enhanced PMA entrance into dead cells at days 15 and 42 but not at 42 h of naturally aging C. coli under oxidative stress. These results indicate that entrance of PMA to dead cells in aged cultures can be facilitated by simple diffusion without Lys/EDTA being present. In contrast, in cultures that were not as old (42 h), the simple osmotic shock treatment with PMA yielded statistically significantly higher viable-cell counts than CBA plate enumeration, indicating that the rigidity of the cell wall was relatively high in the naturally dead cells of this age, resulting in relatively limited PMA entrance. This rigidity seems to be a critical factor during PMA-qPCR and may also vary among different strains, culture media, and niches, such as meat surfaces. Our data support the hypothesis that treatment with Lys/EDTA can address such variations during PMA-qPCR quantification. More precisely, counts obtained at 42 h by PMA-qPCR with spheroplasting (lysozyme and EDTA, 0 to 100 mM) were similar to those obtained by CBA plate enumeration. These similar counts were significantly lower than those obtained by PMA-qPCR utilizing simple PMA addition, whereas the latter method resulted in counts similar to those of qPCR (non-PMA-treated control). The fact that the metabolically active VBNC cells cannot be detected on agar plates may explain the higher Campylobacter counts obtained by PMA-qPCR than by culture-based enumeration, which is in accordance with findings of other studies (7, 13, 18, 45). In the present study, however, the fact that PMA-qPCR with simple PMA addition generated significantly higher counts than PMA-qPCR using spheroplast formation points out that the qPCR signal suppression of dead cells was greater in the latter at all time points. This indicates that spheroplasting is effective in enhancing the entrance of PMA into dead C. coli cells, thus allowing an accurate quantification of viable cells. With regard to the optimum EDTA concentration during spheroplasting, relatively high concentrations should be avoided due to the risk of provoking cell rupture, resulting in DNA leakage, insufficient PMA cross-linking, and higher false-positive qPCR signals. Indeed, spheroplasting with 100 mM EDTA generated signals that resulted in higher viable-cell counts than with lower EDTA concentrations at all tested time points of the naturally aging C. coli cells. Instead, an EDTA concentration between 12.5 and 25 mM is proposed as a safer approach for future relevant studies.
The application of ISPC to aging C. coli cells indicated the presence of false-positive signals during qPCR originating from the inability of PMA to provoke an absolute suppression of DNA amplification. PMA-qPCR with spheroplasting after cell inactivation, except with high EDTA concentrations (50 to 100 mM), resulted in similar false-positive counts. These counts were also similar to those obtained by the negative controls for Lys/EDTA (simple osmotic shock) and for spheroplasting (simple PMA addition) at 42 h and at day 15 but significantly lower at day 42. These results indicate that the biochemical composition of the cell wall of the aged C. coli population at day 42 obstructed the entrance of PMA to a subpopulation of the H2O2-treated cells unless lysozyme and EDTA were present. It has been reported that bacteria in the VBNC state have a cell wall strengthened by increased peptidoglycan cross-linking (48), and the existence of such cells in the aged C. coli population may justify the aforementioned results.
The qPCR signal of each ISPC, which stands for variations in PMA performance generating residual false-positive results, was considered negligible once its CT significantly exceeded the CT of the corresponding noninactivated PMA-treated aliquot (ΔCT > 10). In this study, viable-cell-equivalent counts were not considerably altered by subtracting the corresponding ISPC counts in cases of C. coli cultures with viable cells in relatively high numbers (>4 log10 CFU ml−1). Contrarily, a notably more accurate quantification of the viable fraction of C. coli cells by the application of ISPC was observed when viable cells were <4 log10 CFU ml−1 in the concurrent presence of an equal number or up to 2 log10 CFU ml−1 higher population of dead cells (data not shown). Nevertheless, in the present study, the application of the ISPC has extended the linear range of quantification in populations with low numbers of viable cells when high numbers of dead cells were concurrently present. Therefore, the utility of an ISPC for samples with an unknown composition of a targeted bacterial population can contribute toward a more accurate quantification of its viable-cell fraction.
The final spheroplasting-PMA-qPCR procedure with ISPC presented a high quantification range of C. coli cells in cultures in exponential phase (1.5 to 8.5 log10 viable C. coli cell equivalents ml−1) in the concurrent presence of dead cells from aged cultures, whereas the correlation with counts obtained by plating was high. The application of the same procedure to meat samples artificially inoculated with C. coli revealed no statistical difference between the viable-cell counts obtained by PMA-qPCR and plating on mCCDA, with mCCDA being the selective medium of choice according to the standard method (49). These results indicate that the optimized PMA-qPCR was not hampered either by the presence of organic components (e.g., blood, fats) or by the autochthonous microbiota of meat. The significantly higher C. coli counts obtained by qPCR than by the two aforementioned methods may be justified by the presence of dead C. coli cells on the meat samples, probably originating from the stationary phase of the initial inoculum and the cold stress induced by the storage of the meat samples at 4°C prior to the analysis. Nevertheless, a wider spectrum of applications to meat samples is necessary in order to reinforce the reproducibility of such results.
In conclusion, this study proposes the application of spheroplasting as a pretreatment of Gram-negative bacteria prior to the addition of PMA and ISPC during viable-cell detection irrespective of whether pure cultures or dilutions of food samples are the origin of the tested bacterial suspensions. Although C. coli served as the bacterial model in this study, the proposed pretreatment and ISPC may be combined with any DNA extraction protocol and qPCR assay for the detection of viable cells of other Gram-negative bacteria. In anticipation of a continued future demand for viable-cell detection by PMA-qPCR, the final quantification procedure that was developed in this study may serve as a useful tool, which can be adjusted for the quantitative detection of other foodborne pathogens by addressing different matrix-dependent residual signals from dead cells.
MATERIALS AND METHODS
Bacterial strains and cultivation.
C. coli ATCC 43478 (LGC Standards GmbH, Wesel, Germany) was recovered on CBA plates (bioMérieux, Marcy-l’Étoile, France) from −80°C in nutrient broth no. 2 (Oxoid, Basingstoke, England) supplemented with 5% lysed horse blood (Oxoid) and 20% glycerol (VWR Chemicals BDH, Lutterworth, England), isolated on mCCDA (Oxoid), and then subcultured again on CBA. Plates were incubated at 41.5°C ± 1°C for 24 h under microaerobic conditions (GENbox microaer and GENbox jar; bioMérieux). For the generation of exponential-phase cultures, a pure colony from a CBA plate was inoculated in 100 ml of cation-adjusted Mueller-Hinton broth (CAMHB; BBL BD, Franklin Lakes, NJ) following incubation at 37°C ± 1°C for 17 ± 2 h under microaerobic conditions. The growth of C. coli cells in CAMHB was roughly estimated prior to use by measuring the optical density at 600 nm (OD600) (conversion factor of ∼108 CFU ml−1 for an OD600 of 0.5) using a spectrophotometer (BioPhotometer; Eppendorf, Hamburg, Germany).
DNA extraction and generation of a standard C. coli genome copy number solution.
Genomic DNA was extracted using an in-house-developed protocol as previously described and evaluated for Campylobacter (33). A 24-h pure culture of C. coli on a CBA plate was used for DNA extraction and the generation of a standard DNA solution. The quantity and quality of the C. coli standard DNA were assessed spectrophotometrically (BioPhotometer). C. coli genome copy number per 100 ng of DNA was calculated according to the reported genome size of C. coli (50–52). In all cases, the extracted DNA was eluted with 100 μl Tris-EDTA (TE) buffer (New England Biolabs, Ipswich, MA) and stored at −20°C until further analysis.
Real-time qPCR.
For the detection of C. coli via qPCR, a unique nucleotide region within the C. coli serine hydroxymethyltransferase (glyA) single-copy gene (one genome copy represents one cell equivalent) was targeted (53) because it is identified as specific for this species. The previously reported C. coli-specific primers and TaqMan hydrolysis probe (53) were used with minor modifications of the upstream primer and the probe. The primers amplifying 129 bp of the glyA gene were Cc-F (5′-TGTAAAACCAAAGCTTATCGTGTGC-3′) and Cc-R (5′-AGTCCAGCAATGTGTGCAATG-3′), and the probe was Cc-FAM (5′–6-carboxyfluorescein [FAM]–AGCTCCAACTTCATCCGCAATCTCTCT-BHQ1–3′). All oligonucleotides were compared to sequences in the NCBI database using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) in order to exclude any homology to nontarget sequences. The optimized qPCR (20 μl) was comprised of 1× ThermoPol DF (detergent-free) reaction buffer (New England Biolabs), 0.2 μM each primer, 0.4 μM the TaqMan probe, 0.2 mM each deoxynucleoside triphosphate (dNTP), 2.5 mM MgSO4, 4 U of HotStarTaq DNA polymerase (Qiagen, Hilden, Germany), and 6 μl of DNA extract. For no-template-control (NTC) reactions, 6 μl of nuclease-free water was tested. The following thermal cycling conditions were applied: 95°C for 15 min (Taq polymerase activation), followed by 45 cycles in 2 steps: (i) 95°C for 30 s (denaturation) and (ii) 60°C for 50 s (combined annealing and extension). Fluorescence levels were recorded at the end of each cycle. Reactions were performed on a CFX96 Touch real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA).
The specificity of the primer-probe set for the detection of the C. coli-specific glyA gene by real-time PCR was confirmed by testing pure genomic DNA (approximately 6 log10 genome copies per PCR) of reference strains of Campylobacter (C. coli ATCC 43478 and C. jejuni ATCC 33291), Escherichia coli (ATCC 8739), Salmonella enterica subsp. enterica serovar Typhimurium (ATCC 13311), Pseudomonas aeruginosa (NCTC 10662), Listeria monocytogenes (NCTC 11994), and Staphylococcus aureus (ATCC 25923). Respective genomic DNA preparations of molecularly characterized C. coli (n = 94) and C. jejuni (n = 71) isolates from our in-house collection (33, 34), originally isolated from sheep and goat samples from abattoir and retail settings, were also tested.
Serial 10-fold dilutions of the C. coli ATCC 43478 pure DNA (107 to 100 copies per reaction) were prepared in TE buffer (AppliChem, Darmstadt, Germany) containing 0.04 μg/μl carrier RNA (Qiagen). Each of these calibrator dilutions was run in triplicate, and a standard curve was generated in order to estimate the amplification efficiency and the linearity of the method. For the determination of the limit of detection (LOD), 2-fold serial dilutions representing 32 C. coli cell equivalents down to 1 C. coli cell equivalent per assay were also prepared in TE containing carrier RNA. Each dilution was tested in eight independent replicates, and the LOD was determined with a 95% probability of detection by applying probit regression analysis. The CT value of the LOD was selected as the CT cutoff for the qPCR assay (54). The repeatability of the developed assay was evaluated by testing 20 C. coli ATCC 43478 genome copies in quintuplicate in three separate runs. Mean CT values, standard deviations (SD), and coefficients of variation (CV) were determined within each run (intra-assay) and between the three runs (interassay).
Spheroplast formation.
Prior to the addition of PMA itself, the weakening of the C. coli cell wall using lysozyme digestion of bacterial peptidoglycan, which leads to spheroplast formation, was assessed by modifying a previously described method (55). In brief, 0.5 ml of a hyperosmotic buffer (S1) containing PBS, sucrose, EDTA, and lysozyme (all reagents from Sigma-Aldrich, Steinheim, Germany) was added to each C. coli sample at a ratio of 1:1 in a microcentrifuge tube (2.0 ml; Kisker Biotech GmbH & Co., Steinfurt, Germany). The final concentration was 10 mM for PBS (pH 7.0), 250 mM for sucrose, and 0.5 mg ml−1 for lysozyme, whereas various final concentrations of EDTA (12.5, 25, 50, and 100 mM) were investigated. C. coli cell suspensions were incubated with the S1 buffer at 37°C in order to attain optimum lysozyme activity according to the manufacturer’s instructions. After the incubation with the S1 buffer, a second buffer (S2) containing PBS (10 mM, pH 7.0) and EDTA was added to each sample at a ratio of 1:1 by pipetting gently in order to induce a mild osmotic shock, accompanied by the entrance of water into the cells (spheroplast swelling) (44). Then, samples were further incubated at 37°C so as to prolong the activity of lysozyme. The concentrations of EDTA were always identical in S1 and S2 buffers. Spheroplast formation was assessed by measuring the decrease in the optical density (OD600) of C. coli cultures in CAMHB (cells in exponential phase and heat inactivated at 72°C for 10 min) throughout the incubation with the S1 and S2 buffers every 5 min. Spheroplast formation was regarded as complete when no further reduction in the optical density was observed (39, 41, 44).
PMA illumination conditions.
PMA (Biotium Inc., Hayward, CA) was initially dissolved in 20% dimethyl sulfoxide (DMSO; Sigma-Aldrich) in order to obtain a 2 mM stock solution, which was stored at −20°C in the dark until used on each occasion. In order to cross-link PMA to the DNA (photoactivation), PCR tubes (clear, 0.2 ml; Kisker Biotech GmbH & Co.) containing the samples were exposed to thermally stable light produced by Cree XP-E Blue light-emitting diodes (LEDs) with 465- to 485-nm emission wavelengths (Cree Inc., Durham, NC), coupled with 10.0-mm narrow-spot plain total internal reflection (TIR) lenses (model 10412; Carclo Optics, Aylesbury, United Kingdom). The LED lenses were firmly placed in arrays inside an opaque plastic rack (Kisker Biotech GmbH & Co.) at a 1-cm distance from the bottom of the corresponding tube. Each LED lens was driven at approximately 2 watts using a constant current of 0.6 ampere.
PMA cross-linking to extracellular DNA.
In order to evaluate the DNA-binding capacity of PMA, PMA was added at various final concentrations (50, 100, and 200 μM) to a pure double-stranded DNA solution in quadruplicate (2.3 × 106 C. coli genome copies per sample). Four aliquots from the original pure DNA solution served as quantitative standards. The transparent PCR tubes containing the samples (total volume, 40 μl) were incubated in the dark at room temperature for 1 h, after which they were illuminated for 2 min, as described previously, in order to provoke photo-cross-linking of PMA to the DNA. All samples were then subjected to DNA extraction followed by qPCR. The standard curve equation was used to calculate the number of C. coli copies per assay.
Optimization and evaluation of PMA treatment.
For optimization of sample treatment with PMA, 2 ml of C. coli cells in exponential phase were transferred individually to two microcentrifuge tubes. The culture in one tube was heat inactivated at 72°C for 10 min, and the absence of viable C. coli bacteria was confirmed by plating on CBA plates. After inactivation, both tubes were centrifuged (12,000 × g for 10 min), and the harvested cells were washed with 1 ml of 20 mM PBS, repelleted, and resuspended in 0.5 ml PBS. From these final suspensions of viable and inactivated cells, aliquots of 18 μl were transferred to clear PCR tubes (0.2 ml) in order to generate three sets of triplicate aliquots; the first set was subjected to qPCR (quantitative control), the second to PMA treatment (simple PMA addition) and qPCR, and the third to spheroplast formation and PMA treatment followed by qPCR (spheroplasts and PMA). Eighteen microliters of normal saline (0.9% NaCl) was added to each aliquot of the second set, followed by the addition of PMA to a final concentration of 50 μM. Regarding the third set, 18 μl of double-distilled water (ddH2O) was added to each aliquot, followed by the addition of 39 μl of S1 buffer containing 50 mM EDTA (25 mM final EDTA concentration in the spheroplast suspension) and incubation at 37°C for 20 min. S2 buffer was supplemented with PMA immediately before it was added to each S1 buffer-treated aliquot at a ratio 1:1, so that the final PMA concentration was 50 μM per aliquot. After the addition of PMA, all aliquots were incubated at 37°C under dark conditions for 20 min, followed by a cooling step at 4°C for 10 min, also under dark conditions, so as to minimize any subsequent overheating during illumination. Two illumination times (2 and 4 min) were investigated for optimal assay performance. Finally, all aliquots were subjected to DNA extraction and qPCR. The mean count of the triplicate aliquots was used for the quantification of viable C. coli cells per treatment.
Independent experiments were conducted in order to evaluate whether spheroplasting would compromise the integrity of the cytoplasmic membrane of viable cells, which is a criterion of cell viability and a barrier to PMA entrance. More specifically, C. coli cells in exponential phase were quantified in triplicate both by qPCR and PMA-qPCR after being subjected to spheroplasting using various EDTA concentrations in S1 and S2 buffers (final concentrations, 12.5, 25, and 50 mM). The correlation between the counts obtained by the two methods was calculated, with consideration that any cytoplasmic membrane damage due to spheroplasting would allow the entrance of PMA into originally viable cells, thus resulting in lower counts obtained by PMA-qPCR than by qPCR.
ISPC.
For the ISPC, various inactivation methods were tested in order to identify the one that generated the minimum residual signal (false positive) during PMA-qPCR. Suspensions in CAMHB (1 ml) of overnight (exponential-phase) and aged (a 56-day-old culture on CBA plate stored at 4°C under aerobic conditions) C. coli cultures were individually transferred to a microcentrifuge tube for centrifugation (12,000 × g for 10 min). The harvested cells were washed with 1 ml of 20 mM PBS, repelleted, and resuspended in 0.5 ml of PBS (20 mM). The procedure for developing the ISPC included the following steps: inactivation, spheroplasting (S1 buffer), PMA treatment (S2 buffer), and, finally, DNA extraction followed by qPCR. Three inactivation methods were applied per triplicate aliquot (18 μl each) of the final C. coli suspension in PBS: (i) lethal heat treatment at 95°C for 5 min (13, 18, 45), (ii) lethal heat treatment at 75°C for 30 min with modification of a previously reported temperature-time combination (18), and (iii) oxidative stress (18) induced by the addition of an equal volume of 3.0% H2O2 solution to each aliquot (final concentration, 1.5% H2O2) and incubation at 42°C for 50 min, followed by the addition of 1 μl catalase buffer (33 mg ml−1 catalase, 50 mM PBS, pH 7.0; Sigma-Aldrich) per aliquot to degrade the excess amount of H2O2. The absence of culturable C. coli bacteria in the inactivated samples was assessed by plating them on CBA plates. On each experimental occasion, additional triplicate aliquots (18 ml) that were not subjected to inactivation but instead to DNA extraction and qPCR served as quantitative controls. The mean value from the triplicate aliquots was used for the quantification of C. coli cells per treatment. The selection criterion for the best inactivation method for the development of the ISPC was the relative magnitude of shift between the observed CT values (ΔCT) of the inactivated cells and of the corresponding quantitative controls during qPCR.
Final quantification procedure.
The final procedure for the quantification of culturable, total, and viable C. coli cells in suspensions (pure cultures or swab rinses from inoculated meat samples as described below) is presented in Fig. 6. In brief, C. coli cells were harvested by centrifugation (12,000 × g for 10 min), washed once with 20 mM PBS, repelleted, and resuspended in PBS. The centrifugation steps were applied in order to harvest the bacterial cells and to discard any extracellular DNA present in the supernatant. For the culture-based enumeration, two aliquots (100 μl) were subjected to 10-fold serial dilutions in buffered peptone water (BPW; Merck, Darmstadt, Germany), followed by inoculation on double CBA plates for samples of pure suspensions or on mCCDA plates for inoculated meat samples. The qPCR assay was applied for the confirmation of presumptive Campylobacter colonies on mCCDA plates. The weighted mean was used for the calculation of CFU (56). Nine additional aliquots (18 μl each) in triplicates were transferred to transparent PCR tubes (0.2 ml; Kisker Biotech GmbH & Co.) for the successive treatments. The first set of triplicate aliquots, which were directly subjected to DNA extraction and qPCR, served as quantitative controls (total C. coli cell equivalents). The second and third sets of triplicates corresponded to the optimized spheroplasting-PMA-qPCR procedure with ISPC, namely, PMA-treated cells (spheroplasts) and ISPC (H2O2 inactivation and spheroplasts), respectively, which were finally subjected to DNA extraction followed by qPCR (Fig. 6). For the molecular quantifications, each CT value obtained by qPCR was transformed to the number of cell equivalents per milliliter based on the standard curve and the corresponding dilution factor for the original sample. The false-positive qPCR signals and the variations in PMA performance were estimated by the ISPC. In particular, the mean count of the PMA-qPCR triplicate aliquots minus the mean count of the corresponding ISPC triplicate aliquots was considered the viable-cell population in each sample. Consequently, culture-based enumeration, PMA-qPCR (minus the ISPC value), and qPCR were used to obtain viable and culturable, viable, and total C. coli cell counts in each sample, respectively.
Application of the final quantification procedure in an aging C. coli population.
An aging C. coli culture was used for the comparison of the quantification results obtained by culture on CBA plates, qPCR, and spheroplasting-PMA-qPCR with ISPC according to the final procedure (Fig. 6) using various concentrations of EDTA for spheroplasting. Moreover, a fourth set of triplicate aliquots of 18 μl subjected to simple addition of PMA without spheroplasting was included in the comparisons. In particular, 1 ml of C. coli cells in exponential phase was inoculated in 250 ml of CAMHB, which was further incubated at 37°C ± 1°C for 42 h (long stationary phase) under microaerobic conditions and then stored at room temperature (20°C ± 2°C) under aerobic conditions for up to 42 days. Culturable, viable, and total C. coli cell counts were enumerated at 42 h, 15 days, and 42 days, according to the steps described in the final quantification procedure (Fig. 6). The different quantification methods and S1 buffer compositions are presented in Table 2.
Application of the final quantification procedure in inoculated meat samples.
The final quantification procedure was applied to artificially inoculated meat samples during three independent experiments on different days. Deboned lamb legs (butt portion) were purchased from a retail market, and the C. coli contamination status of the meat was tested by subjecting five aliquots of 1 ml, originating from 50 g of meat diluted in a final volume of 50 ml of BPW, to DNA extraction and qPCR. Once the C. coli-free status of the lamb meat was verified, three pieces (25-cm2 meat surface) were taken for each experiment by means of a sterile lancet and placed individually in sterile petri dishes. No method was applied for the reduction of the autochthonous microbiota of the samples. The meat pieces were inoculated by uniformly placing on their surface 100 μl of a wild C. coli strain suspension in CAMHB (22 ± 2 h culture, OD600 of 0.2), which was originally isolated from a lamb meat surface at an abattoir (in-house collection) (34). Subsequently, the samples were stored at 4°C for 4 ± 2 h to allow adherence of the bacterial cells to the meat surface. A sterile sponge (Whirl-Pak Speci-Sponge; Nasco, NY, USA) that was cut aseptically (0.5 cm by 1 cm) and premoistened with PBS (20 mM) was used to sample the surface of each meat piece (25 cm2). The sponge was then transferred in a microcentrifuge tube containing 2 ml of PBS (20 mM) and homogenized for 1 min in a vortex shaker (IKA-Werke GmbH & Co. KG, Staufen, Germany). One milliliter of the suspension was subjected to the final procedure treatments (Fig. 6). The final concentration of EDTA in each aliquot subjected to spheroplasting was 12.5 mM.
Quantification range of spheroplasting-PMA-qPCR with ISPC and correlation with plating.
The quantification range of spheroplasting-PMA-qPCR with ISPC was explored in C. coli cultures in exponential phase and decimal dilutions thereof in triplicates (1.5 to 8.5 log10 CFU ml−1) during seven independent experiments. Two of these experiments involved mixed viable C. coli cells in exponential phase and dead C. coli populations. In particular, 10-fold serial dilutions in triplicates of an overnight C. coli culture (3.3 to 8.3 log10 CFU ml−1) were mixed individually with a 96-day-old culture in CAMHB (6 log10 dead cells ml−1 as estimated by qPCR) and 10-fold serial dilutions in triplicates of an overnight culture (1.5 to 8.5 log10 CFU ml−1) were mixed individually with a 103-day-old culture in CAMHB (5 log10 dead cells ml−1 as estimated by qPCR). The absence of culturable C. coli bacteria in the old cultures was assessed by plating them on double CBA plates. In the case of 10-fold serial dilutions with estimated counts of <6 log10 CFU ml−1 of an original C. coli suspension, 6 log10 CFU of heat-killed (95°C for 10 min) E. coli ATCC 8739 cells was added to each sample in order to form a clearly visible pellet during centrifugation. On each occasion, the experimental steps of the final procedure (Fig. 6) were followed (initial culture and decimal dilutions). The final concentration of EDTA in each aliquot subjected to spheroplasting was 12.5 mM. The quantification results between the PMA-qPCR and the culture-based enumeration on CBA plates were used for the generation of linear-regression equations.
Statistical analysis.
All the statistical analyses were performed using IBM SPSS version 25 (IBM Corp., Armonk, NY). The assumptions of normality and homogeneity of variances were assessed using the Shapiro-Wilk test and Levene’s test, respectively. On each experimental occasion, the differences between the C. coli cell counts obtained by each method were statistically analyzed using ANOVA. Subsequently, a two-way ANOVA was conducted to examine the effect of the quantification method and experimental occasion on the C. coli counts. The least significant difference (LSD), Tukey honestly significant difference (HSD), and Bonferroni post hoc tests were used, where applicable, to allow for adjustment of multiple comparisons. A P value below 0.05 was considered statistically significant. Microsoft Office Excel 2010 was used for the generation of linear-regression equations.
ACKNOWLEDGMENTS
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
REFERENCES
- 1.Zeng D, Chen Z, Jiang Y, Xue F, Li B. 2016. Advances and challenges in viability detection of foodborne pathogens. Front Microbiol 7:1–12. doi: 10.3389/fmicb.2016.01833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elizaquível P, Aznar R, Sánchez G. 2014. Recent developments in the use of viability dyes and quantitative PCR in the food microbiology field. J Appl Microbiol 116:1–13. doi: 10.1111/jam.12365. [DOI] [PubMed] [Google Scholar]
- 3.Rollins DM, Colwell RR. 1986. Viable but nonculturable stage of Campylobacter jejuni and its role in survival in the natural aquatic environment. Appl Environ Microbiol 52:531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oliver JD. 2010. Recent findings on the viable but nonculturable state in pathogenic bacteria. FEMS Microbiol Rev 34:415–425. doi: 10.1111/j.1574-6976.2009.00200.x. [DOI] [PubMed] [Google Scholar]
- 5.Stingl K, Knüver MT, Vogt P, Buhler C, Krüger NJ, Alt K, Tenhagen BA, Hartung M, Schroeter A, Ellerbroek L, Appel B, Käsbohrer A. 2012. Quo vadis?—Monitoring Campylobacter in Germany. Eur J Microbiol Immunol 2:88–96. doi: 10.1556/EuJMI.2.2012.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L, Mendis N, Trigui H, Oliver JD, Faucher SP. 2014. The importance of the viable but non-culturable state in human bacterial pathogens. Front Microbiol 5:258. doi: 10.3389/fmicb.2014.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duarte A, Botteldoorn N, Coucke W, Denayer S, Dierick K, Uyttendaele M. 2015. Effect of exposure to stress conditions on propidium monoazide (PMA)-qPCR based Campylobacter enumeration in broiler carcass rinses. Food Microbiol 48:182–190. doi: 10.1016/j.fm.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 8.Fittipaldi M, Nocker A, Codony F. 2012. Progress in understanding preferential detection of live cells using viability dyes in combination with DNA amplification. J Microbiol Methods 91:276–289. doi: 10.1016/j.mimet.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Nocker A, Cheung CY, Camper AK. 2006. Comparison of propidium monoazide and ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J Microbiol Methods 67:310–320. doi: 10.1016/j.mimet.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 10.Wagner AO, Malin C, Knapp BA, Illmer P. 2008. Removal of free extracellular DNA from environmental samples by ethidium monoazide and propidium monoazide. Appl Environ Microbiol 74:2537–2539. doi: 10.1128/AEM.02288-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varma M, Field R, Stinson M, Rukovets B, Wymer L, Haugland R. 2009. Quantitative real-time PCR analysis of total and propidium monoazide-resistant fecal indicator bacteria in wastewater. Water Res 43:4790–4801. doi: 10.1016/j.watres.2009.05.031. [DOI] [PubMed] [Google Scholar]
- 12.Chang B, Taguri T, Sugiyama K, Amemura-Maekawa J, Kura F, Watanabe H. 2010. Comparison of ethidium monoazide and propidium monoazide for the selective detection of viable Legionella cells. Jpn J Infect Dis 63:119–123. [PubMed] [Google Scholar]
- 13.Pacholewicz E, Swart A, Lipman LJ, Wagenaar JA, Havelaar AH, Duim B. 2013. Propidium monoazide does not fully inhibit the detection of dead Campylobacter on broiler chicken carcasses by qPCR. J Microbiol Methods 95:32–38. doi: 10.1016/j.mimet.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Luo JF, Lin WT, Guo Y. 2010. Method to detect only viable cells in microbial ecology. Appl Microbiol Biotechnol 86:377–384. doi: 10.1007/s00253-009-2373-1. [DOI] [PubMed] [Google Scholar]
- 15.Soejima T, Schlitt-Dittrich F, Yoshida S. 2011. Polymerase chain reaction amplification length-dependent ethidium monoazide suppression power for heat-killed cells of Enterobacteriaceae. Anal Biochem 418:37–43. doi: 10.1016/j.ab.2011.06.027. [DOI] [PubMed] [Google Scholar]
- 16.Contreras PJ, Urrutia H, Sossa K, Nocker A. 2011. Effect of PCR amplicon length on suppressing signals from membrane-compromised cells by propidium monoazide treatment. J Microbiol Methods 87:89–95. doi: 10.1016/j.mimet.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 17.Nkuipou-Kenfack E, Engel H, Fakih S, Nocker A. 2013. Improving efficiency of viability-PCR for selective detection of live cells. J Microbiol Methods 93:20–24. doi: 10.1016/j.mimet.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Krüger NJ, Buhler C, Iwobi AN, Huber I, Ellerbroek L, Appel B, Stingl K. 2014. “Limits of control”—crucial parameters for a reliable quantification of viable Campylobacter by real-time PCR. PLoS One 9:e88108. doi: 10.1371/journal.pone.0088108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.EFSA (European Food Safety Authority), ECDC (European Centre for Disease Prevention and Control). 2018. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2017. EFSA J 16:5500. doi: 10.2903/j.efsa.2018.5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tam CC, O'Brien SJ, Adak GK, Meakins SM, Frost JA. 2003. Campylobacter coli—an important foodborne pathogen. J Infect 47:28–32. doi: 10.1016/S0163-4453(03)00042-2. [DOI] [PubMed] [Google Scholar]
- 21.Ge B, Wang F, Sjölund-Karlsson M, McDermott PF. 2013. Antimicrobial resistance in Campylobacter: susceptibility testing methods and resistance trends. J Microbiol Methods 95:57–67. doi: 10.1016/j.mimet.2013.06.021. [DOI] [PubMed] [Google Scholar]
- 22.Zhang M, Liu X, Xu X, Gu Y, Tao X, Yang X, Yan G, Zhang J. 2014. Molecular subtyping and antimicrobial susceptibilities of Campylobacter coli isolates from diarrheal patients and food-producing animals in China. Foodborne Pathog Dis 11:610–619. doi: 10.1089/fpd.2013.1721. [DOI] [PubMed] [Google Scholar]
- 23.Torralbo A, Borge C, García-Bocanegra I, Méric G, Perea A, Carbonero A. 2015. Higher resistance of Campylobacter coli compared to Campylobacter jejuni at chicken slaughterhouse. Comp Immunol Microbiol Infect Dis 39:47–52. doi: 10.1016/j.cimid.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 24.EFSA (European Food Safety Authority), ECDC (European Centre for Disease Prevention and Control). 2019. The European Union summary report on antimicrobial resistance in zoonotic and indicator bacteria from humans, animals and food in 2017. EFSA J 17:5598. doi: 10.2903/j.efsa.2019.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du Y, Wang C, Ye Y, Liu Y, Wang A, Li Y, Zhou X, Pan H, Zhang J, Xu X. 2018. Molecular identification of multidrug-resistant Campylobacter species from diarrheal patients and poultry meat in Shanghai, China. Front Microbiol 9:1642. doi: 10.3389/fmicb.2018.01642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.García-Sánchez L, Melero B, Diez AM, Jaime I, Rovira J. 2018. Characterization of Campylobacter species in Spanish retail from different fresh chicken products and their antimicrobial resistance. Food Microbiol 76:457–465. doi: 10.1016/j.fm.2018.07.004. [DOI] [PubMed] [Google Scholar]
- 27.Gillespie IA, O’Brien SJ, Frost JA, Adak GK, Horby P, Swan AV, Painter MJ, Neal KR. 2002. A case-case comparison of Campylobacter coli and Campylobacter jejuni infection: a tool for generating hypotheses. Emerg Infect Dis 8:937–942. doi: 10.3201/eid0809.010817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kärenlampi R, Rautelin H, Schönberg-Norio D, Paulin L, Hänninen ML. 2007. Longitudinal study of Finnish Campylobacter jejuni and C. coli isolates from humans, using multilocus sequence typing, including comparison with epidemiological data and isolates from poultry and cattle. Appl Environ Microbiol 73:148–155. doi: 10.1128/AEM.01488-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Campylobacter MLST Project in Scotland (CaMPS). 2009. The molecular epidemiology of Scottish Campylobacter isolates from human cases of infection using multilocus sequence typing (MLST). Department of Medical Microbiology, University of Aberdeen, Aberdeen, Scotland: https://www.food.gov.uk/sites/default/files/media/document/339-1-595_CaMPS_S14006_Final_Report.pdf. [Google Scholar]
- 30.Roux F, Sproston E, Rotariu O, Macrae M, Sheppard SK, Bessell P, Smith-Palmer A, Cowden J, Maiden MC, Forbes KJ, Strachan NJ. 2013. Elucidating the aetiology of human Campylobacter coli infections. PLoS One 8:e64504. doi: 10.1371/journal.pone.0064504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voidarou C, Alexopoulos A, Plessas S, Stavropoulou E, Fotou K, Tzora A, Skoufos I, Bezirtzoglou E. 2011. Hygienic quality and antibiotic resistance profile of sliced butchery. Anaerobe 17:344–350. doi: 10.1016/j.anaerobe.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 32.Marinou I, Bersimis S, Ioannidis A, Nicolaou C, Mitroussia-Ziouva A, Legakis NJ, Chatzipanagiotou S. 2012. Identification and antimicrobial resistance of Campylobacter species isolated from animal sources. Front Microbiol 3:58. doi: 10.3389/fmicb.2012.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazou T, Dovas C, Houf K, Soultos N, Iossifidou E. 2014. Diversity of Campylobacter in retail meat and liver of lambs and goat kids. Foodborne Pathog Dis 11:320–328. doi: 10.1089/fpd.2013.1678. [DOI] [PubMed] [Google Scholar]
- 34.Lazou T, Houf K, Soultos N, Dovas C, Iossifidou E. 2014. Campylobacter in small ruminants at slaughter: prevalence, pulsotypes and antibiotic resistance. Int J Food Microbiol 173:54–61. doi: 10.1016/j.ijfoodmicro.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 35.European Food Safety Authority (EFSA). 2010. Analysis of the baseline survey on the prevalence of Campylobacter in broiler batches and of Campylobacter and Salmonella on broiler carcasses, in the EU, 2008—part B: analysis of factors associated with Campylobacter colonisation of broiler batches and with Campylobacter contamination of broiler carcasses; and investigation of the culture method diagnostic characteristics used to analyse broiler carcass samples. EFSA J 8:1522. doi: 10.2903/j.efsa.2010.1522. [DOI] [Google Scholar]
- 36.Nobile CGA, Costantino R, Bianco A, Pileggi C, Pavia M. 2013. Prevalence and pattern of antibiotic resistance of Campylobacter spp. in poultry meat in Southern Italy. Food Control 32:715–718. doi: 10.1016/j.foodcont.2013.02.011. [DOI] [Google Scholar]
- 37.Pedonese F, Nuvoloni R, Turchi B, Torracca B, Di Giannatale E, Marotta F, Cerri D. 2017. Prevalence, phenotypic and genetic diversity of Campylobacter in poultry fresh meat and poultry products on retail sale in Tuscany (Italy). Vet Ital 53:29–37. [DOI] [PubMed] [Google Scholar]
- 38.Løvdal T, Hovda MB, Björkblom B, Møller SG. 2011. Propidium monoazide combined with real-time quantitative PCR underestimates heat-killed Listeria innocua. J Microbiol Methods 85:164–169. doi: 10.1016/j.mimet.2011.01.027. [DOI] [PubMed] [Google Scholar]
- 39.Birdsell DC, Cota-Robles EH. 1967. Production and ultrastructure of lysozyme and ethylenediaminetetraacetate-lysozyme spheroplasts of Escherichia coli. J Bacteriol 93:427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Costerton JW, Forsberg C, Matula TI, Buckmire FL, MacLeod RA. 1967. Nutrition and metabolism of marine bacteria. XVI. Formation of protoplasts, spheroplasts, and related forms from a gram-negative marine bacterium. J Bacteriol 94:1764–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Witholt B, Boekhout M, Brock M, Kingma J, Heerikhuizen HV, Leij LD. 1976. An efficient and reproducible procedure for the formation of spheroplasts from variously grown Escherichia coli. Anal Biochem 74:160–170. doi: 10.1016/0003-2697(76)90320-1. [DOI] [PubMed] [Google Scholar]
- 42.Witholt B, Heerikhuizen HV, De Leij L. 1976. How does lysozyme penetrate through the bacterial outer membrane? Biochim Biophys Acta 443:534–544. doi: 10.1016/0005-2736(76)90471-5. [DOI] [PubMed] [Google Scholar]
- 43.Hobb RI, Fields JA, Burns CM, Thompson SA. 2009. Evaluation of procedures for outer membrane isolation from Campylobacter jejuni. Microbiol 155:979–988. doi: 10.1099/mic.0.024539-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson JL, Jesse HE, Hughes B, Lund V, Naylor K, Davidge KS, Cook GM, Mann BE, Poole RK. 2013. Ru(CO)3Cl(glycinate) (CORM-3): a carbon monoxide-releasing molecule with broad-spectrum antimicrobial and photosensitive activities against respiration and cation transport in Escherichia coli. Antioxid Redox Signal 19:497–509. doi: 10.1089/ars.2012.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Josefsen MH, Löfström C, Hansen TB, Christensen LS, Olsen JE, Hoorfar J. 2010. Rapid quantification of viable Campylobacter bacteria on chicken carcasses, using real-time PCR and propidium monoazide treatment, as a tool for quantitative risk assessment. Appl Environ Microbiol 76:5097–5104. doi: 10.1128/AEM.00411-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nocker A, Camper AK. 2009. Novel approaches toward preferential detection of viable cells using nucleic acid amplification techniques. FEMS Microbiol Lett 291:137–142. doi: 10.1111/j.1574-6968.2008.01429.x. [DOI] [PubMed] [Google Scholar]
- 47.Linley E, Denyer SP, McDonnell G, Simons C, Maillard JY. 2012. Use of hydrogen peroxide as a biocide: new consideration of its mechanisms of biocidal action. J Antimicrob Chemother 67:1589–1596. doi: 10.1093/jac/dks129. [DOI] [PubMed] [Google Scholar]
- 48.Signoretto C, Lleò MM, Tafi MC, Canepari P. 2000. Cell wall chemical composition of Enterococcus faecalis in the viable but non-culturable state. Appl Environ Microbiol 66:1953–1959. doi: 10.1128/AEM.66.5.1953-1959.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anonymous. 2017. Microbiology of the food chain—horizontal method for detection and enumeration of Campylobacter spp.—part 2: colony-count technique. ISO 10272–2 International Organization for Standardization, Geneva, Switzerland. [DOI] [PubMed] [Google Scholar]
- 50.Fouts DE, Mongodin EF, Mandrell RE, Miller WG, Rasko DA, Ravel J, Brinkac LM, DeBoy RT, Parker CT, Daugherty SC, Dodson RJ, Durkin AS, Madupu R, Sullivan SA, Shetty JU, Ayodeji MA, Shvartsbeyn A, Schatz MC, Badger JH, Fraser CM, Nelson KE. 2005. Major structural differences and novel potential virulence mechanisms from the genomes of multiple Campylobacter species. PLoS Biol 3:e15. doi: 10.1371/journal.pbio.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lefébure T, Pavinski Bitar PD, Suzuki H, Stanhope MJ. 2010. Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol Evol 2:646–655. doi: 10.1093/gbe/evq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leblanc-Maridor M, Beaudeau F, Seegers H, Denis M, Belloc C. 2011. Rapid identification and quantification of Campylobacter coli and Campylobacter jejuni by real-time PCR in pure cultures and in complex samples. BMC Microbiol 11:113. doi: 10.1186/1471-2180-11-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lagier MJ, Joseph LA, Passaretti TV, Musser KA, Cirino NM. 2004. A real-time multiplexed PCR assay for rapid detection and differentiation of Campylobacter jejuni and Campylobacter coli. Mol Cell Probes 18:275–282. doi: 10.1016/j.mcp.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 54.Caraguel CG, Stryhn H, Gagné N, Dohoo IR, Hammell KL. 2011. Selection of a cutoff value for real-time polymerase chain reaction results to fit a diagnostic purpose: analytical and epidemiologic approaches. J Vet Diagn Invest 23:2–15. doi: 10.1177/104063871102300102. [DOI] [PubMed] [Google Scholar]
- 55.Hill RA, Sillence MN. 1997. Improved membrane isolation in the purification of beta 2-adrenoceptors from transgenic Escherichia coli. Protein Expr Purif 10:162–167. doi: 10.1006/prep.1997.0732. [DOI] [PubMed] [Google Scholar]
- 56.Jarvis B. 2016. Errors associated with colony count procedures, p 122–140. In Tenney S, Versteeg-Buschuman L (ed), Statistical aspects of the microbiological examination of foods, 3rd ed Academic Press, London, United Kingdom. [Google Scholar]