

Abstract
mAbs can induce antibody-dependent cellular cytotoxicity (ADCC) via the innate immune system’s ability to recognize mAb-coated cancer cells and activate immune effector cells. Lenalidomide is an immunomodulatory agent with the capacity to stimulate immune cell cytokine production and ADCC activity. This phase I trial evaluated the combination of cetuximab with lenalidomide for the treatment of advanced colorectal and head and neck squamous cell cancers (HNSCC). This trial included patients with advanced colorectal cancer or HNSCC. Treatment consisted of cetuximab 500 mg/m2 i.v. every two weeks with lenalidomide given orally days 1–21 on a 28-day cycle. Three dose levels of lenalidomide were evaluated (15, 20, 25 mg). Correlative studies included measurement of ADCC, FcγRIIIA polymorphism genotyping, measurement of serum cytokine levels, and flow cytometric analysis of immune cell subtypes. Twenty-two patients were enrolled (19 colorectal cancer, 3 HNSCC). Fatigue was the only dose-limiting toxicity. One partial response was observed and 8 patients had stable disease at least 12 weeks. The recommended phase II dose is cetuximab 500 mg/m2 with lenalidomide 25 mg daily, days 1–21. Correlative studies demonstrated a dose-dependent increase in natural killer cytotoxic activity with increasing doses of lenalidomide. Cetuximab and lenalidomide were well tolerated. There was a lenalidomide dose-dependent increase in ADCC with higher activity in patients enrolled in cohort 3 than those enrolled in cohorts ½. Although response was not a primary endpoint, there was evidence of antitumor activity for the combination therapy. Further investigation of lenalidomide as an immunomodulator in solid tumors is warranted.
Introduction
Characterization of the interactions between the immune system and tumor cells has led to the development of novel therapeutic approaches in cancer immunotherapy, and as such, researchers have developed an arsenal of anticancer tools to exploit the effector mechanisms of the immune system. mAbs targeting tumor-associated antigens have been developed and utilized in a variety of human cancers. In addition to direct binding of mAbs to tumor antigens, each mAb possesses a constant or “Fc” region away from its antigen-binding site that permits the host immune system to recognize and destroy the antibody-coated tumor cells. Innate immune cells, including natural killer (NK) cells, bear specialized fragment C receptors (FcR) for the constant region of mAbs and costimulation of these effector cells with specific immunoactivating compounds can significantly enhance the immune response to antibody-coated tumor cells. Research by our group and others has shown that cytokines, Toll-like receptor agonists, and immunomodulatory agents (such as lenalidomide) can markedly and synergistically enhance the immune response to Ab-coated tumor cells in vitro, in murine models of cancer and in the context of translational phase I clinical trials (1–5). Although multiple immune cells are involved in the clearance of antibody-coated tumor cells, NK cells appear to play a key role (6, 7).
Cetuximab is a chimeric IgG1 mAb directed against the extra-cellular domain of the EGFR. EGFR is a transmembrane protein whose overexpression is associated with a malignant phenotype and the enhanced ability of certain tumors to invade normal tissues and metastasize (8). The receptor consists of an extra-cellular ligand-binding site with a cytoplasmic tyrosine kinase-containing domain. Binding of the appropriate ligand triggers tyrosine kinase phosphorylation resulting in intracellular signaling for growth and proliferation. The activated downstream signaling proteins include P13K-AKT, MAPK, SRC, and STATs 1, 3, 5a, and 5b.
The cytotoxic mechanism of action of cetuximab is two-fold: the antibody binds extracellular EGFR to prevent ligand binding, and it mediates an immunologic antitumor effects via antibody-dependent cellular cytotoxicity (ADCC) conducted by FcR-bearing cells. At least one study specifically investigated the important role of CD56+ NK cells and ADCC activity with cetuximab in colorectal cancer, and this trial demonstrated that the response to cetuximab was positively correlated with tumor infiltration by NK cells (9).
Lenalidomide is an immunomodulatory agent currently approved for use in hematologic malignancies, but it has shown no significant antitumor activity in solid tumors as a single agent (10, 11). Lenalidomide has demonstrated synergistic activity in combination with several mAbs for hematologic malignancies, including rituximab (IgG1 anti-CD20 chimeric mAb) and SGN-40 (IgG1 anti-CD40 humanized mAb; refs. 12–15). In vitro studies have shown that lenalidomide stimulates the proliferation and activation of NK cells, thereby enhancing NK cell–mediated cytotoxicity of Ab-coated tumors (16). Studies in hematologic malignancies (lymphoma and multiple myeloma) also demonstrated a role for the presence of lenalidomide in the activation of natural killer (NK) cells (CD16+ CD56 low; refs. 14, 17). These findings support the role of lenalidomide in enhancing immune cell–mediated cytotoxicity (13, 14, 17). Preclinical studies of lenalidomide in SCID lymphoma mice also demonstrated enhancement of ADCC due to NK cell activity and increased cytokine activity (13, 14).
The present phase I clinical trial evaluated the safety, toxicity, and immunologic activity of the combination of lenalidomide and cetuximab in patients with colorectal and head and neck squamous cell cancer. On the basis of preclinical and clinical work with lenalidomide and mAbs, one of the primary aims was to evaluate the role of NK-cell–mediated ADCC of cetuximab using lenalidomide as an immune stimulant.
Materials and Methods
Patient selection
Patients with metastatic KRAS wild-type colorectal cancer or squamous cell head and neck cancer were eligible to enroll (HNSCC). All patients provided written informed consent. There was no limit to the prior number of therapies that could be received. Prior EGFR-directed therapy (e.g., tyrosine kinase inhibitors and mAbs) including cetuximab, panitumumab, or investigational EGFR-directed mAbs was allowed. No chemotherapy or mAb therapy was permitted within 28 days of trial initiation. Additional eligibility criteria included ECOG performance status 0–2, negative pregnancy test, and use of contraception in women of child-bearing age and preserved organ function as defined by the following parameters: leukocytes >3,000/mcL, absolute neutrophil count >1,500/mcL, platelets >100,000/mcL, total bilirubin < 1.5 mg/dL, AST (SGOT) < 100 U/L, ALT (SGPT) < 120 U/L, creatinine clearance > 60 mL/min/1.73 m2 as calculated using modified Cockcroft–Gault formula. Patients with brain metastases who had received definitive therapy, including radiation, and did not require ongoing medical therapy (i.e., steroids) for brain metastases were eligible. Exclusion criteria included history of allergic reactions attributed to compounds with chemical or biologic composition similar to lenalidomide or cetuximab, deep vein thrombosis/pulmonary embolism requiring therapy within 3 months of enrollment, history of toxicity ≥ grade 3 with prior EGFR-directed therapy, confirmed history of interstitial lung disease, uncontrolled concurrent illness including, but not limited to, ongoing or active infection, symptomatic congestive heart failure, unstable angina pectoris, cardiac arrhythmia, or psychiatric illness/social situations that would limit compliance with study requirements. Pregnant woman and patients less than 18 years of age were excluded. Because of the use of a commercial supply of cetuximab, KRAS-mutant colorectal cancer was specifically excluded in accordance with FDA indication for this drug.
Study design and patient evaluation
Patients received cetuximab 500 mg/m2 i.v. infusion every 2 weeks (days 1 and 15) with lenalidomide orally on days 1–21 of the 28-day cycle. There was no dose escalation for cetuximab, but dose reductions were allowed for toxicity. Three dose levels of lenalidomide were investigated: 15, 20, and 25 mg daily.
A standard phase I 3+3 trial design was utilized. Starting with dose level 1, cohorts of three patients were followed to define dose-limiting toxicity (DLT). An expansion cohort of colorectal cancer patients was recruited at the MTD. All patients enrolled into a given cohort received the same starting dose. DLT was assessed during cycle 1. Dose delays of up to 14 days were permitted for toxicity. Patients with myelosuppression (grade 3 or 4 anemia, thrombocytopenia, or neutropenia without bleeding or infection) or rash were permitted to continue on study if toxicity improved to at least grade 2 within 14 days. Patients were removed from the study if they developed infusion reaction requiring medical intervention (grade 3 or 4), rash with desquamation or blistering, erythema multiforme, or toxic epidermal necrolysis. Patients with other nonhematologic toxicities that did not improve to grade 2 or better within 14 days were also removed from the study. Those patients requiring a delay in dosing secondary to toxicity were dose reduced by 1 dose level. A maximum of 2 dose reductions per patient was allowed. No dose re-escalation was permitted. No intrapatient dose escalation was allowed.
Supportive care measures were utilized at the discretion of the individual treating physician. Recommendations for the treatment of cetuximab-related rash included prophylactic use of minocycline as well as topical clindamycin or bactroban as needed. Lenalidomide increases the risk of thrombotic events in patients who are at high risk or with a history of thrombosis; when combined with other agents such as steroids (e.g., dexamethasone, prednisone) or erythropoietin, the risk of thrombosis is increased. Patients on protocol received aspirin (81 or 325 mg) prophylaxis if the patient had a history of thromboembolic disease, known thrombophilia, erythropoietin use, or ongoing steroid use (>7 days/cycle). No routine thromboembolic prophylaxis was recommended for patients not meeting the above criteria. Patients were assessed for toxicity by the clinical provider at the start of each treatment cycle. Patients were assessed for response with CT imaging every 2 cycles (8 weeks). Treatment was continued until unacceptable toxicity, disease progression, patient withdrawal from study, or investigator discretion. Patients were followed for 6 weeks after removal from study or until death, whichever occurred first. Patients removed from the study for unacceptable adverse events were followed until resolution or stabilization of the adverse event.
Correlative studies
Peripheral blood samples were obtained at study entry and after initiation of lenalidomide therapy at days 15, 30, and 45. Combination therapy (cetuximab and lenalidomide) was continued beyond day 45 unless patients were removed from the study due to intolerance or toxicity. Serum and peripheral blood mononuclear cells (PBMC) were procured and cryo-preserved using standard techniques and used for further analysis (18).
ADCC.
PBMCs from patients at various time points were plated in 96-well V-bottom plates and treated with or without IL12 (10 ng/mL) overnight in RPMI supplemented with 10% human AB serum at 37°C. Eighteen hours later, HT29 human colorectal cancer tumor cells were labeled with 51Cr for 1 hour, followed by incubation with either cetuximab or control IgG (50 μg/mL) for 45 minutes. Tumor cells were then added to NK cells at various effector:target (E:T) ratios. After 4-hour incubation, supernatants were harvested and chromium release was assayed and percent lysis determined as described previously (19). The human colorectal cancer cell line HT29 was acquired from ATCC in 2010 and was validated by karyotyping/cytogenetic analysis prior to use in 2013.
FcγRIIIA genotyping.
DNA was isolated from pretreatment patient PBMCs using a QIAmp kit according to the manufacturer’s instructions (Qiagen) and quantified. Genotyping of the FcγRIIIA-158V/F polymorphism was performed using a nested PCR followed by allele-specific restriction enzyme digestion as described previously (20). The amplified DNA (10 μL) was then digested with 10 U NlaIII (New England Biolabs) at 37°C for 12 hours and separated by electrophoresis on 8% polyacrylamide gel. After staining with ethidium bromide, DNA bands were visualized under UV light. For homozygous FcγRIIIA-158F patients, only one undigested band (94 bp) was visible. Three bands (94 bp, 61 bp, and 33 bp) were seen in heterozygous individuals, whereas for homozygous FcγRIIIA −158V patients only 2 digested bands (61 bp and 33 bp) were obtained. All samples were analyzed in duplicate.
Cytokine analysis.
Patient serum samples from various time points were thawed on ice and analyzed in duplicate for levels of cytokines (IL2, IL12, IFNγ and TNFα) and growth factors (VEGF and FGF-basic) using a Bio-Plex kit following the manufacturer’s specifications (Bio-Rad).
Flow cytometric analysis for immune cell subtypes.
Percentage of T regulatory cells, T cells, NK cells, and monocytes per microliter of whole blood was determined by flow cytometry. Immune cell subtype classification is as follows: T regulatory cells CD4+/CD25+/CD127−; T cells CD3+; NK cells CD3−/CD56+/CD16+; NK cells and monocytes CD16+.
Statistical analysis
A mixed effect model was used to analyze the data (21). The change in ADCC from baseline to posttreatment was compared within cohort 3 or cohort 1 and 2 (combined) and between cohorts. This change was further compared within patients that exhibited clinical benefit versus those with progressive disease. Data analysis was conducted by using SAS, version 9.4 (SAS, Inc).
Results
Patient characteristics
Twenty-two patients (6 women, 16 men) were enrolled. Nineteen patients had colorectal cancer (KRAS wild-type) and 3 patients had squamous cell carcinoma of the head and neck. The average patient age was 61 years old (range 44–71 years); most patients were Caucasian (20 Caucasian, 2 African-American). All but one patient had received prior EGFR-directed therapy: 11 patients had received cetuximab, 6 patients had received panitumumab, and 4 patients received both cetuximab and panitumumab. The patients were heavily pretreated, most patients received at least 4 prior regimens in the metastatic setting (2 prior regimens, 1 patient; 3 prior regimens, 8 patients; 4+ prior regimens, 12 patients). No patients received immunotherapy such as PD/PDL1 or CTLA-4 inhibitors. Patient demographics are summarized in Table 1.
Table 1.
Patient demographics
| Characteristic | Number of patients |
|---|---|
| Male | 16 |
| Female | 6 |
| Colorectal cancer | 19 |
| Head and neck cancer | 3 |
| Prior therapies | |
| Cetuximab | 11 |
| Panitumumab | 6 |
| Cetuximab and panitumumab | 4 |
| Chemotherapy | 19 |
| Bevacizumab | 17 |
| Best response to last EGFR-directed therapy | |
| Stable disease | 3 |
| Progressive disease | 15 |
| Unknown | 3 |
Dose escalation and DLTs
Dose escalation followed the predetermined dosing schedule. Three patients were treated at dose level 1 (cetuximab 500 mg/m2 plus lenalidomide 15 mg daily). Seven patients were treated at dose level 2 (cetuximab 500 mg/m2 plus lenalidomide 20 mg daily). One patient on dose level 2 was removed from the study during cycle 1 due to progressive disease and was replaced. A DLT of fatigue was observed at dose level 2 and this dose was expanded to include a total of 6 evaluable patients. No further toxicity was observed at dose level 2. Twelve patients were treated at dose level 3 (cetuximab 500 mg/m2 plus lenalidomide 25 mg daily) without incident. The only DLT observed was fatigue at dose level 2. Additional common treatment-related toxicities included fatigue, vomiting, cytopenias (particularly lymphopenia/leukopenia), diarrhea, electrolyte abnormalities (hypocalcemia, hypokalemia, hypomagnesemia, hypophosphatemia), and skin toxicity (acneiform rash, dry skin). Grade 4 toxicities included hypokalemia and hypomagnesemia in a patient receiving multiple cycles of cetuximab; electrolyte abnormalities started with cycle 4 and were managed with electrolyte replacement. Of note, 6 thromboembolic events were reported at dose levels 2 and 3. None of these events proceeded to pulmonary embolus and were managed with anticoagulation only. No patients required dose reductions. No treatment-related grade 5 toxicity was observed. Toxicities are further detailed in Table 2.
Table 2.
Treatment-related toxicities
| Toxicity | Grade 1 | Grade 2 | Grade 3 | Grade 4 |
|---|---|---|---|---|
| Fatigue | 5 | 6 | 1 | 0 |
| Rash - acneiform, maculopapular | 13 | 6 | 2 | 0 |
| Dry skin | 7 | 1 | 0 | 0 |
| Skin infection/paronychia | 1 | 2 | 1 | 0 |
| Dehydration | 0 | 0 | 1 | 0 |
| Anorexia | 1 | 1 | 1 | 0 |
| Thromboembolic event | 0 | 6 | 0 | 0 |
| Anemia | 8 | 4 | 2 | 0 |
| Lymphopenia | 11 | 9 | 1 | 0 |
| Leukopenia/neutropenia | 14 | 9 | 0 | 0 |
| Thrombocytopenia | 7 | 0 | 1 | 0 |
| Nausea | 5 | 2 | 0 | 0 |
| Vomiting | 4 | 1 | 0 | 0 |
| Diarrhea | 4 | 2 | 0 | 0 |
| Constipation | 2 | 0 | 0 | 0 |
| Dry mouth/mucositis | 4 | 1 | 0 | 0 |
| Transaminase elevations (ALT/AST) | 7 | 1 | 0 | 0 |
| Hyperbilirubinemia | 1 | 1 | 0 | 0 |
| Renal insufficiency | 1 | 0 | 1 | 0 |
| Hypocalcemia | 6 | 1 | 0 | 0 |
| Hypokalemia | 6 | 1 | 2 | 1 |
| Hypomagnesemia | 8 | 2 | 1 | 1 |
| Hypophosphatemia | 4 | 1 | 1 | 0 |
| Edema of extremities | 2 | 2 | 0 | 0 |
| Fever | 2 | 0 | 0 | 0 |
| Infection | 1 | 2 | 2 | 0 |
| Dyspnea | 0 | 3 | 0 | 0 |
Antitumor activity
Evaluable patients received an average of 4 treatment cycles. One patient had a partial response to treatment that began with cycle 2. They received a total of 8 cycles of therapy prior to progression of disease. Eight patients had stable disease as their best response; 10 patients had progressive disease after 2 cycles. Patients with stable disease received an average of 6 cycles of therapy (range 4–10 cycles). All but one of the patients with clinical benefit (partial response or stable disease) had received prior EGFR-directed therapy. Patient characteristics and response evaluation detailed in Table 3.
Table 3.
Patient outcomes
| Dose level | Number of treatment cycles |
Best response |
Diagnosis | Prior EGFR-directed therapy? |
Fc gamma polymorphism |
|---|---|---|---|---|---|
| 1 | 6 | SD | CRC | Yes | VF |
| 1 | 2 | PD | CRC | Yes | FF |
| 1 | 2 | PD | CRC | Yes | VF |
| 2 | 8 | SD | CRC | Yes | FF |
| 2 | 1 | NE | CRC | Yes | FF |
| 2 | 4 | SD | CRC | Yes | VF |
| 2 | 1 | PD | CRC | Yes | FF |
| 2 | 2 | PD | CRC | Yes | FF |
| 2 | 2 | PD | CRC | Yes | VF |
| 2 | 8 | PR | CRC | Yes | FF |
| 3 | 1 | NE | CRC | Yes | VF |
| 3 | 2 | PD | HEAD | Yes | VF |
| 3 | 6 | SD | HEAD | No | VF |
| 3 | 2 | PD | CRC | Yes | VF |
| 3 | 4 | SD | CRC | Yes | FF |
| 3 | 10 | SD | CRC | Yes | FF |
| 3 | 4 | SD | HEAD | Yes | FF |
| 3 | 2 | PD | CRC | Yes | FF |
| 3 | 1 | NE | CRC | Yes | VF |
| 3 | 2 | PD | CRC | Yes | VF |
| 3 | 2 | PD | CRC | Yes | FF |
| 3 | 6 | SD | CRC | Yes | VF |
Abbreviations: CRC, colorectal cancer; HEAD, head and neck squamous cell cancer; NE, not evaluable for response; PR, partial response; SD, stable disease.
Correlative studies
Because of small patient numbers, cohorts 1 and 2 were combined and compared with cohort 3 for statistical analysis. In cohort 1 and 2, the mean change in ADCC between day 45 posttreatment and baseline samples was −2.45% [95% confidence interval (CI), −7.18–.27; P = 0.305], indicating no significant increase in ADCC activity. In cohort 3, the mean change in ADCC between day 45 posttreatment and baseline samples was 7.53% (95% CI, 1.65–13.4, P = 0.0125), which is significantly greater than that in the combined cohorts 1 and 2 (P = 0.01; Fig. 1). Likewise, there was a trend toward increased ADCC activity in patients with clinical benefit (those patients with either stable disease or response) compared with patients who did not experience clinical benefit. For patients who experienced clinical benefit, the mean change in ADCC at day 45 (compared with baseline) was 6.87% (95% CI, −0.22–13.97; P = 0.0573); in patients with progression after 2 cycles, the mean difference at day 45 (compared with baseline) was 2.69% (95% CI, −3.74–9.63; P = 0.4210) (Fig. 2). Five of 8 patients with clinical benefit and 4 of 9 patients with progressive disease also had increased ADCC (Fig. 3).
Figure 1.
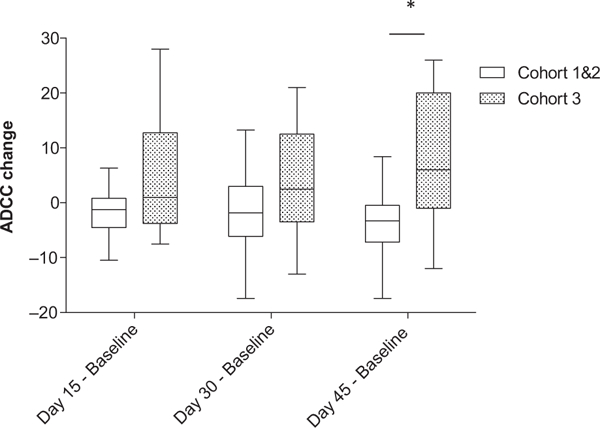
Change in antibody-dependent cellular cytotoxicity between baseline and day 15, day 30, and day 45 for cohort 1 and 2 versus cohort 3. Purified PBMCs from patients enrolled in the trial were incubated overnight with medium supplemented with 10 ng/mL IL12. The lytic activity of patient PBMCs cells was assessed in a standard 4-hour chromium release assay using cetuximab-coated HT29 human colorectal cancer cells. The change in mean percent lysis at 25:1 effector:target ratio for 10 patients enrolled in cohort 1 and 2 and the remaining 12 patients enrolled in cohort 3 between baseline and day 15, baseline and day 30, and baseline and day 45 is shown in box plots, represented as baseline ADCC values subtracted from ADCC values at later time points. The box plots represent the median and interquartile range, with I bars showing the range for each group. *, P = 0.01.
Figure 2.
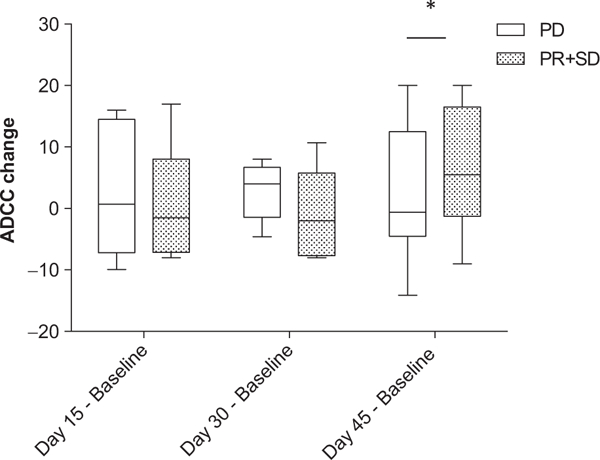
Change in antibody-dependent cellular cytotoxicity in patients with progressive disease versus patients with clinical benefit (response or stable disease). Purified PBMCs from patients enrolled in the trial were incubated overnight with medium supplemented with 10 ng/mL IL12. The lytic activity of patient PBMCs was assessed in a standard 4-hour chromium release assay using cetuximab-coated HT29 human colorectal cancer cells. The change in mean percent lysis at 25:1 effector:target ratio for 9 patients who had progressive disease (PD) and the 8 patients who experienced clinical benefit (partial response and stable disease, PR + SD) at various time points are shown in box plots, represented as baseline ADCC values subtracted from ADCC values at later time points. The box plots represent the median and interquartile range, with I bars showing the range for each group. *, P = 0.05.
Figure 3.
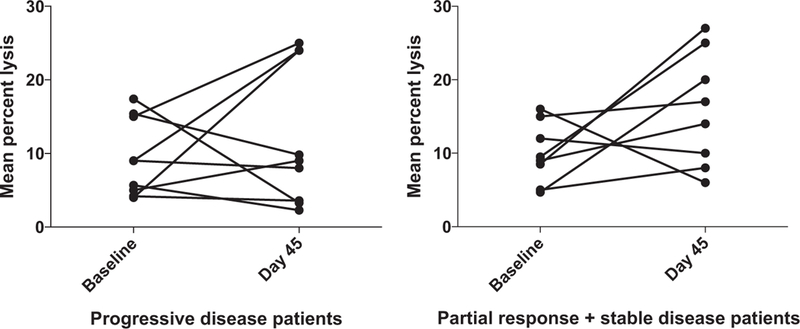
Increased antibody-dependent cellular cytotoxicity is generally correlated to clinical benefit. Purified PBMCs from patients enrolled in the trial were incubated overnight with medium supplemented with 10 ng/mL IL12. The lytic activity of patient PBMCs was assessed in a standard 4-hour chromium release assay using cetuximab-coated HT29 human colorectal cancer cells. The change in mean percent lysis at 25:1 effector:target ratio for 9 patients who had progressive disease (PD) and the 8 patients who experienced clinical benefit (partial response and stable disease, PR + SD) between baseline and day 45 are shown in paired fashion.
No high affinity Fc gamma receptor polymorphisms (VV) were identified. The group was evenly split with 11 VF and 11 FF polymorphisms identified (dns). There was no significant association between polymorphism status and clinical benefit, toxicity, or ADCC activity. In addition, levels of IL2, IL12, IFNγ, TNFα, VEGF, and FGF-basic were measured in serum drawn at baseline and days 15, 30, and 45; however, no significant differences in serum cytokine levels were seen in patients with clinical benefit compared with patients who did not experience clinical benefit (Supplementary Fig. S1A and S1B). The percentage of T regulatory cells, T cells, NK cells, and monocytes was determined by flow cytometry for 10 patients enrolled in cohort 3; however, no significant differences in any of these populations were noted or were indicative of patient outcome (Supplementary Fig. S2A–S2D).
Discussion
This phase I trial of cetuximab and lenalidomide demonstrated that the combination was well tolerated and no excessive toxicities were observed. The observed toxicities were within the expected range for these agents, with the exception of the increased risk of thromboembolic disease (27% in this trial). On the basis of clinical tolerability, the recommended phase II dose for further evaluation of this combination is cetuximab 500 mg/m2 i.v. days 1 and 14 with lenalidomide being dosed at 25 mg on days 1–21 of 28-day cycle.
Thromboembolism is a known risk of lenalidomide therapy. Initial trials leading to drug approval in the United States reported thrombosis in up to 10% of multiple myeloma patients treated with lenalidomide and dexamethasone (6, 7). Studies of lenalidomide in solid tumors demonstrated a variable rate of venous thromboembolism: thyroid cancer (11% pulmonary embolism; ref. 22), pancreatic cancer (18%; ref. 22), and phase I study of multiple cancer types (13%; ref. 10). It is also noted that solid tumor patients have an increased risk of thromboembolic disease compared with the general population. In particular, gastrointestinal tumors carry one of the highest risks of this complication [OR, 16.8 (4.1–69.1); ref. 23]. In this context, the increased risk of venous thrombosis suggests that a more aggressive prophylaxis regimen may be indicated for this population of patients if lenalidomide is utilized with cetuximab.
The observed response rate of combination therapy suggests lenalidomide may play a role in improving single-agent activity of cetuximab, particularly in pretreated patients. Cetuximab was initially approved as a single-agent therapy in colorectal cancer based on a phase III trial demonstrating a partial response rate of 8% and stable disease in 31% of patients (24). When only KRAS wild-type patients were included, the response rate was 12.8% (25). Similarly, a noninferiority trial of cetuximab versus panitumumab demonstrated a response rate of 20%–22% with stable disease in 47%–49% of patients without prior exposure to EGFR inhibitors (26). Likewise, single-agent cetuximab demonstrated response rate of 13% in recurrent/metastatic squamous cell head and neck cancer (27). The potential benefit of rechallenging patients with single-agent EGFR-directed therapy after prior exposure is less clear. One published trial evaluated the efficacy of using panitumumab in patients who had progressed on cetuximab, and a small phase II trial observed no responses, but 45% of patients had stable disease for at least 2 cycles (28). A second small study reported a clinical benefit rate of 15% in patients with cetuximab resistance (29). Thus, the use of immunostimulatory agents including lenalidomide may have the greatest benefit in patients who have had prior EGFR-directed therapies.
Although clinical benefit/response rate was not a primary endpoint in this trial, we did observe clinical benefit in 9 patients (47% of patients completing at least 2 cycles) with one partial response. The patient with a partial response had been previously treated with panitumumab in combination with irinotecan as well as cetuximab. For this patient, the previously observed best response with cetuximab was stable disease. The patient response to the combination of cetuximab plus lenalidomide suggests activity of the combination therapy above and beyond expected response with single-agent treatment. Interestingly, patients with clinical benefit were noted at all three dose level, and the patient with partial response was treated on dose level 2. This finding is consistent with other immunologic therapies—although there is a dose-dependent increase in response, individual patient responses are likely related to intricacies of that individual’s immune response.
The correlative studies focused on the immune response to the combination therapy. Measurement of ADCC was the main marker of immune activation and the results did demonstrate a lenalidomide dose-dependent increase in cytotoxic activity in vitro. As stated above, this effect did not clearly correlate with response rates in individual patients. Comparing patients with clinical benefit to those with progressive disease, however, the ADCC results suggested the existence of a more sustained immunologic enhancement in patients with clinical benefit. These endpoints are exploratory but suggest that immunologic activity may underly the benefit for individual patients.
Cetuximab plus lenalidomide is a well-tolerated combination therapy. Clinical benefit was observed with tolerable toxicity. In addition, immune activity was observed and improved with increasing doses of lenalidomide. Given the limited activity observed in this small patient population, it would be interesting to explore the use of lenalidomide with other EGFR-directed therapies.
Supplementary Material
Acknowledgments
Grant Support
This work was supported by ASCO Conquer Cancer Foundation Young Investigator Award (to E.M. Bertino). Research reported in this publication was also supported by the National Cancer Institute of the NIH under award number U01CA076576 (to M.R. Grever) and P01 CA095426 (to M.A. Caligiuri).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjoumals.org/).
Disclosure of Potential Conflicts of Interest
M. Grever is a consultant/advisory board member for Ascerta. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Parihar R, Dierksheide J, Hu Y, Carson WE. IL-12 enhances the natural killer cell cytokine response to Ab-coated tumor cells. J Clin Invest 2002; 110:983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parihar R, Nadella P, Lewis A, Jensen R, De Hoff C, Dierksheide JE, et al. A phase I study of interleukin 12 with trastuzumab in patients with human epidermal growth factor receptor-2-overexpressing malignancies: analysis of sustained interferon gamma production in a subset of patients. Clin Cancer Res 2004;10:5027–37. [DOI] [PubMed] [Google Scholar]
- 3.Roda JM, Joshi T, Butchar JP, McAlees JW, Lehman A,Tridandapani S, et al. The activation of natural killer cell effector functions by cetuximab-coated, epidermal growth factor receptor positive tumor cells is enhanced by cytokines. Clin Cancer Res 2007;13:6419–28. [DOI] [PubMed] [Google Scholar]
- 4.Ferris RL, Jaffee EM, Ferrone S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: clinical response, cellular immunity, and immunoescape. J Clin Oncol 2010;28:4390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekaii-Saab TS, Roda JM, Guenterberg KD, Ramaswamy B, Young DC, Ferketich AK, et al. A phase I trial of paclitaxel and trastuzumab in combination with interleukin-12 in patients with HER2/neu-expressing malignancies. Mol Cancer Ther 2009;8:2983–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajkumar SV, Hayman SR, Lacy MQ, Dispenzieri A, Geyer SM, Kabat B, et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood 2005;106:4050–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dimopoulos M, Spencer A, Attal M, Prince HM, Harousseau JL, Dmoszynska A, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med 2007;357:2123–32. [DOI] [PubMed] [Google Scholar]
- 8.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005;5:341–54. [DOI] [PubMed] [Google Scholar]
- 9.Marechal R, De Schutter J, Nagy N, Demetter P, Lemmers A, Deviere J, et al. Putative contribution of CD56 positive cells in cetuximab treatment efficacy in first-line metastatic colorectal cancer patients. BMC Cancer 2010;10:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dahut WL, Aragon-Ching JB, Woo S, Tohnya TM, Gulley JL, Arlen PM, et al. Phase I study of oral lenalidomide in patients with refractory metastatic cancer. J Clin Pharma 2009;49:650–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma RA, Steward WP, Daines CA, Knight RD, O’Byrne KJ, Dalgleish AG. Toxicity profile of the immunomodulatory thalidomide analogue, lenalidomide: phase I clinical trial of three dosing schedules in patients with solid malignancies. Eur J Cancer 2006;42:2318–25. [DOI] [PubMed] [Google Scholar]
- 12.Lapalombella R, Yu B, Triantafillou G, Liu Q, Butchar JP, Lozanski G, et al. Lenalidomide down-regulates the CD20 antigen and antagonizes direct and antibody-dependent cellular cytotoxicity of rituximab on primary chronic lymphocytic leukemia cells. Blood 2008;112:5180–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, Qian Z, Cai Z, Sun L, Wang H, Bartlett JB, et al. Synergistic antitumor effects of lenalidomide and rituximab on mantle cell lymphoma in vitro and in vivo. Am J Hematol 2009;84:553–9. [DOI] [PubMed] [Google Scholar]
- 14.Reddy N, Hernandez-Ilizaliturri FJ, Deeb G, Roth M, Vaughn M, Knight J, et al. Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br J Haematol 2008;140:36–45. [DOI] [PubMed] [Google Scholar]
- 15.Tai YT, Li XF, Catley L, Coffey R, Breitkreutz I, Bae J, et al. Immunomodulatory drug lenalidomide (CC-5013, IMiD3) augments anti-CD40 SGN-40-induced cytotoxicity in human multiple myeloma: clinical implications. Cancer Res 2005;65:11712–20. [DOI] [PubMed] [Google Scholar]
- 16.Gribben JG, Fowler N, Morschhauser F. Mechanisms of action of lenalidomide in B-cell non-hodgkin lymphoma. J Clin Oncol 2015;33:2803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu D, Corral LG, Fleming YW, Stein B. Immunomodulatory drugs Revlimid (lenalidomide) and CC-4047 induce apoptosis of both hematological and solid tumor cells through NK cell activation. Cancer Immunol Immunother 2008;57:1849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondadasula SV, Roda JM, Parihar R, Yu J, Lehman A, Caligiuri MA, et al. Colocalization of the IL-12 receptor and FcgammaRIIIato natural killer cell lipid rafts leads to activation of ERK and enhanced production of interferon-gamma. Blood 2008;111:4173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carson WE, Parihar R, Lindemann MJ, Personeni N, Dierksheide J, Meropol NJ, et al. Interleukin-2 enhances the natural killer cell response to Herceptin-coated Her2/neu-positive breast cancer cells. Eur J Immunol 2001;31:3016–25. [DOI] [PubMed] [Google Scholar]
- 20.Farag SS, Flinn IW, Modali R, Lehman TA, Young D, Byrd JC. Fc gamma RIIIa and Fc gamma RIIa polymorphisms do not predict response to rituximab in B-cell chronic lymphocytic leukemia. Blood 2004;103:1472–4. [DOI] [PubMed] [Google Scholar]
- 21.Verbeke G, Molenberghs G. Linear mixed models for longitudinal data. New York, NY: Springer; 2000. [Google Scholar]
- 22.Segler A, Tsimberidou AM. Lenalidomide in solid tumors. Cancer Che-mother Pharmacol 2012;69:1393–406. [DOI] [PubMed] [Google Scholar]
- 23.Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005;293: 715–22. [DOI] [PubMed] [Google Scholar]
- 24.Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007; 357:2040–8. [DOI] [PubMed] [Google Scholar]
- 25.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757–65. [DOI] [PubMed] [Google Scholar]
- 26.Price TJ, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol 2014;15:569–79. [DOI] [PubMed] [Google Scholar]
- 27.Vermorken JB, Trigo J, Hitt R, Koralewski P, Diaz-Rubio E, Rolland F, et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol 2007;25:2171–7. [DOI] [PubMed] [Google Scholar]
- 28.Wadlow RC, Hezel AF, Abrams TA, Blaszkowsky LS, Fuchs CS, Kulke MH, et al. Panitumumab in patients with KRAS wild-type colorectal cancer after progression on cetuximab. Oncologist 2012;17:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metges J, Raoul J, Achour N, Capitain O, Gourlaouen A, Ramee J, et al. PANERB study: Panitumumab after cetuximab-based regimen failure. J Clin Oncol 28, 2010 (suppl; abstr e14000). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.