

Abstract
Native top-down mass spectrometry is a fast, robust biophysical technique that can provide molecular-scale information on the interaction between proteins or peptides and ligands, including metal cations. Here we have analyzed complexes of the full-length amyloid β (1-42) monomer with a range of (patho)physiologically relevant metal cations using native Fourier transform ion cyclotron resonance mass spectrometry and three different fragmentation methods—collision-induced dissociation, electron capture dissociation, and infrared multiphoton dissociation—all yielding consistent results. Amyloid β is of particular interest as its oligomerization and aggregation are major events in the etiology of Alzheimer’s disease, and it is known that interactions between the peptide and bioavailable metal cations have the potential to significantly damage neurons. Those metals which exhibited the strongest binding to the peptide (Cu2+, Co2+, Ni2+) all shared a very similar binding region containing two of the histidine residues near the N-terminus (His6, His13). Notably, Fe3+ bound to the peptide only when stabilized toward hydrolysis, aggregation, and precipitation by a chelating ligand, binding in the region between Ser8 and Gly25. We also identified two additional binding regions near the flexible, hydrophobic C-terminus, where other metals (Mg2+, Ca2+, Mn2+, Na+, and K+) bound more weakly—one centered on Leu34, and one on Gly38. Unexpectedly, collisional activation of the complex formed between the peptide and [CoIII(NH3)6]3+ induced gas-phase reduction of the metal to CoII, allowing the peptide to fragment via radical-based dissociation pathways. This work demonstrates how native mass spectrometry can provide new insights into the interactions between amyloid β and metal cations.
Electronic supplementary material
The online version of this article (10.1007/s13361-019-02283-7) contains supplementary material, which is available to authorized users.
Keywords: Native mass spectrometry, Amyloid beta, Peptide-metal complex, Native top-down, Mass spectrometry, Electron capture dissociation, Collision-induced dissociation, Infrared multiphoton dissociation, Radical-directed dissociation
Introduction
Gaining insight into protein-protein, protein-ligand, and protein-metal interactions at the molecular level is critical for understanding the biological function of proteins. For example, it has been estimated that metalloproteins alone make up more than a third of all proteins [1, 2]. Widely used methods for elucidating such interactions include nuclear magnetic resonance (NMR) spectroscopy and X-ray crystallography. However, these biophysical techniques typically are limited in terms of sample throughput, and NMR in particular often samples ensemble averages if multiple interaction regions/stoichiometries coexist, depending on the timescales of exchange.
Native mass spectrometry (MS) is a fast, robust method that allows study of intact noncovalent protein-protein and protein-ligand/metal complexes in the gas phase [3–5]. While the lack of a hydrophobic effect in the absence of water originally led to speculation that protein ions in vacuo would turn “inside out” [6], it is currently believed that instead they retain many aspects of their native structure, including overall shape, complex stoichiometry, and binding of ligands, even those that are hydrophobically bound [7–10]. Combined with top-down fragmentation, native MS provides a powerful tool for obtaining information about higher-order structure, including subunit connectivity and ligand binding site(s) [11–13]. Often, binding sites can be determined with single-residue specificity, although this requires extensive backbone cleavage in the region of interest, which is often not achieved in practice. This technique is also ideally and uniquely suited for systems where multiple states coexist, as a specific precursor is almost always selected based on its mass-to-charge (m/z) ratio.
In this work, we have used high-resolution Fourier transform ion cyclotron resonance (FTICR) mass spectrometry to gain insight into the sequence regions involved in peptide-metal interactions. We have used electron capture dissociation (ECD), infrared multiphoton dissociation (IRMPD), and collision-induced dissociation (CID), to determine the peptide-metal interaction sites. In this type of “native top-down” study [14], it is always a concern whether the binding site remains the same during the dissociation process. It has been shown that the increase in internal energy during CID can lead not only to loss but also to migration of a noncovalently bound ligand [15]. ECD has been used successfully to study binding sites of anticancer complexes to biomolecules [16–21]; however, the charge reduction inherent to ECD in some cases is capable of disrupting ion-dipole interactions and this mechanism can also induce ligand migration [22]. While, therefore, there are indications that results from each of our three dissociation methods in isolation should not be fully accepted a priori, consistent results from all three methods allow confident identification of the true gas-phase binding region, given the fundamentally different fragmentation mechanisms involved. An important objective of this study was to investigate to what extent results from the three methods differ. This is because consistency between all three across the relatively broad range of metals tested here would be a strong indication that each method in isolation can be used to identify metal cation binding regions, and that it would generally suffice in such a study to use the method most conveniently available and/or that would lead to the most efficient dissociation.
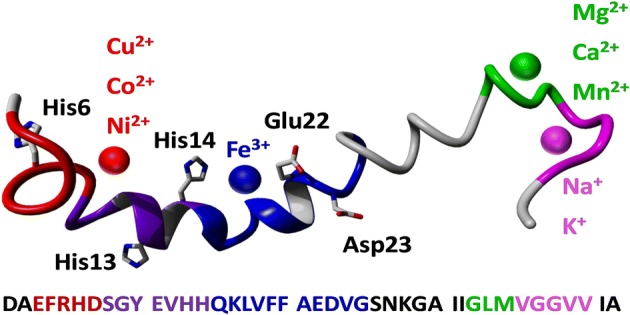
Amyloid β (1-42) was used as a model peptide in this work. Misfolding and accumulation of this peptide, particularly the 40- and 42-residue isoforms, are widely believed to play a major role in the pathogenesis of Alzheimer’s disease, the most prevalent neurodegenerative disease worldwide. It is less widely recognized that interactions between certain trace metals and the peptide play an important role in disease etiology [23–27]. Such interactions have been studied in both in vitro and ex vivo samples, using a range of techniques, including fluorescence assays [27–29], NMR [28–30], Raman [26], and synchrotron X-ray spectromicroscopy [31–33]. In the current work, the metal ions Na+, K+, Mg2+, Ca2+, Mn2+, Co2+, [Co(NH3)6]3+, Ni2+, Cu2+, and Fe3+ were reacted with buffered aqueous solutions of the full-length peptide. These metal cations were mainly chosen because they easily and reproducibly lead to the formation and detection of amyloid-metal complexes under the in vitro conditions used here, but with the exception of [Co(NH3)6]3+, they are also all physiologically or pathophysiologically relevant [24, 31, 33–41]. Although there is widespread interest in the possible involvement of Zn2+ and Al3+ in neurodegeneration on account of their known or suspected dysregulation functions in Alzheimer’s disease, we were unable to obtain consistent MS data for binding of these metal cations to amyloid β (1-42), and so no results are reported here. We believe this is due to their complicated hydrolytic chemistry at physiological pH (formation of hydroxide-bridged oligomers, etc.), as we will illustrate for Fe3+. Although rarely mentioned in the context of AD, Co is included and potentially of interest following a reported increase in brain Co in AD patients versus healthy controls [42]. Interest in Ca has primarily focussed on signalling, and in the context of amyloid accumulation, we highlight our recent observation of diverse calcium biomineral phases within amyloid plaques isolated from humans with AD [33].
Experimental
MS experiments were performed on a 12 T solariX quadrupole/FTICR instrument (Bruker Daltonik GmbH, Bremen, Germany) equipped with an infinity cell and advanced dissociation methods, including electron capture dissociation and infrared multiphoton dissociation. Ions were introduced using nano-ESI in positive ion mode and externally accumulated in a hexapole collision cell before being transferred to the ICR cell. The ESI source used 1.2-mm thin-walled glass capillaries (World Precision Instruments, Hitching, UK) that were pulled in-house to obtain tips of ca. 1-μm orifice diameter with a P97 Flaming/Brown type micropipette puller (Sutter Instrument Co., Novato, CA, USA). For CID and ECD experiments, precursor ions were selected in the quadrupole of the solariX instrument. For IRMPD, in-cell isolation was performed using the correlated harmonic excitation fields (CHEF) method [43], with a notch width of 3 to 5 m/z, and excitation energy of 55%. ECD was performed by generating electrons from a heated hollow cathode, using a current of 1.5 A. IRMPD was performed using a Synrad 48-2 25 W CO2 laser with a 10.6-μm wavelength operated at 50% power. Data analysis was performed using Bruker Compass DataAnalysis 4.1, and peaks were assigned manually. Detailed peak assignment tables can be found in Supporting Information S7. To preserve native peptide-metal interactions, samples were prepared in Milli-Q H2O containing 25 mM ammonium acetate (pH 7.4). Amyloid β (1-42) (monoisotopic mass 4511.27 Da) was purchased from Bachem (Bubendorf, Switzerland). The concentration of the peptide was kept constant at 12.5 μM. NaCl, KCl, MgSO4, CaCl2, MnCl2, [Co(NH3)6]Cl3, CoCl2, CuSO4, NiCl2, FeCl3, and nitrilotriacetic acid were purchased from Sigma (Dorset, UK). Transmission electron microscopy (TEM) images were acquired using a JEOL 2010 microscope operated at 200 kV, and negative staining was performed using uranyl acetate.
Results and Discussion
The non-traditional buffer required for native MS of amyloid-metal complexes did not prevent normal fibril formation
As a first step, we briefly investigated the use of different buffers to study amyloid-metal interactions by mass spectrometry. Most literature reports do not employ MS and have used conventional molecular biology buffers, such as phosphate-buffered saline (PBS), and performing MS using these buffers would have the benefit of eliminating one potential source of discrepancy when comparing our results to the literature. Unfortunately, these conventional buffers contain significant levels of non-volatile cations, such as Na+, which are generally considered incompatible with (nano-)electrospray mass spectrometry. Recently, Williams et al. have demonstrated an approach, based on the use of sub-μM ESI emitters, that seems to allow observation of native-like charge state distributions from a range of non-volatile buffers [44–47]. There have been concerns that use of these emitters leads to significant interaction between the protein and the negatively charged glass surface [46] as well as a decrease in pH at the emitter tip [48], possibly causing denaturation [48, 49]. Furthermore, as a stable spray over the time scale of the experiment—typically ca. 45 min, up to several hours in some cases—was needed for tandem MS, we wanted to avoid the previously acknowledged clogging issues associated with these ultrafine emitters [45], especially as these issues are likely to be exacerbated when spraying an aggregation-prone peptide such as amyloid β. As such, we opted to use more conventional nano-ESI emitters (described in the “Experimental” section) when comparing different buffers.
To avoid excessive Na+ concentrations, a buffer was prepared based on ammonium acetate and ammonium phosphate, and the pH was adjusted to 7.4 by dropwise addition of 35% aqueous NH3. Similarly, an analog for Krebs-Henseleit (KH) buffer, which we have used in previous—synchrotron-based—studies of amyloid/metal interactions [50, 51] was made based on ammonium acetate and piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES). Performance of these buffers was compared to pure water and aqueous ammonium acetate. While some peptide signal was detected using this phosphate-containing buffer, signal intensity was low and significant adduct formation of the peptide with H3PO4 clusters was observed (data not shown). In the KH buffer analog, no peptide signal was detected and it was found that PIPES even at a concentration as low as 1 mM completely suppressed peptide ionization. The use of pure H2O led to very good signal intensity; however, significant peptide binding to trace amounts of Na+ was observed, whereas this was mostly abolished by adding ammonium acetate. As a result, it was decided to carry out the rest of these experiments using 25 mM aqueous ammonium acetate. It could be questioned as to what extent results obtained in (non-physiological) ammonium acetate buffer are relevant for understanding the peptide’s normal behavior. To verify that amyloid β (1-42) did not lose its capability to aggregate under these conditions, peptide solutions were prepared in aqueous ammonium acetate, pure H2O, and KH buffer, and incubated for 375 h (approximately 15 days) at 37 °C. Subsequent TEM imaging of these samples in all cases revealed the typical fibril networks expected for this peptide (see Supporting Information S1).
Formation of amyloid-metal complexes was straightforward in most cases, but Fe3+ required stabilization toward hydrolysis and aggregation by a chelating agent at physiological pH
For mass spectrometry, we initially added 12.5 μM of metal salt to the peptide solution, to provide a 1:1 molar ratio, and measured the degree of adduct formation with native MS. This way, abundant adduct formation with Cu2+, Ni2+, and Co2+ was observed (see Supporting Information S2). The other metals showed no or only limited binding under these conditions, so their concentration was increased to 125 μM (i.e., a metal:peptide molar ratio of 10:1), leading to significant adduct formation in most cases (Supporting Information S2). For Na+ and K+, adduct formation was still quite limited even under these conditions, likely a result of these alkali metals competing for binding sites with the far more abundant (25 mM) singly charged NH4+ ion from ammonium acetate. The fact that adducts with the NH4+ ions were not observed is easily explained by the facile loss of NH3 in the gas phase [52]. As addition of NaCl or KCl did not significantly affect the solution pH, it was decided to test binding of these cations in pure H2O, without addition of ammonium acetate. Under these conditions, sufficient binding for tandem MS was observed at a metal cation concentration of 12.5 μM (Supporting Information S2). Based on their ability to bind most of the available peptide in solution when added in a 1:1 ratio, even in the presence of ammonium acetate, we conclude that, of the metals tested here, Cu2+, Ni2+, and Co2+ bind most strongly to amyloid β (1-42).
Increasing the FeCl3 concentration or incubation time did not lead to any observed binding of Fe3+ to the peptide, in agreement with previous work by Messori et al. [53]. The most likely explanation for this is the hydrolysis and acidity of aqua adducts, and aggregation and precipitation of bridged hydroxide/oxo species leading to poor solubility of this ion at physiological pH. To overcome this issue, we solubilized the Fe3+ using a chelating agent, an approach which has been successfully applied previously [54–56]. Briefly, nitrilotriacetic acid (H3NTA) was added to an aqueous 10 mM solution of FeCl3 in order to give a 1:1 mol ratio. The pH was subsequently adjusted to 7.4 by dropwise addition of 1 M aqueous NH3. Under these conditions, nitrilotriacetate (NTA3-) can act as a tetradentate ligand, with H2O and OH− coordinating to the Fe3+ center, bringing the total coordination number to six [57]. This resulted in a clear solution with no visible precipitate, which was added to the 12.5 μM amyloid β solution. As was done for the other metals, we initially chose our dilution to obtain a 1:1 ratio of amyloid to the [Fe-NTA] complex. As this resulted in only very limited observation of the ternary [amyloid-Fe-NTA] complex, we increased the iron-NTA concentration to 500 μM, corresponding to a 40:1 iron:peptide ratio. This resulted in the observation of a sufficiently intense [Aβ + Fe3+ + NTA3− + 4H]4+ signal to allow interrogation by tandem MS (see Supporting Information S3). No mass increase due to H2O or OH− was observed in this complex; therefore, it seems likely that these were replaced by side chains with a free electron pair. Activation of the ternary complex using either collisions with argon atoms or irradiation by infrared laser resulted in the loss of neutral H3NTA and the observation of [Aβ + Fe + H]4+ (Figure 1). The fact that the chelating agent is lost as a protonated species under these conditions is perhaps surprising; however, loss of a 3− anion from a 7+ cation is likely to be energetically unfavorable. It has been shown previously that multiply charged protein cations can act as surprisingly strong proton donors in the gas phase due to electrostatic repulsion between protons [58]. Interestingly, further activation of this ion resulted in backbone cleavage rather than the observation of any iron-free amyloid (vide infra), indicating that the gas-phase interaction between Fe3+ and the amyloid was strong, consistent with our hypothesis that solubility was the limiting factor in the initial experiments. It has been suggested in the past that ferritin—a water-soluble protein complex and the primary Fe3+ reservoir in the brain—might be the origin of the mixed oxidation state iron phases that have been identified in plaque core material extracted from Alzheimer’s disease brain tissue [33, 59]. Therefore, improved understanding of the interaction between amyloid β (1-42) and Fe3+ could provide insight into an important event in the etiology of Alzheimer’s disease.
Figure 1.
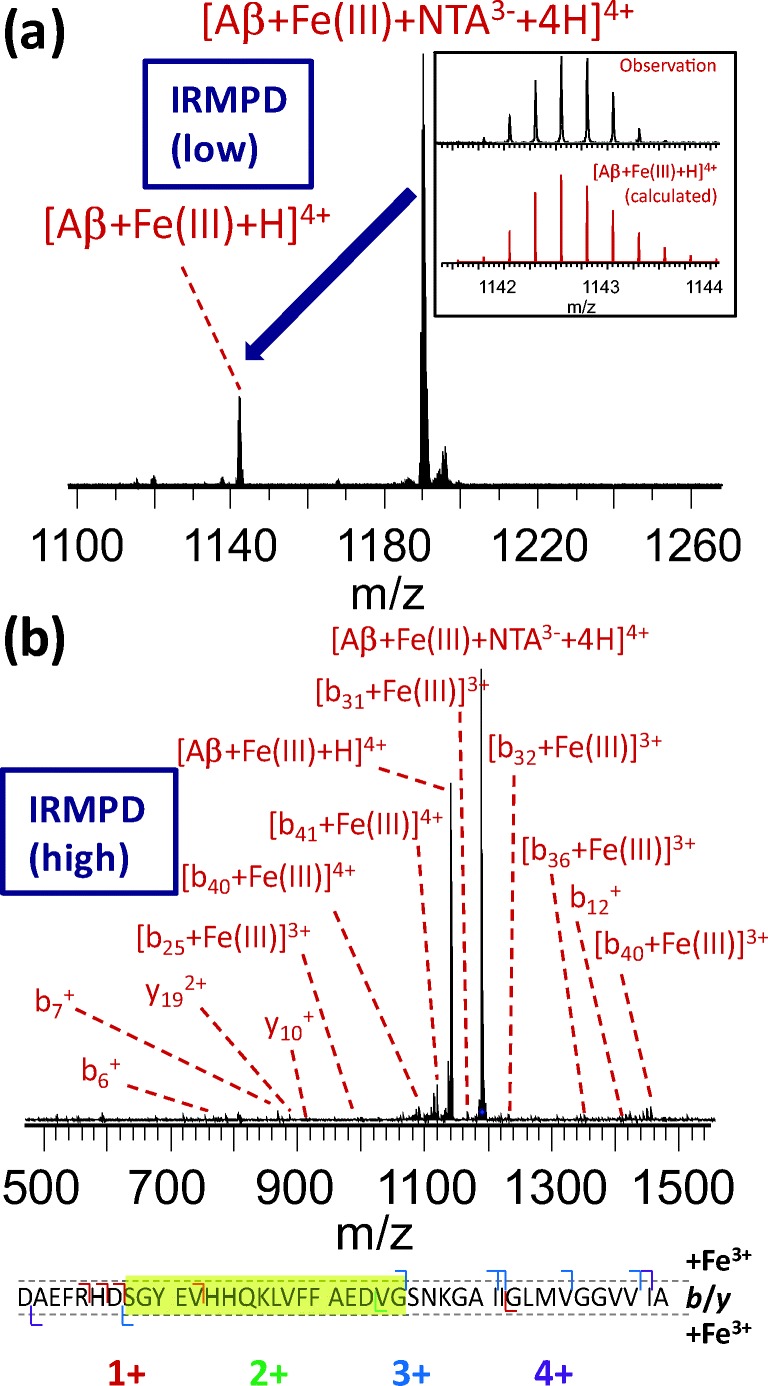
(a) Formation of [Aβ + Fe3+ + H]4+ after IR activation of the ternary complex between amyloid β and [Fe-NTA]. Inset shows the observed and calculated isotope distribution of the product ion, confirming the 3+ oxidation state of Fe. Longer exposure to the IR laser induced backbone fragmentation, as shown in (b). The observed b/y fragment ions both with and without bound Fe3+ are summarized below the fragment spectrum, and the apparent binding region is indicated with a yellow highlight. Fragment charge states are indicated using a color code
As shown in Supporting Information S3, despite our attempts to eliminate sodium contamination from these experiments, a few Na+-related “satellite” peaks were observed, likely due to the presence of trace amounts of Na+ in the H3NTA. Using the quadrupole mass filter of the solariX instrument, isolation of the [Aβ + Fe3+ + NTA3− + 4H]4+ ion with a sufficiently narrow window (approximately 4 m/z) to eliminate these satellite signals resulted in a significant decrease in the precursor ion signal, complicating tandem MS experiments. As a result, in-cell isolation using CHEF was performed, resulting in near-complete elimination of Na+-related signals with minimal signal loss (see Supporting Information S3). As collisional activation within the ICR cell is not trivial, subsequent ion activation was performed using IRMPD. ECD of the [Aβ + Fe + H]4+ complex only resulted in non-dissociative charge reduction, which we attribute to the fact that three of the four protons in the peptide ion were replaced with the metal cation in this case. The IRMPD results will be discussed in detail later. First, we turn our focus toward an in-depth analysis of tandem MS results for the other amyloid-metal complexes, which were successfully interrogated with all three fragmentation methods.
CID, ECD, and IRMPD all resulted in consistent determination of binding regions for eight (patho)physiologically relevant metals, indicating that gas-phase ligand migration did not occur to a significant extent in these experiments
For the complexes of amyloid β with Na+, K+, Mg2+, Ca2+, Mn2+, Co2+, Ni2+, and Cu2+, the 4+ charge state was isolated and subjected to fragmentation using CID (30 V collision energy), IRMPD, and ECD. For ECD, limited concomitant infrared activation [60–62] improved fragmentation efficiency without inducing the formation of the b/y ions that are characteristic for IRMPD fragmentation, and this supplemental activation was used in all cases. Regardless of the choice of fragmentation technique, numerous fragments carrying the metal were observed, providing information on the gas-phase binding region. Briefly, observation of a metal-bound N- (or C-)terminal fragment consisting of n residues provides strong evidence that at least one residue of the first (or last) n coordinates the metal ion. Conversely, observation of a metal-free fragment consisting of m residues can be taken as evidence that the first (or last) m residues do not interact strongly with the metal, although this does not necessarily hold if loss or migration of the metal is energetically more favorable than backbone cleavage and this therefore provides weaker evidence. From the above, it follows that, if multiple residues are involved in coordinating the metal cation, this can result in a “gap” where no fragmentation is observed. As metal cations are generally coordinated by several amino acid residues simultaneously, this method allows the determination of a binding region rather than detailed characterization of the metal binding site in the metal-amyloid complexes studied here.
For any given complex, the identified binding region was relatively consistent, regardless of the dissociation method used. While ECD selectively cleaves the N-Cα bond in the peptide backbone, and has therefore long been established [9, 13, 63–68] as a method for identifying noncovalent ligand binding sites, this result was somewhat unexpected for CID and IRMPD, as these are both considered “slow-heating” methods [69]. Indeed, for most noncovalent ligands, the gradual increase of internal energy, and distribution of this energy across the ion’s internal degrees of freedom, often leads to loss of these ligands prior to backbone fragmentation [64]. However, due to the ca. 80-fold lower dielectric permittivity of vacuum compared with water, electrostatic interactions were significantly strengthened, explaining why metal binding survived the CID process, while cleavage of backbone amide bonds was observed [70–73].
The identified metal binding regions for all metals are summarized in Table 1. A detailed overview of the identified fragments in tandem MS of complexes with Cu2+, Na+, and Ca2+ is shown in Figures 2d, 3d, and 4d, respectively. A similar overview for Co2+, Ni2+, K+, Mn2+, and Mg2+ can be found in Supporting Information S4. Co2+ and Ni2+ bound in a very similar region to Cu2+; K+ bound in a similar way to Na+; and Mn2+ and Mg2+ bound in the same region as Ca2+. Interestingly, the three metal ions that bound strongly at a low concentration (Ni2+, Cu2+, and Co2+) all did so in the N-terminal region. In IRMPD and CID of the Cu2+ adduct (Figure 2), all metal-bound fragments contained the [Asp1–His13] region. In these spectra, the isotope distribution of a number of copper-bound fragment peaks was shifted upward by one hydrogen mass (as indicated in Figure 2d), most likely due to gas-phase reduction of Cu2+ to Cu+. Also, a c7+ fragment and a small number of a fragments were observed. This behavior is consistent with radical-directed dissociation, which will be discussed in detail when discussing results for top-down CID of the amyloid-[Co(NH3)6] complex (vide infra). Details of the in vitro and ex vivo interaction between amyloid β and copper are considered in a forthcoming paper. The IR-ECD spectrum of native [Aβ + 2H + Cu]4+ revealed that the [Arg5–Gln15] region—which includes His6, His13, and His14—contained the Cu2+ binding site (Figure 2b, d). Combined, these data suggest that the metal ion was coordinated by ligands in the N-terminal region, likely including His6 and His13. Importantly, this finding matches literature results obtained using other (solution-phase) methods [30, 34]. This match, along with the remarkable consistency between CID, IRMPD, and ECD results, provides compelling evidence that the metal binding site remained more or less unchanged during ionization and transfer into the gas phase, and hence that the method used here accurately probed these sites on amyloid β.
Table 1.
Overview of Identified Binding Regions for the Metal Cations Investigated in This Study. Dotted Lines Are Used to Divide the Metals into Groups with Similar Binding Regions

Figure 2.
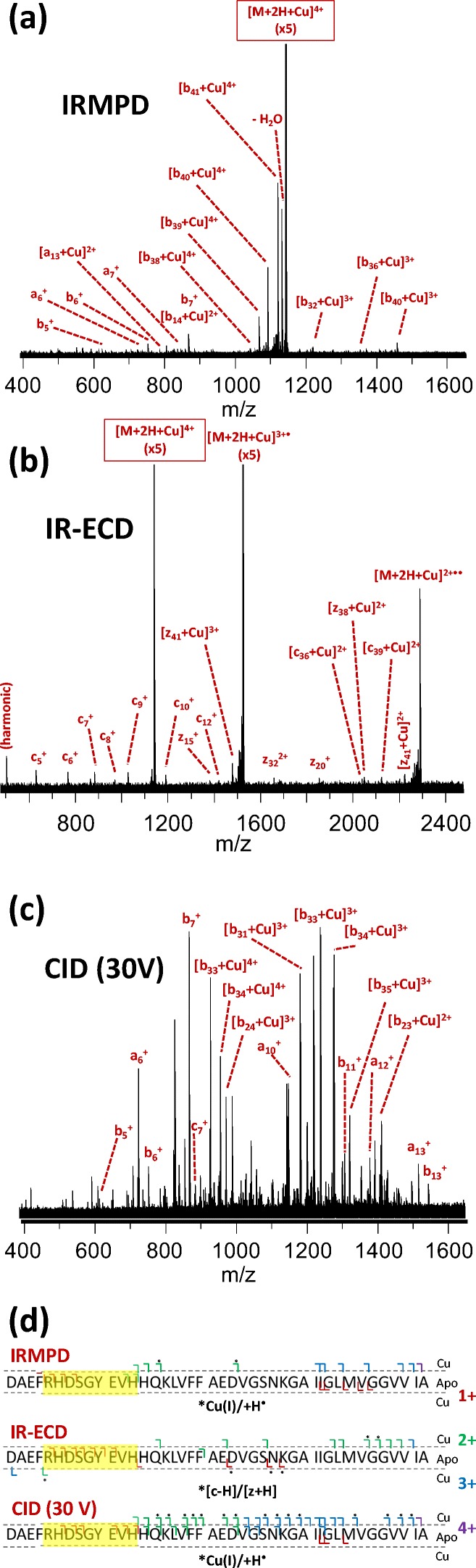
“Native top-down” fragmentation of native [Aβ + 2H + Cu]4+, using (a) IRMPD, (b) IR-ECD, and (c) CID. Fragments with and without a bound metal ion are summarized outside and inside the dotted lines in panel (d), respectively, and charge states are indicated using a color code. In the summary of the CID results in this panel, Cu-bound fragments with isotope distributions shifted upward by one hydrogen mass (most likely due to gas-phase reduction of Cu2+ to Cu+) are indicated with an asterisk. For IR-ECD, fragments formed through hydrogen abstraction from the c fragment by the corresponding z fragment are indicated in the same way. The “consensus” binding region that is consistent with all data obtained from the three fragmentation methods is highlighted in yellow
Figure 3.
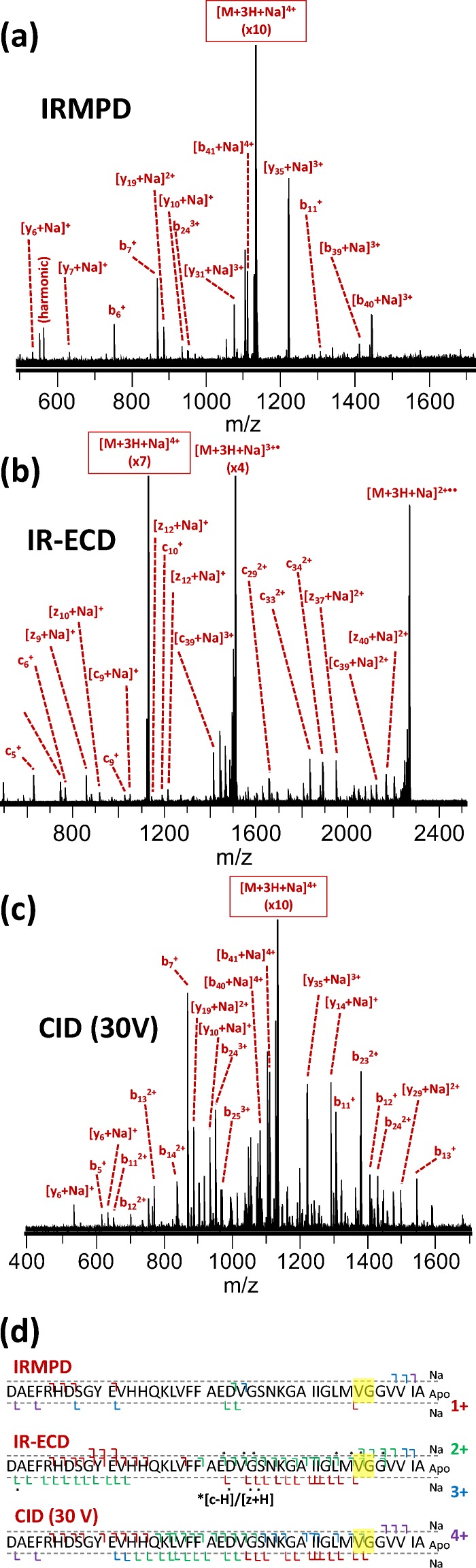
“Native top-down” fragmentation of native [Aβ + 3H + Na]4+, using (a) IRMPD, (b) IR-ECD, and (c) CID. Fragments with and without a bound metal ion are summarized outside and inside the dotted lines in panel (d), respectively, and charge states are indicated using a color code. For IR-ECD, fragments formed through hydrogen abstraction from the c fragment by the corresponding z fragment are indicated with an asterisk. The “consensus” binding region that is consistent with all data obtained from the three fragmentation methods is highlighted in yellow
Figure 4.
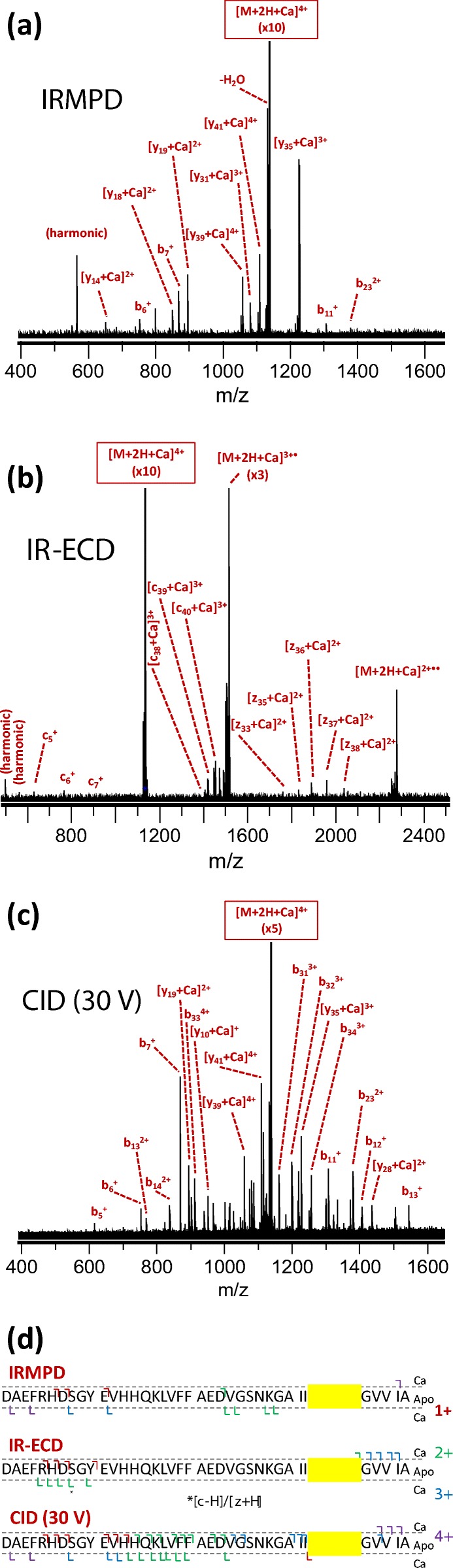
“Native top-down” fragmentation of native [Aβ + 2H + Ca]4+, using (a) IRMPD, (b) IR-ECD, and (c) CID. Fragments with and without a bound metal ion are summarized outside and inside the dotted lines in panel (d), respectively, and charge states are indicated with a color code. For IR-ECD, fragments formed through hydrogen abstraction from the c fragment by the corresponding z fragment are indicated with an asterisk. The “consensus” binding region that is consistent with all data obtained from the three fragmentation methods is highlighted in yellow
The hypothesis that several residues were involved in coordinating these metals is supported by the fragmentation of the Ni2+-bound peptide (Supporting Information S4). In both CID and IRMPD, a metal-bound b6 fragment was observed, demonstrating an interaction between the metal and (at least one of) the first six N-terminal residues of the peptide. Metal-bound b7 and b12 fragments were exclusively found in the CID spectrum, and signal intensity increased significantly for fragments larger than b12, which contain both His6 and His13. At the same time, the observation of metal-bound z30, z32, z33, and z34 in ECD provided evidence that binding of Ni2+ involves at least one of the residues in the [His13–Ala42] region. Therefore, our data indicated that Ni2+ is coordinated by multiple ligands, including His6 and His13. This is consistent with findings reported by Sóvágó and colleagues, who used UV-Vis and circular dichroism spectroscopy to study the complex formed between Ni2+ and amyloid β (1-16) in solution [34]. Similarly, for the Co2+ adduct, the observation of metal-bound b13 (CID) and z40 (IR-ECD) fragments restricted the possible binding site to [Glu3–His13], nearly identical to what was observed for Cu2+.
In contrast to the strongly binding metals discussed so far, the metals that bound more weakly to the peptide did so in the C-terminal region. Na+ was found to bind in the [Val36–Val39] region using IRMPD, the [Gly37–Gly38] region using CID, and at Val36 with ECD (see Table 1 and Figure 3). Similarly, K+ appeared to bind in the [Val39–Val40] region using a combination of the three dissociation methods (see Table 1 and Supporting Information S4). While it might be expected that these ions would bind at either a basic or a negatively charged (deprotonated) site, no polar side chains are found in the region between Val36 and Val40. As such, these ions were most likely coordinated purely by backbone amide oxygens, as has been observed previously in other peptides [74, 75]. The selectivity for this particular region was most likely due to the very high backbone flexibility and side chain hydrophobicity of this part of the peptide. The discrepancy between CID and ECD results for Na+ is likely due to minor variability in binding site, with multiple residues being involved as seen for Ni2+. Interestingly, three N-terminal ECD fragments (c9+, c10+, c11+) were observed which were bound to Na+. With or without addition of Na+, no “zIc”-type internal fragments (i.e., possessing a z-type N-terminus and a c-type C-terminus [76]) or obvious neutral losses would give rise to signals close enough to those of these c fragments to be misidentified. Therefore, we conclude that some—presumably very weak—interaction did in fact occur between Na+ and certain N-terminal residues, either to complete the coordination sphere around the metal center or as a minor, co-existing binding site. Evidently, native top-down ECD was sensitive enough to detect this minor interaction. The K+-bound analogs of these N-terminal fragments were not observed in ECD of [M + 3H + K]4+. It is also noteworthy that cleavage coverage was very good for the sodiated and potassiated peptides, similar to previous observations on small peptoids made by Ren and colleagues [77].
Based on the discussion so far, one might assume that the charge state of the metal cation determines the binding strength and site to amyloid β (1-42)—doubly charged Cu2+, Ni2+, and Co2+ bound easily to the N-terminal histidine residues of the peptide, while singly charged Na+ and K+ bound weakly to a site near the C-terminus. Ca2+, Mg2+, and Mn2+, however, also bound fairly weakly, and did so at a slightly different site compared with the alkali metals. As backbone cleavage for the adduct with Ca2+ was fairly limited, the binding region could not be specified further than [Gly33–Gly37] using a combination of our three dissociation methods (Figure 4). Similarly, the residues binding Mg2+ were located within the region [Ile32–Gly37], while the Mn2+ adduct showed more extensive fragmentation and the binding region in this case was narrowed down to [Gly33–Met35] (see Table 1 and Supporting Information S4). It has long been established that the Mn2+ ion has very similar coordination preferences to those of Mg2+, resulting in complexes with similar geometries [78]. Therefore, although the Mg2+ and Ca2+ binding regions could not be determined as specifically as that for Mn2+ in our MS experiments, it is likely that all three metals shared the same binding site. Warmlander and colleagues, based on solution NMR, concluded that Mn2+ binds primarily to the N-terminal histidine side chains of amyloid β (1-40) [39], although it was noted that Mn2+ does not compete for N-terminal histidine binding sites with Cu2+. Conversely, this could merely reflect the much greater stability of the Cu2+ complex compared with the Mn2+ analog [79]. These authors also identified a second binding site involving Glu22 and Asp23. Presumably, completing the coordination sphere around Mn2+ required involvement of backbone amide groups and we note that, as with the alkali metals Na+ and K+, the region found to bind the metal is very hydrophobic and possesses a relatively flexible structure. Recently, binding of Ca2+ to α-synuclein was investigated by Kaminski-Schierle and colleagues using NMR, and binding was also found to involve a relatively hydrophobic, flexible part of the peptide [80].
For all three fragmentation methods used here, fragmentation efficiency, cleavage coverage, and fragment charge states for the amyloid-metal complexes were broadly similar to those observed for the metal-free peptide (Supporting Information S5), implying that metal bridge formation did not significantly affect fragmentation. It has been observed previously that reducing the number of mobile protons generally leads to reduced fragmentation efficiency in CID, particularly for intermediate and large proteins [81, 82]. For this reason, fragmentation might be expected to be inhibited in the amyloid-metal complexes studied here. That such inhibited fragmentation was not observed is potentially due to the relatively small size of the amyloid β peptide.
IRMPD of the ternary [Aβ-Fe-NTA] complex resulted in limited fragmentation, and indicated binding of Fe3+ in the [His13–Asp23] region
As noted earlier, analysis of the ternary [Aβ + Fe3+ + NTA3− + 4H]4+ complex proved particularly challenging. IRMPD provided some insight, as limited activation resulted in loss of H3NTA and the observation of [Aβ + Fe + H]4+ at m/z 1142.053. Upon further irradiation, no loss of iron was observed but limited backbone fragmentation occurred (Figure 1). Based on the excellent consistency between results from CID, IRMPD, and ECD for the other eight complexes described in the previous section, it can be assumed that the IRMPD fragments accurately reflect the gas-phase structure of the [amyloid-Fe3+] complex. The observation of Fe3+-bound b22 and y35 fragments shows that the binding region is located between Ser8 and Gly25, a region containing Tyr, Glu, His, and Asp residues, all potential Fe3+ binding sites. Due to the complete absence of signal corresponding to iron-free peptide, the observation of metal-free fragments b12+ and y192+ and absence of fragments spanning the [His13–Asp23] region can be seen as evidence that the main interaction region with Fe3+ was likely situated in this stretch of the peptide sequence. As NTA3− coordinates Fe3+ via three acetate moieties, and its loss was coupled to the loss of three protons from the peptide, it is very likely that binding of the iron to the peptide was mediated through several residues in this region. The precise charge sites of gas-phase amyloid β (1-42) are unknown at this point, and it is known that protonation can occur at non-basic amino acid residues such as glutamine in addition to lysine, arginine, and histidine [83, 84]. Therefore, our data do not allow us at this point to ascertain whether the Fe3+ “replaced” three protons and was coordinated by the (formerly) protonated residues, or if proton loss and coordination of Fe3+ occur at different sites in a more complex rearrangement. The amount of information on the interaction of Fe3+ with amyloid β that is available in the literature is limited, as the poor solubility of the metal ion and aggregation of the peptide make this system highly challenging to investigate using solution-phase techniques [85]. More detailed studies of iron binding to amyloid β are therefore underway; however, we note that His13, Glu22, and Asp23 have all been suggested previously to be involved in this interaction [54, 86, 87], in good agreement with our findings.
Gas-phase metal reduction and radical-directed dissociation were observed in CID of the amyloid-[Co(NH3)6] complex
As discussed, binding of Co2+ involved the same N-terminal histidines as Cu2+ and Ni2+. Interesting behavior was observed when cobalt was added as [Co(NH3)6]Cl3, a highly stable Co3+ complex, rather than Co(II)Cl2. This complex bound to amyloid β as strongly as Ca2+ or Mn2+ (see Supporting Information S2), and the theoretical isotope distribution of [M + H + Co(NH3)6]4+ is compared with the observed distribution in Figure 5a. Upon CID, the NH3 ligands dissociated and cobalt was transferred to the N-terminus of the amyloid β peptide. Unexpectedly, rather than [M + H + Co]4+, the observed isotope distribution of the resulting peptide-cobalt complex matched that of [M + 2H + Co]4+ (Figure 5b), which indicates that, during ligand dissociation, Co3+ was reduced to Co2+. This is somewhat similar to what was observed by O’Hair and colleagues, using three different hexapeptides and [Co(salen)]OAc (where salen = N,N‘-ethylenebis(salicylideneaminato)) [88]. This process introduced a radical site in the peptide, leading to a mix of collision-induced and radical-directed dissociation (RDD; Figure 5c). The latter was evidenced by the presence of predominantly a-ions in addition to the b- and y-type fragments typical for CID, with c-ions occurring N-terminal to serine, in agreement with the RDD literature [89, 90].
Figure 5.
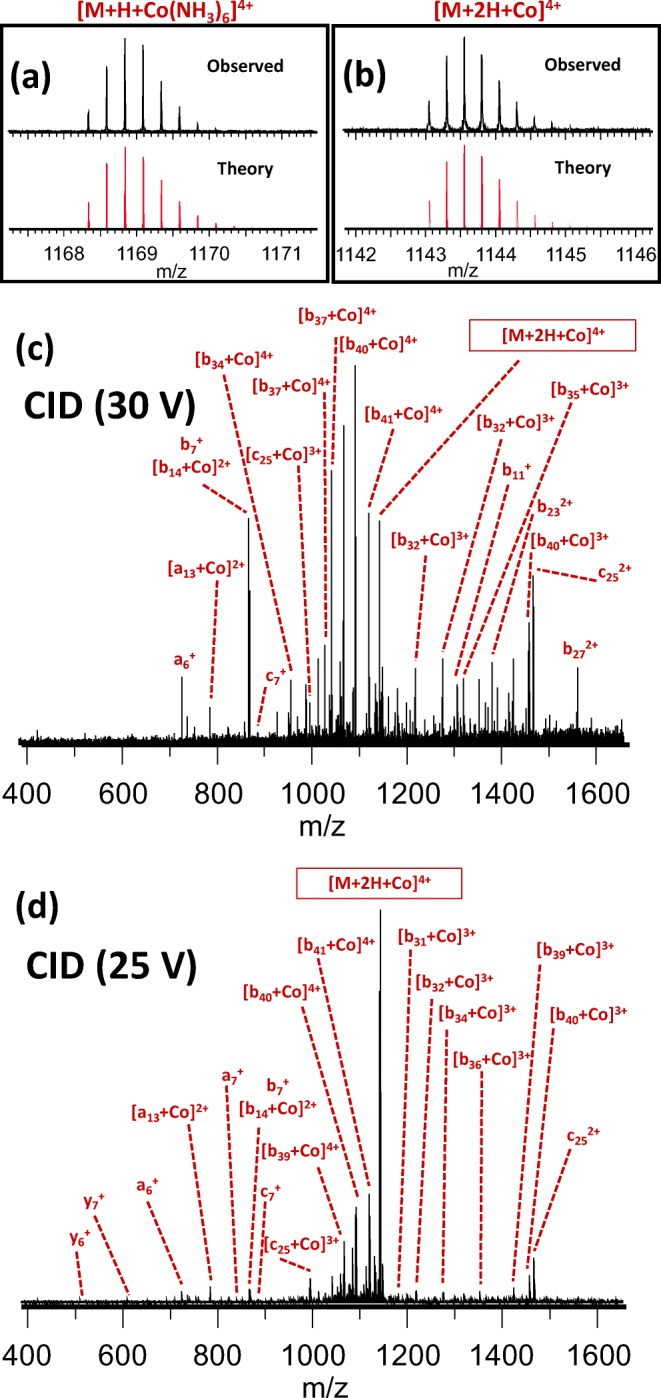
Observed versus calculated isotope distribution (a) for [Aβ + H + Co(NH3)6]4+ and (b) for the [Aβ + 2H + Co]4+ ion observed upon collisional activation of the complex; the bottom two panels show top-down fragmentation of native [Aβ + H + Co(NH3)6]4+, using CID at (c) 30 V, and (d) 25 V. An overview of all fragments identified in (c) and (d) can be found in Supporting Information S6
An a-ion, by definition, is exactly one CO unit lower in mass than the corresponding b-ion. As such, it is also plausible that a-ions were formed by CO loss from a b-ion. It is unlikely that the a-ions observed in our experiments were formed this way, as they did not appear in CID spectra of the metal-free peptide, nor in CID of adducts with other metals besides [Co(NH3)6]3+ and Cu2+. To further rule out secondary fragmentation of “normal” b-ions as the source of the observed a fragments, we repeated the CID experiment using a collision energy of 25 V. As shown in Figure 5d, this still resulted in observation of both a- and c-ions, making the secondary fragmentation mechanism even more unlikely. Furthermore, while formation of a-ions can be explained through an even-electron mechanism (i.e., loss of CO), the presence of c-ions in the spectra is difficult to rationalize without invoking a radical-based mechanism. We therefore conclude that it is far more likely that these fragments were indeed formed by RDD. While radical-directed dissociation has so far only received fairly limited attention from the MS community, it should be noted that inclusion of the a and c fragments in the analysis significantly increased cleavage coverage. A great deal of excellent research on RDD of peptides and intact proteins has been performed by Julian et al., who used homolytic ultraviolet photodissociation of C–I bonds (either in a lariat crown ether [91], or formed through iodination of specific amino acid side chains [92–96]) to generate radicals. However, to the best of our knowledge, this is the first time that a relatively large peptide has been analyzed in an RDD experiment where the radical was generated through collisional activation of a peptide-metal complex, as well as the first reported use of Co(NH3)6Cl3 as an RDD reagent.
Conclusions
Complexes of amyloid β (1-42) with nine bioavailable and potentially (patho)physiologically relevant metals have been studied with native FTICR mass spectrometry, with the aim of preserving the metal-peptide interaction. For each metal, the gas-phase binding region was successfully determined using tandem MS via three different activation techniques. Consistently, and owing to the low dielectric permittivity of vacuum, the metal-peptide interactions were sufficiently strong in the gas phase that determination of the binding region was possible using collision-induced dissociation, leading to results matching those from more “gentle” ECD and IRMPD. This indicates that ligand migration in the gas phase was not a major issue in tandem MS of peptide-metal complexes using any of these dissociation techniques, and that the method that is most conveniently available and/or leads to the most efficient dissociation can be used by itself to confidently identify gas-phase binding regions. Formation of an [amyloid-Fe3+] complex at physiological pH was observed only when a chelating agent was used to minimize aggregation and precipitation of hydroxide/oxido Fe3+ species, followed by mild gas-phase activation. This observation is important and has implications for the design of future studies of peptide interactions with metal ions which readily form hydroxide and oxido species at physiological pH, e.g., Al3+ and Zn2+.More extensive activation of the Fe3+ complex suggested interaction with amyloid β (1-42) in the region between residues His13 and Asp23.
The binding regions identified in the gas phase using tandem MS are consistent with previous studies using solution-phase methods. The metals that exhibited the strongest binding to the peptide all share a very similar binding region involving the N-terminal histidine residues. The metals studied display a strong preference for a particular region, although not the same region. This was observed even for the alkali metals Na+ and K+, which are often assumed to engage in non-specific binding in (native) mass spectrometry. Collisional activation of the complex with [Co(NH3)6]3+ induced unexpected gas-phase redox chemistry and radical-directed peptide dissociation, and inclusion of the resulting fragments in the data analysis significantly increased the observed cleavage coverage. The insights gained from this fast, robust MS method, into the interactions between metal cations and amyloid β, are likely to contribute to improved understanding and treatment of Alzheimer’s disease.
Electronic supplementary material
(DOCX 2603 kb)
Acknowledgements
This work was supported by EPSRC grants EP/N033191/1 and EP/N033140/1. We are grateful to Mr. Ian Hands-Portman and Dr. Saskia Bakker for their help acquiring TEM images. The authors thank the reviewers for their valuable comments.
Footnotes
Spectral data for this paper will be accessible through the University of Warwick open access research repository (WRAP) at http://wrap.warwick.ac.uk/122328/
Peter B. O’Connor and Joanna F. Collingwood contributed equally to this work.
References
- 1.Shi W, Chance MR. Metalloproteomics: forward and reverse approaches in metalloprotein structural and functional characterization. Curr. Opin. Chem. Biol. 2011;15:144–148. doi: 10.1016/j.cbpa.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou R, Wang Q, Wu J, Wu J, Schmuck C, Tian H. Peptide self-assembly triggered by metal ions. Chem. Soc. Rev. 2015;44:5200–5219. doi: 10.1039/c5cs00234f. [DOI] [PubMed] [Google Scholar]
- 3.Loo JA. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 1997;16:1–23. doi: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 4.Benesch JL, Ruotolo BT. Mass spectrometry: come of age for structural and dynamical biology. Curr. Opin. Struct. Biol. 2011;21:641–649. doi: 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konijnenberg A, Butterer A, Sobott F. Native ion mobility-mass spectrometry and related methods in structural biology. Biochim. Biophys. Acta. 2013;1834:1239–1256. doi: 10.1016/j.bbapap.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 6.Wolynes PG. Biomolecular folding in vacuo!!!(?) Proc. Natl. Acad. Sci. U. S. A. 1995;92:2426–2427. doi: 10.1073/pnas.92.7.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruotolo BT, Robinson CV. Aspects of native proteins are retained in vacuum. Curr. Opin. Chem. Biol. 2006;10:402–408. doi: 10.1016/j.cbpa.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 8.Benesch JL, Ruotolo BT, Simmons DA, Robinson CV. Protein complexes in the gas phase: technology for structural genomics and proteomics. Chem. Rev. 2007;107:3544–3567. doi: 10.1021/cr068289b. [DOI] [PubMed] [Google Scholar]
- 9.Xie Y, Zhang J, Yin S, Loo JA. Top-down ESI-ECD-FT-ICR mass spectrometry localizes noncovalent protein-ligand binding sites. J. Am. Chem. Soc. 2006;128:14432–14433. doi: 10.1021/ja063197p. [DOI] [PubMed] [Google Scholar]
- 10.Liu L, Bagal D, Kitova EN, Schnier PD, Klassen JS. Hydrophobic protein-ligand interactions preserved in the gas phase. J. Am. Chem. Soc. 2009;131:15980–15981. doi: 10.1021/ja9060454. [DOI] [PubMed] [Google Scholar]
- 11.Zhou M, Wysocki VH. Surface induced dissociation: dissecting noncovalent protein complexes in the gas phase. Acc. Chem. Res. 2014;47:1010–1018. doi: 10.1021/ar400223t. [DOI] [PubMed] [Google Scholar]
- 12.Brodbelt JS. Photodissociation mass spectrometry: new tools for characterization of biological molecules. Chem. Soc. Rev. 2014;43:2757–2783. doi: 10.1039/c3cs60444f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lermyte F, Valkenborg D, Loo JA, Sobott F. Radical solutions: principles and application of electron-based dissociation in mass spectrometry-based analysis of protein structure. Mass Spectrom. Rev. 2018;37:750–771. doi: 10.1002/mas.21560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lermyte F, Tsybin YO, O’Connor PB, Loo JA. Top or middle? Up or down? Toward a standard lexicon for protein top-down and allied mass spectrometry approaches. J. Am. Soc. Mass Spectrom. 2019;30:1149–1157. doi: 10.1007/s13361-019-02201-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Deng L, Kitova EN, Klassen JS. Dissociation of multisubunit protein-ligand complexes in the gas phase. Evidence for ligand migration. J. Am. Soc. Mass Spectrom. 2013;24:1573–1583. doi: 10.1007/s13361-013-0712-z. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Snelling JR, Barrow MP, Scrivens JH, Sadler PJ, O’Connor PB. Mass spectrometric strategies to improve the identification of Pt(II)-modification sites on peptides and proteins. J. Am. Soc. Mass Spectrom. 2014;25:1217–1227. doi: 10.1007/s13361-014-0877-0. [DOI] [PubMed] [Google Scholar]
- 17.Wills RH, Habtemariam A, Lopez-Clavijo AF, Barrow MP, Sadler PJ, O’Connor PB. Insights into the binding sites of organometallic ruthenium anticancer compounds on peptides using ultra-high resolution mass spectrometry. J. Am. Soc. Mass Spectrom. 2014;25:662–672. doi: 10.1007/s13361-013-0819-2. [DOI] [PubMed] [Google Scholar]
- 18.Qi Y, Liu Z, Li H, Sadler PJ, O’Connor PB. Mapping the protein-binding sites for novel iridium(III) anticancer complexes using electron capture dissociation. Rapid Commun. Mass Spectrom. 2013;27:2028–2032. doi: 10.1002/rcm.6643. [DOI] [PubMed] [Google Scholar]
- 19.Wootton CA, Sanchez-Cano C, Liu HK, Barrow MP, Sadler PJ, O’Connor PB. Binding of an organo-osmium(II) anticancer complex to guanine and cytosine on DNA revealed by electron-based dissociations in high resolution top-down FT-ICR mass spectrometry. Dalton Trans. 2015;44:3624–3632. doi: 10.1039/c4dt03819c. [DOI] [PubMed] [Google Scholar]
- 20.Wootton CA, Sanchez-Cano C, Lopez-Clavijo AF, Shaili E, Barrow MP, Sadler PJ, O’Connor PB. Sequence-dependent attack on peptides by photoactivated platinum anticancer complexes. Chem. Sci. 2018;9:2733–2739. doi: 10.1039/c7sc05135b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Lin TY, Van Orden SL, Zhao Y, Barrow MP, Pizarro AM, Qi Y, Sadler PJ, O’Connor PB. Use of top-down and bottom-up Fourier transform ion cyclotron resonance mass spectrometry for mapping calmodulin sites modified by platinum anticancer drugs. Anal. Chem. 2011;83:9507–9515. doi: 10.1021/ac202267g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heath BL, Jockusch RA. Ligand migration in the gaseous insulin-CB7 complex--a cautionary tale about the use of ECD-MS for ligand binding site determination. J. Am. Soc. Mass Spectrom. 2012;23:1911–1920. doi: 10.1007/s13361-012-0470-3. [DOI] [PubMed] [Google Scholar]
- 23.Adlard PA, Bush AI. Metals and Alzheimer’s disease: how far have we come in the clinic? J. Alzheimers Dis. 2018;62:1369–1379. doi: 10.3233/JAD-170662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnham KJ, Bush AI. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 2014;43:6727–6749. doi: 10.1039/c4cs00138a. [DOI] [PubMed] [Google Scholar]
- 25.Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U. S. A. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR. Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry. 2003;42:2768–2773. doi: 10.1021/bi0272151. [DOI] [PubMed] [Google Scholar]
- 27.House E, Collingwood J, Khan A, Korchazkina O, Berthon G, Exley C. Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Abeta42 in a manner which may have consequences for metal chelation therapy in Alzheimer’s disease. J. Alzheimers Dis. 2004;6:291–301. doi: 10.3233/jad-2004-6310. [DOI] [PubMed] [Google Scholar]
- 28.Guilloreau L, Damian L, Coppel Y, Mazarguil H, Winterhalter M, Faller P. Structural and thermodynamical properties of CuII amyloid-beta16/28 complexes associated with Alzheimer’s disease. J. Biol. Inorg. Chem. 2006;11:1024–1038. doi: 10.1007/s00775-006-0154-1. [DOI] [PubMed] [Google Scholar]
- 29.Syme CD, Nadal RC, Rigby SE, Viles JH. Copper binding to the amyloid-beta (Abeta) peptide associated with Alzheimer’s disease: folding, coordination geometry, pH dependence, stoichiometry, and affinity of Abeta-(1-28): insights from a range of complementary spectroscopic techniques. J. Biol. Chem. 2004;279:18169–18177. doi: 10.1074/jbc.M313572200. [DOI] [PubMed] [Google Scholar]
- 30.Hureau C, Coppel Y, Dorlet P, Solari PL, Sayen S, Guillon E, Sabater L, Faller P. Deprotonation of the Asp1-Ala2 peptide bond induces modification of the dynamic copper(II) environment in the amyloid-beta peptide near physiological pH. Angew. Chem. Int. Ed. Eng. 2009;48:9522–9525. doi: 10.1002/anie.200904512. [DOI] [PubMed] [Google Scholar]
- 31.Collingwood JF, Davidson MR. The role of iron in neurodegenerative disorders: insights and opportunities with synchrotron light. Front. Pharmacol. 2014;5:191. doi: 10.3389/fphar.2014.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Telling ND, Everett J, Collingwood JF, Dobson J, van der Laan G, Gallagher JJ, Wang J, Hitchcock AP. Iron biochemistry is correlated with amyloid plaque morphology in an established mouse model of Alzheimer’s disease. Cell Chem. Biol. 2017;24:1205–1215 e1203. doi: 10.1016/j.chembiol.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 33.Everett J, Collingwood JF, Tjendana-Tjhin V, Brooks J, Lermyte F, Plascencia-Villa G, Hands-Portman I, Dobson J, Perry G, Telling ND. Nanoscale synchrotron X-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer’s disease subjects. Nanoscale. 2018;10:11782–11796. doi: 10.1039/c7nr06794a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jozsa E, Osz K, Kallay C, de Bona P, Damante CA, Pappalardo G, Rizzarelli E, Sovago I. Nickel(II) and mixed metal complexes of amyloid-beta N-terminus. Dalton Trans. 2010;39:7046–7053. doi: 10.1039/c0dt00189a. [DOI] [PubMed] [Google Scholar]
- 35.Andrasi E, Suhajda M, Saray I, Bezur L, Ernyei L, Reffy A. Concentration of elements in human brain: glioblastoma multiforme. Sci. Total Environ. 1993;139-140:399–402. doi: 10.1016/0048-9697(93)90036-6. [DOI] [PubMed] [Google Scholar]
- 36.Isaacs AM, Senn DB, Yuan M, Shine JP, Yankner BA. Acceleration of amyloid beta-peptide aggregation by physiological concentrations of calcium. J. Biol. Chem. 2006;281:27916–27923. doi: 10.1074/jbc.M602061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garai K, Sengupta P, Sahoo B, Maiti S. Selective destabilization of soluble amyloid beta oligomers by divalent metal ions. Biochem. Biophys. Res. Commun. 2006;345:210–215. doi: 10.1016/j.bbrc.2006.04.056. [DOI] [PubMed] [Google Scholar]
- 38.Roberts BR, Ryan TM, Bush AI, Masters CL, Duce JA. The role of metallobiology and amyloid-beta peptides in Alzheimer’s disease. J. Neurochem. 2012;120(Suppl 1):149–166. doi: 10.1111/j.1471-4159.2011.07500.x. [DOI] [PubMed] [Google Scholar]
- 39.Wallin C, Kulkarni YS, Abelein A, Jarvet J, Liao Q, Strodel B, Olsson L, Luo J, Abrahams JP, Sholts SB, Roos PM, Kamerlin SC, Graslund A, Warmlander SK. Characterization of Mn(II) ion binding to the amyloid-beta peptide in Alzheimer’s disease. J. Trace Elem. Med. Biol. 2016;38:183–193. doi: 10.1016/j.jtemb.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 40.Collingwood JF, Adams F. Chemical imaging analysis of the brain with X-ray methods. Spectrochim. Acta B At. Spectrosc. 2017;130:101–118. [Google Scholar]
- 41.Chellan, P., Sadler, P.J.: The elements of life and medicines. Philos Transact. A Math. Phys. Eng. Sci. 373, 20140182 (2015) [DOI] [PMC free article] [PubMed]
- 42.Wenstrup D, Ehmann WD, Markesbery WR. Trace element imbalances in isolated subcellular fractions of Alzheimer’s disease brains. Brain Res. 1990;533:125–131. doi: 10.1016/0006-8993(90)91804-p. [DOI] [PubMed] [Google Scholar]
- 43.de Koning LJ, Nibbering NMM, Van Orden SL, Laukien FH. Mass selection of ions in a Fourier transform ion cyclotron resonance trap using correlated harmonic excitation fields (CHEF) Int. J. Mass Spectrom. Ion Process. 1997;165:209–219. [Google Scholar]
- 44.Susa AC, Xia Z, Williams ER. Small emitter tips for native mass spectrometry of proteins and protein complexes from nonvolatile buffers that mimic the intracellular environment. Anal. Chem. 2017;89:3116–3122. doi: 10.1021/acs.analchem.6b04897. [DOI] [PubMed] [Google Scholar]
- 45.Susa AC, Xia Z, Williams ER. Native mass spectrometry from common buffers with salts that mimic the extracellular environment. Angew. Chem. Int. Ed. Eng. 2017;56:7912–7915. doi: 10.1002/anie.201702330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xia Z, Williams ER. Protein-glass surface interactions and ion desalting in electrospray ionization with submicron emitters. J. Am. Soc. Mass Spectrom. 2018;29:194–202. doi: 10.1007/s13361-017-1825-6. [DOI] [PubMed] [Google Scholar]
- 47.Susa AC, Lippens JL, Xia Z, Loo JA, Campuzano IDG, Williams ER. Submicrometer emitter ESI tips for native mass spectrometry of membrane proteins in ionic and nonionic detergents. J. Am. Soc. Mass Spectrom. 2018;29:203–206. doi: 10.1007/s13361-017-1793-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu J, Guan QY, Wang J, Jiang XX, Wu ZQ, Xia XH, Xu JJ, Chen HY. Effect of nanoemitters on suppressing the formation of metal adduct ions in electrospray ionization mass spectrometry. Anal. Chem. 2017;89:1838–1845. doi: 10.1021/acs.analchem.6b04218. [DOI] [PubMed] [Google Scholar]
- 49.Mortensen DN, Williams ER. Surface-induced protein unfolding in submicron electrospray emitters. Anal. Chem. 2016;88:9662–9668. doi: 10.1021/acs.analchem.6b02499. [DOI] [PubMed] [Google Scholar]
- 50.Everett J, Cespedes E, Shelford LR, Exley C, Collingwood JF, Dobson J, van der Laan G, Jenkins CA, Arenholz E, Telling ND. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide beta-amyloid (1-42) J. R. Soc. Interface. 2014;11:20140165. doi: 10.1098/rsif.2014.0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Everett J, Cespedes E, Shelford LR, Exley C, Collingwood JF, Dobson J, van der Laan G, Jenkins CA, Arenholz E, Telling ND. Evidence of redox-active iron formation following aggregation of ferrihydrite and the Alzheimer’s disease peptide beta-amyloid. Inorg. Chem. 2014;53:2803–2809. doi: 10.1021/ic402406g. [DOI] [PubMed] [Google Scholar]
- 52.Kebarle P, Verkerk UH. Electrospray: from ions in solution to ions in the gas phase, what we know now. Mass Spectrom. Rev. 2009;28:898–917. doi: 10.1002/mas.20247. [DOI] [PubMed] [Google Scholar]
- 53.Valensin D, Migliorini C, Valensin G, Gaggelli E, La Penna G, Kozlowski H, Gabbiani C, Messori L. Exploring the reactions of beta-amyloid (Abeta) peptide 1-28 with Al(III) and Fe(III) ions. Inorg. Chem. 2011;50:6865–6867. doi: 10.1021/ic201069v. [DOI] [PubMed] [Google Scholar]
- 54.Pramanik D, Dey SG. Active site environment of heme-bound amyloid beta peptide associated with Alzheimer’s disease. J. Am. Chem. Soc. 2011;133:81–87. doi: 10.1021/ja1084578. [DOI] [PubMed] [Google Scholar]
- 55.Jiang D, Li X, Williams R, Patel S, Men L, Wang Y, Zhou F. Ternary complexes of iron, amyloid-beta, and nitrilotriacetic acid: binding affinities, redox properties, and relevance to iron-induced oxidative stress in Alzheimer’s disease. Biochemistry. 2009;48:7939–7947. doi: 10.1021/bi900907a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu H, Alexeev D, Hunter DJ, Campopiano DJ, Sadler PJ. Oxo-iron clusters in a bacterial iron-trafficking protein: new roles for a conserved motif. Biochem. J. 2003;376:35–41. doi: 10.1042/BJ20031283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bunescu A, Besse-Hoggan P, Sancelme M, Mailhot G, Delort AM. Comparison of microbial and photochemical processes and their combination for degradation of 2-aminobenzothiazole. Appl. Environ. Microbiol. 2008;74:2976–2984. doi: 10.1128/AEM.01696-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zenaidee MA, Leeming MG, Zhang F, Funston TT, Donald WA. Highly charged protein ions: the strongest organic acids to date. Angew. Chem. Int. Ed. Eng. 2017;56:8522–8526. doi: 10.1002/anie.201702781. [DOI] [PubMed] [Google Scholar]
- 59.Collingwood JF, Chong RK, Kasama T, Cervera-Gontard L, Dunin-Borkowski RE, Perry G, Posfai M, Siedlak SL, Simpson ET, Smith MA, Dobson J. Three-dimensional tomographic imaging and characterization of iron compounds within Alzheimer’s plaque core material. J. Alzheimers Dis. 2008;14:235–245. doi: 10.3233/jad-2008-14211. [DOI] [PubMed] [Google Scholar]
- 60.Lin C, O’Connor PB, Cournoyer JJ. Use of a double resonance electron capture dissociation experiment to probe fragment intermediate lifetimes. J. Am. Soc. Mass Spectrom. 2006;17:1605–1615. doi: 10.1016/j.jasms.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 61.Lin C, Cournoyer JJ, O’Connor PB. Probing the gas-phase folding kinetics of peptide ions by IR activated DR-ECD. J. Am. Soc. Mass Spectrom. 2008;19:780–789. doi: 10.1016/j.jasms.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsybin YO, He H, Emmett MR, Hendrickson CL, Marshall AG. Ion activation in electron capture dissociation to distinguish between N-terminal and C-terminal product ions. Anal. Chem. 2007;79:7596–7602. doi: 10.1021/ac071165u. [DOI] [PubMed] [Google Scholar]
- 63.Yin S, Loo JA. Elucidating the site of protein-ATP binding by top-down mass spectrometry. J. Am. Soc. Mass Spectrom. 2010;21:899–907. doi: 10.1016/j.jasms.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 64.Goth M, Lermyte F, Schmitt XJ, Warnke S, von Helden G, Sobott F, Pagel K. Gas-phase microsolvation of ubiquitin: investigation of crown ether complexation sites using ion mobility-mass spectrometry. Analyst. 2016;141:5502–5510. doi: 10.1039/c6an01377e. [DOI] [PubMed] [Google Scholar]
- 65.Liu H, Hakansson K. Divalent metal ion-peptide interactions probed by electron capture dissociation of trications. J. Am. Soc. Mass Spectrom. 2006;17:1731–1741. doi: 10.1016/j.jasms.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 66.Flick TG, Donald WA, Williams ER. Electron capture dissociation of trivalent metal ion-peptide complexes. J. Am. Soc. Mass Spectrom. 2013;24:193–201. doi: 10.1007/s13361-012-0507-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen X, Liu G, Elaine Wong YL, Deng L, Wang Z, Li W, Dominic Chan TW. Dissociation of trivalent metal ion (Al(3+), Ga(3+), In(3+) and Rh(3+))--peptide complexes under electron capture dissociation conditions. Rapid Commun. Mass Spectrom. 2016;30:705–710. doi: 10.1002/rcm.7502. [DOI] [PubMed] [Google Scholar]
- 68.Dong J, Vachet RW. Coordination sphere tuning of the electron transfer dissociation behavior of Cu(II)-peptide complexes. J. Am. Soc. Mass Spectrom. 2012;23:321–329. doi: 10.1007/s13361-011-0299-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McLuckey SA, Goeringer DE. Slow heating methods in mass spectrometry. J. Mass Spectrom. 1997;32:461–474. [Google Scholar]
- 70.Loo JA, Hu P, Smith RD. Interaction of angiotensin peptides and zinc metal ions probed by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 1994;5:959–965. doi: 10.1016/1044-0305(94)80014-6. [DOI] [PubMed] [Google Scholar]
- 71.Hu P, Loo JA. Gas-phase coordination properties of Zn2+, Cu2+, Ni2+, and Co2+ with histidine-containing peptides. J. Am. Chem. Soc. 1995;117:11314–11317. [Google Scholar]
- 72.Nemirovskiy OV, Gross ML. Determination of calcium binding sites in gas-phase small peptides by tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 1998;9:1020–1028. doi: 10.1016/S1044-0305(98)00071-3. [DOI] [PubMed] [Google Scholar]
- 73.Wongkongkathep P, Han JY, Choi TS, Yin S, Kim HI, Loo JA. Native top-down mass spectrometry and ion mobility MS for characterizing the cobalt and manganese metal binding of alpha-synuclein protein. J. Am. Soc. Mass Spectrom. 2018;29:1870–1880. doi: 10.1007/s13361-018-2002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Teesch LM, Adams J. Fragmentations of gas-phase complexes between alkali metal ions and peptides: metal ion binding to carbonyl oxygens and other neutral functional groups. J. Am. Chem. Soc. 1991;113:812–820. [Google Scholar]
- 75.Feng WY, Gronert S, Fletcher KA, Warres A, Lebrilla CB. The mechanism of C-terminal fragmentations in alkali metal ion complexes of peptides. Int. J. Mass Spectrom. 2003;222:117–134. [Google Scholar]
- 76.Harvey SR, Porrini M, Tyler RC, MacPhee CE, Volkman BF, Barran PE. Electron capture dissociation and drift tube ion mobility-mass spectrometry coupled with site directed mutations provide insights into the conformational diversity of a metamorphic protein. Phys. Chem. Chem. Phys. 2015;17:10538–10550. doi: 10.1039/c4cp05136j. [DOI] [PubMed] [Google Scholar]
- 77.Morishetti KK, Russell SC, Zhao X, Robinson DB, Ren J. Tandem mass spectrometry studies of protonated and alkali metalated peptoids: enhanced sequence coverage by metal cation addition. Int. J. Mass Spectrom. 2011;308:98–108. [Google Scholar]
- 78.Bock CW, Katz AK, Markham GD, Glusker JP. Manganese as a replacement for magnesium and zinc: functional comparison of the divalent ions. J. Am. Chem. Soc. 1999;121:7360–7372. [Google Scholar]
- 79.Irving, H., Williams, R.J.P.: The stability of transition-metal complexes. J Chem Soc. 3192–3210 (1953)
- 80.Lautenschlager J, Stephens AD, Fusco G, Strohl F, Curry N, Zacharopoulou M, Michel CH, Laine R, Nespovitaya N, Fantham M, Pinotsi D, Zago W, Fraser P, Tandon A, St George-Hyslop P, Rees E, Phillips JJ, De Simone A, Kaminski CF, Schierle GSK. C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018;9:712. doi: 10.1038/s41467-018-03111-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Greer, S.M., Holden, D.D., Fellers, R., Kelleher, N.L., Brodbelt, J.S.: Modulation of protein fragmentation through carbamylation of primary amines. J Am Soc Mass Spectrom. 28, 1587–1599 (2017) [DOI] [PMC free article] [PubMed]
- 82.Bashyal A, Sanders JD, Holden DD, Brodbelt JS. Top-down analysis of proteins in low charge states. J. Am. Soc. Mass Spectrom. 2019;30:704–717. doi: 10.1007/s13361-019-02146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schnier PD, Gross DS, Williams ER. On the maximum charge state and proton transfer reactivity of peptide and protein ions formed by electrospray ionization. J. Am. Soc. Mass Spectrom. 1995;6:1086–1097. doi: 10.1016/1044-0305(95)00532-3. [DOI] [PubMed] [Google Scholar]
- 84.Morrison LJ, Brodbelt JS. Charge site assignment in native proteins by ultraviolet photodissociation (UVPD) mass spectrometry. Analyst. 2016;141:166–176. doi: 10.1039/c5an01819f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bousejra-ElGarah F, Bijani C, Coppel Y, Faller P, Hureau C. Iron(II) binding to amyloid-beta, the Alzheimer’s peptide. Inorg. Chem. 2011;50:9024–9030. doi: 10.1021/ic201233b. [DOI] [PubMed] [Google Scholar]
- 86.Miura T, Suzuki K, Takeuchi H. Binding of iron(III) to the single tyrosine residue of amyloid β-peptide probed by Raman spectroscopy. J. Mol. Struct. 2001;598:79–84. [Google Scholar]
- 87.Nair NG, Perry G, Smith MA, Reddy VP. NMR studies of zinc, copper, and iron binding to histidine, the principal metal ion complexing site of amyloid-beta peptide. J. Alzheimers Dis. 2010;20:57–66. doi: 10.3233/JAD-2010-1346. [DOI] [PubMed] [Google Scholar]
- 88.Barlow CK, McFadyen WD, O’Hair RA. Formation of cationic peptide radicals by gas-phase redox reactions with trivalent chromium, manganese, iron, and cobalt complexes. J. Am. Chem. Soc. 2005;127:6109–6115. doi: 10.1021/ja043088f. [DOI] [PubMed] [Google Scholar]
- 89.Turecek F, Julian RR. Peptide radicals and cation radicals in the gas phase. Chem. Rev. 2013;113:6691–6733. doi: 10.1021/cr400043s. [DOI] [PubMed] [Google Scholar]
- 90.Chu IK, Rodriquez CF, Lau T, Hopkinson AC, Siu KW. Molecular radical cations of oligopeptides. J. Phys. Chem. B. 2000;104:3393–3397. [Google Scholar]
- 91.Sun Q, Nelson H, Ly T, Stoltz BM, Julian RR. Side chain chemistry mediates backbone fragmentation in hydrogen deficient peptide radicals. J. Proteome Res. 2009;8:958–966. doi: 10.1021/pr800592t. [DOI] [PubMed] [Google Scholar]
- 92.Ly T, Julian RR. Residue-specific radical-directed dissociation of whole proteins in the gas phase. J. Am. Chem. Soc. 2008;130:351–358. doi: 10.1021/ja076535a. [DOI] [PubMed] [Google Scholar]
- 93.Ly T, Julian RR. Elucidating the tertiary structure of protein ions in vacuo with site specific photoinitiated radical reactions. J. Am. Chem. Soc. 2010;132:8602–8609. doi: 10.1021/ja910665d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sun Q, Yin S, Loo JA, Julian RR. Radical directed dissociation for facile identification of iodotyrosine residues using electrospray ionization mass spectrometry. Anal. Chem. 2010;82:3826–3833. doi: 10.1021/ac100256v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang X, Julian RR. Investigating the gas phase structure of KIX with radical directed dissociation and molecular dynamics: retention of the native structure. Int. J. Mass Spectrom. 2011;308:225–231. [Google Scholar]
- 96.Zhang X, Li H, Moore B, Wongkongkathep P, Ogorzalek Loo RR, Loo JA, Julian RR. Radical-directed dissociation of peptides and proteins by infrared multiphoton dissociation and sustained off-resonance irradiation collision-induced dissociation with Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 2014;28:2729–2734. doi: 10.1002/rcm.7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 2603 kb)