

Abstract
Alzheimer’s disease is the most common cause of dementia and one of the most complex human neurodegenerative diseases. Numerous studies have demonstrated a critical role of the environment in the pathogenesis and pathophysiology of the disease, where daily life stress plays an important role. A lot of epigenetic studies have led to the conclusion that chronic stress and stress-related disorders play an important part in the onset of neurodegenerative disorders, and an enormous amount of research yielded valuable discoveries but has so far not led to the development of effective treatment strategies for Alzheimer’s disease. Corticotropin-releasing factor (CRF) is one of the major hormones and at the same time a neuropeptide acting in stress response. Deregulation of protein levels of CRF is involved in the pathogenesis of Alzheimer’s disease, but little is known about the precise roles of CRF and its binding protein, CRF-BP, in neurodegenerative diseases. In this review, we summarize the key evidence for and against the involvement of stress-associated modulation of the CRF system in the pathogenesis of Alzheimer’s disease and discuss how recent findings could lead to new potential treatment possibilities in Alzheimer’s disease by using CRF-BP as a therapeutic target.
Subject terms: Molecular neuroscience, Pharmacology
Release of CRF in response to stress
Stress is a life-saving mechanism that has been shaped and refined throughout evolution1. Acute stress leads to an increase in attention and to memory consolidation2,3. For example, anxiety is a normal reaction to stress and, if not excessive, is crucial for homeostasis. However, chronic or excessive stress leads to a decrease in performance and cognition, and hence limited adaptation to the stressor4,5. Individual kinetics and magnitude of stress response determine its outcome in terms of resilience or development of stress-related disorders6. This can occur in every stage of life5,7. Already before birth, stressful situations can have a major impact on the future life span of the organism as well as in newborn, young adult, and older stages of life7,8.
Stress response is a highly orchestrated mechanism whereby the body rapidly activates the autonomic nervous system and the hypothalamic-pituitary-adrenocortical (HPA) axis9,10. By activation of the HPA axis and the autonomic nervous system, an enormous number of hormones, neurotransmitters, and neuropeptides are released as adaptive reactions to restore homeostasis10. Corticotropin-releasing factor (CRF) and CRF family peptides (Fig. 1) are major regulators of stress response due to their ability to integrate physiological responses to react against a stressor, and due to their dual roles as hormones and as neuromodulators10–12.
Fig. 1. The CRF family and its relation with CRF-BP.
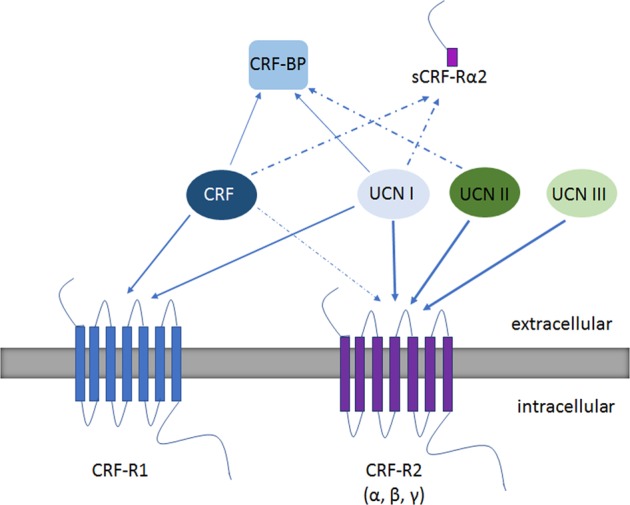
The CRF family consists of two different CRF receptors: CRF-R1 (one functional isoform) and CRF-R2 (three functional isoforms: α, β, γ). The ligands, CRF and UCN I, UCN II and UCN III will bind and will induce G-protein-coupled signaling via CRF-Rs. The arrows represent the affinity between ligand and receptor or ligand and binding protein. The different affinities are represented by the pattern and thickness of the arrow lines; dashed lines will represent lower affinity as compared with solid arrow lines. The weight of the solid lines will give even more detail about the affinity between both ligand and receptor. CRF displays a relatively high affinity for CRF-R1 and CRF-R2. CRF has a comparatively lower affinity for CRF-R2 compared with its affinity for CRF-R1. UCN I has an approximately equal affinity for both receptors, and UCN II and UCN III seem to be selective for CRF-R2. The signaling cascade also includes CRF-BP and the recently identified soluble α-isoform of CRF-R2 (sCRF-Rα2). CRF-BP binds to CRF and UCN I with high affinity to modulate the biological activities of the ligands. Both CRF-BP and CRF-Rα2 are able to sequester CRF and UCN I, whereas CRF-BP exerts a low affinity for UCN II. Abbreviations; corticotropin-releasing factor (CRF), urocortin (UCN), CRF receptor 1 (CRF-R1), CRF receptor 2 (CRF-R2), CRF-binding protein (CRF-BP), α-soluble isoform of CRF-R2 (sCRF-Rα2)
As a hormone, the 41-amino acid polypeptide CRF is secreted at the onset of stress in the paraventricular nucleus of the hypothalamus13. CRF is delivered via the bloodstream to the anterior pituitary, where it binds to its receptors and stimulates adrenocorticotropic hormone (ACTH) release. ACTH release activates synthesis of corticosteroids in the adrenal cortex: glucocorticoids such as cortisol in humans and corticosterone in rodents13,14. Glucocorticoids can exert profound modulatory effects on a variety of brain functions from early sensitive developmental stages to late adulthood. Fetal exposure to exogenous glucocorticoids or prenatal stress can lead to permanent alteration of HPA function and stress-related performance15. At the adult stage, high levels of glucocorticoids have been associated with reduced cognitive ability, including poor memory and decreased mental flexibility and processing speed16. After crossing the blood–brain barrier, glucocorticoids activate two types of receptors, the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR), which mediate stress response in the brain6,17. MRs are active under basal conditions and have high affinity to glucocorticoids. GRs have low affinity to glucocorticoids and are activated in response to high levels of the hormone during stress18,19. To summarize, endogenous corticosteroid secretion from the adrenal cortex is mainly under the control of ACTH produced by the pituitary gland. ACTH secretion is controlled mostly by the hypothalamic CRF. The aforementioned phenomena rely on transcriptional regulation and occur within time frames ranging from hours to weeks20.
Roles of CRF as a neuromodulator
In contrast to hormones, neurotransmitters and neuromodulators exhibit fast action, allowing neurons to react to stressful situations on the synaptic level, within milliseconds to minutes20. CRF acts as the principal hormone in the HPA axis21, as discussed above, and triggers a number of secondary stress-related events such as secretion of the corticosteroids in the adrenal cortex. However, in addition to its action to regulate the HPA axis at the level of the pituitary, CRF is also expressed in different parts of the brain, where it can act as a neuromodulator or neurotransmitter in response to autonomic and behavioral responses22. Here, CRF synergizes with corticosteroids to fine-tune stress responses in a short time frame11,23,24.
CRF binds to two receptors, CRF-R1 and CRF-R2, with a higher affinity for CRF-R123 (Fig. 1). Both are seven-span G-coupled receptors, are expressed in different regions in the brain, and share ~70% identity with each other25–28. In addition to CRF, these receptors can bind three other ligands (Fig. 1): urocortin (UCN) I, UCN II (stresscopin-related peptide), and UCN III (stresscopin). UCN I can bind to both receptors with similar affinity29, UCN II shows preferential binding to CRF-R2, and UCN III selectively binds CRF-R223. Binding of CRF and UCN I to CRF-R1 and CRF-R2 is modulated in vivo and in vitro by the CRF-binding protein (CRF-BP)30, a 37 kDa glycoprotein that was first detected in human plasma31–33. CRF-BP is structurally unrelated to CRF receptors34. In vertebrates, CRF-BP binds to both CRF (Ki = 0.2 nM) and UCN I (Ki = 2 nM) with sub-nanomolar affinity (Fig. 1 and Table 1), which is greater than the affinity between CRF and its receptors. The affinity of CRF-BP for the other CRF family members is substantially lower: for UCN II, the IC50 is 4.4 nM and UCN III has no affinity for CRF-BP, only for CRF-R230,35. This suggests that CRF-BP could differentially modulate the activity of several CRF family members.
Table 1.
A summary of the most common agonists and antagonists of the CRF family
| Agonist | Antagonist | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | Peptide/nonpeptide | Peptide sequence | Binding affinity Ki (nM) | References | Name | Peptide/nonpeptide | Peptide sequence | Binding affinity Ki (nM) | References | |
| CRF-R1 | h/r CRF | Peptide | SEEPPISLDLT FHLLREVLEM ARAEQLAQQA HSNRKLMEII | 1.0 (0.2–4.6) | 109, 131 | Alpha-helical CRF fragment 9–41 | Peptide | DLT FHLLREMLEM AKAEQEAEQA ALNRLLLEEA | 19 (5.5–66) | 108, 129, 130 |
| Astressin | Peptide | fHLLREVLEZ ARAEQLAQEA HKNRKLZEII | 0.7 (0.3–1.8) | 101, 129 | ||||||
| Antalarmin | Non-peptide | NA | 1 | 110, 132 | ||||||
| CP-154,526 | Non-peptide | NA | 2.7 | 110, 132 | ||||||
| CP-316,311 | Non-peptide | NA | 6.8 | 110, 111 | ||||||
| h UCN I | Peptide | DNPSLSIDLT FHLLRTLLEL ARTQSQRERA EQNRIIFDSV | 0.1 (0.1–0.2) | 109, 131 | CRA-0450 | Non-peptide | NA | 40–60 | 110, 133 | |
| NBI-30775/R121919 | Non-peptide | NA | 3.5 | 114 , 134 , 135 | ||||||
| Pexacerfont | Non-peptide | NA | 7.2 ± 0.9 | 113 , 136 | ||||||
| Verucerfont | Non-peptide | NA | 6.1 | 136 | ||||||
| CRF-R2 | h/r CRF | Peptide | SEEPPISLDLT FHLLREVLEM ARAEQLAQQA HSNRKLMEII | 6.2 (2.0–19) | 109, 131 | Antisauvagine 30 | Peptide | fHLLRKMIEI EKQEKEKQQA ANNRLLLDTI | 1.4 | 130 |
| h UCN I | Peptide | DNPSLSIDLT FHLLRTLLEL ARTQSQRERA EQNRIIFDSV | 0.5 (0.3–0.7) | 109, 131 | ||||||
| h UCN II | Peptide | IVLSLDV IGLLQILLEQ ARARAAREQA TTNARILARV IGLLQILLEQ ARARAAREQA TTNARILARV
|
0.5 (0.2–1.2) | 109, 131 | Astressin2-B | Peptide | DLS FHLLRKXIEI EKQEKEKQQA ENNKLLLDLI | 1.3 | 130 | |
| h UCN III | Peptide | FTLSLDV TNIMNLLFNI AKAKNLRAQA AANAHLMAQI TNIMNLLFNI AKAKNLRAQA AANAHLMAQI
|
13.5 (9.2–19.7) | 109, 131 | ||||||
| CRF-BP | CRF | Peptide | SEEPPISLDLT FHLLREVLEM ARAEQLAQQA HSNRKLMEII | 0.2 | 109, 131 | h/r CRF6-33 | Peptide | ISLDLTFH LLREVLEMAR AEQLAQQAHS | 0.2–5 | 40 |
| UCN I | Peptide | DNPSLSIDLT FHLLRTLLEL ARTQSQRERA EQNRIIFDSV | 2 | 109, 131 | h/r CRF9-33 | Peptide | DLTFH LLREVLEMAR AEQLAQQAHS | 0.2–5 | 40 | |
In bold, receptor activation; underlined, ECD1-binding domain; blue letter, CRF-R2 specific; double underlined, CRF-BP sequence; Ki, binding affinity
The discovery of a rodent-specific splice variant α-isoform of CRF-R2 (sCRF-Rα2), which encodes the soluble ligand-binding domain of CRF-R236, suggests an even greater complexity of the CRF family. Similar to CRF-BP, sCRF-Rα2 binds CRF and UCN I but has a distinct distribution in comparison with CRF-BP36,37 (Fig. 1). sCRF-Rα2 is highly expressed in the olfactory bulb, cortex, midbrain, and the pituitary, and lower levels are found in the hypothalamus, pons, medulla, and spinal cord, and shows high overlap with the cellular distribution of CRF-R136,38. In the CNS, CRF-BP is highly expressed in the cortex, olfactory bulb, hippocampus, and amygdala, and is known to inhibit CRF actions in the HPA axis39,40. In the brain, CRF-BP function depends on the tissue and cellular context41. Here, CRF-BP and CRF exhibit limited co-expression at the cellular level36,38. CRF-BP is localized at the CRF target sites such as the prefrontal cortex and amygdala, regions important in stress-related conditions including anxiety and addiction41. Notably, there is a difference in expression of the CRF-BP between male and female mice. In females, CRF-BP expression is positively regulated by estrogen and gonadotropin-releasing hormone in pituitary cell types. This increased expression positively correlates with several conditions such as anorexia, mood disorders, and anxiety disorders, which are more prevalent in females42.
In addition to its functions in stress, CRF exerts time- and dose-dependent direct actions during learning and memory formation. The exposure of neurons to acute CRF in vitro induces adaptation by spine increase and spine maturation, but exposure to high dosages of the peptide results in spine retraction and loss of synapses in the hippocampus20,43. Opposite effects were seen in the cerebellum, where an increase of spines and dendritic complexity was observed in response to physiological concentrations of CRF in organotypic slice cultures44,45. Clearly, effects of CRF on cellular plasticity and connectivity are highly dependent on the context, i.e., the administered dose, duration of stimulation, and cellular composition. It was further shown that chronic exposure or high levels of CRF lead to the activation of CRF-R1, and subsequently actin cytoskeleton rearrangements and atrophy of the spines and dendrites over longer time periods46,47. This decrease in spines and in dendritic complexity, and changes in synaptic function are cellular hallmarks of cognitive impairment20, characteristic, among others, for late stages of neurodegenerative disorders such as Alzheimer’s disease48–50.
Stress as an environmental risk factor in Alzheimer’s disease
Alzheimer’s disease (AD) is an age-dependent neurodegenerative disorder that causes progressive cognitive impairment and structural alterations in the brain. The brains of patients with AD are characterized by the deposition of extracellular amyloid plaques consisting of β-amyloid (Aβ) peptides, the product of the proteolytic processing of the amyloid precursor protein (APP), and by abnormal hyperphosphorylation of the Tau protein, which precipitates in the form of neurofibrillary tangles inside the neurons48,49. A hallmark of the disease is the progressive neuronal loss in the hippocampus and cortex, brain structures that are extremely important in memory formation50–52. The most common form of AD is sporadic AD without a familial link, but genetically the disease presents as both familial and sporadic cases53. Despite many years of research, whether all forms of AD have a common underlying etiology remains poorly understood. A major effort in the research field has been put into the investigation of genetic factors, although environmental factors could be similarly or even more important in the onset of the disease54.
For example, in an AD mouse model, carrying a mutated form of APP, accumulation of Aβ and hyperphosphorylated Tau has been shown to occur as a result of exposure to high stress levels by acute restrained stress, chronic isolation, and social stress paradigms55–57. A reduction in the number of mature dendritic spines in a triple-transgenic mouse model of AD (3 × Tg-AD: PS1 (M146V), APP (Swe), tau (P301L)) and increased Aβ levels due to the augmentation of amyloid-β protein precursor processing58 have been observed in response to short-term stress.
Exposure to stress early in life can contribute to the later development of AD59–62. High and chronic stress levels lead to a hyper-activation of the HPA axis, which correlates with the rate of AD progression63,64. At the same time, chronic stress and stress-dependent disorders, such as posttraumatic stress, and major depression disorders have been linked to the hyper-activation of HPA axis and are directly correlated to a dysregulation in the CRF system11,13 and AD5,14,15.
Chronic stress-dependent disorders can trigger and/or facilitate AD. This effect is specific to the onset of depression late in adulthood, but not for early-age depression65,66. However, stress-dependent disorders such as depression, anxiety disorders, and mood disorders are often concurrent with AD. Patients with early AD display alterations in mood before or concurrently with initial signs of minor cognitive impairment or memory loss67,68. Recently, a unique transgenic rat model of AD, TgF344-AD (APP (Swe), (PS1ΔE9)), has been used to investigate emotional components of AD69. Young TgF344-AD rats were tested for spatial memory functions in a Morris water maze and for anxiety-like behavior in an elevated plus maze. TgF344-AD rats showed increased anxiety-like behavior without a significant decline in spatial memory. These results indicate that enhanced anxiety-like behavior correlates with an early stage of AD and can be used as an early marker58. Furthermore, depression-like symptoms can accelerate the loss of brain tissue shown in magnetic resonance imagingstudies in patients with early stages of AD70–72. To summarize, stress can lead to AD and accelerate neurodegeneration, but disease progression can in turn cause disruption of stress response circuits, which can exacerbate stress-like symptoms, forming a vicious cycle66. On the other hand, AD is caused by a combination of different factors and not only linked to high and/or chronic stress levels.
The link between CRF and AD
As discussed above, the effect of chronic or high levels of stress leads to the hyperactivation of the HPA axis. Besides the elevation of glucocorticoids73, clinical studies have documented the CRF dysregulation in patients with AD74–76. Significant reduction of the CRF immunoreactivity (CRF-IR) was observed in postmortem tissue in the cerebrocortical areas and in the cerebrospinal fluid of patients with AD compared with that in control tissue75,77,78. Together with the decrease in CRF-IR, increase in the concentration of the CRF receptors (CRF-Rs) in the cortex has been directly linked to AD74,79. CRF-expressing neurons in the paraventricular nucleus of the hypothalamus show elevated levels of CRF mRNA in postmortem brain from AD patients compared with age-matched healthy controls48; however, the number of CRF-expressing cells is not altered. Calcium flux measurements and neuronal death assays in culture suggested a neuroprotective role for CRF in response to Aβ toxicity in AD brain, although this hypothesis was never confirmed by in vivo studies80. It has been hypothesized that CRF might stimulate non-amyloidogenic APP cleavage, exerting a beneficial effect on Aβ accumulation81. In vivo animal studies identified CRF as a factor that enhances cognition and memory. Upon injection in the amygdala and the ventricles of the rat brain, CRF dose-dependently improved retention of the inhibitory avoidance response and improved acquisition of a visual discrimination task22,82. In addition, blockage of CRF in the basolateral amygdala in rats and restoring free levels of CRF afterwards showed improved memory83. Notably, these are all aspects of memory and cognition that are significantly impacted in AD patients84.
In contrast to these findings, multiple studies have demonstrated that exposure to stress, along with elevated levels of CRF, provoked Aβ neuropathology85–87. For example, in the 3 × Tg-AD mice88, enhanced CRF signaling associated with short-term life stress in adults enhanced the progression of Aβ neuropathology58. In parallel, infusion of CRF directly into the hippocampus increased the concentration of two major isoforms of Aβ, namely Aβ40 and Aβ4285,86, therefore facilitating AD. Park et al.89 reported a link between this Aβ-enhancing potential of stress-induced CRF and γ-secretase activity as the underlying mechanism. Further, reduced CRF signaling in CRF-R1-deficient mice or treatment with the antagonist NBI-30775 (also known as R121919) both alleviated the stress-induced aggravation in Aβ pathology in a stress-like paradigm in AD mouse models90,91. In addition to increasing Aβ concentration, CRF can also enhance Tau pathology56,92. Enhancement of Tau phosphorylation at specific epitopes was observed in CRF-overexpressing mice and could be reversed by the specific CRF-R1 antagonist NBI-3077593.
To summarize, chronic stress in combination with high CRF levels can cause stress-dependent disorders, increase the risk of AD, and can lead to other diseases94. On the other hand, in neurodegenerative diseases, low CRF concentrations are observed78,95,96. Chronic stress leads to HPA axis overstimulation, followed by the generation of high CRF and GRs levels in the blood, which can cause detrimental effects to neurons and their connections. These detrimental effects can lower CRF levels in the brain leading to a decrease in its neuromodulatory functions in memory and cognition6,58,97.
As of today, AD remains incurable. There is a clear potential to investigate the link between CRF and AD with a focus on developing CRF-centric therapeutic approaches. Targeting CRF signaling, particularly CRF receptors, in stress disorders has been considered as a valid approach for a considerable time10.
Signaling and pharmacology of CRF receptors
CRF-Rs belong to the family of G-protein-coupled receptors (GPCRs). These GPCRs mainly activate Gs, which will in turn increase adenylcyclase and stimulate enzymatic activity. Activation of the CRF-Rs, by CRF or by their other ligands, follows a two-step mechanism. First, the C-terminus of CRF binds to the N-terminus of the CRF-Rs. This first interaction causes a conformational change in the CRF-R, which stabilizes the CRF–CRF-Rs interaction, whereupon the interaction with the N-terminus of CRF initiates, and thereby triggers, cellular signaling by activating G-protein signaling98–102. Different ligands cause different conformational changes and consequently lead to binding of different G-proteins that will each activate distinct signaling pathways103. For example, in primary cells of pregnant myometrium, upon binding of UCN I with CRF-R1, there is an activation of mitogen-activated protein, which stimulates Gq for activation of the IP3-protein kinase signaling pathway; however, CRF binding does not activate the same signaling cascade104. CRF-R1 peptide agonists, such as astressin (Ki = 0,7), lack the N-terminus of the CRF (Table 1) but bind with high affinity to the extracellular binding domain to prevent binding of a CRF-R agonist. The lack of the N-terminus prevents G-protein interactions and thus inhibits downstream signaling101,105. Conversely, ligands such as α-helical CRF fragment 9–41 that lack the C-terminus (Table 1) can bind with low affinity (Ki = 19) and still will be able to activate the receptor106,107. Non-peptide antagonists or small molecule agonists, such as NBI-30775, antalarmin, and CRA-0450, bind almost exclusively to the juxtamembrane region, which partially inhibits CRF-R1 ligand binding by blocking the interaction of a ligand with the juxtamembrane domain, but cannot prevent the binding between the ligand and the extracellular domain of the receptor. NBI-30775, antalarmin, and CRA-0450 are allosteric inhibitors; they do not bind to the N-terminus to activate the receptor, but instead bind to the J-domain where they will cause a conformational change, leading to the inactivation of the receptor (Table 1).
To date, there are no specific non-peptide or small-molecule antagonists for CRF-R210,108. CRF-R1 antagonists such as NBI-30775, pexacerfont, and CP-316,311 were the first allosteric GPCR inhibitors in clinical trials against depression and anxiety disorders. However, none of these compounds showed efficacy in advanced trials109–111. More recent pre-clinical discoveries present a more detailed view on the interaction between a ligand and the CRF-Rs. The observation of receptor association and dissociation revealed a significant difference between lead compounds. These differences could explain the disappointing outcome of previous clinical trials. Future trials could be improved by focusing on maximizing the receptor residence time or adjusting the dosage of antagonist to ensure CRF-R1 in the brain will remain occupied over longer periods112. In addition, novel technologies, such as positron emission tomography, genetic screening, and local viral CRF overexpression tools open up new opportunities to develop new compounds that will be more effective to influence CRF-R1 downstream signaling10,113.
CRF-BP and treatment of AD
A growing body of evidence suggests a prominent role of CRF-BP in the CRF system. CRF-BP regulates CRF bioavailability in the HPA axis and can restore normal levels of CRF. It is therefore tempting to consider targeting CRF-BP as a potential therapeutic route to control CRF levels, which are disrupted in AD, to improve memory and to delay AD progression.
CRF-BP is highly conserved in gene structure throughout the evolution from invertebrates to humans114. Studies have indicated its importance in the regulation of CRF activity39. CRF-BP expression is increased in the pituitary and the brain, predominantly by stress115. This increased expression of the CRF-BP, specifically in the HPA axis, results in lower levels of free, i.e., CRF-BP-unbound, CRF and suppresses CRF-R1 activation and downstream signaling. CRF-BP is expressed in a highly tissue-specific pattern and differs between species. Human CRF-BP is detected in the plasma, placenta, synovial fluid, amniotic fluid, liver, the pituitary, and the brain116. In other species, notably in rodents, CRF-BP expression is exclusively restricted to the brain and the pituitary32,39. These species-specific expression patterns represent an important limiting factor in behavioral and in vivo studies for evaluating the full physiological functions of the protein in rodent stress models. In the past, three different mouse models with altered CRF-BP expression were created: two transgenic mouse lines that overexpress CRF-BP and one CRF-BP knockout model117–120 (Table 2). Phenotypical studies showed increased anxiety in mice with a CRF-BP-null mutation117,121. Unfortunately, no study has so far shown changes in memory consolidation or cognition in these transgenic CRF-BP models. In the human brain, a large portion of CRF appears to be complexed with CRF-BP and therefore likely unavailable for activating CRF-Rs and downstream events122. The effective concentration of free CRF is not directly related to total CRF abundance. Instead, the effective concentration is a function of both CRF and CRF-BP levels116. Several clinical trials that investigated the inhibition of CRF by blocking its receptor CRF-R1 as a potential treatment for CRF-dependent disorders had negative outcomes, as discussed above. The clinical studies either demonstrated multiple side effects, such as in the case of compound R121919, which caused elevated liver enzymes, or reported a complete lack of efficacy, such as in the case of CP-316,311, verucerfont, and pexacerfont10,123. Accordingly, such drugs that directly target the binding of CRF-BP to CRF and indirectly inactivate CRF-R1 may represent attractive next-generation candidates for the treatment of stress-dependent disorders, potentially including AD.
Table 2.
An overview of studies to observe the role of CRF-BP by genetic or non-genetic manipulations in functional animal models
| Model | Molecular changes | References | ||
|---|---|---|---|---|
| Genetic modifications | Overexpression models | Overexpression of CRF-BP in the pituitary | Compensatory increase of CRF and arginine vasopressin in the paraventricular nucleus | 137 |
| Phenotype | ||||
| Increased locomotor activity and a non-significant decrease in anxiety | ||||
| Overexpression of CRF-BP in the brain, the pituitary, and peripheral tissue | Attenuation of ACTH secretion after lipopolysaccharide treatment | 119 | ||
| Phenotype | ||||
| Increased body weight | ||||
| Knockout models | CRF-BP-null mutation (CRF-BP KO) | Levels of corticosterone and ACTH were unchanged | 117, 121 | |
| Phenotype | ||||
| Reduced body weight (male mice), increased anxiety (sex dependent) | ||||
| Impairment of maternal aggression but no change of inter male aggression | ||||
| Non-genetic modifications | Acute stress condition and/or removal from adrenal glands in rats (adrenalectomy) | The acute stress condition: increased CRF-BP expression Adrenalectomy condition: decreased CRF-BP expression | 138 | |
| Induced stress by food deprivation in rats | Increased CRF-BP in basolateral amygdala. Decreased CRF-BP expression in the pituitary and elevated plasma corticosterone | 139 | ||
| The effect of acute and chronic restraint stress, specific on basolateral amygdala and dorsal hippocampus in rats | Acute stress condition: increased CRF-BP mRNA expression. Chronic stress condition: no change in CRF-BP mRNA levels in basolateral amygdala. CRF-BP mRNA levels in the hippocampus were unaltered | 140 | ||
| The effect of acute restraint stress in basolateral and central amygdala in rats | Increased expression in the basolateral amygdala. No change in CFR-BP expression in the central amygdala. No changes in CRF or CRF-Rs mRNA in both regions | 141 | ||
| Social defeat in male rats | There may be an increase in free available CRF | 142 | ||
| Phenotype | ||||
| Impairment of social approach behavior. After adding CRF6-33 into the bed nuclei of the stria terminals, restored social approach |
Using the CRF-BP antagonist ligand CRF6-33, Behan et al.124 showed a restoration of normal brain levels of CRF in AD models. The application of CRF6-33 increased free levels of CRF and demonstrated cognition-enhancement properties in the Morris water maze paradigm of learning and memory in AD mice. This indirect approach to activate CRF-Rs does not require CRF-R agonists and is consequently unlikely to result in no characteristic side effects of CRF-R agonisms, such as disturbance in anxiety and arousal, which are characteristic to CRF-R1 and not CRF-R2124,125. In AD, there is no change in CRF-BP, so the total amount of hCRF does provide a readout of free levels of CRF. Therefore, releasing CRF from its binding protein would elevate the levels of available CRF in the aging brain. Consequently, CRF-BP ligands could potentially be sufficient to alleviate some of the symptoms and memory deficits in AD patients. In addition, the critical role of astrocytes in AD and other neurodegenerative diseases such as amyotrophic lateral sclerosis and multiple sclerosis is currently emerging126,127. Astrocytes express and release CRF-BP, and display the highest amount of membrane-bound CRF-BP. The astrocyte-associated CRF-BP reservoir might potentially play a causative role in AD and represent a therapeutic target. Astrocytes have attracted attention due to their role in disease127 and until now their roles as an important source of CRF-BP have not been broadly investigated in stress-related diseases or neurodegenerative disorders.
Conclusions
Stress and AD are strongly linked to each other. Ligands and receptors of the CRF system have been investigated as therapeutic targets in neuropsychiatric disorders in numerous studies and clinical trials. Interpretation of these data is not always straightforward. As discussed above, effects of CRF as neuropeptide are highly variable depending on the location, dose, and exposure time. Further, a clear distinction must be made between functions as hormone and as neuropeptide, particularly with regard to local vs. systemic and primary vs. secondary effects. Here we provide a rationale for targeting CRF-BP as an alternative approach in AD. Admittedly, there is still a shortage of mechanistic data about CRF-BP and its possible function and distribution in the brain. Two studies investigating the potential roles of CRF-BP in AD focused on its inactivating impact/effect on CRF and UCN I interactions with their receptors124,125. In an animal model, by targeting the CRF-BP it was possible to achieve restoration of memory and learning the brain functions impaired in AD125. These findings could lead to new treatment possibilities in AD by using CRF-BP as a therapeutic target. The aforementioned studies did not investigate any other possible roles of the binding protein, its possible brain region-specific functions, or its cellular and subcellular localization124,125. Furthermore, one needs to consider the difference in AD prevalence between male and female subjects, and the fact that females are more susceptible to develop stress-dependent disorders, correlating with and potentially due to the different distribution pattern of CRF-BP in the brain42,128. Nevertheless, CRF-BP appears to be an attractive target for symptomatic treatment in AD. Inhibition of CRF-BP has the potential to boost free CRF levels, which are decreased in AD. Unlike CRF-R1 antagonists, targeting CRF-BP is unlikely to interfere with the HPA axis and to cause pertinent side effects. Clearly, more fundamental research is required prior to any progression to the clinic.
Acknowledgements
We thank Evgenia Salta (VIB-KU Leuven) and Vasily Rybakin (REGA Institute, KU Leuven) for critical reading of the manuscript, and all members of the Gounko laboratory for discussion and comments. D.V. is supported by a Methusalem grant from KU Leuven and the Flemish Government awarded to Professor Bart De Strooper (METH/14/07).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nesse R. M., Bhatnagar S., Young E. A. in Encyclopedia of Stress p. 965–970 (Elsevier, 2007).
- 2.Baram TZ, Joëls M. The neuro-symphony of stress. Nat. Rev. Neurosci. 2009;10:459–466. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McEwen BS, Gianaros PJ. Stress- and allostasis-induced brain plasticity. Annu Rev. Med. 2010;62:431–445. doi: 10.1146/annurev-med-052209-100430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McEwen BS. Allostasis and allostatic load: Implications for neuropsychopharmacology. Neuropsychopharmacology. 2000;22:108–124. doi: 10.1016/S0893-133X(99)00129-3. [DOI] [PubMed] [Google Scholar]
- 5.McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol. Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 6.Bisht K, Sharma K, Tremblay MÈ. Chronic stress as a risk factor for Alzheimer’s disease: roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobiol. Stress. 2018;9:9–21. doi: 10.1016/j.ynstr.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marin MF, et al. Chronic stress, cognitive functioning and mental health. Neurobiol. Learn Mem. 2011;96:583–595. doi: 10.1016/j.nlm.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 8.Charil A, Laplante DP, Vaillancourt C, King S. Prenatal stress and brain development. Brain Res. Rev. 2010;65:56–79. doi: 10.1016/j.brainresrev.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Herman JP, et al. Neural regulation of endocrine and ANS responses. Clin. Auton. Res. 2010;20:121. [Google Scholar]
- 10.Deussing JM, Chen AA. The corticotropin-releasing factor family: physiology of the stress response. Physiol. Rev. 2018;98:2225–2286. doi: 10.1152/physrev.00042.2017. [DOI] [PubMed] [Google Scholar]
- 11.Boorse GC, Denver RJ. Widespread tissue distribution and diverse functions of corticotropin-releasing factor and related peptides. Gen. Comp. Endocrinol. 2006;146:9–18. doi: 10.1016/j.ygcen.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 12.Koob GF, Heinrichs SC. A role for corticotropin releasing factor and urocortin in behavioral responses to stressors. Brain Res. 1999;848:141–152. doi: 10.1016/S0006-8993(99)01991-5. [DOI] [PubMed] [Google Scholar]
- 13.Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- 14.Juruena MF. Early-life stress and HPA axis trigger recurrent adulthood depression. Epilepsy Behav. 2014;38:148–159. doi: 10.1016/j.yebeh.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 15.Kapoor Amita, Dunn Elizabeth, Kostaki Alice, Andrews Marcus H., Matthews Stephen G. Fetal programming of hypothalamo-pituitary-adrenal function: prenatal stress and glucocorticoids. The Journal of Physiology. 2006;572(1):31–44. doi: 10.1113/jphysiol.2006.105254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reynolds RM, et al. Morning cortisol levels and cognitive abilities in people with type 2 diabetes: the Edinburgh type 2 diabetes study. Diabetes Care. 2010;33::714–720. doi: 10.2337/dc09-1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheuer DA. Regulation of the stress response in rats by central actions of glucocorticoids. Exp. Physiol. 2010;95:26–31. doi: 10.1113/expphysiol.2008.045971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Kloet ER, et al. Brain mineralocorticoid receptors and centrally regulated functions. Kidney Int. 2000;57:1329–1336. doi: 10.1046/j.1523-1755.2000.00971.x. [DOI] [PubMed] [Google Scholar]
- 19.Groeneweg FL, Karst H, de Kloet ER, Joëls M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol. Cell Endocrinol. 2012;350:299–309. doi: 10.1016/j.mce.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 20.Maras PM, Baram TZ. Sculpting the hippocampus from within: stress, spines, and CRH. Trends Neurosci. 2012;35:315–324. doi: 10.1016/j.tins.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spiess J, Rivier J, Rivier C, Vale W. Primary structure of corticotropin-releasing factor from ovine hypothalamus. Proc. Natl Acad. Sci. USA. 1981;78:6517–6521. doi: 10.1073/pnas.78.10.6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koob GFBF. Corticotropin-releasing factor and behavior. Fed. Proc. 1985;44:259–263. [PubMed] [Google Scholar]
- 23.Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu. Rev. Pharm. Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- 24.Dunn Adrian J., Berridge Craig W. Physiological and behavioral responses to corticotropin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Research Reviews. 1990;15(2):71–100. doi: 10.1016/0165-0173(90)90012-D. [DOI] [PubMed] [Google Scholar]
- 25.Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol. Disord. Drug Targets. 2006;5:453–479. doi: 10.2174/187152706777950684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen R, Lewis KA, Perrin MH, Vale WW. Expression cloning of a human corticotropin-releasing-factor receptor. Proc. Natl Acad. Sci. USA. 1993;90:8967–8971. doi: 10.1073/pnas.90.19.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perrin MH, Donaldson CJ, Chen R, Lewis KA, Vale WW. Cloning and functional expression of a rat brain corticotropin releasing factor (CRF) receptor. Endocrinology. 1993;133:3058–3061. doi: 10.1210/endo.133.6.8243338. [DOI] [PubMed] [Google Scholar]
- 28.Chang CP, Pearse RV, O’Connell S, Rosenfeld MG. Identification of a seven transmembrane helix receptor for corticotropin-releasing factor and sauvagine in mammalian brain. Neuron. 1993;11:1187–1195. doi: 10.1016/0896-6273(93)90230-O. [DOI] [PubMed] [Google Scholar]
- 29.Perrin M, et al. Identification of a second corticotropin-releasing factor receptor gene and characterization of a cDNA expressed in heart. Proc. Natl Acad. Sci. USA. 1995;92:2969–2973. doi: 10.1073/pnas.92.7.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jahn O, Eckart K, Tezval H, Spiess J. Characterization of peptide-protein interactions using photoaffinity labeling and LC/MS. Anal. Bioanal. Chem. 2004;378:1031–1036. doi: 10.1007/s00216-003-2353-8. [DOI] [PubMed] [Google Scholar]
- 31.Suda T, et al. Glucocorticoids decrease a binding of corticotropin-releasing hormone-binding protein in human plasma. J. Clin. Endocrinol. Metab. 1990;71:913–917. doi: 10.1210/jcem-71-4-913. [DOI] [PubMed] [Google Scholar]
- 32.Potter E, et al. Cloning and characterization of the cDNAs for human and rat corticotropin releasing factor-binding proteins. Nature. 1991;349:423–426. doi: 10.1038/349423a0. [DOI] [PubMed] [Google Scholar]
- 33.Petraglia F, et al. Corticotropin-releasing factor-binding protein is produced by human placenta and intrauterine tissues. J. Clin. Endocrinol. Metab. 1993;77:919–924. doi: 10.1210/jcem.77.4.8408466. [DOI] [PubMed] [Google Scholar]
- 34.Behan DP, Linton EA, Lowry PJ. Isolation of the human plasma corticotrophin-releasing factor-binding protein. J. Endocrinol. 1989;122:23–31. doi: 10.1677/joe.0.1220023. [DOI] [PubMed] [Google Scholar]
- 35.Haass-Koffler CL. The corticotropin releasing factor binding protein: a strange case of Dr. Jekyll and Mr. Hyde in the stress system? Alcohol. 2018;72:3–8. doi: 10.1016/j.alcohol.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen AM, et al. A soluble mouse brain splice variant of type 2 corticotropin-releasing factor (CRF) receptor binds ligands and modulates their activity. Proc. Natl Acad. Sci. USA. 2005;102:2620–2625. doi: 10.1073/pnas.0409583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan RKW, Vale WW, Sawchenko PE. Paradoxical activational effects of a corticotropin-releasing factor-binding protein “ligand inhibitor” in rat brain. Neuroscience. 2000;101:115–129. doi: 10.1016/S0306-4522(00)00322-5. [DOI] [PubMed] [Google Scholar]
- 38.Peto Ca, Arias C, Vale WW, Sawchenko PE. Ultrastructural localization of the corticotropin-releasing factor-binding protein in rat brain and pituitary. J. Comp. Neurol. 1999;413:241–254. doi: 10.1002/(SICI)1096-9861(19991018)413:2<241::AID-CNE6>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 39.Potter E, et al. The central distribution of a corticotropin-releasing factor (CRF)-binding protein predicts multiple sites and modes of interaction with CRF. Proc. Natl Acad. Sci. USA. 1992;89:4192–4196. doi: 10.1073/pnas.89.9.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sutton SW, et al. Ligand requirements of the human corticotropin-releasing factor-binding protein. Endocrinology. 1995;136:1097–1102. doi: 10.1210/endo.136.3.7867564. [DOI] [PubMed] [Google Scholar]
- 41.Ketchesin K. D., Huang N. S., Seasholtz A. F. Cell type-specific expression of corticotropin-releasing hormone-binding protein in GABAergic interneurons in the prefrontal cortex. Front Neuroanat. 11, 90 (2017). [DOI] [PMC free article] [PubMed]
- 42.Westphal NJ, Seasholtz AF. CRH-BP: the regulation and function of a phylogenetically conserved binding protein. Front. Biosci. 2006;11:1878–1891. doi: 10.2741/1931. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y., Andres A. L., Frotscher M., Baram T. Z. Tuning synaptic transmission in the hippocampus by stress: the CRH system. Front. Cell Neurosci. 6, 13 (2012). [DOI] [PMC free article] [PubMed]
- 44.Gounko NV, et al. Corticotropin-releasing factor and urocortin regulate spine and synapse formation: structural basis for stress-induced neuronal remodeling and pathology. Mol. Psychiatry. 2013;18:86–92. doi: 10.1038/mp.2012.43. [DOI] [PubMed] [Google Scholar]
- 45.Swinny JD, et al. Corticotropin-releasing factor and urocortin differentially modulate rat Purkinje cell dendritic outgrowth and differentiation in vitro. Eur. J. Neurosci. 2004;19:1749–1758. doi: 10.1111/j.1460-9568.2004.03279.x. [DOI] [PubMed] [Google Scholar]
- 46.Chen Y, Dube CM, Rice CJ, Baram TZ. Rapid loss of dendritic spines after stress involves derangement of spine dynamics by corticotropin-releasing hormone. J. Neurosci. 2008;28:2903–2911. doi: 10.1523/JNEUROSCI.0225-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y, et al. Impairment of synaptic plasticity by the stress mediator CRH involves selective destruction of thin dendritic spines via RhoA signaling. Mol. Psychiatry. 2013;18:485–496. doi: 10.1038/mp.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Annaert W, De Strooper B. A cell biological perspective on Alzheimer’s disease. Annu. Rev. Cell Dev. Biol. 2002;18:25–51. doi: 10.1146/annurev.cellbio.18.020402.142302. [DOI] [PubMed] [Google Scholar]
- 49.Querfurth HW, LaFerla FM. Alzheimer’s disease: mechanism of disease. N. Engl. J. Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 50.Weiner Michael W., Veitch Dallas P., Aisen Paul S., Beckett Laurel A., Cairns Nigel J., Cedarbaum Jesse, Donohue Michael C., Green Robert C., Harvey Danielle, Jack Clifford R., Jagust William, Morris John C., Petersen Ronald C., Saykin Andrew J., Shaw Leslie, Thompson Paul M., Toga Arthur W., Trojanowski John Q. Impact of the Alzheimer's Disease Neuroimaging Initiative, 2004 to 2014. Alzheimer's & Dementia. 2015;11(7):865–884. doi: 10.1016/j.jalz.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Izquierdo I, Medina JH. Memory formation: the sequence of biochemical events in the hippocampus and its connection to activity in other brain structures. Neurobiol. Learn Mem. 1997;68:285–316. doi: 10.1006/nlme.1997.3799. [DOI] [PubMed] [Google Scholar]
- 52.Lavenex P, Banta Lavenex P. Building hippocampal circuits to learn and remember: Insights into the development of human memory. Behav. Brain Res. 2013;254:8–21. doi: 10.1016/j.bbr.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 53.Golde TE, Schneider LS, Koo EH. Anti-Aβ therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;69:203–213. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011;7:137–152. doi: 10.1038/nrneurol.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rothman SM, et al. 3xTgAD mice exhibit altered behavior and elevated Aβ after chronic mild social stress. Neurobiol. Aging. 2012;33:830.e1–830.e12. doi: 10.1016/j.neurobiolaging.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carroll JC, et al. Chronic stress exacerbates Tau pathology, neurodegeneration, and cognitive performance through a mechanism in a transgenic mouse model of tauopathy. J. Neurosci. 2011;31:14436–14449. doi: 10.1523/JNEUROSCI.3836-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong H, et al. Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience. 2004;127:601–609. doi: 10.1016/j.neuroscience.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 58.Baglietto-Vargas D, et al. Short-term modern life-like stress exacerbates Aβ-pathology and synapse loss in 3xTg-AD mice. J. Neurochem. 2015;134:915–926. doi: 10.1111/jnc.13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stern Y, et al. Influence of education and occupation on the incidence of Alzheimer’s disease. JAMA. 1994;271:1004–1010. doi: 10.1001/jama.1994.03510370056032. [DOI] [PubMed] [Google Scholar]
- 60.Räihä I, Kaprio J, Koskenvuo M, Rajala T, Sourander L. Environmental differences in twin pairs discordant for Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. 1998;65:785–787. doi: 10.1136/jnnp.65.5.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moceri VM, et al. Using census data and birth certificates to reconstruct the early-life socioeconomic environment and the relation to the development of Alzheimer’s disease. Epidemiology. 2001;12:383–389. doi: 10.1097/00001648-200107000-00007. [DOI] [PubMed] [Google Scholar]
- 62.Miller DB, O’Callaghan JP. Do early-life insults contribute to the late-life development of Parkinson and Alzheimer diseases? Metabolism. 2008;57(SUPL.2):S44–S49. doi: 10.1016/j.metabol.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 63.Pomara N, Greenberg WM, Branford MD, Doraiswamy PM. Therapeutic implications of HPA axis abnormalities in Alzheimer’s disease: review and update. Psychopharmacol. Bull. 2003;37:120–134. [PubMed] [Google Scholar]
- 64.Csernansky JG, et al. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am. J. Psychiatry. 2006;163:2164–2169. doi: 10.1176/ajp.2006.163.12.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Singh-Manoux A, et al. Trajectories of depressive symptoms before diagnosis of dementia: a 28-year follow-up study. JAMA Psychiatry. 2017;74:712–718. doi: 10.1001/jamapsychiatry.2017.0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Justice NJ. The relationship between stress and Alzheimer’s disease. Neurobiol. Stress. 2018;8:127–133. doi: 10.1016/j.ynstr.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feldman H, et al. Behavioral symptoms in mild cognitive impairment. Neurology. 2004;62:1199–1201. doi: 10.1212/01.WNL.0000118301.92105.EE. [DOI] [PubMed] [Google Scholar]
- 68.Gabryelewicz T, et al. Prevalence of major and minor depression in elderly persons with mild cognitive impairment - MADRS factor analysis. Int J. Geriatr. Psychiatry. 2004;19:1168–1172. doi: 10.1002/gps.1235. [DOI] [PubMed] [Google Scholar]
- 69.Pentkowski NS, et al. Anxiety-like behavior as an early endophenotype in the TgF344-AD rat model of Alzheimer’s disease. Neurobiol. Aging. 2018;61:169–176. doi: 10.1016/j.neurobiolaging.2017.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee GJ, et al. Depressive symptoms in mild cognitive impairment predict greater atrophy in Alzheimer’s disease-related regions. Biol. Psychiatry. 2012;71:814–821. doi: 10.1016/j.biopsych.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mah L, Binns MA, Steffens DC. Anxiety symptoms in amnestic mild cognitive impairment are associated with medial temporal atrophy and predict conversion to Alzheimer disease. Am. J. Geriatr. Psychiatry. 2015;23:466–476. doi: 10.1016/j.jagp.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lebedeva A. K., et al. MRI-based classification models in prediction of mild cognitive impairment and dementia in late-life depression. Front. Aging Neurosci. 9, 13 (2017). [DOI] [PMC free article] [PubMed]
- 73.Swaab DF, et al. Increased cortisol levels in aging and Alzheimer’s disease in postmortem cerebrospinal fluid. J. Neuroendocrinol. 1994;6:681–687. doi: 10.1111/j.1365-2826.1994.tb00635.x. [DOI] [PubMed] [Google Scholar]
- 74.De Souza Errol B., Whitehouse Peter J., Kuhar Michael J., Price Donald L., Vale Wylie W. Reciprocal changes in corticotropin-releasing factor (CRF)-like immunoreactivity and CRF receptors in cerebral cortex of Alzheimer's disease. Nature. 1986;319(6054):593–595. doi: 10.1038/319593a0. [DOI] [PubMed] [Google Scholar]
- 75.May C, Rapoport SI, Tomai TP, Chrousos GP, Gold PW. Cerebrospinal fluid concentrations of corticotropin-releasing hormone (CRH) and corticotropin (ACTH) are reduced in patients with Alzheimer’s disease. Neurology. 1987;37:535–538. doi: 10.1212/WNL.37.3.535. [DOI] [PubMed] [Google Scholar]
- 76.Raadsheer FC, Oorschot DE, Verwer RWH, Tilders FJH, Swaab DF. Age‐related increase in the total number of corticotropin‐releasing hormone neurons in the human paraventricular nucleus in controls and alzheimer’s disease: comparison of the disector with an unfolding method. J. Comp. Neurol. 1994;339:447–457. doi: 10.1002/cne.903390311. [DOI] [PubMed] [Google Scholar]
- 77.Bissette G, Reynolds GP, Kilts CD, Widerlöv E, Nemeroff CB. Corticotropin-releasing factor-like immunoreactivity in senile dementia of the Alzheimer type: reduced cortical and striatal concentrations. JAMA. 1985;254:3067–3069. doi: 10.1001/jama.1985.03360210083036. [DOI] [PubMed] [Google Scholar]
- 78.Mouradian MM, et al. Spinal fluid CRF reduction in Alzheimer’s disease. Neuropeptides. 1986;8:393–400. doi: 10.1016/0143-4179(86)90010-7. [DOI] [PubMed] [Google Scholar]
- 79.Souza EBDe. CRH defects in Alzheimer’s and other neurologic diseases. Hosp. Pract. 1988;23:59–71. doi: 10.1080/21548331.1988.11703535. [DOI] [PubMed] [Google Scholar]
- 80.Pedersen WA, et al. Corticotropin-releasing hormone protects neurons against insults relevant to the pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2001;8:492–503. doi: 10.1006/nbdi.2001.0395. [DOI] [PubMed] [Google Scholar]
- 81.Lezoualc’h F, Engert S, Berning B, Behl C. Corticotropin-releasing hormone-mediated neuroprotection against oxidative stress is associated with the increased release of non-amyloidogenic amyloid beta precursor protein and with the suppression of nuclear factor-kappaB. Mol. Endocrinol. 2000;14:147–159. doi: 10.1210/mend.14.1.0403. [DOI] [PubMed] [Google Scholar]
- 82.Liang KC, Lee EHY. Intra-amygdala injections of corticotropin releasing factor facilitate inhibitory avoidance learning and reduce exploratory behavior in rats. Psychopharmacology. 1988;96:232–236. doi: 10.1007/BF00177566. [DOI] [PubMed] [Google Scholar]
- 83.Roozendaal B, Schelling G, McGaugh JL. Corticotropin-releasing factor in the basolateral amygdala enhances memory consolidation via an interaction with the β-adrenoceptor-cAMP pathway: dependence on glucocorticoid receptor activation. J. Neurosci. 2008;28:6642–6651. doi: 10.1523/JNEUROSCI.1336-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Canter RG, Penney J, Tsai L-H. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature. 2016;539:187–196. doi: 10.1038/nature20412. [DOI] [PubMed] [Google Scholar]
- 85.Kang JE, Cirrito JR, Dong H, Csernansky JG, Holtzman DM. Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proc. Natl Acad. Sci. USA. 2007;104:10673–10678. doi: 10.1073/pnas.0700148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dong H., Csernansky J. G. Effects of stress and stress hormones on amyloid-beta protein and plaque deposition. J. Alzheimers Dis. 18, 459–469 (2009). [DOI] [PMC free article] [PubMed]
- 87.Dong H, et al. Corticotrophin releasing factor accelerates neuropathology and cognitive decline in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2012;28:579–592. doi: 10.3233/JAD-2011-111328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oddo S, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 89.Park H-J, et al. The stress response neuropeptide CRF increases amyloid-β production by regulating γ-secretase activity. EMBO J. 2015;34:1674–1686. doi: 10.15252/embj.201488795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang C, et al. Corticotropin-releasing factor receptor-1 antagonism mitigates beta amyloid pathology and cognitive and synaptic deficits in a mouse model of Alzheimer’s disease. Alzheimer’s. Dement. 2016;12:527–537. doi: 10.1016/j.jalz.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Campbell SN, et al. Impact of CRFR1 ablation on amyloid-β production and accumulation in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2015;45:1175–1184. doi: 10.3233/JAD-142844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rissman RA, et al. Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J. Neurosci. 2007;27:6552–6562. doi: 10.1523/JNEUROSCI.5173-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Campbell SN, et al. Increased tau phosphorylation and aggregation in the hippocampus of mice overexpressing corticotropin-releasing factor. J. Alzheimer’s. Dis. 2015;43:967–976. doi: 10.3233/JAD-141281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mariotti A. The effects of chronic stress on health: new insights into the molecular mechanisms of brain–body communication. Future Sci. OA. 1, FSO23 (2015). [DOI] [PMC free article] [PubMed]
- 95.De Souza EB. Chapter 23. Role of corticotropin-releasing factor in neuropsychiatric disorders and neurodegenerative diseases. Annu. Rep. Med. Chem. 1990;25(C):215–224. [Google Scholar]
- 96.Nemeroff CB, Kizer JS, Reynolds GP, Bissette G. Neuropeptides in Alzheimer’s disease: a postmortem study. Regul. Pept. 1989;25:123–130. doi: 10.1016/0167-0115(89)90254-1. [DOI] [PubMed] [Google Scholar]
- 97.Piirainen S, et al. Psychosocial stress on neuroinflammation and cognitive dysfunctions in Alzheimer’s disease: the emerging role for microglia? Neurosci. Biobehav. Rev. 2017;77:148–164. doi: 10.1016/j.neubiorev.2017.01.046. [DOI] [PubMed] [Google Scholar]
- 98.Grace CRR, et al. Structure of the N-terminal domain of a type B1 G protein-coupled receptor in complex with a peptide ligand. Proc. Natl Acad. Sci. USA. 2007;104:4858–4863. doi: 10.1073/pnas.0700682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Perrin MH, Sutton S, Bain DL, Berggren WT, Vale WW. The first extracellular domain of corticotropin releasing factor-R1 contains major binding determinants for urocortin and astressin. Endocrinology. 1998;139:566–570. doi: 10.1210/endo.139.2.5757. [DOI] [PubMed] [Google Scholar]
- 100.Grace CRR, et al. NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc. Natl Acad. Sci. USA. 2004;101:12836–12841. doi: 10.1073/pnas.0404702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rivier J, Rivier C, Vale W. Synthetic competitive antagonists of corticotropin-releasing factor: effect on ACTH secretion in the rat. Science. 1984;224:889–891. doi: 10.1126/science.6326264. [DOI] [PubMed] [Google Scholar]
- 102.Rijkers DTS, et al. Structure-activity studies on the corticotropin releasing factor antagonist astressin, leading to a minimal sequence necessary for antagonistic activity. ChemBioChem. 2004;5:340–348. doi: 10.1002/cbic.200300769. [DOI] [PubMed] [Google Scholar]
- 103.Flock T, et al. Selectivity determinants of GPCR-G-protein binding. Nature. 2017;545:317–322. doi: 10.1038/nature22070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grammatopoulos DK, Randeva HS, Levine MA, Katsanou ES, Hillhouse EW. Urocortin, but not corticotropin-releasing hormone (CRH), activates the mitogen-activated protein kinase signal transduction pathway in human pregnant myometrium: an effect mediated via R1alpha and R2beta CRH receptor subtypes and stimulation of Gq-protei. Mol. Endocrinol. 2000;14:2076–2091. doi: 10.1210/mend.14.12.0574. [DOI] [PubMed] [Google Scholar]
- 105.Berger H, Heinrich N, Wietfeld D, Bienert M, Beyermann M. Evidence that corticotropin-releasing factor receptor type 1 couples to Gs- and Gi-proteins through different conformations of its J-domain. Br. J. Pharmacol. 2006;149:942–947. doi: 10.1038/sj.bjp.0706926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hoare SRJ, et al. Conformational states of the corticotropin releasing factor 1 (CRF1) receptor: detection, and pharmacological evaluation by peptide ligands. Peptides. 2003;24:1881–1897. doi: 10.1016/j.peptides.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 107.Hoare SRJ, et al. Ligand affinity for amino-terminal and juxtamembrane domains of the corticotropin releasing factor type I receptor: regulation by G-protein and nonpeptide antagonists. Biochemistry. 2004;43:3996–4011. doi: 10.1021/bi036110a. [DOI] [PubMed] [Google Scholar]
- 108.Kehne JH, Cain CK. Therapeutic utility of non-peptidic CRF1receptor antagonists in anxiety, depression, and stress-related disorders: evidence from animal models. Pharm. Ther. 2010;128:460–487. doi: 10.1016/j.pharmthera.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zobel AW, et al. Effects of high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J. Psychiatr. Res. 2000;34:171–181. doi: 10.1016/S0022-3956(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 110.Binneman B, et al. A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am. J. Psychiatry. 2008;165:617–620. doi: 10.1176/appi.ajp.2008.07071199. [DOI] [PubMed] [Google Scholar]
- 111.Coric V, et al. Multicenter, randomized, double-blind, active comparator and placebo-controlled trial of a corticotropin-releasing factor receptor-1 antagonist in generalized anxiety disorder. Depress Anxiety. 2010;27:417–425. doi: 10.1002/da.20695. [DOI] [PubMed] [Google Scholar]
- 112.Fleck BA, Hoare SRJ, Pick RR, Bradbury MJ, Grigoriadis DE. Binding kinetics redefine the antagonist pharmacology of the corticotropin-releasing factor type 1 receptor. J. Pharm. Exp. Ther. 2012;341:518–531. doi: 10.1124/jpet.111.188714. [DOI] [PubMed] [Google Scholar]
- 113.Devigny C, et al. Biomimetic screening of class-B G protein-coupled receptors. J. Am. Chem. Soc. 2011;133:8927–8933. doi: 10.1021/ja200160s. [DOI] [PubMed] [Google Scholar]
- 114.Ketchesin KD, Stinnett GS, Seasholtz AF. Corticotropin-releasing hormone-binding protein and stress: from invertebrates to humans. Stress. 2017;20:449–464. doi: 10.1080/10253890.2017.1322575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Van Den Eede F, Van Broeckhoven C, Claes SJ. Corticotropin-releasing factor-binding protein, stress and major depression. Ageing Res. Rev. 2005;4:213–239. doi: 10.1016/j.arr.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 116.Behan DP, et al. Corticotropin releasing factor (CRF) binding protein: a novel regulator of CRF and related peptides. Front. Neuroendocr. 1995;16:362–382. doi: 10.1006/frne.1995.1013. [DOI] [PubMed] [Google Scholar]
- 117.Karolyi IJ, et al. Altered anxiety and weight gain in corticotropin-releasing hormone-binding protein-deficient mice. Proc. Natl Acad. Sci. USA. 1999;96:11595–11600. doi: 10.1073/pnas.96.20.11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Camper Sa, et al. Excess corticotropin releasing hormone-binding protein in the hypothalamic-pituitary-adrenal axis in transgenic mice. J. Clin. Invest. 2008;101:1439–1447. doi: 10.1172/JCI1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lovejoy DA, et al. Ectopic expression of the CRF-binding protein: minor impact on HPA axis regulation but induction of sexually dimorphic weight gain. J. Neuroendocrinol. 1998;10:483–491. doi: 10.1046/j.1365-2826.1998.00206.x. [DOI] [PubMed] [Google Scholar]
- 120.Contarino A, Heinrichs SC, Gold LH. Understanding corticotropin releasing factor neurobiology: contributions from mutant mice. Neuropeptides. 1999;33:1–12. doi: 10.1054/npep.1999.0001. [DOI] [PubMed] [Google Scholar]
- 121.Gammie SC, Seasholtz AF, Stevenson SA. Deletion of corticotropin-releasing factor binding protein selectively impairs maternal, but not intermale aggression. Neuroscience. 2008;157:502–512. doi: 10.1016/j.neuroscience.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jahn O, Eckart K, Brauns O, Tezval H, Spiess J. The binding protein of corticotropin-releasing factor: ligand-binding site and subunit structure. Proc. Natl Acad. Sci. USA. 2002;99:12055–12060. doi: 10.1073/pnas.192449299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Spierling SR, Zorrilla EP. Don’t stress about CRF: assessing the translational failures of CRF1antagonists. Psychopharmacology. 2017;234:1467–1481. doi: 10.1007/s00213-017-4556-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Behan DP, et al. Displacement of corticotropin releasing factor from its binding protein as a possible treatment for Alzheimer’s disease. Nature. 1995;378:284–287. doi: 10.1038/378284a0. [DOI] [PubMed] [Google Scholar]
- 125.Behan DP, et al. Corticotropin-releasing factor (CRF), CRF-binding protein (CRF-BP), and CRF/CRF-BP complex in Alzheimer’s disease and control postmortem human brain. J. Neurochem. 1997;68:2053–2060. doi: 10.1046/j.1471-4159.1997.68052053.x. [DOI] [PubMed] [Google Scholar]
- 126.González-Reyes RE, Nava-Mesa MO, Vargas-Sánchez K, Ariza-Salamanca D, Mora-Muñoz L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017;10:427. doi: 10.3389/fnmol.2017.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat. Clin. Pr. Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- 128.Brinton RD. A women’s health issue: Alzheimer’s disease and strategies for maintaining cognitive health. Int J. Fertil. Womens Med. 1999;44:174–185. [PubMed] [Google Scholar]
- 129.Takao T, et al. Corticotrophin‐releasing factor antagonist [alpha helical CRF(9–41)] blocks central noradrenaline‐induced ACTH secretion. J. Neuroendocrinol. 1989;1:77–78. doi: 10.1111/j.1365-2826.1989.tb00082.x. [DOI] [PubMed] [Google Scholar]
- 130.Ruhmann A, Bonk I, Lin CR, Rosenfeld MG, Spiess J. Structural requirements for peptidic antagonists of the corticotropin-releasing factor receptor (CRFR): development of CRFR2-selective antisauvagine-30. Proc. Natl Acad. Sci. USA. 1998;95:15264–15269. doi: 10.1073/pnas.95.26.15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rivier JE, Rivier CL. Corticotropin-releasing factor peptide antagonists: Design, characterization and potential clinical relevance. Front. Neuroendocrinol. 2014;35:161–170. doi: 10.1016/j.yfrne.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Seymour PA, Schmidt AW, Schulz DW. The pharmacology of CP-154,526, a non-peptide antagonist of the CRH1 receptor: a review. CNS Drug Rev. 2003;9:57–96. doi: 10.1111/j.1527-3458.2003.tb00244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chaki S, et al. Anxiolytic- and antidepressant-like profile of a new CRF1receptor antagonist, R278995/CRA0450. Eur. J. Pharmacol. 2004;485:145–158. doi: 10.1016/j.ejphar.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 134.Held K, et al. Treatment with the CRH1-receptor-antagonist R121919 improves sleep-EEG in patients with depression. J. Psychiatr. Res. 2004;38:129–136. doi: 10.1016/S0022-3956(03)00076-1. [DOI] [PubMed] [Google Scholar]
- 135.Künzel HE, et al. Treatment of depression with the CRH-1-receptor antagonist R121919: endocrine changes and side effects. J. Psychiatr. Res. 2003;37:525–533. doi: 10.1016/S0022-3956(03)00070-0. [DOI] [PubMed] [Google Scholar]
- 136.Zorrilla EP, Heilig M, de Wit H, Shaham Y. Behavioral, biological, and chemical perspectives on targeting CRF1 receptor antagonists to treat alcoholism. Drug Alcohol Depend. 2013;128:175–186. doi: 10.1016/j.drugalcdep.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Burrows HL, et al. Excess corticotropin releasing hormone-binding protein in the hypothalamic-pituitary-adrenal axis in transgenic mice. J. Clin. Invest. 1998;101:1439–1447. doi: 10.1172/JCI1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mcclennen SJ, Cortright DN, Seasholtz AF. Regulation of pituitary corticotropin-releasing hormone-binding protein messenger ribonucleic acid levels by restraint stress and adrenalectomy. Endocrinology. 1998;139:4435–4441. doi: 10.1210/endo.139.11.6311. [DOI] [PubMed] [Google Scholar]
- 139.Timofeeva E, Deshaies Y, Picard F, Richard D. Corticotropin-releasing hormone-binding protein in brain and pituitary of food-deprived obese (fa/fa) Zucker rats. Am. J. Physiol. Integr. Comp. Physiol. 1999;277:R1749–R1759. doi: 10.1152/ajpregu.1999.277.6.R1749. [DOI] [PubMed] [Google Scholar]
- 140.Lombardo KA, et al. Effects of acute and repeated restraint stress on corticotropin-releasing hormone binding protein mRNA in rat amygdala and dorsal hippocampus. Neurosci. Lett. 2001;302:81–84. doi: 10.1016/S0304-3940(01)01680-9. [DOI] [PubMed] [Google Scholar]
- 141.Herringa RJ, Nanda SA, Hsu DT, Roseboom PH, Kalin NH. The effects of acute stress on the regulation of central and basolateral amygdala CRF-binding protein gene expression. Mol. Brain Res. 2004;131:17–25. doi: 10.1016/j.molbrainres.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 142.Vasconcelos M, Stein DJ, Albrechet-Souza L, Miczek KA, de Almeida RMM. Recovery of stress-impaired social behavior by an antagonist of the CRF binding protein, CRF6−33, in the bed nucleus of the stria terminalis of male rats. Behav. Brain Res. 2019;357–358:104–110. doi: 10.1016/j.bbr.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]