

Trimeric autotransporter adhesins (TAAs) are specialized bacterial outer membrane proteins consisting of three identical subunits. TAAs contain large extracellular domains that trimerize and promote virulence, but the mechanism by which they are secreted is poorly understood. We found that the extracellular domains of a native TAA were secreted rapidly but that disordered and artificially folded polypeptides fused to native passenger domains were secreted in a slow, sequential fashion. Our results strongly suggest that the efficient secretion of native extracellular domains is driven by their trimerization following export but that alternative energy sources can be harnessed to secrete nonnative polypeptides. Furthermore, we obtained evidence that TAA extracellular domains are secreted before the assembly of the linked membrane spanning domain is completed.
KEYWORDS: Bam complex, membrane proteins, outer membrane, protein folding, protein secretion, trimeric autotransporters
ABSTRACT
Trimeric autotransporter adhesins (TAAs) are a family of bacterial outer membrane (OM) proteins that are comprised of three identical subunits. Each subunit contains an N-terminal extracellular (“passenger”) domain and a short C-terminal segment that contributes four β strands to a single 12-stranded β barrel. The mechanism by which the passenger domains are translocated across the OM and the energetics of the translocation reaction are poorly understood. To address these issues, we examined the secretion of modified versions of the passenger domain of UpaG, a TAA produced by Escherichia coli CFT073. Using the SpyTag-SpyCatcher system to probe passenger domain localization, we found that both intrinsically disordered polypeptides fused to the UpaG passenger domain and artificially disulfide-bonded polypeptides were secreted effectively but relatively slowly. Surprisingly, we also found that in some cases, the three nonnative passenger domain segments associated with a single trimer were secreted sequentially. Photo-cross-linking experiments indicated that incompletely assembled UpaG derivatives remained bound to the barrel assembly machinery (Bam) complex until all three passenger domains were fully secreted. Taken together, our results strongly suggest that the secretion of polypeptides through the TAA pathway is coordinated with the assembly of the β barrel domain and that the folding of passenger domains in the extracellular space maximizes the rate of secretion. Furthermore, our work provides evidence for an unprecedented sequential mode of protein translocation, at least under specific experimental conditions.
INTRODUCTION
Autotransporters are a distinctive group of outer membrane proteins (OMPs) that are produced by a wide range of Gram-negative bacteria (1, 2). Like the vast majority of integral OMPs, autotransporters are anchored to the membrane by a β sheet that folds into a closed cylindrical structure known as a “β barrel.” Because OMP β barrels expose a hydrophobic exterior, they partition readily into the lipid environment of the outer membrane (OM). In addition to the β barrel domain, which is located at the C terminus, “classical” autotransporters contain an N-terminal extracellular (“passenger”) domain. Although passenger domains differ considerably in sequence and function, they often exceed 100 kDa in size and almost always fold into a repetitive structure known as a β helix (3–7). The passenger domain is connected to the C terminus by an α-helical “linker” that traverses the β barrel. Trimeric autotransporter adhesins (TAAs) are thematically related to classical autotransporters but differ in that they are composed of three identical subunits (8). Each subunit contains an N-terminal passenger domain and an ∼70-residue C-terminal segment that contributes 4 β strands to a single 12-stranded β barrel domain that, remarkably, is nearly identical in structure to the β barrel domains of classical autotransporters (9–13). TAA passenger domains are highly diverse in sequence and often very large but, unlike the passenger domains of classical autotransporters, fold into a coiled-coil “stalk.” This extended trimeric structure is capped and often interspersed with β prism or β roll “head” domains that mediate adhesive functions and play important roles in virulence (14–19). The passenger domains are each connected to the C terminus by an α helix that emerges from the β barrel domain and initiates the coiled-coil structure.
Despite their unique trimeric structure, the β barrel domains of TAAs—like those of essentially all other OMPs—are inserted into the OM by a heterooligomer called the barrel assembly machinery (Bam) complex (20–22). The Bam complex in Escherichia coli consists of BamA, an integral OMP that contains a β barrel domain, and five periplasmic POTRA (polypeptide transport-associated) domains that mediate interactions with four lipoprotein subunits (BamB to BamE). The composition of the Bam complex varies in different Gram-negative bacteria, and only BamA and BamD are highly conserved and essential for viability (21, 23, 24). Although the structure of the Bam holocomplex was recently solved (25–28), the mechanism by which it catalyzes the membrane insertion of β barrels is unknown. On the basis of the BamA crystal structure and molecular dynamics simulations, it has been proposed that substrates enter the pore of the β barrel in an unfolded conformation and then form hairpins that insert into the lipid bilayer sequentially through a lateral gate produced by the transient separation of the first and last β strands (29). Consistent with this “budding” model, artificial disulfide bonds that lock the BamA β barrel into a closed conformation create lethal phenotypes and inhibit Bam complex function in vitro (28, 30). A variety of other observations, however, are consistent with an alternative proposal (the “assisted” model) in which the Bam complex facilitates the membrane integration of folded or partially folded β barrels by perturbing the lipid bilayer (31–33). Interestingly, strong support for this model has emerged from studies on autotransporters. Evidence that the linker segment of a classical autotransporter rapidly becomes protected from proteolysis and chemical modification in vivo and accelerates folding in vitro suggests that it is incorporated into a barrel-like structure in the periplasm (34, 35). Furthermore, cross-linking experiments have clearly shown that TAA β barrels begin to assemble prior to membrane integration (36).
The need to localize a large polypeptide on the cell surface is a feature that distinguishes autotransporters from most other OMPs. While significant insights into the mechanism by which the passenger domains of classical autotransporters are secreted have emerged, the secretion of TAA passenger domains is still poorly understood. The name “autotransporter” arose from the proposal that bacterial proteins that have a large extracellular segment are self-contained protein translocation systems in which the passenger domain is secreted through a channel formed solely by the covalently linked β barrel domain (37). Since this idea was elaborated in 1987, the discovery of the Bam complex and new experimental evidence have strongly challenged the self-transport hypothesis. The finding that small folded polypeptides fused to the passenger domain of a classical autotransporter are secreted effectively even though the β barrel pore is too narrow to accommodate polypeptides that have tertiary structure suggested that additional cellular factors play a role in the transport reaction (38–40). Subsequent cross-linking experiments and evidence that the passenger domain is secreted before the folding of the β barrel domain is completed have strongly suggested that the Bam complex catalyzes the membrane integration of the β barrel domain and the secretion of the passenger domain in a concerted reaction (41–44). On the basis of the available information, it has been proposed that the passenger domain is secreted through a hybrid channel in which the covalently linked β barrel domain and the BamA β barrel are both in an open conformation (45). Although the periplasm is devoid of ATP, there is now considerable evidence that the vectorial folding of the β helix in the extracellular space helps to drive passenger domain translocation (5, 46–50). Because both disordered and small folded polypeptides can be secreted efficiently, however, it appears that other energy sources can be harnessed to drive translocation (51). The finding that large N-terminal deletions do not affect TAA assembly (52, 53) shows that the passenger domains—like those of classical autotransporters—are secreted in a C- to N-terminal direction (presumably through the formation of a hairpin), but the mechanistic relationship between the secretion of the passenger domains of classical autotransporters and TAAs is otherwise unknown.
To gain insight into the mechanism by which the passenger domains of TAAs are transported across the OM, we examined the secretion of both native and modified passenger domain fragments of a TAA produced by E. coli CFT073 (UpaG) in vivo. In these experiments, we used the SpyCatcher-SpyTag method (54) to assess the kinetics of secretion of individual passenger domains. We found that the secretion of the native polypeptide was extremely rapid and was independent of the length of the passenger domain fragment. In contrast, we found that the secretion of a chimeric passenger domain that contains an intrinsically disordered segment was initiated rapidly but completed very slowly. Even more strikingly, we found that other chimeric passenger domains and passenger domains that contain an artificial disulfide bond were secreted in a sequential fashion. Crosslinking experiments revealed that the UpaG β barrel domain remained bound to the Bam complex until all of the passenger domains were fully secreted. Our results not only suggest that the mechanism of TAA passenger domain secretion is similar to that of classical autotransporters but also provide evidence for an unprecedented mode of protein secretion that has potential implications for our understanding of the function of the Bam complex.
RESULTS
Native TAA passenger domain fragments are secreted rapidly.
We chose UpaG, a TAA produced by a uropathogenic strain of E. coli (CFT073), as a model protein to study TAA biogenesis. UpaG consists of three identical 1,778-amino-acid subunits that contain conserved sequence motifs often associated with bacterial adhesins (55). In a detailed model of the structure of UpaG, these motifs were proposed to contribute to the formation of multiple internal head domains and other β-stranded elements (19). As previously noted, full-length UpaG promotes autoaggregation in laboratory strains of E. coli and is toxic (36, 55). To circumvent this problem, we used N-terminally truncated derivatives of UpaG that were missing significant portions of the passenger domains that are dispensable for assembly in our experiments. These derivatives are relatively nontoxic and, like similar derivatives of other TAAs, form stable trimers that insert into the OM with the correct topology (36, 52, 53, 56).
To gain insight into the mechanism of translocation, we first examined the kinetics of the translocation reaction. We previously found that a hemagglutinin (HA)-tagged version of UpaGΔ2, a 170-residue derivative of UpaG that contains ∼90 residues of the native passenger domain (Fig. 1A), assembles into SDS- and heat-resistant trimers very rapidly (t1/2 of <1 min) in E. coli K-12 strain AD202 (MC4100 ompT::kan) (36). On the basis of the common assumption that SDS- and heat-resistant trimers represent the fully assembled form of a TAA (57), it would follow that all three of the passenger domains of UpaGΔ2 are completely secreted in the stable trimeric form of the protein. To probe the status of the passenger domain directly, we utilized the recently developed SpyTag-SpyCatcher method (54). This technique is based on the observation that a spontaneous intramolecular isopeptide bond that forms between two residues in the CnaB2 domain of the Streptococcus pyogenes FbaB protein can be reconstituted after the domain is split into a 138-amino-acid protein (SpyCatcher) and a 13-amino-acid peptide (SpyTag [ST]). An irreversible reaction occurs under diverse experimental conditions when the SpyCatcher protein is added to a protein of interest that is tagged with the SpyTag peptide and results in an ∼15-kDa shift on SDS-PAGE.
FIG 1.
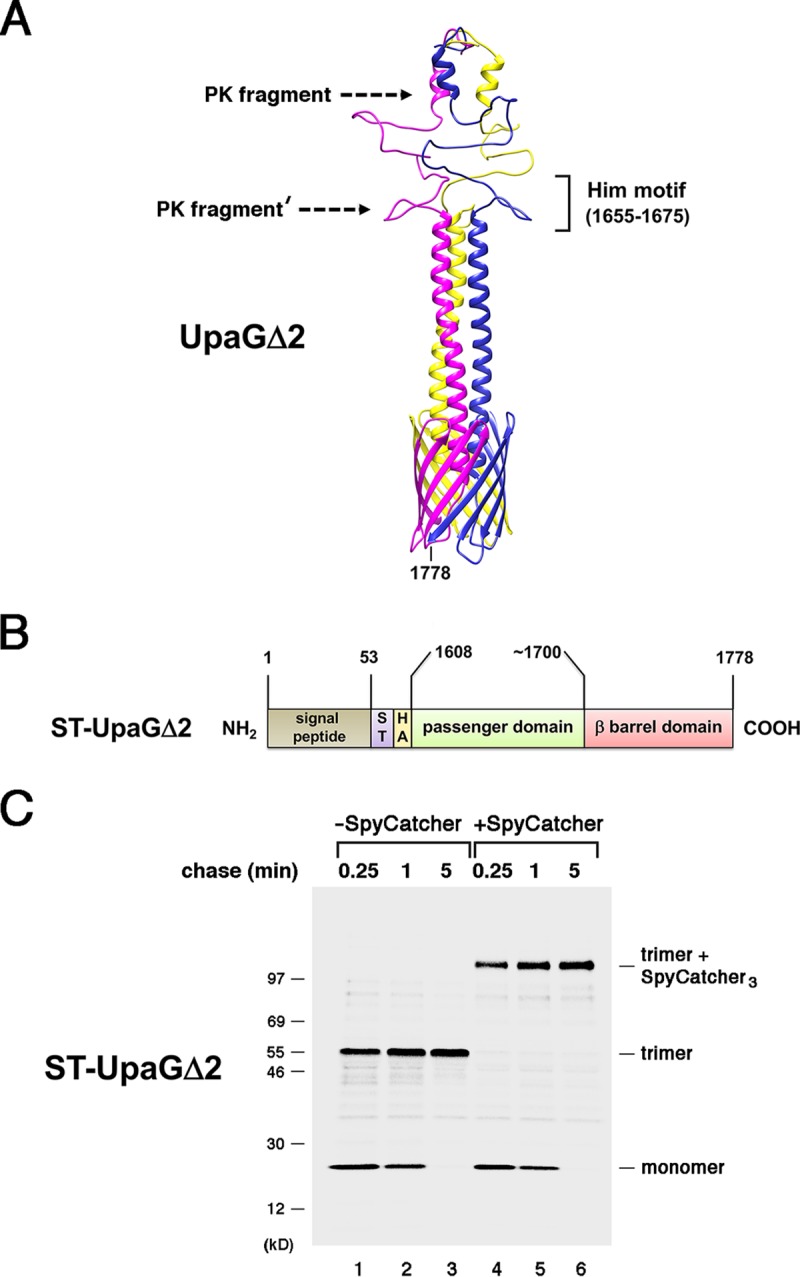
The passenger domains of UpaGΔ2 are secreted rapidly. (A) Homology-based model of the structure of UpaGΔ2 generated using PHYRE2 and GalaxyHOMOMER software. The approximate locations of proteinase K (PK) cleavage sites and a previously identified Him motif (55) are shown. The larger C-terminal fragment was produced by the treatment of native UpaGΔ2 trimer with PK (PK fragment), and the slightly smaller fragment (PK fragment′) was produced by PK treatment of incompletely folded derivatives. The numbers shown here and throughout refer to positions in the full-length UpaG sequence. (B) Illustration of the ST-UpaGΔ2 protein. HA, HA tag; ST, SpyTag. (C) AD202 cells transformed with a plasmid encoding ST-UpaGΔ2 (pRS31) were subjected to pulse-chase labeling. After cells were either incubated with SpyCatcher or mock treated, immunoprecipitations were conducted using an anti-UpaG antiserum and proteins were resolved by SDS-PAGE.
To monitor the rate at which the secretion reaction is completed, we fused the SpyTag peptide to the N terminus of the UpaGΔ2 passenger domain. AD202 cells were transformed with a plasmid encoding the resulting UpaG derivative (ST-UpaGΔ2; Fig. 1B) and grown to mid-log phase at 37°C in minimal medium. Cells were then subjected to pulse-chase labeling, collected by centrifugation, and divided into two equal aliquots. The SpyCatcher protein was added to one aliquot, and all samples were incubated at 4°C. Proteins were collected by trichloroacetic acid (TCA) precipitation, and immunoprecipitations were performed using a polyclonal antiserum against UpaGΔ2. Consistent with previous results (36), most of the ST-UpaGΔ2 monomer was assembled into a heat- and SDS-resistant trimer that migrated at ∼55 kDa within 1 min (Fig. 1C, lanes 1 to 3). Upon incubation with the SpyCatcher protein, all of the trimer was converted into an ∼100-kDa adduct (Fig. 1C, lanes 4 to 6). The ∼45-kDa shift (15 kDa × 3) implies that the new species resulted from the formation of a covalent bond between the SpyCatcher protein and all three of the SpyTag peptides associated with the trimer. The observation that none of the residual ST-UpaGΔ2 monomer that was present at early time points was modified by the SpyCatcher protein indicates that the cells remained intact during the incubation and that the reactive SpyTag peptides were surface exposed.
We next wished to determine if secretion kinetics correlate with the size of the passenger domain. To this end, we created ST-UpaGΔ1, an UpaG derivative that contains nearly 450 residues of the native passenger domain as well as N-terminal HA and SpyTag peptides (Fig. 2A). To monitor the biogenesis of ST-UpaGΔ1, we transformed AD202 with a plasmid that encodes this derivative under the control of a rhamnose-inducible promoter and grew cells to mid-log phase in minimal medium. Cells were pulse-chase radiolabeled after the addition of inducer and collected by centrifugation. One half of each sample was then treated with proteinase K (PK) to cleave surface-exposed ST-UpaGΔ1, proteins were TCA precipitated, and immunoprecipitations were conducted using the anti-UpaG antiserum. Our analysis showed that ST-UpaGΔ1 assembled into a stable trimer essentially as rapidly as ST-UpaGΔ2 (Fig. 2B, lanes 1 to 5). The finding that the addition of PK converted all of the ∼180-kDa trimer to an ∼100-kDa C-terminal fragment (Fig. 2B, lanes 6 to 10) indicated that at least a significant portion of each passenger domain was exposed on the cell surface. To determine if the passenger domains in the stable trimer were completely secreted, we used the SpyTag-SpyCatcher method as described above. Despite the fact that the passenger domain of ST-UpaGΔ1 is much longer than that of ST-UpaGΔ2, the SpyCatcher protein completely modified all three subunits of the trimer (Fig. 2C). Lengthening the passenger domain therefore did not significantly increase the time required to complete the translocation reaction or lead to the creation of detectable stalled translocation intermediates.
FIG 2.

The length of the passenger domain does not affect the kinetics of UpaG assembly. (A) Illustration of the ST-UpaGΔ1 protein. HA, HA tag; ST, SpyTag. (B) AD202 cells transformed with a plasmid encoding ST-UpaGΔ1 (pRS37) were subjected to pulse-chase labeling. After cells were either incubated with PK or mock treated, immunoprecipitations were conducted using an anti-UpaG antiserum and proteins were resolved by SDS-PAGE. (C) The experiment represented in panel part B was repeated, except that cells were incubated with SpyCatcher instead of PK. In panels B and C, unidentified bands that appear to have been derived from ST-UpaGΔ1 are labeled with an asterisk.
The finding that UpaGΔ1 and UpaGΔ2 assemble into fully folded structures at similar rates strongly suggests that passenger domain secretion is a fast step and that the rate-limiting step in TAA biogenesis precedes the initiation of translocation. In this regard, it is noteworthy that very short UpaG derivatives that have only a few amino acids exposed on the cell surface (HA-UpaGΔ3) or that consist of the β barrel plus a portion of the embedded linker segment (UpaGΔ4) appear to assemble more rapidly than UpaGΔ2. Almost all of the HA-UpaGΔ3 was assembled into a stable trimer in <15 s, and no UpaGΔ4 monomer could be observed unless assembly was slowed by incubating cells at 25°C (see Fig. S1 in the supplemental material). In contrast, the monomeric form of an UpaG derivative that completely lacks the linker (UpaGΔ5) was rapidly degraded (Fig. S1B, bottom gel). These results suggest that the rate-limiting step is either the incorporation of a hairpin into a trimeric structure in the periplasm or the insertion of a trimeric assembly intermediate into the OM in a translocation-active state.
UpaG derivatives that contain only the β barrel and the embedded linker segment are assembled extremely rapidly. (A) Illustration of HA-UpaGΔ3, UpaGΔ4, and UpaGΔ5 constructs. HA, HA tag. The site of each deletion is shown on a homology-based model of the C terminus of an UpaG monomer. (B) Strain AD202 cells transformed with a plasmid encoding HA-UpaGΔ3 (pRS18), UpaGΔ4 (pRS19), or UpaGΔ5 (pRS20) were subjected to pulse-chase labeling. After the cells were either incubated with PK or subjected to mock treatment, immunoprecipitations were conducted using a polyclonal anti-HA antiserum (for HA-UpaGΔ3) or an anti-UpaG antiserum (for all constructs). Proteins were resolved by SDS-PAGE on either 8% to 16% Tris-glycine minigels (for HA-UpaGΔ3) or 12% Bis-Tris minigels using a morpholineethanesulfonic acid (MES)-based buffer system (Thermo Fisher Scientific) (for UpaGΔ4 and UpaGΔ5). (C) The experiment performed with UpaGΔ4 was repeated except that the cells were transferred to a prewarmed 25°C water bath for 15 min prior to pulse-chase labeling at the same temperature. Immunoprecipitations were conducted using an anti-UpaG antiserum, and proteins were resolved by SDS-PAGE on an 8% to 16% Tris-glycine minigel. Unidentified background bands are labeled with an asterisk in panels B and C. Download FIG S1, PDF file, 0.9 MB (1.1MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Chimeric and disulfide-bonded TAA passenger domains are secreted slowly or in a stepwise fashion.
The finding that native TAA passenger domain fragments are translocated across the OM rapidly is consistent with the possibility that the secretion reaction is driven by the formation of a coiled-coil structure in the extracellular space. Indeed, the presence of a surprisingly large number of polar residues in the core of TAA coiled-coils has been proposed to promote a rapid transition from an unstructured state in the periplasm to a folded state following translocation (58). Furthermore, the folding of at least some coiled-coil structures appears to approach the diffusion-limited value (59). On the basis of the vectorial folding model, we hypothesized that disordered polypeptides fused to a TAA would either remain trapped in the periplasm or be secreted relatively inefficiently. To test this idea, we first fused a 224-amino-acid fragment of the receptor binding domain of the Bordetella pertussis CyaA toxin (the “ΔRD” fragment described in reference 51) to the passenger domain of UpaGΔ2. This highly acidic fragment contains multiple repeat-in-toxin (RTX) motifs that fold into a stable β-roll structure in the presence of calcium but that remain intrinsically disordered in its absence (60). AD202 cells transformed with a plasmid that encodes an HA-tagged version of the chimera (HA–RTX-UpaGΔ2; Fig. 3A) were subjected to pulse-chase labeling, and the exposure of the passenger domain was monitored by PK treatment as described above. Most but not all of the monomer was converted to one of two different trimeric forms of the protein (Fig. 3B, lanes 1 to 5). On the basis of the finding that the faster-migrating species were not observed in the presence of calcium (Fig. S2), this polypeptide likely corresponds to a form of the trimer in which the RTX repeats remain disordered. The form that migrates much more slowly is produced in the presence of calcium (but not effectively exposed on the cell surface) and appears to correspond to a trimer in which the RTX repeats adopt an alternative conformation.
FIG 3.

The presence of an intrinsically disordered segment delays the completion of passenger domain secretion. (A) Illustration of the HA–RTX-UpaGΔ2 protein. HA, HA tag. (B) AD202 cells transformed with a plasmid encoding HA–RTX-UpaGΔ2 (pRS42) were subjected to pulse-chase labeling. After cells were either incubated with PK or mock treated, immunoprecipitations were conducted using an anti-UpaG antiserum and proteins were resolved by SDS-PAGE. (C) AD202 cells were transformed with a plasmid encoding ST–RTX-UpaGΔ2 (pRS43). The experiment represented in panel B was repeated, except that cells were incubated with SpyCatcher instead of PK. The OM of half of the cells was permeabilized prior to the addition of SpyCatcher. (D) Model for the secretion of the ST–RTX-UpaGΔ2 passenger domain. The secretion of the segment of the chimeric passenger domain derived from UpaG is fast and is potentially driven by the formation of a coiled-coil structure. Because the RTX segment cannot fold, its rate of secretion and the level of concomitant surface exposure of the N-terminal SpyTag are considerably lower.
The faster-migrating form of an RTX-UpaGΔ2 trimer contains a disordered RTX domain. AD202 cells transformed with a plasmid encoding HA–RTX-UpaGΔ2 (pRS42) were grown in MOPS minimal media to mid-log phase and induced with 0.2% l-rhamnose. Cultures were divided in half, and 2 mM CaCl2 was added to one half 5 min prior to pulse-chase labeling. After the cells were either incubated with PK or mock treated, immunoprecipitations were conducted using an anti-UpaG antiserum and proteins were resolved by SDS-PAGE. The observation that the level of the faster-migrating (∼110-kDa) form of the RTX-UpaGΔ2 trimer was greatly reduced when calcium was added to induce folding indicates that it contained an unfolded RTX moiety. Download FIG S2, PDF file, 0.4 MB (372.7KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
An analysis of the chimeric protein provided intriguing evidence that unfolded polypeptide segments can be secreted by the TAA pathway but more slowly than native passenger domain segments. At least at first glance, the assembly of HA–RTX-UpaGΔ2 seemed to resemble that of UpaGΔ2 in that the formation of a heat- and SDS-resistant trimer was rapid (a considerable amount of stable trimer was seen at the earliest time point) and all of the passenger domains associated with the trimer were exposed on the cell surface (Fig. 3B). Curiously, however, the accumulation of a characteristic ∼42-kDa C-terminal fragment that is observed when cells that produce UpaGΔ2 are treated with PK (36) (Fig. 1) was very slow and correlated with the disappearance of a unique ∼36-kDa band (Fig. 3B, lanes 6 to 10). Because the ∼42-kDa band contains the UpaG β barrel domain plus a passenger domain segment that is resistant to PK digestion (Fig. 1A), the pattern of PK fragments provided a clue that the presence of the RTX repeats perturbs assembly. Consistent with this possibility, most of the faster-migrating form of the stable trimer formed by a RTX-UpaGΔ2 derivative that contained an N-terminal SpyTag (ST–RTX-UpaGΔ2) did not react with SpyCatcher after a 0.25-min chase (Fig. 3C, top gel, lane 4). By 5 min, however, all of the SpyTags were modified by the protein (Fig. 3C, top gel, lane 6). To confirm that the SpyTags that were unreactive at early time points were trapped in the periplasm, we permeabilized the OM before adding SpyCatcher. As expected, this manipulation exposed all of the SpyTags to the SpyCatcher protein and led to nearly 100% bond formation at all time points (Fig. 3C, bottom gel; see also Fig. S3A). Taken together, the results indicate that the translocation of the chimeric passenger domain was initiated rapidly but completed only very slowly. Indeed, the data suggest a two-step process in which the portion of the chimeric passenger domain derived from UpaGΔ2 is secreted rapidly through the formation of a coiled-coil but the disordered RTX repeats are secreted slowly via a less efficient mechanism (Fig. 3D). Importantly, the results also show that the appearance of a heat- and SDS-resistant trimer does not necessarily imply that its assembly has been completed.
Properties of monomeric and slower-migrating trimeric forms of ST–RTX-UpaGΔ2. (A) Regions of the gels presented in Fig. 3C that contained the RTX-UpaGΔ2 monomer (which is located entirely in the periplasm) are shown. The results indicate that the periplasm of cells that produce ST–RTX-UpaGΔ2 remained inaccessible to the SpyCatcher protein unless the OM was permeabilized. Perhaps due to the use of a cell permeabilization buffer, a greater amount of a polypeptide that likely corresponded to the precursor form of the ST–RTX-UpaGΔ2 monomer (*) was reproducibly immunoprecipitated from samples generated from permeabilized cells than from intact cells. (B) Regions of the gels presented in Fig. 3C that contained the slower-migrating (alternative) form of the ST–RTX-UpaGΔ2 trimer are shown. The results indicate that the secretion of the passenger domains of the slower-migrating form of the ST–RTX-UpaGΔ2 trimer was completed in a stepwise fashion. Download FIG S3, PDF file, 0.2 MB (230.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Further examination of RTX-UpaGΔ2 as well as other chimeras that harbor unfolded passenger domain segments demonstrated that another mode of discontinuous secretion can be observed during TAA assembly. After a 0.25-min chase, almost none of the very slowly migrating form of trimeric ST–RTX-UpaGΔ2 formed a covalent bond with SpyCatcher, but a small amount of a singly modified species could be detected (Fig. S3B, top gel, lane 4). Remarkably, increasing amounts of doubly and triply modified species accumulated at later time points, and by 5 min, nearly half of the protein was completely modified (Fig. S3B, lanes 5 and 6). Although some of the ST–RTX-UpaGΔ2 trimers contained one or more passenger domains that were not fully secreted by the last time point, the results nevertheless suggest that at least the RTX moieties could be secreted in an asynchronous or stepwise fashion. In subsequent experiments, we analyzed a chimera in which a mutant form of the cholera toxin B subunit [CtxB (C9S/C86S) or CtxB*] was fused to the N terminus of UpaGΔ2. The mutations prevented the CtxB moiety from folding into its native conformation by preventing disulfide bond formation. Like HA–RTX-UpaGΔ2, much of the HA-CtxB*-UpaGΔ2 fusion rapidly formed a stable trimer that was cleaved by PK (Fig. S4A and B). A slight delay in the accumulation of the ∼42-kDa PK fragment that correlated with the disappearance of an ∼36-kDa band was also observed (Fig. S4B, lanes 6 to 10). Consistent with the evidence of a slight assembly defect, most of a SpyTagged version of the protein (ST-CtxB*-UpaGΔ2) was triply modified by SpyCatcher after a 0.25-min chase, but small amounts of unmodified, singly modified, and doubly modified forms were also detected (Fig. S4C, lane 4). The conversion of the unmodified protein to modified forms after longer chase times provided evidence of stepwise secretion (Fig. S4C, lanes 5 and 6).
The secretion of chimeric passenger domains that contain an unfolded segment is completed in a stepwise fashion. (A) Illustration of the HA-CtxB*-UpaGΔ2 protein. HA, HA tag. (B) AD202 cells transformed with a plasmid encoding HA-CtxB*-UpaGΔ2 (pRS39) were subjected to pulse-chase labeling. After cells were either incubated with PK or mock treated, immunoprecipitations were conducted using an anti-UpaG antiserum and proteins were resolved by SDS-PAGE. (C) AD202 cells were transformed with a plasmid encoding ST-CtxB*-UpaGΔ2 (pRS41). The experiment represented in panel B was repeated, except that cells were incubated with SpyCatcher instead of PK. Download FIG S4, PDF file, 0.3 MB (344.1KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
We noted a similar phenomenon in the analysis of a chimera that contains a disordered ∼100-residue passenger domain fragment comprised of 33 glycine-glycine-serine repeats [(GGS)33]. A construct in which (GGS)33 was fused to the N terminus of UpaGΔ2 [(GGS)33-UpaGΔ2] was assembled as efficiently as UpaGΔ2 (Fig. S5). Another chimera that contains the same fragment fused to the smaller UpaGΔ4 derivative also formed heat- and SDS-resistant trimers very rapidly (Fig. 4A and B, lanes 1 to 5). Presumably because the passenger domain of this construct consists entirely of a disordered polypeptide, PK treatment converted the stable trimer to an ∼25-kDa fragment that corresponds to the β barrel domain alone (Fig. 4B, lanes 6 to 10). Curiously, the (GGS)33-UpaGΔ4 trimer was degraded at the first time point and no ∼25-kDa fragment was observed. The results suggested that the (GGS)33 segment caused an assembly defect that initially exposed the β barrel domain to proteolysis. Consistent with this possibility, pulse-chase analysis of the SpyTagged version of the protein [(ST-GGS33-UpaGΔ4)] revealed a slight delay in the completion of secretion. After a 0.25-min chase, most of the ST–(GGS)33-UpaGΔ4 trimer was only doubly modified by SpyCatcher, but at later time points almost all of the chimera was triply modified (Fig. 4C). These results are noteworthy not only because they provide another example of stepwise secretion but also because they demonstrate that the presence of a folding-competent segment of a native TAA passenger domain is not required to drive the secretion of a disordered polypeptide.
FIG 4.

The secretion of disordered passenger domains is completed in a stepwise fashion. (A) Illustration of the HA-(GGS)33-UpaGΔ4 protein. HA, HA tag. (B) AD202 cells transformed with a plasmid encoding HA-(GGS)33-UpaGΔ4 (pRS22) were subjected to pulse-chase labeling. After cells were either incubated with PK or mock treated, immunoprecipitations were conducted using an anti-UpaG antiserum and proteins were resolved by SDS-PAGE. (C) AD202 cells were transformed with a plasmid encoding ST–(GGS)33-UpaGΔ4 (pRS36). The experiment represented in panel B was repeated, except that cells were incubated with SpyCatcher instead of PK. The OM of an equal number of cells was permeabilized prior to the addition of SpyCatcher.
Rapid secretion of chimeric passenger domains that contain a disordered segment. (A) AD202 cells transformed with a plasmid encoding HA-(GGS)33-UpaGΔ2 (pRS21) were subjected to pulse-chase labeling. After cells were either incubated with PK or mock treated, immunoprecipitations were conducted using an anti-UpaG antiserum and proteins were resolved by SDS-PAGE. (B) AD202 cells were transformed with a plasmid encoding ST–(GGS)33-UpaGΔ2 (pRS35). The experiment represented in panel A was repeated, except that cells were incubated with SpyCatcher instead of PK. Download FIG S5, PDF file, 0.3 MB (308.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The clearest evidence for the stepwise secretion of TAA passenger domains emerged from experiments analyzing derivatives of UpaGΔ2 that contain an engineered disulfide bond. Because disulfide bonds form in the periplasm, they were originally introduced into UpaGΔ2 to determine if passenger domains that have tertiary structure can be secreted by the TAA pathway. Pairs of serine residues in the UpaGΔ2 passenger domain were mutated to cysteine to create UpaGΔ2 (S1624C/S1652C) and UpaGΔ2 (S1620C/S1678C). To confirm that the two cysteine residues formed a disulfide bond, AD202 and RI2, an isogenic dsbA deletion strain deficient in disulfide bond formation, were transformed with a plasmid that encodes one of the double mutants, subjected to pulse-chase labeling, and collected by centrifugation. Half of each sample was then incubated with a cysteine-specific biotinylation reagent, and biotinylated protein was isolated on NeutrAvidin-agarose. As expected, the cysteine residues remained largely inaccessible to the biotinylation reagent unless the mutants were produced in the dsbA deletion strain (Fig. S6). Despite the presence of the disulfide bonds, both double mutants assembled into stable trimers as rapidly as UpaGΔ2 (Fig. 5A, lanes 1 to 5). Like UpaGΔ2, the stable trimers were converted into typical ∼42-kDa fragments by PK, but there was a noticeable lag in their accumulation (especially in the case of the UpaGΔ2 [S1624C/S1652C] mutant) that suggested an assembly defect (Fig. 5A, lanes 6 to 10; for comparison, see reference 36, Fig. 1). Interestingly, although most of a SpyTagged version of UpaGΔ2 (S1624C/S1652C) was left unmodified or modified only partially by SpyCatcher after a 0.25-min chase, a clear progression from partially to triply modified forms of the protein was seen over 5 min (Fig. 5B, top gel). The progressive modification appeared to be independent of the buffer or pH (Fig. S7, top gel). Furthermore, the finding that the protein was completely modified at all time points when the OM was permeabilized (Fig. 5B, middle gel; Fig. S7, bottom gel) confirmed that unreactive SpyTags were trapped in the periplasm. A SpyTagged version of UpaGΔ2 (S1620C/S1678C) exhibited a milder secretion defect, but partially modified forms could also be observed at early time points (Fig. 5B, bottom gel). Taken together, the results suggest a model in which small folded polypeptides jam the TAA pathway but can be secreted sequentially during transient conformational changes in the β barrel domain (Fig. 5C).
FIG 5.
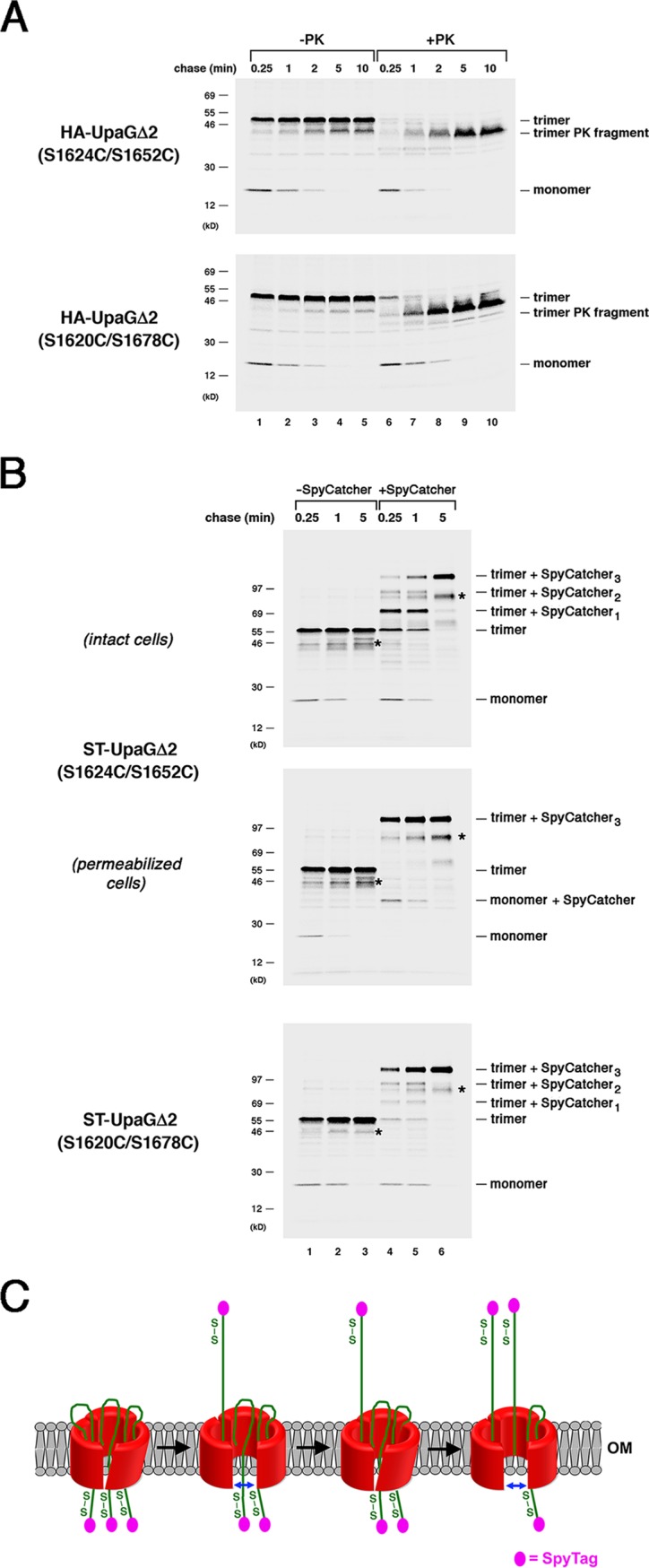
The secretion of passenger domains that contain a nonnative disulfide bond is completed in a stepwise fashion. (A) AD202 cells transformed with a plasmid encoding HA-UpaGΔ2 (S1624C/S1652C) or HA-UpaGΔ2 (S1620C/S1678C) (pRS29 or pRS30) were subjected to pulse-chase labeling. After cells were either incubated with PK or mock treated, immunoprecipitations were conducted using an anti-UpaG antiserum and proteins were resolved by SDS-PAGE. (B) AD202 cells were transformed with a plasmid encoding ST-UpaGΔ2 (S1624C/S1652C) or ST-UpaGΔ2 (S1620C/S1678C) (pRS33 or pRS34). The experiment represented in panel A was repeated, except that cells were incubated with SpyCatcher instead of PK. The OM of an equal number of cells that produced ST-UpaGΔ2 (S1624C/S1652C) was permeabilized prior to the addition of SpyCatcher. Unidentified bands that might have been breakdown products are labeled with an asterisk. (C) Model for the stepwise secretion of UpaGΔ2 passenger domain derivatives that contain a disulfide bond. Passenger domain translocation stalls because the disulfide-bonded segments cannot pass readily through the linked β barrel domain. We propose that as a result of a series of conformational changes, the β barrel expands transiently and enables each passenger domain to be fully secreted.
Nonnative cysteine residues introduced into the passenger domain of UpaGΔ2 form disulfide bonds. AD202 and RI2 (AD202 dsbA::cm) cells transformed with a plasmid encoding HA-UpaGΔ2 (S1624C/S1652C) or HA-UpaGΔ2 (S1620C/S1678C) (pRS29 or pRS30) were subjected to pulse-chase labeling. Samples were divided in half, and one aliquot was incubated with EZ-Link maleimide-PEG2-biotin to biotinylate free cysteine residues. After immunoprecipitations were conducted using a monoclonal anti-HA antibody, biotinylated proteins were isolated from the treated aliquots using NeutrAvidin-agarose beads. Proteins were subsequently resolved by SDS-PAGE. Download FIG S6, PDF file, 0.5 MB (540.9KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The efficiency of the SpyTag/SpyCatcher reaction is not increased by the use of a Tris-based buffer. AD202 cells transformed with a plasmid encoding HA-UpaGΔ2 (S1624C/S1652C) (pRS29) were subjected to pulse-chase labeling. Cells were resuspended in spheroplast buffer (33 mM Tris [pH 7.0], 40% sucrose) and divided in half (intact cells) or incubated with lysozyme to permeabilize the OM before they were divided in half (permeabilized cells). SpyCatcher was then added to one of the two halves of each sample. Immunoprecipitations were conducted using an anti-UpaG antiserum, and proteins were resolved by SDS-PAGE. Unidentified bands that might represent breakdown products are labeled with an asterisk. Download FIG S7, PDF file, 0.3 MB (289.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
A secretion defect prolongs the interaction of a TAA with the Bam complex.
Because the stepwise translocation of the UpaGΔ2 (S1624C/S1652C) passenger domains suggested that the β barrel domain might need to remain highly dynamic, we hypothesized that it stays bound to the Bam complex (presumably in an incompletely assembled state) until all three passenger domains are secreted. To test this possibility, we used a site-specific photo-cross-linking approach in which the photoactivatable amino acid analog p-benzoyl l-phenylalanine (Bpa) was incorporated at the C-terminal residue (W1778; see Fig. 1A) of both HA-UpaGΔ2 and HA-UpaGΔ2 (S1624C/S1652C) by amber suppression (61). This position was chosen because the C-terminal residue of a classical autotransporter was shown to interact with BamA in a previous cross-linking study (43). AD202 cells were collected following pulse-chase labeling, and half of the cells were subjected to UV irradiation. Immunoprecipitations were then performed using a monoclonal anti-HA antibody. As in previous experiments, stable trimers accumulated very rapidly (Fig. 6, top gels). A small amount of a high-molecular-weight cross-linking product was seen in cells that produced HA-UpaGΔ2 (W1778am) after 0.25 min, but it rapidly disappeared at later time points (Fig. 6, top left gel). In contrast, a much larger amount of the product was seen in cells that produced HA-UpaGΔ2 (S1624C/S1652C/W1778am) after both the 0.25-min and 1-min time points (Fig. 6, top right gel). A small amount of the cross-linking product could be detected even after 5 min. On the basis of its size, we surmised that this polypeptide corresponds to the UpaGΔ2 trimer cross-linked to BamA. Consistent with our prediction, the cross-linking product was immunoprecipitated with an anti-BamA antiserum (Fig. 6, bottom gels). The same polypeptide was also pulled down in sequential anti-HA and anti-BamA immunoprecipitations (Fig. S8). Given that the accumulation of triply modified ST-UpaGΔ2 and ST-UpaGΔ2 (S1624C/S1652C) parallels the disappearance of the cross-linking product, these results strongly suggest that the β barrel domain of UpaGΔ2 is released from the Bam complex only after the passenger domain translocation reaction has been completed. Consistent with the notion that ST-UpaGΔ2 (S1624C/S1652C) forms a prolonged interaction with the Bam complex (and thereby reduces its availability), overproduction of the TAA mutant in a strain that contains only a low level of BamA created a synthetic growth defect (Fig. S9).
FIG 6.

UpaGΔ2 remains associated with the Bam complex until passenger domain secretion is complete. AD202 cells transformed with plasmids encoding pDULE-Bpa and HA-UpaGΔ2 (W1778am) or HA-UpaGΔ2 (S1624C/S1652C/1778am) (pRS45 or pRS46) were subjected to pulse-chase labeling. Half of the cells were irradiated with UV light, and the other half were left untreated. Immunoprecipitations were then conducted with a monoclonal anti-HA antibody or a polyclonal antiserum raised against a BamA C-terminal peptide, and proteins were resolved by SDS-PAGE. An unidentified cross-linking product that served as an internal gel loading control is labeled with an asterisk.
Isolation of a high-molecular-weight crosslinking product by sequential immunoprecipitation. AD202 cells transformed with plasmids encoding pDULE-Bpa and HA-UpaGΔ2 (W1778am) or HA-UpaGΔ2 (S1624C/S1652C/1778am) (pRS45 or pRS46) were subjected to pulse-chase labeling and UV irradiation, and immunoprecipitations were conducted with a monoclonal anti-HA antibody. Immunoprecipitated proteins were then released by resuspending protein A beads in 50 μl 100 mM Tris base–8 mM EDTA–2% SDS–7% glycerol and heating them at 99°C for 5 min. After the beads were pelleted, 10 μl of each sample was placed on ice and the remaining 40 μl was mixed with 800 μl RIPA buffer and used for a second round of immunoprecipitations with a polyclonal antiserum raised against a BamA C-terminal peptide. Proteins isolated after both single and sequential immunoprecipitations were then resolved by SDS-PAGE. A larger portion of each sample was used for the sequential immunoprecipitations because the anti-BamA antiserum binds to BamA relatively weakly. Download FIG S8, PDF file, 1.1 MB (1.1MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Expression of ST-UpaGΔ2 (S1624C/S1652C) in cells that produce a low level of BamA creates a synthetic lethal phenotype. AD202 and RS959 (bamA101) cells were transformed with a plasmid encoding either ST-UpaGΔ2 or ST-UpaGΔ2 (S1624C/S1652C) under the control of a rhamnose-inducible promoter (pRS47 or pRS48). Cells were then streaked on LB agar plates with or without 0.2% l-rhamnose and incubated overnight at 37°C. Download FIG S9, PDF file, 1.9 MB (1.9MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DISCUSSION
In this study, we monitored the translocation of both native and nonnative polypeptides across the OM via the TAA pathway to gain insights into the mechanism of protein secretion and to define the limits of the secretion reaction. An examination of N-terminally truncated derivatives of UpaG showed that all three passenger domain fragments were secreted more rapidly than we could measure given the limited time resolution of our pulse-chase methodology even in cases in which the fragments were relatively long (∼450 residues). The results are compatible with the notion that the formation of a coiled-coil structure in the extracellular space drives rapid secretion (58). Although it is unclear if the folding of the globular segments that are interspersed between the coiled-coil regions also promotes rapid secretion, the results imply that the presence of these segments does not significantly slow or stall translocation. In contrast, nonnative passenger domains that contained disordered regions or that formed disulfide bonds in the periplasm were generally secreted considerably less efficiently. While the secretion of the three chimeric passenger domains of RTX-UpaGΔ2 was slow but appeared to be completed simultaneously, the secretion of the three polypeptides of other chimeras that contain disordered passenger domain segments was completed sequentially. The presence of a disulfide bond in UpaGΔ2 also caused the passenger domains to be secreted in a slow, stepwise fashion. Given that the translocation of all of the passenger domains was initiated normally, it seems likely that translocation was rapid until the nonnative segments approached the OM. In any case, the results show that both unfolded polypeptides and polypeptides that contain small regions of tertiary structure can be secreted by the TAA pathway, possibly by drawing energy from the Donnan potential across the OM (62) or as a result of the effects of molecular crowding in the periplasm. Interestingly, our analysis of UpaGΔ2 (S1624C/S1652C) also provided evidence that the β barrel domain of UpaGΔ2 remains bound to the Bam complex (presumably in an incompletely folded state) until all three passenger domains are fully secreted.
Taken together with the results of an earlier study (36), the results presented here suggest a general model for TAA biogenesis. Using a cross-linking approach, we previously obtained strong evidence that the three polypeptide segments that form the β barrel domain of UpaG fold into an asymmetrical trimer in the periplasm (Fig. 7, step i). The finding that the deletion of the linker region prevents trimerization and destabilizes the protein suggests that the linker is embedded into the middle of the trimer at this stage and may even nucleate trimer assembly. Presumably, the linker folds into a hairpin that facilitates passenger domain translocation and thereby prevents the trimer from forming a closed structure. Subsequently, the partially assembled β barrel domain is targeted to the Bam complex, which catalyzes its insertion into the OM (Fig. 7, step ii). The observation that a population of incompletely assembled trimers whose passenger domains are inaccessible to PK can be detected in the OM (36) suggests that a conformational change that exposes the passenger domains on the cell surface precedes the initiation of translocation. Translocation then proceeds through a hybrid channel composed of open forms of the β barrel domain and the BamA β barrel. This proposal is consistent with the observation that polypeptide segments that contain tertiary structure are secreted through the TAA pathway even though they are too large to fit inside the narrow pore formed by the β barrel domain (38–40). Translocation is then driven by the folding of the passenger domain in the extracellular space (Fig. 7, step iii). Following the completion of translocation and the resolution of the hairpin structures, the β barrel domain closes and dissociates from the Bam complex (Fig. 7, step iv). This aspect of the model is consistent with the finding that the release of the UpaG β barrel domain from BamA is linked to the full translocation of all three passenger domains. Note that although the translocation of the native UpaG passenger domain is very rapid (and is clearly faster than the initial formation of a trimeric structure in the periplasm), our results reveal that passenger domain modifications that convert translocation into a slow, rate-limiting step can be accommodated by the Bam complex.
FIG 7.

Model for the assembly of TAAs. The three subunits of a TAA form an asymmetric trimer in the periplasm (step i). The partially folded trimer is then targeted to the OM, where the Bam complex catalyzes the membrane integration of the β barrel domain (step ii). A conformational change in the β barrel domain may be required to expose the passenger domains on the cell surface and to initiate translocation. Translocation proceeds through a hybrid channel comprised of open forms of the linked β barrel domain and the BamA β barrel (step iii). Translocation is driven at least in part by the folding of the passenger domains into a coiled-coil structure. Following the completion of the translocation reaction, the β barrel domain folds completely and dissociates from the Bam complex.
Our model has two significant implications. First, it challenges other models (63) by suggesting that, like classical autotransporters, TAAs are not self-contained secretion systems in which the fully folded β barrel domain catalyzes the secretion of the covalently linked passenger domain after dissociation from the Bam complex. Instead, our model holds that the passenger domains are secreted before the β barrel domain is fully assembled and that the Bam complex plays at least an indirect role in the secretion reaction. Indeed, in many respects our model for TAA assembly is similar to a model that we proposed previously for the assembly of classical autotransporters (45). Second, our model, together with the proposal that the stepwise secretion of artificially disulfide-bonded UpaGΔ2 passenger domains requires multiple conformational changes in the β barrel domain, raises the intriguing possibility that the Bam complex induces dynamic changes in client proteins. While this potentially novel attribute of the Bam complex was revealed through the analysis of a mutant form of a distinct class of OMPs, it is conceivable that it plays a critical role in the assembly of a broad range of substrates. Interestingly, transient stalling of the translocation of classical autotransporter passenger domains has been observed previously (41), but the possibility that a change in the conformation of the β barrel domain might be required to restart the movement of the passenger domain was not apparent.
The finding that three identical polypeptides can be secreted by the TAA pathway in an asynchronous fashion also provides insight into the mechanism of translocation. To be clear, we cannot determine from our results if the translocation of the unnatural passenger domains that we examined involves stalling followed by sequential movement of individual polypeptide chains or by continuous movement of all three polypeptide chains at different rates. Nevertheless, the results invoke a scenario in which the polypeptide chains exhibit a remarkable ability to slide through the channel and past each other without becoming trapped. Presumably, enough energy can be applied to the system to offset any nonproductive interactions that might strongly inhibit the movement of the passenger domains across the OM. One might certainly imagine an alternative scenario in which the progression of the translocation reaction would require continuous assembly of the three polypeptides into a structure that moves as a single entity. From a practical perspective, the finding that a wide range of unfolded polypeptides (and perhaps of slowly folding polypeptides that remain unfolded in the periplasm) can be secreted by the TAA pathway suggests that modified TAAs might represent a valuable tool for the development of a novel cell surface display technology. Indeed, the production and surface localization of polypeptides that are naturally occurring trimers such as the influenza hemagglutinin and TRAIL proteins might have considerable utility.
Finally, our results led to the curious discovery that TAA trimers that are resistant to heat denaturation and SDS denaturation are not necessarily completely assembled. On the basis of first principles, it would certainly be expected that the exceptionally strong network of interactions that define the native state would be required to prevent the three subunits from dissociating under such harsh conditions. Indeed, the presence of stable TAA trimers has been taken as evidence that TAAs can assemble correctly in unnatural environments (e.g., mitochondria) and that two different TAAs can form completely assembled heterotrimers (64–66). We cannot determine if partially assembled derivatives of UpaG migrated as heat- and SDS-resistant trimers because β barrel domains that contain one or more unresolved hairpins are unexpectedly stable or because structural changes occurred during sample preparation. In either case, our results suggest that the presence of stable trimers should be interpreted with caution and that further experimental analysis is required to assess their status.
MATERIALS AND METHODS
Reagents, bacterial strains, and growth conditions.
E. coli strain AD202 (MC4100 ompT::kan; see reference 67), was used in almost all experiments, but RI2 (AD202 dsbA::cm; see reference 34) and RS959, a strain that contains the bamA101 allele (DPR959 ompT::spc; see reference 68), were also used. Unless otherwise noted, all cultures were grown at 37°C in M9 minimal medium containing 0.2% glycerol and all the l-amino acids (40 μg/ml) except methionine and cysteine. In some experiments, MOPS (morpholinepropanesulfonic acid) minimal medium (diluted from Teknova 10× stock; catalog number M2101) containing 1.32 mM KH2PO4 in addition to 0.2% glycerol and the same concentration of l-amino acids was used. Ampicillin (100 μg/ml), tetracycline (5 μg/ml), or trimethoprim (50 μg/ml) was added as necessary. A rabbit polyclonal antiserum (sc-805; Y-11) and a mouse monoclonal antibody (sc-7392; F-7) against the HA epitope tag were obtained from Santa Cruz Biotechnology. Rabbit antisera raised against purified UpaGΔ2 and a BamA C-terminal peptide have been described previously (36, 69).
Plasmid construction.
All plasmids used in this study were derivatives of pTrc99a (70) or pSCRhaB2 (71) and have been listed in Table S1A in the supplemental material. The oligonucleotides used for PCRs and for site-directed mutagenesis are listed in Table S1B. The PCR products used for plasmid construction and the construction methods are described in Table S1C and D. A DNA fragment encoding GGS33 was synthesized by Genewiz and provided in pUC57. All site-directed mutagenesis experiments were performed using a previously described protocol (72).
Plasmids and oligonucleotides used in this study. Download Table S1, DOCX file, 0.03 MB (32.9KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Pulse-chase labeling, photo-cross-linking, and immunoprecipitation of labeled proteins.
For experiments in which cells transformed with pTrc99a derivatives were grown in M9 medium, overnight cultures were washed and diluted in fresh medium to an optical density at 550 nm (OD550) of 0.02 to 0.03. Cultures were grown to an OD550 of 0.2 to 0.3, and cells were subjected to pulse-chase labeling 30 min later as previously described (41). Because radiolabeled protein produced as a result of leaky transcription from the trc promoter was readily detected, to avoid any possible effects of overexpression, no IPTG (isopropyl-β-d-thiogalactopyranoside) was added. For experiments in which cells transformed with pSCRhaB2 derivatives were grown in M9 medium, overnight cultures were washed and diluted in fresh medium to an OD550 of 0.03. Cultures were grown to an OD550 of 0.3, and 0.2% (wt/vol) l (+)-rhamnose was added to induce expression. Cells were radiolabeled 15 min later. All radiolabeled samples were collected by centrifugation (3,000 × g, 10 min, 4°C) and resuspended in fresh M9 medium. In some experiments, the populations of resuspended cells were divided in half, and one half was treated with 200 μg/ml proteinase K (PK) on ice for 20 min. The protease reaction was stopped by the addition of 2 mM phenylmethylsulfonyl fluoride (PMSF). Photo-cross-linking was performed essentially as described previously (41, 61) except that 10-ml samples were obtained at each time point. In all experiments, 10% (wt/vol) trichloroacetic acid (TCA) was added to both treated and untreated samples to precipitate proteins. Immunoprecipitations were performed as previously described (73), and, unless otherwise, the noted proteins were resolved by SDS-PAGE on 8% to 16% Tris-glycine minigels (Thermo Fisher Scientific). Radiolabeled proteins were visualized using a Fujifilm FLA-9000 phosphorimager.
SpyTag-SpyCatcher reactions.
Cells harboring plasmids that encode UpaG derivatives with an N-terminal SpyTag were grown and subjected to pulse-chase radiolabeling as described above, except that 4-ml volumes of cells were collected at each time point. Cells were pelleted by centrifugation (3,000 × g, 10 min, 4°C) and resuspended in 500 μl phosphate-buffered saline (PBS; pH 7.2). The cell populations were divided in half, and 10 μM purified SpyCatcher protein (Kerafast) was added to one aliquot. Both untreated and SpyCatcher-treated samples were incubated in a 4°C water bath for 40 min. Samples were then transferred to ice, and proteins were precipitated with 10% TCA. In some experiments, equal volumes of radiolabeled cells were collected by centrifugation (3,000 × g, 10 min, 4°C) and resuspended in 500 μl spheroplast buffer (33 mM Tris [pH 7.0], 40% sucrose). The cells were then incubated for 20 min on ice with lysozyme (200 μg/ml) and EDTA (pH 8.0) (2 mM) to permeabilize the OM. SpyCatcher protein (10 μM) was added to one half of each sample, and both treated and untreated samples were processed as described above.
Biotinylation and sequential purification of biotinylated proteins.
Biotinylation reactions were performed to monitor the accessibility of cysteine residues introduced into the passenger domain of UpaGΔ2. Radiolabeled cells were collected by centrifugation (3,000 × g, 10 min, 4°C) and resuspended in 1 ml PBS (pH 7.2). One half of each sample was immediately mixed with TCA to precipitate proteins, and the other half was incubated with 1 mM EZ-Link maleimide-PEG2-biotin (Thermo Fisher Scientific) for 18 h on ice prior to TCA precipitation. UpaGΔ2 derivatives were then immunoprecipitated using a monoclonal anti-HA antibody as described above. Following immunoprecipitations, the protein A-Sepharose beads were washed once with radioimmunoprecipitation assay (RIPA) buffer (74) containing 500 mM NaCl and once with RIPA buffer containing 150 mM NaCl. The beads added to biotinylated samples were heated in a buffer containing 100 mM Tris base, 2% SDS, and 7.5% glycerol at 99°C for 5 min to release bound proteins and then pelleted. The supernatants were removed, diluted 1:10 into cold RIPA buffer containing 150 mM NaCl, and incubated with NeutrAvidin-agarose beads (Pierce) on a rotary shaker at 4°C for 1 h. The NeutrAvidin-agarose beads were then washed with RIPA buffer containing 150 mM NaCl. Finally, the protein A-Sepharose and NeutrAvidin-agarose beads used to isolate proteins from the nonbiotinylated and biotinylated samples, respectively, and resuspended in sample buffer, and proteins were resolved on SDS-PAGE as described above.
Homology-based modeling of the UpaGΔ2 structure.
In the absence of a crystal structure of UpaG, we used the PHYRE2 (Protein Homology/analogY Recognition Engine version 2.0) server (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index; see reference 4) to generate a structure of the UpaGΔ2 monomer from its primary amino acid sequence. A structure of the UpaGΔ2 trimer was generated by submitting the structure of the monomer to the GalaxyHOMOMER server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=HOMOMER; see reference 75).
ACKNOWLEDGMENTS
We thank Janine Peterson for providing technical assistance and Matt Doyle for providing helpful comments on the manuscript.
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Citation Sikdar R, Bernstein HD. 2019. Sequential translocation of polypeptides across the bacterial outer membrane through the trimeric autotransporter pathway. mBio 10:e01973-19. https://doi.org/10.1128/mBio.01973-19.
REFERENCES
- 1.Henderson IR, Navarro-Garcia F, Desvaux M, Fernandez RC, Ala'Aldeen D. 2004. Type V protein secretion pathway: the autotransporter story. Microbiol Mol Biol Rev 68:692–744. doi: 10.1128/MMBR.68.4.692-744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Celik N, Webb CT, Leyton DL, Holt KE, Heinz E, Gorrell R, Kwok T, Naderer T, Strugnell RA, Speed TP, Teasdale RD, Likić VA, Lithgow T. 2012. A bioinformatic strategy for the detection, classification and analysis of bacterial autotransporters. PLoS One 7:e43245. doi: 10.1371/journal.pone.0043245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emsley P, Charles IG, Fairweather NF, Isaacs NW. 1996. Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature 381:90–92. doi: 10.1038/381090a0. [DOI] [PubMed] [Google Scholar]
- 4.Otto BR, Sijbrandi R, Luirink J, Oudega B, Heddle JG, Mizutani K, Park S-Y, Tame JRH. 2005. Crystal structure of hemoglobin protease, a heme binding autotransporter protein from pathogenic Escherichia coli. J Biol Chem 280:17339–17345. doi: 10.1074/jbc.M412885200. [DOI] [PubMed] [Google Scholar]
- 5.Junker M, Schuster CC, McDonnell AV, Sorg KA, Finn MC, Berger B, Clark PL. 2006. Pertactin α-helix folding mechanism suggests common themes for the secretion and folding of autotransporter proteins. Proc Natl Acad Sci U S A 103:4918–4923. doi: 10.1073/pnas.0507923103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gangwer KA, Mushrush DJ, Stauff DL, Spiller B, McClain MS, Cover TL, Lacy DB. 2007. Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc Natl Acad Sci U S A 104:16293–16298. doi: 10.1073/pnas.0707447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heras B, Totsika M, Peters KM, Paxman JJ, Gee CL, Jarrott RJ, Perugini MA, Whitten AE, Schembri MA. 2014. The antigen 43 structure reveals a molecular Velcro-like mechanism of autotransporter-mediated bacterial clumping. Proc Natl Acad Sci U S A 111:457–462. doi: 10.1073/pnas.1311592111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linke D, Riess T, Autenrieth IB, Lupas A, Kempf VA. 2006. Trimeric autotransporter adhesins: variable structure, common function. Trends Microbiol 14:264–270. doi: 10.1016/j.tim.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Meng G, Surana NK, St Geme JII, Waksman G. 2006. Structure of the outer membrane translocator domain of the Haemophilus influenzae Hia trimeric autotransporter. EMBO J 25:2297–2304. doi: 10.1038/sj.emboj.7601132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shahid SA, Bardiaux B, Franks WT, Krabben L, Habeck M, van Rossum B-J, Linke D. 2012. Membrane-protein structure determination by solid-state NMR spectroscopy of microcrystals. Nat Methods 9:1212–1217. doi: 10.1038/nmeth.2248. [DOI] [PubMed] [Google Scholar]
- 11.Oomen CJ, van Ulsen P, van Gelder P, Feijen M, Tommassen J, Gros P. 2004. Structure of the translocator domain of a bacterial autotransporter. EMBO J 23:1257–1266. doi: 10.1038/sj.emboj.7600148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnard TJ, Dautin N, Lukacik P, Bernstein HD, Buchanan SK. 2007. Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat Struct Mol Biol 14:1214–1220. doi: 10.1038/nsmb1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhai Y, Zhang K, Huo Y, Zhu Y, Zhou Q, Lu J, Black I, Pang X, Roszak AW, Zhang X, Isaacs NW, Sun F. 2011. Autotransporter passenger domain secretion requires a hydrophobic cavity at the extracellular entrance of the α-domain pore. Biochem J 435:577–587. doi: 10.1042/BJ20101548. [DOI] [PubMed] [Google Scholar]
- 14.Nummelin H, Merckel MC, Leo JC, Lankinen H, Skurnik M, Goldman A. 2004. The Yersinia adhesin YadA collagen-binding domain structure is a novel left-handed parallel β-roll. EMBO J 23:701–711. doi: 10.1038/sj.emboj.7600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szczesny P, Linke D, Ursinus A, Bär K, Schwarz H, Riess TM, Kempf VAJ, Lupas AN, Martin J, Zeth K. 2008. Structure of the head of the Bartonella adhesin BadA. PLoS Pathog 4:e1000119. doi: 10.1371/journal.ppat.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edwards TE, Phan I, Abendroth J, Dieterich SH, Masoudi A, Guo W, Hewitt SN, Kelley A, Leibly D, Brittnacher MJ, Staker BL, Miller SI, Van Voorhis WC, Myler PJ, Stewart LJ. 2010. Structure of a Burkholderia pseudomallei trimeric autotransporter adhesin head. PLoS One 5:e12803. doi: 10.1371/journal.pone.0012803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agnew C, Borodina E, Zaccai NR, Conners R, Burton NM, Vicary JA, Cole DK, Antognozzi M, Virji M, Brady RL. 2011. Correlation of in situ mechanosensitive responses of the Moraxella catarrhalis adhesin UspA1 with fibronectin and receptor CEACAM1 binding. Proc Natl Acad Sci U S A 108:15174–15178. doi: 10.1073/pnas.1106341108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leo JC, Lyskowski A, Hattula K, Hartmann MD, Schwarz H, Butcher SJ, Linke D, Lupas AN, Goldman A. 2011. The structure of E. coli IgG-binding protein D suggests a general model for bending and binding in trimeric autotransporter adhesins. Structure 19:1021–1030. doi: 10.1016/j.str.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 19.Hartmann MD, Grin I, Dunin-Horkawicz S, Deiss S, Linke D, Lupas AN, Hernandez Alvarez B. 2012. Complete fiber structures of complex trimeric autotransporter adhesins conserved in enterobacteria. Proc Natl Acad Sci U S A 109:20907–20912. doi: 10.1073/pnas.1211872110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voulhoux R, Bos MP, Geurtsen J, Mols M, Tommassen J. 2003. Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299:262–266. doi: 10.1126/science.1078973. [DOI] [PubMed] [Google Scholar]
- 21.Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Lehr U, Schütz M, Oberhettinger P, Ruiz-Perez F, Donald JW, Palmer T, Linke D, Henderson IR, Autenrieth IB. 2010. C-terminal amino acid residues of the trimeric autotransporter adhesin YadA of Yersinia enterocolitica are decisive for its recognition and assembly by BamA. Mol Microbiol 78:932–946. doi: 10.1111/j.1365-2958.2010.07377.x. [DOI] [PubMed] [Google Scholar]
- 23.Malinverni JC, Werner J, Kim S, Sklar JG, Kahne D, Misra R, Silhavy TJ. 2006. YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol Microbiol 61:151–164. doi: 10.1111/j.1365-2958.2006.05211.x. [DOI] [PubMed] [Google Scholar]
- 24.Webb CT, Heinz E, Lithgow T. 2012. Evolution of the β-barrel assembly machinery. Trends Microbiol 20:612–620. doi: 10.1016/j.tim.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Bakelar J, Buchanan SK, Noinaj N. 2016. The structure of the β-barrel assembly machinery complex. Science 351:180–186. doi: 10.1126/science.aad3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu Y, Li H, Dong H, Zeng Y, Zhang Z, Paterson NG, Stansfeld PJ, Wang Z, Zhang Y, Wang W, Dong C. 2016. Structural basis of outer membrane protein insertion by the BAM complex. Nature 531:64–69. doi: 10.1038/nature17199. [DOI] [PubMed] [Google Scholar]
- 27.Han L, Zheng J, Wang Y, Yang X, Liu Y, Sun C, Cao B, Zhou H, Ni D, Lou J, Zhao Y, Huang Y. 2016. Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat Struct Mol Biol 23:192–196. doi: 10.1038/nsmb.3181. [DOI] [PubMed] [Google Scholar]
- 28.Iadanza MG, Higgins AJ, Schiffrin B, Calabrese AN, Brockwell DJ, Ashcroft AE, Radford SE, Ranson NA. 2016. Lateral opening in the intact β-barrel assembly machinery captured by cryo-EM. Nat Commun 7:12865. doi: 10.1038/ncomms12865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noinaj N, Kuszak AJ, Gumbart JC, Lukacik P, Chang H, Easley NC, Lithgow T, Buchanan SK. 2013. Structural insight into the biogenesis of β-barrel membrane proteins. Nature 501:385–390. doi: 10.1038/nature12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noinaj N, Kuszak AJ, Balusek C, Gumbart JC, Buchanan SK. 2014. Lateral opening and exit pore formation are required for BamA function. Structure 22:1055–1062. doi: 10.1016/j.str.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gessmann D, Chung YH, Danoff EJ, Plummer AM, Sandlin CW, Zaccai NR, Fleming KG. 2014. Outer membrane β-barrel protein folding is physically controlled by periplasmic lipid head groups and BamA. Proc Natl Acad Sci U S A 111:5878–5883. doi: 10.1073/pnas.1322473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schiffrin B, Calabrese AN, Higgins AJ, Humes JR, Ashcroft AE, Kalli AC, Brockwell DJ, Radford SE. 2017. Effects of periplasmic chaperones and membrane thickness on BamA-catalyzed outer-membrane protein folding. J Mol Biol 429:3776–3792. doi: 10.1016/j.jmb.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doerner PA, Sousa MC. 2017. Extreme dynamics in the BamA β-barrel seam. Biochemistry 56:3142–3149. doi: 10.1021/acs.biochem.7b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ieva R, Skillman KM, Bernstein HD. 2008. Incorporation of a polypeptide segment into the β domain pore during the assembly of a bacterial autotransporter. Mol Microbiol 67:188–201. doi: 10.1111/j.1365-2958.2007.06048.x. [DOI] [PubMed] [Google Scholar]
- 35.Hussain S, Bernstein HD. 2018. The Bam complex catalyzes efficient insertion of bacterial outer membrane proteins into membrane vesicles of variable lipid composition. J Biol Chem 293:2959–2973. doi: 10.1074/jbc.RA117.000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sikdar R, Peterson JH, Anderson DE, Bernstein HD. 2017. Folding of a bacterial integral outer membrane protein is initiated in the periplasm. Nat Commun 8:1309. doi: 10.1038/s41467-017-01246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pohlner J, Halter R, Beyreuther K, Meyer TF. 1987. Gene structure and extracellular secretion of Neisseria gonorrhoeae IgA protease. Nature 325:458–462. doi: 10.1038/325458a0. [DOI] [PubMed] [Google Scholar]
- 38.Veiga E, de Lorenzo V, Fernández LA. 2004. Structural tolerance of bacterial autotransporters for folded passenger protein domains. Mol Microbiol 52:1069–1080. doi: 10.1111/j.1365-2958.2004.04014.x. [DOI] [PubMed] [Google Scholar]
- 39.Skillman KM, Barnard TJ, Peterson JH, Ghirlando R, Bernstein HD. 2005. Efficient secretion of a folded protein domain by a monomeric bacterial autotransporter. Mol Microbiol 58:945–958. doi: 10.1111/j.1365-2958.2005.04885.x. [DOI] [PubMed] [Google Scholar]
- 40.Sauri A, ten Hagen-Jongman C, van Ulsen P, Luirink J. 2012. Estimating the size of the active translocation pore of an autotransporter. J Mol Biol 416:335–345. doi: 10.1016/j.jmb.2011.12.047. [DOI] [PubMed] [Google Scholar]
- 41.Ieva R, Bernstein HD. 2009. Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc Natl Acad Sci U S A 106:19120–19125. doi: 10.1073/pnas.0907912106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sauri A, Soprova Z, Wickström D, de Gier J-W, Van der Schors RC, Smit AB, Jong WSP, Luirink J. 2009. The Bam (Omp85) complex is involved in secretion of the autotransporter haemoglobin protease. Microbiology 155:3982–3991. doi: 10.1099/mic.0.034991-0. [DOI] [PubMed] [Google Scholar]
- 43.Ieva R, Tian P, Peterson JH, Bernstein HD. 2011. Sequential and spatially restricted interactions of assembly factors with an autotransporter β domain. Proc Natl Acad Sci U S A 108:E383–E391. doi: 10.1073/pnas.1103827108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peterson JH, Hussain S, Bernstein HD. 2018. Identification of a novel post-insertion step in the assembly of a bacterial outer membrane protein. Mol Microbiol 110:143–159. doi: 10.1111/mmi.14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernstein HD. 2015. Looks can be deceiving: recent insights into the mechanism of protein secretion by the autotransporter pathway. Mol Microbiol 97:205–215. doi: 10.1111/mmi.13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Renn JP, Clark PL. 2008. A conserved stable core structure in the passenger domain β-helix of autotransporter virulence proteins. Biopolymers 89:420–427. doi: 10.1002/bip.20924. [DOI] [PubMed] [Google Scholar]
- 47.Peterson JH, Tian P, Ieva R, Dautin N, Bernstein HD. 2010. Secretion of a bacterial virulence factor is driven by the folding of a C-terminal segment. Proc Natl Acad Sci U S A 107:17739–17744. doi: 10.1073/pnas.1009491107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soprova Z, Sauri A, van Ulsen P, Tame JRH, den Blaauwen T, Jong WSP, Luirink J. 2010. A conserved aromatic residue in the autochaperone domain of the autotransporter Hbp is critical for initiation of outer membrane translocation. J Biol Chem 285:38224–38233. doi: 10.1074/jbc.M110.180505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drobnak I, Braselmann E, Clark PL. 2015. Multiple driving forces required for efficient secretion of autotransporter virulence proteins. J Biol Chem 290:10104–10116. doi: 10.1074/jbc.M114.629170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baclayon M, Ulsen PV, Mouhib H, Shabestari MH, Verzijden T, Abeln S, Roos WH, Wuite GJL. 2016. Mechanical unfolding of an autotransporter passenger protein reveals the secretion starting point and processive transport intermediates. ACS Nano 10:5710–5719. doi: 10.1021/acsnano.5b07072. [DOI] [PubMed] [Google Scholar]
- 51.Kang'ethe W, Bernstein HD. 2013. Charge-dependent secretion of an intrinsically disordered protein via the autotransporter pathway. Proc Natl Acad Sci U S A 110:E4246–E4255. doi: 10.1073/pnas.1310345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roggenkamp A, Ackermann N, Jacobi CA, Truelzsch K, Hoffmann H, Heesemann J. 2003. Molecular analysis of transport and oligomerization of the Yersinia enterocolitica adhesin YadA. J Bacteriol 185:3735–3744. doi: 10.1128/JB.185.13.3735-3744.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chauhan N, Hatlem D, Orwick-Rydmark M, Schneider K, Floetenmeyer M, van Rossum B, Leo JC, Linke D. 2019. Insights into the autotransport process of a trimeric autotransporter, Yersinia adhesin A (YadA). Mol Microbiol 111:844–862. doi: 10.1111/mmi.14195. [DOI] [PubMed] [Google Scholar]
- 54.Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M. 2012. Peptide tag formed through a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc Natl Acad Sci U S A 109:E690–E697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valle J, Mabbett AN, Ulett GC, Toledo-Arana A, Wecker K, Totsika M, Schembri MA, Ghigo J-M, Beloin C. 2008. UpaG, a new member of the trimeric autotransporter family of adhesins in uropathogenic Escherichia coli. J Bacteriol 190:4147–4161. doi: 10.1128/JB.00122-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wollmann P, Zeth K, Lupas AN, Linke D. 2006. Purification of the YadA membrane anchor for secondary structure analysis and crystallization. Int J Biol Macromol 39:3–9. doi: 10.1016/j.ijbiomac.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 57.Cotter SE, Surana NK, Grass S, St Geme JW III. 2006. Trimeric autotransporters require trimerization of the passenger domain for stability and adhesive activity. J Bacteriol 188:5400–5407. doi: 10.1128/JB.00164-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hartmann MD, Ridderbusch O, Zeth K, Albrecht R, Testa O, Woolfson DN, Sauer G, Dunin-Horkawicz S, Lupas AN, Alvarez BH. 2009. A coiled-coil motif that sequesters ions to the hydrophobic core. Proc Natl Acad Sci U S A 106:16950–16955. doi: 10.1073/pnas.0907256106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meisner WK, Sosnick TR. 2004. Fast folding of a helical protein initiated by the collision of unstructured chains. Proc Natl Acad Sci U S A 101:13478–13482. doi: 10.1073/pnas.0404057101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chenal A, Karst JC, Sotomayor Pérez AC, Wozniak AK, Baron B, England P, Ladant D. 2010. Calcium-induced folding and stabilization of the intrinsically disordered RTX domain of the CyaA toxin. Biophys J 99:3744–3753. doi: 10.1016/j.bpj.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Farrell IS, Toroney R, Hazen JL, Mehl RA, Chin JW. 2005. Photo-cross-linking interacting proteins with a genetically encoded benzophenone. Nat Methods 2:377–384. doi: 10.1038/nmeth0505-377. [DOI] [PubMed] [Google Scholar]
- 62.Stock JB, Rauch B, Roseman S. 1977. Periplasmic space in Salmonella typhimurium and Escherichia coli. J Biol Chem 252:7850–7861. [PubMed] [Google Scholar]
- 63.Fan E, Chauhan N, Udatha DB, Leo JC, Linke D. 2016. Type V secretion systems in bacteria. Microbiol Spectr 4(1):VMBF-0009-2015. doi: 10.1128/microbiolspec.VMBF-0009-2015. [DOI] [PubMed] [Google Scholar]
- 64.Müller JEN, Papic D, Ulrich T, Grin I, Schütz M, Oberhettinger P, Tommassen J, Linke D, Dimmer KS, Autenrieth IB, Rapaport D. 2011. Mitochondria can recognize and assemble fragments of a β-barrel structure. Mol Biol Cell 22:1638–1647. doi: 10.1091/mbc.E10-12-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mikula KM, Leo JC, Łyskowski A, Kedracka-Krok S, Pirog A, Goldman A. 2012. The translocation domain in trimeric autotransporter adhesins is necessary and sufficient for trimerization and autotransportation. J Bacteriol 194:827–838. doi: 10.1128/JB.05322-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schmidgen T, Kaiser PO, Ballhorn W, Franz B, Gottig S, Linke D, Kempf VAJ. 2014. Heterologous expression of Bartonella adhesin A in Escherichia coli by exchange of trimeric autotransporter adhesin domains results in enhanced adhesion properties and a pathogenic phenotype. J Bacteriol 196:2155–2165. doi: 10.1128/JB.01461-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Akiyama Y, Ito K. 1990. SecY protein, a membrane-embedded secretion factor of E. coli, is cleaved by the OmpT protease in vitro. Biochem Biophys Res Commun 167:711–715. doi: 10.1016/0006-291X(90)92083-C. [DOI] [PubMed] [Google Scholar]
- 68.Ricci DP, Hagan CL, Kahne D, Silhavy TJ. 2012. Activation of the Escherichia coli β-barrel assembly machinery (Bam) is required for essential components to interact properly with substrate. Proc Natl Acad Sci U S A 109:3487–3491. doi: 10.1073/pnas.1201362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pavlova O, Peterson JH, Ieva R, Bernstein HD. 2013. Mechanistic link between β barrel assembly and the initiation of autotransporter secretion. Proc Natl Acad Sci U S A 110:E938–E947. doi: 10.1073/pnas.1219076110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Amann E, Ochs B, Abel K. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69:301–315. doi: 10.1016/0378-1119(88)90440-4. [DOI] [PubMed] [Google Scholar]
- 71.Cardona ST, Valvano MA. 2005. An expression vector containing a rhamnose-inducible promoter provides tightly regulated gene expression in Burkholderia cenocepacia. Plasmid 54:219–228. doi: 10.1016/j.plasmid.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 72.Zheng L, Baumann U, Reymond JL. 2004. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ulbrandt ND, Newitt JA, Bernstein HD. 1997. The E. coli signal recognition particle is required for the insertion of a subset of inner membrane proteins. Cell 88:187–196. doi: 10.1016/S0092-8674(00)81839-5. [DOI] [PubMed] [Google Scholar]
- 74.Harlow E, Lane D. 1999. Using antibodies: a laboratory manual. Cold Spring Harbor Press, Cold Spring Harbor, NY. [Google Scholar]
- 75.Baek M, Park T, Heo L, Park C, Seok C. 2017. GalaxyHomomer: a Web server for protein homo-oligomer structure prediction from a monomer sequence or structure. Nucleic Acids Res 45:W320–W324. doi: 10.1093/nar/gkx246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
UpaG derivatives that contain only the β barrel and the embedded linker segment are assembled extremely rapidly. (A) Illustration of HA-UpaGΔ3, UpaGΔ4, and UpaGΔ5 constructs. HA, HA tag. The site of each deletion is shown on a homology-based model of the C terminus of an UpaG monomer. (B) Strain AD202 cells transformed with a plasmid encoding HA-UpaGΔ3 (pRS18), UpaGΔ4 (pRS19), or UpaGΔ5 (pRS20) were subjected to pulse-chase labeling. After the cells were either incubated with PK or subjected to mock treatment, immunoprecipitations were conducted using a polyclonal anti-HA antiserum (for HA-UpaGΔ3) or an anti-UpaG antiserum (for all constructs). Proteins were resolved by SDS-PAGE on either 8% to 16% Tris-glycine minigels (for HA-UpaGΔ3) or 12% Bis-Tris minigels using a morpholineethanesulfonic acid (MES)-based buffer system (Thermo Fisher Scientific) (for UpaGΔ4 and UpaGΔ5). (C) The experiment performed with UpaGΔ4 was repeated except that the cells were transferred to a prewarmed 25°C water bath for 15 min prior to pulse-chase labeling at the same temperature. Immunoprecipitations were conducted using an anti-UpaG antiserum, and proteins were resolved by SDS-PAGE on an 8% to 16% Tris-glycine minigel. Unidentified background bands are labeled with an asterisk in panels B and C. Download FIG S1, PDF file, 0.9 MB (1.1MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The faster-migrating form of an RTX-UpaGΔ2 trimer contains a disordered RTX domain. AD202 cells transformed with a plasmid encoding HA–RTX-UpaGΔ2 (pRS42) were grown in MOPS minimal media to mid-log phase and induced with 0.2% l-rhamnose. Cultures were divided in half, and 2 mM CaCl2 was added to one half 5 min prior to pulse-chase labeling. After the cells were either incubated with PK or mock treated, immunoprecipitations were conducted using an anti-UpaG antiserum and proteins were resolved by SDS-PAGE. The observation that the level of the faster-migrating (∼110-kDa) form of the RTX-UpaGΔ2 trimer was greatly reduced when calcium was added to induce folding indicates that it contained an unfolded RTX moiety. Download FIG S2, PDF file, 0.4 MB (372.7KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Properties of monomeric and slower-migrating trimeric forms of ST–RTX-UpaGΔ2. (A) Regions of the gels presented in Fig. 3C that contained the RTX-UpaGΔ2 monomer (which is located entirely in the periplasm) are shown. The results indicate that the periplasm of cells that produce ST–RTX-UpaGΔ2 remained inaccessible to the SpyCatcher protein unless the OM was permeabilized. Perhaps due to the use of a cell permeabilization buffer, a greater amount of a polypeptide that likely corresponded to the precursor form of the ST–RTX-UpaGΔ2 monomer (*) was reproducibly immunoprecipitated from samples generated from permeabilized cells than from intact cells. (B) Regions of the gels presented in Fig. 3C that contained the slower-migrating (alternative) form of the ST–RTX-UpaGΔ2 trimer are shown. The results indicate that the secretion of the passenger domains of the slower-migrating form of the ST–RTX-UpaGΔ2 trimer was completed in a stepwise fashion. Download FIG S3, PDF file, 0.2 MB (230.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The secretion of chimeric passenger domains that contain an unfolded segment is completed in a stepwise fashion. (A) Illustration of the HA-CtxB*-UpaGΔ2 protein. HA, HA tag. (B) AD202 cells transformed with a plasmid encoding HA-CtxB*-UpaGΔ2 (pRS39) were subjected to pulse-chase labeling. After cells were either incubated with PK or mock treated, immunoprecipitations were conducted using an anti-UpaG antiserum and proteins were resolved by SDS-PAGE. (C) AD202 cells were transformed with a plasmid encoding ST-CtxB*-UpaGΔ2 (pRS41). The experiment represented in panel B was repeated, except that cells were incubated with SpyCatcher instead of PK. Download FIG S4, PDF file, 0.3 MB (344.1KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Rapid secretion of chimeric passenger domains that contain a disordered segment. (A) AD202 cells transformed with a plasmid encoding HA-(GGS)33-UpaGΔ2 (pRS21) were subjected to pulse-chase labeling. After cells were either incubated with PK or mock treated, immunoprecipitations were conducted using an anti-UpaG antiserum and proteins were resolved by SDS-PAGE. (B) AD202 cells were transformed with a plasmid encoding ST–(GGS)33-UpaGΔ2 (pRS35). The experiment represented in panel A was repeated, except that cells were incubated with SpyCatcher instead of PK. Download FIG S5, PDF file, 0.3 MB (308.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Nonnative cysteine residues introduced into the passenger domain of UpaGΔ2 form disulfide bonds. AD202 and RI2 (AD202 dsbA::cm) cells transformed with a plasmid encoding HA-UpaGΔ2 (S1624C/S1652C) or HA-UpaGΔ2 (S1620C/S1678C) (pRS29 or pRS30) were subjected to pulse-chase labeling. Samples were divided in half, and one aliquot was incubated with EZ-Link maleimide-PEG2-biotin to biotinylate free cysteine residues. After immunoprecipitations were conducted using a monoclonal anti-HA antibody, biotinylated proteins were isolated from the treated aliquots using NeutrAvidin-agarose beads. Proteins were subsequently resolved by SDS-PAGE. Download FIG S6, PDF file, 0.5 MB (540.9KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The efficiency of the SpyTag/SpyCatcher reaction is not increased by the use of a Tris-based buffer. AD202 cells transformed with a plasmid encoding HA-UpaGΔ2 (S1624C/S1652C) (pRS29) were subjected to pulse-chase labeling. Cells were resuspended in spheroplast buffer (33 mM Tris [pH 7.0], 40% sucrose) and divided in half (intact cells) or incubated with lysozyme to permeabilize the OM before they were divided in half (permeabilized cells). SpyCatcher was then added to one of the two halves of each sample. Immunoprecipitations were conducted using an anti-UpaG antiserum, and proteins were resolved by SDS-PAGE. Unidentified bands that might represent breakdown products are labeled with an asterisk. Download FIG S7, PDF file, 0.3 MB (289.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Isolation of a high-molecular-weight crosslinking product by sequential immunoprecipitation. AD202 cells transformed with plasmids encoding pDULE-Bpa and HA-UpaGΔ2 (W1778am) or HA-UpaGΔ2 (S1624C/S1652C/1778am) (pRS45 or pRS46) were subjected to pulse-chase labeling and UV irradiation, and immunoprecipitations were conducted with a monoclonal anti-HA antibody. Immunoprecipitated proteins were then released by resuspending protein A beads in 50 μl 100 mM Tris base–8 mM EDTA–2% SDS–7% glycerol and heating them at 99°C for 5 min. After the beads were pelleted, 10 μl of each sample was placed on ice and the remaining 40 μl was mixed with 800 μl RIPA buffer and used for a second round of immunoprecipitations with a polyclonal antiserum raised against a BamA C-terminal peptide. Proteins isolated after both single and sequential immunoprecipitations were then resolved by SDS-PAGE. A larger portion of each sample was used for the sequential immunoprecipitations because the anti-BamA antiserum binds to BamA relatively weakly. Download FIG S8, PDF file, 1.1 MB (1.1MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Expression of ST-UpaGΔ2 (S1624C/S1652C) in cells that produce a low level of BamA creates a synthetic lethal phenotype. AD202 and RS959 (bamA101) cells were transformed with a plasmid encoding either ST-UpaGΔ2 or ST-UpaGΔ2 (S1624C/S1652C) under the control of a rhamnose-inducible promoter (pRS47 or pRS48). Cells were then streaked on LB agar plates with or without 0.2% l-rhamnose and incubated overnight at 37°C. Download FIG S9, PDF file, 1.9 MB (1.9MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Plasmids and oligonucleotides used in this study. Download Table S1, DOCX file, 0.03 MB (32.9KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.