

Abstract
Background
The majority of people with epilepsy have a good prognosis and their seizures are controlled by a single antiepileptic drug. However, up to 20% of patients from population‐based studies, and up to 30% from clinical series (not population‐based), develop drug‐resistant epilepsy, especially those with focal‐onset seizures. In this review, we summarise the current evidence regarding topiramate, an antiepileptic drug first marketed in 1996, when used as an add‐on treatment for drug‐resistant focal epilepsy.
This is an update of a Cochrane Review first published in 1999, and last updated in 2014.
Objectives
To evaluate the efficacy and tolerability of topiramate when used as an add‐on treatment for people with drug‐resistant focal epilepsy.
Search methods
For the latest update of this review we searched the following databases on 2 July 2018: Cochrane Register of Studies (CRS Web), which includes the Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL); MEDLINE (Ovid, 1946‐ ); ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform (ICTRP). We imposed no language restrictions. We also contacted the manufacturers of topiramate and researchers in the field to identify any ongoing or unpublished studies.
Selection criteria
Randomised, placebo‐controlled add‐on trials of topiramate, recruiting people with drug‐resistant focal epilepsy.
Data collection and analysis
Two review authors independently selected trials for inclusion and extracted the relevant data. We assessed the following outcomes: (1) 50% or greater reduction in seizure frequency; (2) seizure freedom; (3) treatment withdrawal (any reason); (4) adverse effects. Primary analyses were intention‐to‐treat (ITT), and summary risk ratios (RRs) with 95% confidence intervals (95% CIs) are presented. We evaluated dose‐response in regression models. We carried out a 'Risk of bias' assessment for each included study using the Cochrane 'Risk of bias' tool and assessed the overall certainty of evidence using the GRADE approach.
Main results
We included 12 trials, representing 1650 participants. Baseline phases ranged from four to 12 weeks and double‐blind phases ranged from 11 to 19 weeks. The RR for a 50% or greater reduction in seizure frequency with add‐on topiramate compared to placebo was 2.71 (95% CI 2.05 to 3.59; 12 studies; high‐certainty evidence). Dose regression analysis showed increasing effect with increasing topiramate dose demonstrated by an odds ratio (OR) of 1.45 (95% CI 1.28 to 1.64; P < 0.001) per 200 mg/d increase in topiramate dosage. The proportion of participants achieving seizure freedom was also significantly increased with add‐on topiramate compared to placebo (RR 3.67, 95% CI 1.79 to 7.54; 8 studies; moderate‐certainty evidence). Treatment withdrawal was significantly higher for add‐on topiramate compared to placebo (RR 2.37, 95% CI 1.66 to 3.37; 12 studies; high‐certainty evidence). The RRs for the following adverse effects indicate that they are significantly more prevalent with topiramate, compared to placebo: ataxia 2.29 (99% CI 1.10 to 4.77; 4 studies); concentration difficulties 7.81 (99% CI 2.08 to 29.29; 6 studies; moderate‐certainty evidence); dizziness 1.52 (99% CI 1.07 to 2.16; 8 studies); fatigue 2.08 (99% CI 1.37 to 3.15; 10 studies); paraesthesia 3.65 (99% CI 1.58 to 8.39; 7 studies; moderate‐certainty evidence); somnolence 2.44 (99% CI 1.61 to 3.68; 9 studies); 'thinking abnormally' 5.70 (99% CI 2.26 to 14.38; 4 studies; high‐certainty evidence); and weight loss 3.99 (99% CI 1.82 to 8.72; 9 studies; low‐certainty evidence). Evidence of publication bias for the primary outcome was found (Egger test, P = 0.001). We rated all studies included in the review as having either low or unclear risk of bias. Overall, we assessed the evidence as moderate to high certainty due to the evidence of publication bias, statistical heterogeneity and imprecision, which was partially compensated for by large effect sizes.
Authors' conclusions
Topiramate has efficacy as an add‐on treatment for drug‐resistant focal epilepsy as it is almost three times more effective compared to a placebo in reducing seizures. The trials reviewed were of relatively short duration and provided no evidence for the long‐term efficacy of topiramate. Short‐term use of add‐on topiramate was shown to be associated with several adverse events. The results of this review should only be applied to adult populations as only one study included children. Future research should consider further examining the effect of dose.
Plain language summary
Topiramate add‐on for drug‐resistant focal epilepsy
Background
Epilepsy is a disorder where recurrent seizures are caused by abnormal electrical discharges from the brain. Most seizures can be controlled by a single antiepileptic drug. Unfortunately, some people require more than one antiepileptic medication to control their seizures, especially if these originate from one area of the brain (focal epilepsy), instead of affecting the entire brain (generalised epilepsy). These people are said to have drug‐resistant epilepsy. Topiramate can be used in addition to other antiepileptic drugs, called an add‐on treatment, to try to control drug‐resistant epilepsy.
Aim of this review
This review investigated the effectiveness and tolerability of topiramate when used as an add‐on treatment for people with drug‐resistant focal epilepsy.
Results
We found 12 trials that investigated topiramate as an add‐on treatment. They included a total of 1650 people with drug‐resistant focal epilepsy. These trials compared the antiepileptic drug topiramate to a placebo drug (an inactive, dummy drug which should not show any effect) for a period of up to 18 weeks. Taking all the evidence of the trials into account, the review found that topiramate is almost three times more effective, when used with other drugs, at reducing the number of seizures in drug‐resistant focal epilepsy than placebo. Adding topiramate to people's usual treatment was, however, associated with an increase in adverse effects such as problems with co‐ordination (ataxia), concentration, dizziness, drowsiness (somnolence), fatigue, 'thinking abnormally', tickling or numbness of the skin (paraesthesia) and weight loss. People taking add‐on topiramate were also more than twice as likely to withdraw from treatment than those taking placebo, most likely due to adverse effects.
Certainty of the evidence
We assessed the trials with regards to potential bias and certainty. Overall, we rated the certainty of the evidence as moderate to high which means that we are fairly certain that the findings that we have reported are accurate. The trials included in this review did not examine the long‐term effects of topiramate as an add‐on treatment and only one study investigated the use of add‐on topiramate in children. The findings should, therefore, only be applied to adults with drug‐resistant focal epilepsy. Future research should test which dose is most effective.
The evidence is current to July 2018.
Summary of findings
Summary of findings for the main comparison. Topiramate compared to placebo for drug‐resistant focal epilepsy.
| Add‐on topiramate compared to placebo for drug‐resistant focal epilepsy | ||||||
| Patients or population: people with drug‐resistant focal epilepsy Setting: outpatients Intervention: add‐on topiramate Comparison: add‐on placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI; Adverse effects: 99% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with topiramate | |||||
|
50% or greater reduction in seizure frequency (ITT analysis) Follow‐up: range 11 to 19 weeks |
Study population | RR 2.71 (2.05 to 3.59) | 1650 (12 RCTs) | ⊕⊕⊕⊕ Higha,b,c,f,h | Topiramate increases the proportion of participants attaining a 50% or greater reduction in seizure frequency. | |
| 163 per 1000 | 441 per 1000 (333 to 584) | |||||
|
Seizure freedom Follow‐up: range 11 to 19 weeks |
Study population | RR 3.67 (1.79 to 7.54) | 1177 (8 RCTs) | ⊕⊕⊕⊝ Moderatea,e,f | Topiramate likely increases the proportion of participants achieving seizure freedom. | |
| 17 per 1000 | 61 per 1000 (30 to 125) | |||||
|
Treatment withdrawal Follow‐up: range 11 to 19 weeks |
Study population | RR 2.37 (1.66 to 3.37) | 1650 (12 RCTs) | ⊕⊕⊕⊕ Higha,d,f | Topiramate increases the incidence of treatment withdrawal. | |
| 61 per 1000 | 144 per 1000 (101 to 204) | |||||
|
Adverse effects ‐ weight loss/decrease Follow‐up: range 11 to 18 weeks |
Study population | RR 3.99 (1.82 to 8.72) | 1070 (9 RCTs) | ⊕⊕⊝⊝ Lowa,c,e,f | Topiramate may produce a large increase in the proportion of participants experiencing weight loss. | |
| 20 per 1000 | 81 per 1000 (37 to 177) | |||||
|
Adverse effects ‐ paraesthesia Follow‐up: range 11 to 18 weeks |
Study population | RR 3.65 (1.58 to 8.39) | 1071 (7 RCTs) | ⊕⊕⊕⊝ Moderatea,e,f | Topiramate likely increases the proportion of participants experiencing paraesthesia. | |
| 25 per 1000 | 91 per 1000 (40 to 210) | |||||
|
Adverse effects ‐ 'thinking abnormally' Follow‐up: range 12 to 19 weeks |
Study population | RR 5.70 (2.26 to 14.38) | 640 (4 RCTs) | ⊕⊕⊕⊕ Higha,d,g | Topiramate increases the proportion of participants reporting that they are 'thinking abnormally'. | |
| 43 per 1000 | 243 per 1000 (96 to 614) | |||||
|
Adverse effects ‐ difficulty with concentration/concentration impaired/concentration‐attention difficulties Follow‐up: range 11 to 18 weeks |
Study population | RR 7.81 (2.08 to 29.29) | 702 (6 RCTs) | ⊕⊕⊕⊝ Moderatea,c,e,g | Topiramate likely increases the number of participants who experience difficulty with concentration. | |
| 11 per 1000 | 88 per 1000 (23 to 330) | |||||
| *The risk in the intervention group (and its 95% or 99% confidence interval, dependent on the outcome) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% or 99% CI, dependent on the outcome). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
aWe did not downgrade evidence for risk of bias; we judged risk of bias across studies to be low. Ten studies did not describe the blinding of outcome assessors, one study failed to explain randomisation and allocation concealment and another did not describe any method of blinding. We, however, reasoned that the blinding of outcome assessors would minimally impact the estimated effect size due to the self‐reported nature of the review outcomes. bWe downgraded the evidence once for inconsistency: we detected significant statistical heterogeneity. cWe downgraded the evidence once for publication bias: examination of funnel plot and Egger test indicates the possibility of publication bias. dWe downgraded the evidence once for imprecision: the number of events (< 400) did not suffice optimal information size. eWe downgraded the evidence twice for imprecision: the number of events (< 100) did not suffice optimal information size. fWe upgraded the evidence once for large effect: RR > 2.00. gWe upgraded the evidence twice for large effect: RR > 5.00. hWe upgraded the evidence once for dose‐response gradient: logistic regression demonstrated a significant dose‐response relationship.
Background
This is an update of a Cochrane Review first published in 1999 (Jette 1999b), and last updated in 2014 (Pulman 2014). The purpose of this update is to synthesise the current data in order to understand the role of topiramate as an add‐on treatment in drug‐resistant focal epilepsy. For the purpose of this review, people with drug‐resistant focal epilepsy have been defined as having focal‐onset seizures (simple focal and/or complex focal seizures and/or secondary generalised tonic‐clonic seizures) that have failed to respond to at least one monotherapy treatment with a standard antiepileptic drug.
Description of the condition
The majority of people given a diagnosis of epilepsy have a good prognosis and their seizures are controlled by treatment with a single antiepileptic drug (Kwan 2000; Reynolds 1981), but up to 20% of patients from population‐based studies, and up to 30% from clinical (non population‐based) series, will develop drug‐resistant epilepsy (Cockerell 1995; Kwan 2000), often requiring treatment with combinations of antiepileptic drugs. This presents a significant therapeutic problem when approximately 1% of the general population will suffer from epilepsy at some point in their lifetime (Fiest 2017).
Description of the intervention
Over the past decade, there has been renewed interest in the development of newer antiepileptic drugs. This is largely because several of the standard antiepileptic drugs are not always successful at controlling seizures, and because some have been associated with certain adverse effects. In the first instance, new antiepileptic drugs are tested as an add‐on treatment for people with drug‐resistant focal epilepsy in randomised controlled trials (RCTs). Having demonstrated a therapeutic effect in these trials, new antiepileptic drugs tend to be licensed for add‐on use before monotherapy trials have been undertaken, in which new antiepileptic drugs are compared with standard ones.
How the intervention might work
There are an ever increasing number of licensed antiepileptic drugs from which to choose from, for people with drug‐resistant focal epilepsy. This review focuses upon the effects of topiramate, a drug whose mechanisms of action include a modulatory effect on voltage‐dependent sodium conductance (Coulter 1993), enhancement of gamma‐aminobutyric acid‐A (GABAA)‐mediated chloride flux (Brown 1993), antagonism of kainate receptor‐mediated excitatory currents (Gryder 2003), and inhibition of carbonic anhydrase, an enzyme necessary for GABAergic neurotransmission (Herrero 2002). Topiramate was licensed for add‐on use in the UK in 1996.
Why it is important to do this review
In this review, we summarise evidence from RCTs which have investigated the effects of topiramate in people with drug‐resistant focal epilepsy in order to aid clinical decision‐making when prescribing antiepileptic drug treatments within this population.
Objectives
To evaluate the efficacy and tolerability of topiramate when used as an add‐on treatment for people with drug‐resistant focal epilepsy.
Methods
Criteria for considering studies for this review
Types of studies
Studies had to meet all of the following criteria.
RCTs
Double‐ or single‐blinded trials
Placebo‐controlled
Parallel‐group or cross‐over studies
Minimum treatment period of eight weeks
Types of participants
People of any age with drug‐resistant focal epilepsy (i.e. experiencing simple focal, complex focal or secondarily generalised tonic‐clonic seizures).
Types of interventions
The active treatment group received treatment with topiramate in addition to conventional antiepileptic drug treatment.
The control group received a matched placebo or an alternative dose of topiramate in addition to conventional antiepileptic drug treatment.
Types of outcome measures
Primary outcomes
50% or greater reduction in seizure frequency
The primary outcome is the proportion of people with a 50% or greater reduction in seizure frequency in the treatment period compared to the pre‐randomisation baseline period. This outcome was chosen as it is commonly reported in this type of study. Furthermore, this outcome can also be calculated for studies which report baseline and follow‐up seizure frequencies.
Secondary outcomes
Seizure freedom
The proportion of people with complete cessation of seizures during the treatment period.
Treatment withdrawal
The proportion of people having treatment withdrawn during the course of the treatment period was used as a measure of 'global effectiveness'. Treatment is likely to be withdrawn due to adverse effects, lack of efficacy or a combination of both, and this is an outcome to which participants make a direct contribution. In trials of short duration, it is likely that adverse effects will be the most common reason for withdrawal.
Adverse effects
The proportion of people experiencing:
ataxia;
dizziness;
headache;
nausea/vomiting;
paraesthesias;
weight loss/decrease;
fatigue;
somnolence;
concentration impairment;
speech difficulty;
thinking abnormally.
We chose these adverse effects as we considered them to be common and important adverse effects of antiepileptic drugs.
Search methods for identification of studies
Electronic searches
We ran searches for the original review on 18 June 2013, and subsequent searches in August 2016 and November 2016. For the latest update, we searched the following databases on 2 July 2018.
Cochrane Register of Studies (CRS Web), which includes the Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL), using the strategy outlined in Appendix 1.
MEDLINE (Ovid, 1946‐ ) using the strategy outlined in Appendix 2.
ClinicalTrials.gov using the strategy outlined in Appendix 3.
WHO International Clinical Trials Registry Platform (ICTRP) using the strategy outlined in Appendix 4.
There were no language restrictions.
Previously we also searched SCOPUS (1823 to 18 June 2013) as an alternative to Embase, using the strategy outlined in Appendix 5. This is no longer necessary, because randomised and quasi‐randomised controlled trials in Embase are now included in CENTRAL, so the SCOPUS search has not been updated.
Searching other resources
We reviewed reference lists of included studies to search for additional reports of relevant studies and contacted Johnson and Johnson for information about any unpublished or ongoing studies.
Data collection and analysis
Selection of studies
For the update, two review authors (RB and JH) independently assessed trials for inclusion. Any disagreements were resolved by discussion with a third review author (AGM). Two review authors (RB and JH) extracted data and assessed risk of bias; disagreements again were resolved by discussion.
Data extraction and management
We extracted the following information for each trial using a data extraction form.
Methodological/trial design
Method of randomisation and allocation concealment
Method of blinding
Whether any participants had been excluded from reported analyses
Length of baseline period
Length of treatment period
Dose(s) of topiramate tested
Patient/demographic information
Total number of participants allocated to each treatment group
Age/sex
Number with focal/generalised epilepsy
Seizure types
Seizure frequency during the baseline period
Number of background drugs
Most of the trials found were sponsored by Johnson and Johnson who were asked to confirm the following information.
Method of randomisation
Total number randomised to each group
Number of participants in each group achieving a 50% or greater reduction in seizure frequency per treatment group
Number of participants having treatment withdrawn post‐randomisation per treatment group
-
For those excluded:
the reason for exclusion;
whether any of those excluded completed the treatment phase;
whether any of those excluded had a 50% or greater reduction in seizure frequency during the treatment phase.
Outcomes
We recorded the number of people experiencing each outcome (see Types of outcome measures) per randomised group. We contacted authors of trials for any missing information.
Assessment of risk of bias in included studies
Two review authors (RB and JH) independently made an assessment of the risk of bias for each trial using the Cochrane 'Risk of bias' tool as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We discussed and resolved any disagreements. We rated the included studies as high risk, low risk or unclear risk for the six domains applicable to RCTs: randomisation method, allocation concealment, blinding methods, incomplete outcome data, selective outcome reporting and other sources of bias.
We created 'Summary of findings' tables, and employed the GRADE approach for assessing certainty of evidence.
Measures of treatment effect
We presented the primary outcome, 50% or greater reduction in seizure frequency, and the secondary outcomes, seizure freedom and treatment withdrawal, as risk ratios (RRs) with 95% confidence intervals (CIs). For the reporting of the individual adverse effects, we again presented the results as RRs, but instead quoted 99% CIs to compensate for multiple testing. By contrast, we reported the results from the dose regression model as odds ratios (ORs) with 95% CIs.
Unit of analysis issues
There were no unit of analysis issues.
Dealing with missing data
We sought any missing data from the study authors. We carried out intention‐to‐treat (ITT), best‐case and worst‐case analysis on the primary outcome to account for any missing data. All analyses are presented in the main report.
Assessment of heterogeneity
We assessed clinical heterogeneity by comparing the distribution of important individual participant factors among trials (for example, age, seizure type, duration of epilepsy, number of antiepileptic drugs taken at the time of randomisation) and trial factors (for example, randomisation concealment, blinding, losses to follow‐up). We examined statistical heterogeneity using a Chi2 test and the I2 statistic for heterogeneity and, providing no heterogeneity was present (P > 0.10), we employed a fixed‐effect model. In the event that heterogeneity was found, we planned to use a random‐effects model analysis using the inverse‐variance method.
Assessment of reporting biases
We requested all protocols from study authors to enable a comparison of outcomes of interest. Outcome reporting bias was to be investigated using the ORBIT matrix system (Kirkham 2010). We also undertook visual examination of funnel plots as well as the Egger test, a statistical test to determine publication bias in meta‐analyses, to establish any publication bias.
Data synthesis
We employed a fixed‐effect model meta‐analysis to synthesise the data for the comparison, topiramate versus placebo, for the outcomes:
50% or greater reduction in seizure frequency;
seizure freedom;
treatment withdrawal;
adverse effects.
Each outcome was to be stratified by study characteristics to ensure the appropriate combination of study data.
Our preferred estimator was the Mantel‐Haenszel RR. For the outcomes 50% or greater reduction in seizure frequency and treatment withdrawal, we used 95% CIs. For individual adverse effects we used 99% CIs to make an allowance for multiple testing.
Our analyses included all participants in the treatment group to which they had been allocated. For the efficacy outcome, 50% or greater reduction in seizure frequency, we undertook three analyses, two of which were sensitivity analyses:
-
Primary (ITT) analysis: participants from both treatment groups not completing follow‐up or with inadequate seizure data were assumed to be non‐responders.
Worst‐case analysis (sensitivity analysis): participants not completing follow‐up or with inadequate seizure data were assumed to be non‐responders in the intervention (topiramate) group, but were assumed to be responders in the placebo group.
Best‐case analysis (sensitivity analysis): participants not completing follow‐up or with inadequate seizure data were assumed to be responders in the intervention (topiramate) group, but were assumed to be non‐responders in the placebo group.
The best‐case and worst‐case analyses served to determine whether the assumption made during ITT analysis (i.e. that all participants not completing follow‐up or with inadequate seizure data are non‐responders) grossly affected the estimated effect size.
Dose regression analysis
For the primary outcome, we examined the dose‐response relationship using a generalised linear mixed model with the xtmelogit in STATA SE version 14. The binary outcome was defined with the value '0' for patients who did not achieve a 50% or greater reduction in seizure frequency and was defined as the value '1' for patients who did. Study and dose were included as a fixed‐effect and a random‐effect was included for the treatment (no random‐effect for the constant term), as described in Turner 2000. Dose was standardised by its standard deviation (351.9 mg/d). This method estimated an OR as opposed to a RR. We then used the command, meqrlogit, to predict the probabilities for the following: (i) the percentage of patients having a 50% response at differing doses; (ii) the difference in the percentage of patients responding to each dose compared to placebo.
Subgroup analysis and investigation of heterogeneity
We undertook subgroup analysis for adverse effects. We intended to investigate heterogeneity using sensitivity analysis if deemed appropriate.
Sensitivity analysis
We conducted five forms of sensitivity analyses in total during the data synthesis. Two such sensitivity analyses were highlighted and explained earlier, best‐ and worst‐case analysis (see Data synthesis). We also intended to carry out sensitivity analysis if peculiarities were found between study quality, characteristics of participants, interventions and outcomes. Specifically, we detected heterogeneity in the outcome, 50% or greater reduction in seizure frequency, and, therefore, we applied the following sensitivity analysis according to: study quality (we excluded studies with either high or unclear risk of bias) and age (we excluded one study because it included paediatric participants). Finally, we conducted a sensitivity according to dosage. We excluded three studies because all three only assessed dose of 200 mg/d topiramate.
Summarising and interpreting results using GRADE
We used the GRADE approach, as outlined in the GRADE Handbook (Schünemann 2013), to assess the certainty of evidence and to interpret findings. We used GRADEpro GDT software (GRADEpro GDT 2015), which imports data from Review Manager 5 software (Review Manager 2014), to create a 'Summary of findings' table for the following outcomes: 50% or greater reduction in seizure frequency, seizure freedom, treatment withdrawal, weight loss/decrease, paraesthesia, thinking abnormally, and difficulty with concentration.
Results
Description of studies
Results of the search
The search revealed 1346 records identified from the databases, outlined in Electronic searches. After removing 541 duplicates, 805 records remained and we screened all for inclusion in the review. We excluded 758 records at this point due to irrelevance, leaving 46 records to be assessed for eligibility at the full‐text stage of screening. Following this, we excluded five studies, linked to seven individual records (see Figure 1 and Characteristics of excluded studies for reasons of exclusion). We included a total of 12 studies (linked to 37 individual records) in the review, all of which we included in meta‐analyses. We identified two abstracts (two records) as studies awaiting classification and contacted the authors of these studies for more information. At the time of publication, no correspondence had been received.
1.
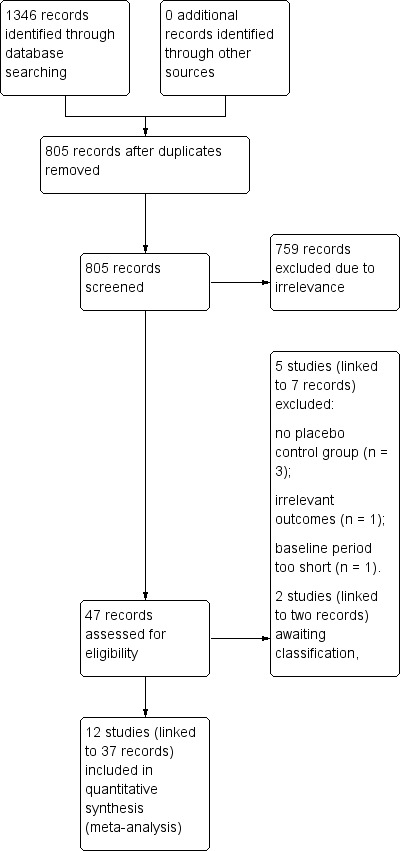
Study flow diagram.
Included studies
The 12 RCTs identified by the screening process recruited a total of 1650 participants and between them tested doses of 200 mg, 300 mg, 400 mg, 600 mg, 800 mg and 1000 mg topiramate per day. For further information on each trial, see Characteristics of included studies.
Overall, there were 10 randomised controlled trials (RCTs) which compared topiramate to placebo in adults aged 18 to 75 years (Ben‐Menachem 1996; Chung 2014; Faught 1996; Guberman 2002; Korean Topiramate Study Group 1999; Privitera 1996; Rosenfeld 1996;Sharief 1996; Tassinari 1996; Yen 2000), one RCT which examined topiramate versus a placebo in children (Elterman 1999), and one other RCT which examined the same comparison within the elderly (Zhang 2011). Eight of these trials were sponsored by Johnson and Johnson. In all of the trials, participants were eligible to take part in the double‐blind part of the trials if they were found to experience a minimum number of focal seizures (range 3 to 12 seizures) and were currently taking one to two or one to three antiepileptic drug treatments. See Table 2 for comparison of study characteristics.
1. Study characteristics.
| Study name | Country | Participants | No. of previous AEDs | No. of participants | Baseline period | Treatment period |
| Ben‐Menachem 1996 | Sweden, Norway, Denmark, Germany | Adults | 1 to 2 | 56 | 8 weeks | 13 weeks |
| Chung 2014 | Argentina, Australia, Belgium, Canada, Chile, Germany, Greece, Hungary, India, Israel, New Zealand, Poland, Russia, South Africa, Spain, USA | Adults | 1 to ≥7 | 249 | 8 weeks | 11 weeks |
| Elterman 1999 | USA, Costa Rica | Children | 1 to 2 | 86 | 8 weeks | 16 weeks |
| Faught 1996 | USA | Adults | 1 to 2 | 181 | 12 weeks | 16 weeks |
| Guberman 2002 | Hungary, Poland, Israel, Canada, Russia, Czech Republic | Adults | 1 to 2 | 263 | 4 weeks | 12 weeks |
| Korean Topiramate Study Group 1999 | Korea | Adults | 1 to 2 | 177 | 12 weeks | 18 weeks |
| Privitera 1996 | USA | Adults | 1 to 2 | 190 | 12 weeks | 18 weeks |
| Rosenfeld 1996 | USA | Adults | 1 | 209 | 8 weeks | 19 weeks |
| Sharief 1996 | Sweden, Spain, UK, France | Adults | 1 to 2 | 47 | 8 weeks | 11 weeks |
| Tassinari 1996 | UK, Italy, France, Norway, Denmark | Adults | 1 to 2 | 60 | 8 weeks | 12 weeks |
| Yen 2000 | China | Adults | 1 to 4 | 46 | 8 weeks | 14 weeks |
| Zhang 2011 | China | Elderly | 1 to 3 | 86 | 8 weeks | 12 weeks |
AED: antiepileptic drug
One multicentre, parallel trial had a pre‐randomisation period of eight weeks and a treatment period of 13 weeks (Ben‐Menachem 1996), randomising 56 adults to one of two treatment arms: 800 mg of topiramate (n = 28) or placebo (n = 28). The study medication was administered twice daily or to the maximal tolerated dose.
Similarly, a multicentre, parallel trial had a pre‐randomisation baseline period of eight weeks (Chung 2014). This trial featured a shorter treatment period of 11 weeks and consisted of two treatment arms. Two hundred and forty‐nine participants were randomised to either placebo (n = 125) or 200 mg/d topiramate (n = 124).
The Elterman 1999 multicentre (USA, Costa Rica), parallel trial, randomised 41 children aged one to 16 years to topiramate and 45 to placebo. Participants were eligible if they experienced six or more seizures during baseline and were taking one or two antiepileptic drugs. The baseline period was eight weeks in duration with a treatment period of 16 weeks.
Another parallel trial from the USA had three different treatment arms and a placebo arm (Faught 1996). Forty‐five adults aged 18 to 65 years were randomised to 200 mg per day of topiramate, 45 mg to 400 mg per day topiramate or 46 mg to 600 mg per day topiramate and 45 were randomised to the placebo group. This trial had a baseline period of 12 weeks and a treatment period of 16 weeks.
A further multicentre, parallel trial (Guberman 2002), randomised adults to one of three treatment arms: topiramate escalated weekly by 25 mg to 200 mg per day (n = 85), topiramate escalated weekly by 50 mg to 200 mg per day (n = 83), and placebo (n = 91). This trial had a baseline period of four weeks and a titration period of eight weeks for the 25 mg group or four weeks for the 50 mg group. The double‐blind treatment phase was 12 weeks in duration.
The parallel trial run by the Korean Topiramate Study Group (Korean Topiramate Study Group 1999), investigated adults only and consisted of two main treatment arms including 600 mg topiramate (n = 91) and placebo (n = 86). The baseline period was 12 weeks in duration followed by a titration period of 10 weeks and then a stabilisation period of eight weeks.
A multicentre, parallel trial had a 12‐week baseline period followed by an 18‐week double‐blind treatment period (Privitera 1996). Within this phase was a six‐week titration period and 12‐week stabilisation period. Adults were randomised to one of four treatment arms: 48 mg to 600 mg per day topiramate, 48 mg to 800 mg per day topiramate, 47 mg to 1000 mg per day topiramate and 47 were randomised to placebo.
Another multicentre, parallel trial had an eight‐week baseline period and a 19‐week treatment period (Rosenfeld 1996). Two hundred and nine adults were randomised to one of two treatment arms in a ratio of 1:3, resulting in 42 patients being randomised to placebo and 167 patients being randomised to 1000 mg per day topiramate.
Another multicentre, parallel trial had a pre‐randomisation period of eight weeks and a treatment period of 11 weeks (Sharief 1996), where 23 adults were randomised to 400 mg per day topiramate and 24 were randomised to placebo. At the target dose, medication was administered twice daily.
The parallel trial by Tassinari 1996 was a multicentre trial in which adults were randomised to one of two treatments arms: 600 mg topiramate (n = 30) and placebo (n = 30). The baseline period was eight weeks followed by a treatment period of 12 weeks.
One single‐centre, parallel trial in China had a baseline period of eight weeks and a treatment period of 14 weeks (Yen 2000), and randomised 23 adults to 300 mg per day topiramate and 23 to placebo.
One study included elderly patients (aged over 65) only and had two treatment arms (Zhang 2011): 200 mg per day topiramate (n = 46) or placebo (n = 40). The baseline period for this study was eight weeks in duration followed by a titration phase of eight weeks and then a treatment period of 12 weeks.
Excluded studies
We excluded five RCTs for the following reasons: two studies had no placebo control group (Christensen 2003; Ramsay 2008), one study was an active comparator‐controlled trial (Chung 2009), one study had a baseline period of 48 hours (Novotny 2010), and one study did not investigate any of the outcomes under review (Coles 1999). For further information regarding exclusion, see Characteristics of excluded studies.
Studies awaiting classification
We were unable to include a further two studies in the review as we unsure about their eligibility for inclusion (Aranguiz 1991; Kazibutowska 2000). We attempted to contact the authors for additional information to allow for the classification of the studies, but no correspondence had been received at the time of publication. For details regarding these studies, see Characteristics of studies awaiting classification.
Risk of bias in included studies
We allocated each study a rating for each risk of bias domain. All studies included in the review were rated as having either low risk of bias or unclear risk of bias across all domains. Below, the specific domain ratings are explained.
See 'Risk of bias' tables within the Characteristics of included studies tables for further details. See Figure 2 for a summary of the risk of bias in each included study and Figure 3 for a summary of each domain across studies.
2.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
3.
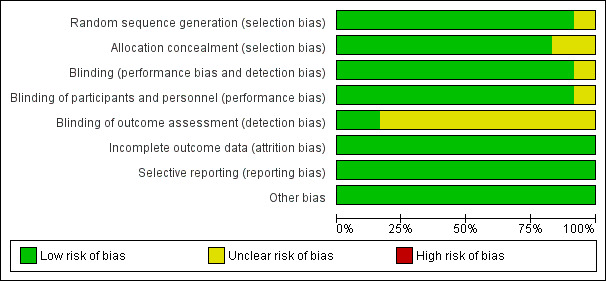
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
For the domain of random sequence generation, we rated 11 studies as low risk of bias due to the use of a computer‐generated randomisation schedule or the use of random number tables/random permuted blocks (Ben‐Menachem 1996; Chung 2014; Elterman 1999; Faught 1996; Korean Topiramate Study Group 1999; Privitera 1996; Rosenfeld 1996; Sharief 1996; Tassinari 1996; Yen 2000; Zhang 2011). We rated one study as unclear due to a lack of detail provided regarding the methods used (Guberman 2002).
In addition, we rated the methods by which allocation was concealed in 10 of the included studies as low risk of bias (Ben‐Menachem 1996; Chung 2014; Elterman 1999; Faught 1996; Korean Topiramate Study Group 1999; Privitera 1996; Rosenfeld 1996; Sharief 1996; Tassinari 1996; Yen 2000). Nine of the studies used sealed, numbered packages, allocated sequentially, to conceal allocation (Ben‐Menachem 1996; Elterman 1999; Faught 1996; Korean Topiramate Study Group 1999; Privitera 1996; Rosenfeld 1996; Sharief 1996; Tassinari 1996; Yen 2000), and one more recent trial used an interactive voice response system (Chung 2014). Two trials did not provide clear methods and we thus rated them as having unclear risk of bias for this domain (Guberman 2002; Zhang 2011).
Blinding
In all but one study, successful blinding of participants was achieved by using identical medication within the topiramate and placebo groups (Ben‐Menachem 1996; Chung 2014; Elterman 1999; Faught 1996; Guberman 2002; Korean Topiramate Study Group 1999; Privitera 1996; Rosenfeld 1996; Sharief 1996; Tassinari 1996; Yen 2000); we judged these 11 studies at low risk of performance bias. There were no details reported by Zhang 2011, and so we rated this study as having unclear risk of bias. The blinding of the outcome assessor was difficult to judge due to the lack of detail in 10 of the publications (Ben‐Menachem 1996; Chung 2014; Elterman 1999; Faught 1996; Guberman 2002; Korean Topiramate Study Group 1999; Privitera 1996; Rosenfeld 1996; Tassinari 1996; Zhang 2011) and, therefore, we rated these studies as having unclear detection bias. We rated the other two studies at low risk of bias for this particular domain (Sharief 1996; Yen 2000), as the two studies specified that outcome assessors remained blinded.
Incomplete outcome data
We rated all studies at low risk of bias for this domain due to the ITT analyses undertaken by the study authors.
Selective reporting
We requested the protocols for all included studies to compare a priori methods and outcomes to the published report but the majority of these were unavailable. We rated all included studies as low risk of bias for this domain as there was no suspicion of selective outcome reporting bias; all expected outcomes were reported in each of the publications.
Other potential sources of bias
We rated all studies at low risk of bias for this domain as we did not detect any further bias in any of the included studies.
Effects of interventions
See: Table 1
See Table 1 for a summary and GRADE assessment of the primary and secondary outcomes.
50% or greater reduction in seizure frequency
Intention‐to‐treat (ITT) analysis
Data from all 12 studies and including 1650 participants contributed to this outcome. A Chi2 test for heterogeneity in a response to topiramate showed significant heterogeneity between trials (Chi² = 18.29, df = 11, P = 0.07, I² = 40%), therefore we employed a random‐effects model. The overall risk ratio (RR) for a response to topiramate compared to placebo using the random‐effects model was 2.71 (95% confidence interval (CI) 2.05 to 3.59; 12 studies, 1650 participants; high‐certainty evidence; Analysis 1.2), indicating that participants receiving add‐on topiramate were nearly three times more likely to have a 50% or greater reduction in seizure frequency than those receiving add‐on placebo.
1.2. Analysis.
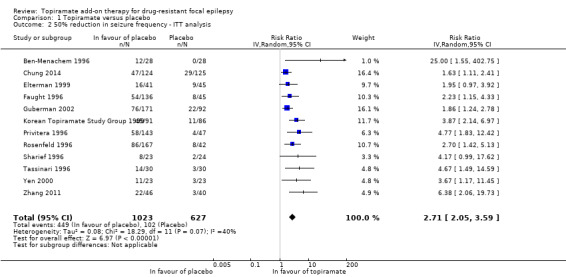
Comparison 1 Topiramate versus placebo, Outcome 2 50% reduction in seizure frequency ‐ ITT analysis.
As a result of the significant heterogeneity detected, we conducted three separate sensitivity analyses. Firstly, we noted that the studies included in the analysis for this outcome varied with regards to the overall risk of bias awarded. We thus completed a sensitivity analysis by study quality by only including studies associated with low risk of bias overall. Following this sensitivity analysis, the RR increased to 4.36 (95% CI 2.24 to 8.50) and there was no longer any detectable statistical heterogeneity (Chi² = 0.13, df = 2, P = 0.94, I² = 0%; Analysis 1.1). We similarly conducted a sensitivity analysis according to patient characteristics. For this, we excluded one study from the analysis because it studied a paediatric study population and all other studies were conducted in adults (Elterman 1999). The exclusion of this study did not resolve the detected heterogeneity (Chi² = 17.94, df = 10, P = 0.06, I² = 44%) and did not greatly impact the estimated RR (RR 2.85, 95% CI 2.09 to 3.88; Analysis 1.3). Finally, we conducted a sensitivity analysis according to intervention, during which we excluded three studies (Chung 2014; Guberman 2002; Zhang 2011). The three excluded studies only assessed the lowest dosage of topiramate (200 mg/d). Again, heterogeneity was no longer detected amongst the data set (Chi² = 7.29, df = 8, P = 0.51, I² = 0%), however, the effect estimate remained statistically significant and was unsurprisingly slightly higher than that detected when all studies were included (RR 3.32, 95% CI 2.51 to 4.38; Analysis 1.4). We did not conduct a sensitivity analysis according to outcome as this was not deemed appropriate.
1.1. Analysis.
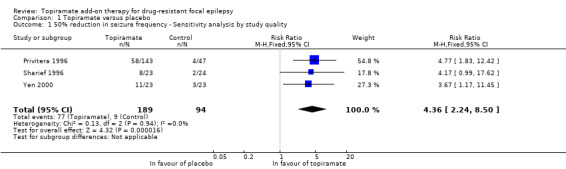
Comparison 1 Topiramate versus placebo, Outcome 1 50% reduction in seizure frequency ‐ Sensitivity analysis by study quality.
1.3. Analysis.
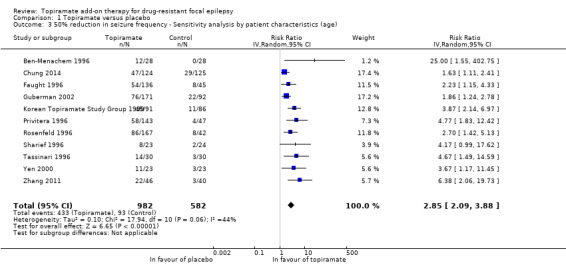
Comparison 1 Topiramate versus placebo, Outcome 3 50% reduction in seizure frequency ‐ Sensitivity analysis by patient characteristics (age).
1.4. Analysis.
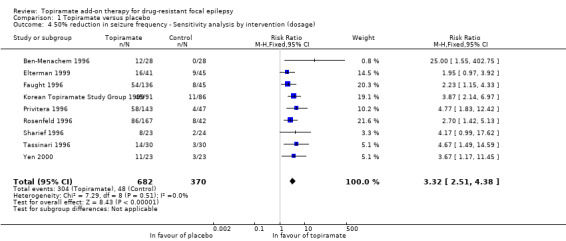
Comparison 1 Topiramate versus placebo, Outcome 4 50% reduction in seizure frequency ‐ Sensitivity analysis by intervention (dosage).
Best‐ and worst‐case scenarios
We again used a random‐effects model for both the best‐case and worst‐case analyses due to the significant heterogeneity detected (Chi² = 20.93, df = 11, P = 0.03, I² = 47%; Analysis 1.5; and Chi² = 18.97, df = 11, P = 0.06, I² = 42%; Analysis 1.6, respectively). The overall best‐case RR scenario for a response to topiramate was RR 3.54 (95% CI 2.67 to 4.69), whilst the worst‐case RR scenario was 1.96 (95% CI 1.54 to 2.50). Importantly, all three analyses for the outcome, 50% or greater seizure reduction, suggest a significant treatment effect for add‐on topiramate compared to add‐on placebo (P < 0.00001).
1.5. Analysis.
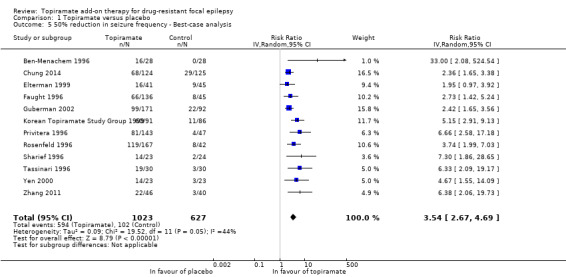
Comparison 1 Topiramate versus placebo, Outcome 5 50% reduction in seizure frequency ‐ Best‐case analysis.
1.6. Analysis.
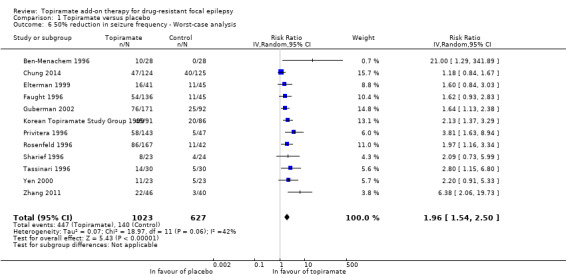
Comparison 1 Topiramate versus placebo, Outcome 6 50% reduction in seizure frequency ‐ Worst‐case analysis.
Dose‐response regression
We fitted a generalised linear mixed model to the data provided in Analysis 1.2 to estimate the effect of dose on the primary outcome, 50% or greater reduction in seizure frequency (details in Data synthesis). To include the data extracted from the study by Elterman 1999, which only recruited children, the daily dose was approximated by assuming an average adult weight of 75 kg, multiplied by the study dose of 6 mg/kg/day, to give 450 mg/day topiramate. It was not necessary to conduct any more dose adjustments for the other included studies. The study by Zhang 2011 only recruited elderly patients, above 65 years of age, and the remaining studies all recruited patients from adult populations. Doses ranged between 200 mg/day and 1000 mg/day topiramate. In order to use this model, the dose was standardised by its standard deviation (352 mg). Importantly, the method used estimated an OR as opposed to a RR.
The OR for a 50% or greater reduction in seizure frequency per 200 mg/d increase in topiramate dose was 1.45 (95% CI 1.28 to 1.64; P < 0.001), with estimated between‐study standard deviation of 0.54 (standard error (SE) 0.16), thus revealing that patients were significantly more likely to attain a 50% or greater seizure reduction if their dose was increased by 200 mg/d (P < 0.001). Notably, the odds of a patient achieving a 50% or greater reduction in seizure frequency were nearly doubled (OR 1.92, 95% CI 1.54 to 2.40; P < 0.001) when their dose of topiramate was increased by 350 mg/d, the standard deviation of the included doses (P < 0.001).
The estimated response rate (the percentage of participants achieving a 50% or greater reduction in seizure frequency) per dose, as well as the estimated increase in response rate compared to placebo for each dose are given in Table 3. The estimated response rate on placebo was 17.8% (95% CI 6.3% to 29.4%), whereas the estimated response rate for topiramate ranged from 37.6% to 51.1%, dependent on dose (Table 3). Accordingly, the estimated response rate per dose compared to placebo ranged from 19.7% for participants randomised to 200 mg/day topiramate up to 33.3% for participants randomised to 1000 mg/day. Both the estimated fitted response rate and the difference in response rate compared to placebo generally increased with dosage, as would be expected from the dose‐response relationship demonstrated.
2. Estimated response rates per dose and percentage difference in responders per dose compared to placebo.
| Dose (mg) | Fitted response rate (%) | 95% CI | Dose (mg) | Difference in response rate from placebo (%) | 95% CI | ||
| Lower | Upper | Lower | Upper | ||||
| Placebo | 17.8 | 6.3 | 29.4 | ||||
| 200 | 37.6 | 20.2 | 55.0 | 200 | 19.7 | ‐1.2 | 40.6 |
| 300 | 38.1 | ‐28.1 | 104.2 | 300 | 20.2 | ‐46.9 | 87.3 |
| 400 | 37.5 | 7.8 | 67.3 | 400 | 19.7 | ‐12.2 | 51.6 |
| 450 | 38.1 | ‐16.1 | 92.4 | 450 | 20.3 | ‐35.2 | 75.8 |
| 600 | 42.2 | 23.5 | 61.0 | 600 | 24.4 | 2.4 | 46.5 |
| 800 | 42.2 | 13.8 | 70.6 | 800 | 24.4 | ‐6.3 | 55.1 |
| 1000 | 51.1 | 29.5 | 72.7 | 1000 | 33.3 | 8.8 | 57.8 |
CI: confidence intervals
Seizure freedom
A Chi2 test for heterogeneity showed no significant heterogeneity between trials (Chi² = 1.84, df = 6, P = 0.93, I² = 0%). The overall RR for seizure freedom was 3.67 (95% CI 1.79 to 7.54; 8 studies, 1177 participants; moderate‐certainty evidence; Analysis 1.7), indicating that participants randomised to add‐on topiramate were over three times more likely to attain seizure freedom than participants randomised to add‐on placebo.
1.7. Analysis.
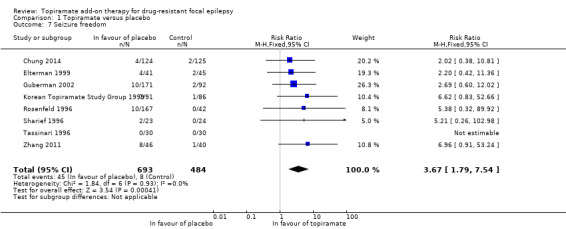
Comparison 1 Topiramate versus placebo, Outcome 7 Seizure freedom.
Treatment withdrawal
Data from 12 studies, consisting of 1650 participants, contributed to this outcome. A Chi2 test for heterogeneity demonstrated no significant statistical heterogeneity (Chi² = 8.76, df = 10, P = 0.55, I² = 0%). The overall RR (95% CI) for withdrawal for any reason was 2.37 (95% CI 1.66 to 3.37; 12 studies, 1650 participants; high‐certainty evidence; Analysis 1.8), thus highlighting that participants were significantly more likely to withdraw from topiramate than placebo.
1.8. Analysis.
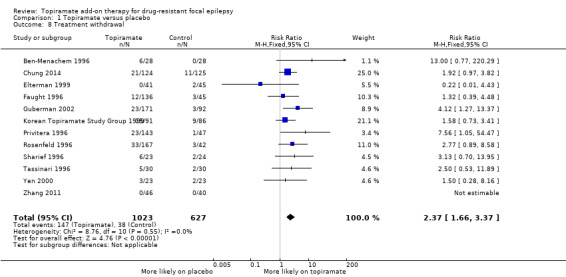
Comparison 1 Topiramate versus placebo, Outcome 8 Treatment withdrawal.
Adverse effects
The RRs for adverse effects were as follows (Analysis 1.9).
1.9. Analysis.
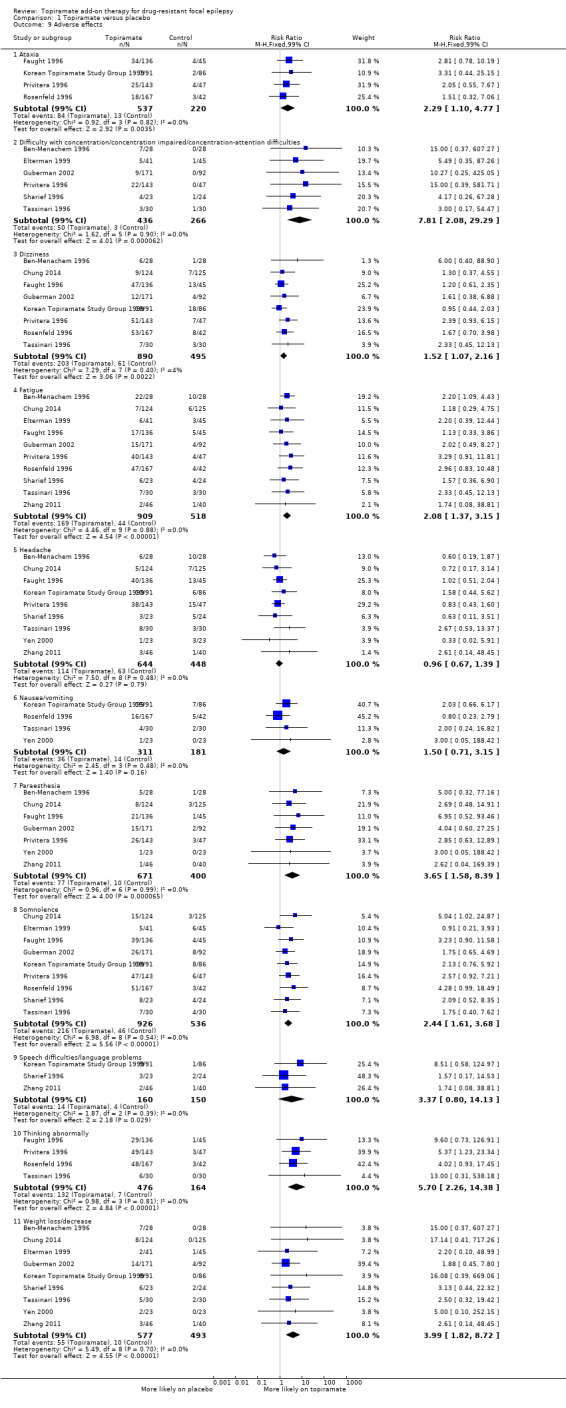
Comparison 1 Topiramate versus placebo, Outcome 9 Adverse effects.
Ataxia: 2.29 (99% CI 1.10 to 4.77; 4 studies; 757 participants)
Concentration difficulties: 7.81 (99% CI 2.08 to 29.29; 6 studies; 702 participants)
Dizziness: 1.52 (99% CI 1.07 to 2.16; 8 studies; 1385 participants)
Fatigue: 2.08 (99% CI 1.37 to 3.15; 9 studies; 1092 participants)
Headache: 0.96 (99% CI 0.67 to 1.39; 10 studies; 1427 participants
Nausea/vomiting: 1.50 (99% CI 0.71 to 3.15; 4 studies; 492 participants)
Paraesthesia: 3.65 (99% CI 1.58 to 8.39; 7 studies; 1071 participants)
Somnolence: 2.44 (99% CI 1.61 to 3.68; 9 studies; 1462 participants)
Speech difficulty: 3.37 (99% CI 0.80 to 14.13; 3 studies; 310 participants)
Thinking abnormally: 5.70 (99% CI 2.26 to 14.38; 4 studies; 640 participants)
Weight loss/decrease: 3.99 (99% CI 1.82 to 8.72; 9 studies; 1070 participants)
Collectively, the results imply that receiving add‐on topiramate is associated with a higher incidence rate for many adverse effects compared to add‐on placebo. Specifically, add‐on topiramate was associated with a significantly higher incidence rate for the following adverse effects: ataxia (P = 0.003), concentration difficulties (P < 0.001), dizziness (P = 0.002), fatigue (P < 0.001) , paraesthesia (P < 0.001), somnolence (P < 0.001), speech difficulty (P = 0.03), 'thinking abnormally' (P < 0.001), and weight loss (P < 0.001), although to differing degrees of effect. For example, the incidence rate for dizziness was increased by a modest 50% when receiving add‐on topiramate, whereas the incidence rate for 'thinking abnormally' was almost six times greater with add‐on topiramate than with placebo. Notably, however, add‐on topiramate did not incur a statistically significant increase in the risk of experiencing headache (RR 0.96, 99% CI 0.67 to 1.39; 9 studies; 1092 participants; P = 0.79) or nausea (RR 1.50, 99% CI 0.71 to 3.15; 4 studies; 492 participants; P = 0.16) compared to placebo.
Discussion
Summary of main results
The 12 trials included in this review were all double‐blinded and the majority expressly used adequate methods of allocation concealment. All analyses were by intention‐to‐treat (ITT). Results from the overall efficacy analysis show that topiramate is effective in reducing seizure frequency by at least 50% when used as an add‐on drug for people with drug‐resistant focal epilepsy. Topiramate was found to be almost three times more effective than a placebo drug. Even the lowest dose, 200 mg per day, results in nearly 20% more patients achieving a 50% reduction in seizure rate than placebo. The dose regression model shows increasing effect with increasing dose, though the increase is quite modest. For people taking 200 mg to 1000 mg per day, results indicate a response rate of between 38% (95% confidence interval (CI) 20.2 to 55.0) and 51% (95% CI 29.5 to 72.7), an improvement over placebo of 20% (CI ‐1.2 to 40.6) to 33% (CI 8.8 to 57.8).
We suspected evidence of publication bias on examination of a funnel plot for the outcome, 50% or greater reduction in seizure frequency, plus we detected statistical heterogeneity in the data set. The heterogeneity could be explained by both study quality (excluding studies associated with unclear risk of bias) and by topiramate dosage. Specifically, statistical heterogeneity was no longer detected when studies which only investigated the lowest dosage of topiramate (200 mg) were excluded from the analysis. This generates further support for the observed dose‐response relationship as it suggests that lower dosage give a significantly different effect estimate compared to higher dosages, such that it generates methodological heterogeneity within the data set.
For seizure freedom, the results estimate that topiramate is over three times more likely to be effective in stopping seizures completely, than a placebo drug. Only half of the included studies reported data on this outcome, however, and, as a result, this finding must be interpreted with caution.
Results for the outcome, treatment withdrawal, showed that participants are significantly more likely to withdraw from treatment with add‐on topiramate than add‐on placebo. In trials of relatively short duration, such as those reviewed here, treatment withdrawal is likely to represent problems with tolerability rather than poor seizure control. With respect to adverse events, most of those investigated were significantly more likely to occur in the topiramate‐treated group, except for headache and nausea and vomiting. The risk ratio (RR) for the significant adverse effects ranged from a low of 1.52 for dizziness to a high of 7.81 for impaired concentration. Although many of the adverse effects were significantly more likely with add‐on topiramate, there was not sufficient information to determine whether this could be attributed to studies using higher doses.
Overall completeness and applicability of evidence
Primarily, this review aimed to determine the efficacy of topiramate as add‐on treatment for people of any age with drug‐resistant focal epilepsy. Markedly, however, the majority of the data were obtained from adults aged 16 to 65 years old. Only one study specifically evaluated the use of add‐on topiramate in children, aged 2 to 16 years (Elterman 1999), and, equally, only one study investigated its use in the elderly (over 65 years old; Zhang 2011). Consequently, the findings presented here cannot be easily generalised to all age groups and are, instead, largely representative of the effect of add‐on topiramate in adults.
Similarly, although the results of this review indicate that topiramate is likely an effective add‐on treatment with regard to seizure reduction, this review cannot tell us how topiramate compares with other antiepileptic drugs in this scenario. This is an extremely important issue for clinicians who are faced with an ever‐increasing number of antiepileptic drugs to choose from. Head‐to‐head trials are needed to provide the evidence that is necessary to enable clinicians to make an evidence‐based choice between antiepileptic drugs and their inclusion should be considered for future review updates.
Moreover, with the possibility of publication bias, we cannot be completely certain that we have identified all placebo‐controlled trials evaluating add‐on topiramate. The suspected publication bias suggests that there could be several small studies, possibly with negative results, which have remained unpublished and that may not have been highlighted by our searches. This generates uncertainty in our finding that add‐on topiramate is more effective than placebo. The implications of this are addressed further in the subsequent subsections.
Additionally, this review focuses on the use of topiramate in drug‐resistant focal epilepsy. The results cannot, therefore, be generalised to add‐on treatment for generalised epilepsies. Likewise, no inference can be made about the effects of topiramate when used as monotherapy.
Certainty of the evidence
Out of the 12 included studies, we rated all but three of the studies as having unclear risk of bias. We rated the other three studies as having low risk of bias. Taking all the studies together, we rated the overall risk of bias as low and considered the evidence to be methodologically sound. The most common issue to generate risk of bias was the blinding of outcome assessors. Nine of the included studies did not provide explicit details on the blinding of outcome assessors, however, given the self‐report aspect of the outcomes measured, i.e. seizure frequency and adverse effects experienced, the blinding of outcome assessors was deemed to have minimal impact on the effect size estimated, hence the overall assessment of low risk of bias.
We employed the GRADE approach to rate the level of certainty of evidence per outcome, the results of which are presented in Table 1. We did not downgrade any of the outcomes for risk of bias, as reasoned above. For the main outcome of 50% or greater seizure reduction, we rated the certainty of evidence as high. We downgraded the evidence for 50% or greater seizure reduction once due to the influence that publication bias may have had on the overall effect estimate. Specifically, a lack of symmetry was noted in the funnel plot for the primary outcome during visual examination (Figure 4). This observation was then supported by the Egger test which detected significant publication bias within the meta‐analysis (P = 0.001). We downgraded the evidence once more due to inconsistency resulting from significant statistical heterogeneity detected within the data set. We then upgraded the evidence once for a large effect size (RR > 2.00), and once again due to the dose‐response relationship demonstrated for the outcome.
4.
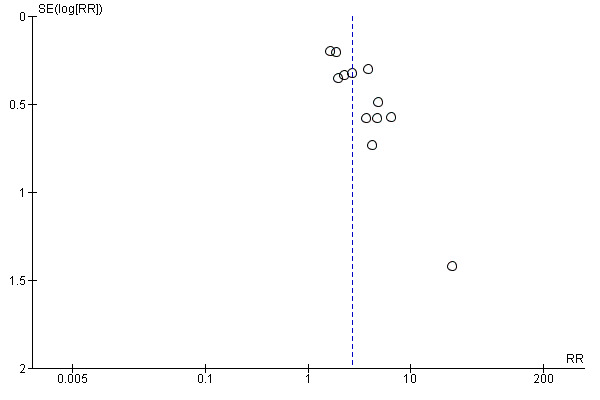
Funnel plot of comparison: 1 Topiramate versus placebo, outcome: 1.2 50% reduction in seizure frequency ‐ ITT analysis.
We judged the evidence for the alternative efficacy outcome, seizure freedom, to be of moderate certainty. We downgraded the evidence twice due to the very low number of events contributing to the meta‐analysis, but then upgraded once because of the large effect size (RR > 2.00) observed. Similarly, we downgraded the certainty of evidence for the adverse effects: weight loss, paraesthesia, and difficulties with concentration, twice because these outcomes, likewise, featured an extremely low number of events (< 100 events). For the outcome, weight loss, we downgraded the evidence once again as a result of suspected publication bias (Egger test: P = 0.009). We then upgraded the evidence for weight loss once due to the large effect size observed, thus producing an overall judgement of low‐certainty of evidence for this outcome. We downgraded the evidence for the outcome, concentration difficulties, once for suspected publication bias (Egger test: P = 0.019) but then upgraded this twice due to the very large effect size recognised (RR > 5.00). This led to an overall judgement of moderate‐certainty of evidence for concentration difficulties. We also judged the evidence for paraesthesia as being of moderate certainty. We did not detect any publication bias in the data set (Egger test: P = 0.478), however, we could only upgrade the evidence once back to moderate certainty as the observed effect size was large, as opposed to very large.
In contrast, we only downgraded the evidence for the other two outcomes, treatment withdrawal and abnormal thinking, once for imprecision. Although the number of events was insufficient to satisfy the optimal information size, there were more events than were noted for the other outcomes (> 100 events). We then upgraded the evidence for both outcomes back to high certainty due to the large (treatment withdrawal) and very large (abnormal thinking) effect size revealed.
Potential biases in the review process
We strongly suspect that publication bias has impacted this review. We specifically suspect publication bias for three of the GRADE‐assessed outcomes (50% or greater seizure reduction, concentration difficulties, and weight loss) after examining the resultant funnel plots and conducting the Egger test. Importantly, publication bias was also suspected for the outcome, seizure freedom, however, the low number of studies contributing data for this outcome prevented us from making a conclusive judgement regarding the funnel plot (Figure 5), and this was reflected in the results of the Egger test (P = 0.09).
5.
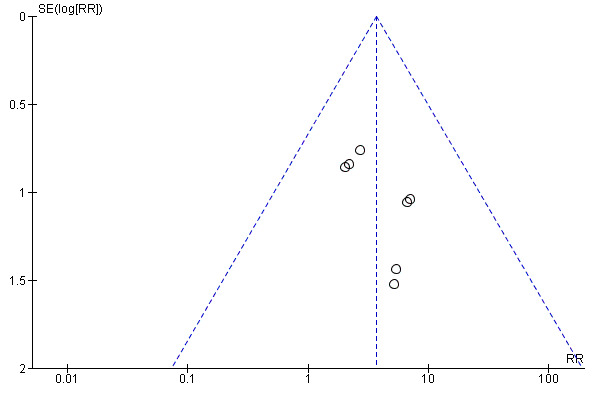
Funnel plot of comparison: 1 Topiramate versus placebo, outcome: 1.7 Seizure freedom.
Studies showing positive results are more likely to be published than those expressing negative results and this consequently leads to an overestimation of the effect size (Murad 2018). It is thus possible that, in the instance of this particular review, we have overestimated the effectiveness of topiramate compared to placebo as an add‐on therapy. Expanding the search terms in future updates could aid us in identifying any additional unpublished data sets to help resolve this issue. Additionally, we could contact more individuals that might have knowledge of any unpublished trials. We do, however, feel that, for the purposes of this current review update, we have exhausted all potential sources of data. This will, however, be reassessed and further pursued for the next review update.
Agreements and disagreements with other studies or reviews
The observations made in this review update are consistent with the previous versions of this review (Jette 2002; Jette 2008; Marson 1996; Marson 1997; Pulman 2014). Specifically, this review has indicated that topiramate is efficacious as an add‐on treatment for drug‐resistant focal epilepsy. This review has, however, also emphasised that treatment with topiramate remains significantly associated with certain adverse effects, including: concentration difficulties, paraesthesia, 'thinking abnormally', and weight loss. As with the earlier versions of this review, we are still unable to extrapolate the results to other types of epilepsy and cannot comment on the long‐term effectiveness or tolerability of topiramate as an add‐on treatment.
The results of this review are also consistent with other reviews (Perucca 1997; Privitera 1997), conducted independently of Cochrane, however, these reviews are now outdated. Notably, Perucca 1997 similarly reported that doses of topiramate between 200 mg/day to 1000 mg/day are effective at managing drug‐resistant focal epilepsy, emphasising that even doses below 400 mg/day are likely to be beneficial. The other review identified (Privitera 1997), likewise noted a two‐ to three‐fold difference in the number of people achieving 50% reduction in seizure frequency when allocated add‐on topiramate compared to placebo. Both reviews also described ataxia, concentration difficulties, paraesthesias, fatigue, dizziness, somnolence and weight loss in their participant‐reported adverse effects (Perucca 1997; Privitera 1997).
Of further significance, two of the trials included in this review explored dose‐response relationships (Faught 1996; Privitera 1996). Faught 1996 considered responses at doses of 200 mg, 400 mg and 600 mg per day, whilst Privitera 1996 evaluated responses at 600 mg, 800 mg and 1000 mg per day. Results from Faught 1996 suggest that 400 mg/day topiramate increases the likelihood of a response compared to a 200 mg/day dose, but that no further increase is obtainable at doses of 600 mg/day. Results from Privitera 1996 also suggest little difference in response rates between doses of 600 mg/day to 1000 mg/day. Although neither study observed an increased response rate with doses in excess of 600 mg/day, this current review successfully demonstrated a significant dose‐response relationship whilst including all doses from 200 mg/day to 1000 mg/day. Importantly, although the response rates were very similar for 600 mg/day and 800 mg/day, the response rate continued to increase again when participants were randomised to the highest dose, 1000 mg/day, contrary to the reports in these two studies.
Authors' conclusions
Implications for practice.
Topiramate has efficacy as an add‐on treatment for people with drug‐resistant focal epilepsy. A daily dose of 200 mg is the lowest dose tested in the trials included in this review, and this would seem a reasonable starting dose. The dose regression model implied additional benefit with increased dosage, however, the increase was fairly modest. It is likely that higher doses will result in greater issues with adverse effects.
Implications for research.
To evaluate further the place of topiramate in the armamentarium of available antiepileptic drugs, further studies are required addressing the following.
The long‐term effects of add‐on topiramate.
How topiramate compares with other add‐on treatments in drug‐resistant focal epilepsy.
The role of topiramate in childhood epilepsies.
How topiramate compares with standard antiepileptic drugs, such as monotherapy for focal or generalised epilepsies.
The dose‐response relationship with adverse effects.
What's new
| Date | Event | Description |
|---|---|---|
| 2 July 2018 | New citation required but conclusions have not changed | Conclusions are unchanged. |
| 2 July 2018 | New search has been performed | Searches updated 2 July 2018; one new study has been included (Chung 2014). |
History
Protocol first published: Issue 1, 1999 Review first published: Issue 3, 1999
| Date | Event | Description |
|---|---|---|
| 18 June 2013 | New citation required but conclusions have not changed | One new study added to the review; authors of review changed. |
| 18 June 2013 | New search has been performed | Search re‐run to include head‐to‐head trials; new 'Summary of findings' table; methods sections updated. |
| 31 August 2012 | New citation required but conclusions have not changed | Updated search, added additional study and re‐ran analyses including additional side effect analyses. Additional figure to better demonstrate the dose‐response analysis. Completed 'Risk of bias' assessment for all included studies. Completed 'Summary of findings' table. Studies added to excluded studies. MEDLINE search strategy included. |
| 24 October 2008 | Amended | Search strategy amended to comply with RevMan 5 format. |
| 24 April 2008 | Amended | Converted to new review format. |
| 24 April 2008 | New search has been performed | We re‐ran our searches on 10 May 2007 ‐ several potentially relevant studies were found. |
| 24 April 2008 | New citation required but conclusions have not changed | New studies have been added to the 'included', 'excluded' and 'ongoing assessment' sections. Analysis has been re‐run and the text of the study has been updated to take into account the newly included studies. |
| 22 April 2002 | New citation required and conclusions have changed | Substantive amendment. |
Acknowledgements
We would like to acknowledge Zak Kadir for the contribution he made to the initial review.
We would like to acknowledge Sarah Nevitt for her statistical support for the current review update.
We would like to acknowledge Jonathan Dykeman, Karla Hemming, and Jennifer Pulman (previously Jennifer Weston) for their contributions to the previous versions of this review.
Appendices
Appendix 1. CRS Web search strategy
1. Topiram* or Tipiram* or Topamax or TPM or Qudexy AND CENTRAL:TARGET
2. (monotherap* not (adjunct* or "add‐on" or "add on" or adjuvant* or combination* or polytherap*)):TI AND CENTRAL:TARGET
3. #1 NOT #2
4. MESH DESCRIPTOR Epilepsy EXPLODE ALL AND CENTRAL:TARGET
5. MESH DESCRIPTOR Seizures EXPLODE ALL AND CENTRAL:TARGET
6. (epilep* OR seizure* OR convuls*):AB,KW,MC,MH,TI AND CENTRAL:TARGET
7. #4 OR #5 OR #6 AND CENTRAL:TARGET
8. #7 AND #3
Appendix 2. MEDLINE search strategy
This strategy is based on the Cochrane Highly Sensitive Search Strategy for identifying randomized trials (Lefebvre 2011).
1. (Topiram$ or Tipiram$ or Topamax or TPM or Qudexy).tw.
2. exp Epilepsy/
3. exp Seizures/
4. (epilep$ or seizure$ or convuls$).tw.
5. 2 or 3 or 4
6. exp *Pre‐Eclampsia/ or exp *Eclampsia/
7. 5 not 6
8. (randomized controlled trial or controlled clinical trial or pragmatic clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
9. clinical trials as topic.sh.
10. trial.ti.
11. 8 or 9 or 10
12. exp animals/ not humans.sh.
13. 11 not 12
14. 1 and 7 and 13
15. (monotherap$ not (adjunct$ or "add‐on" or "add on" or adjuvant$ or combination$ or polytherap$)).ti.
16. 14 not 15
17. remove duplicates from 16
Appendix 3. ClinicalTrials.gov search strategy
Interventional Studies | Epilepsies, Partial | Topiramate OR Topamax or TPM or Qudexy
Appendix 4. WHO International Clinical Trials Registry Platform (ICTRP) search strategy
partial epilepsy OR focal epilepsy in the Condition AND
Topiramate OR Topamax OR TPM OR Qudexy in the Intervention
Appendix 5. SCOPUS search strategy
((TITLE(Topiramate or Topamax) OR ABS(Topiramate or Topamax)) AND (TITLE((randomiz* OR randomis* OR controlled OR placebo OR blind* OR unblind* OR "parallel group" OR crossover OR "cross over" OR cluster OR "head to head") PRE/2 (trial OR method OR procedure OR study)) OR ABS((randomiz* OR randomis* OR controlled OR placebo OR blind* OR unblind* OR "parallel group" OR crossover OR "cross over" OR cluster OR "head to head") PRE/2 (trial OR method OR procedure OR study))) AND ((TITLE‐ABS‐KEY(epilep* OR "infantile spasm" OR seizure OR convuls* OR (syndrome W/2 (aicardi OR angelman OR doose OR dravet OR janz OR jeavons OR "landau kleffner" OR "lennox gastaut" OR ohtahara OR panayiotopoulos OR rasmussen OR rett OR "sturge weber" OR tassinari OR "unverricht lundborg" OR west)) OR "ring chromosome 20" OR "R20" OR "myoclonic encephalopathy" OR "pyridoxine dependency") AND NOT (TITLE(*eclampsia) OR INDEXTERMS(*eclampsia))) OR (TITLE‐ABS‐KEY(lafora* W/4 (disease OR epilep*)) AND NOT (TITLE(dog OR canine) OR INDEXTERMS(dog OR canine))))) AND NOT (TITLE(monotherap* AND NOT (adjunct* OR "add‐on" OR "add on" OR adjuvant* OR combination* OR polytherap*)))
Data and analyses
Comparison 1. Topiramate versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 50% reduction in seizure frequency ‐ Sensitivity analysis by study quality | 3 | 283 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.36 [2.24, 8.50] |
| 2 50% reduction in seizure frequency ‐ ITT analysis | 12 | 1650 | Risk Ratio (IV, Random, 95% CI) | 2.71 [2.05, 3.59] |
| 3 50% reduction in seizure frequency ‐ Sensitivity analysis by patient characteristics (age) | 11 | 1564 | Risk Ratio (IV, Random, 95% CI) | 2.85 [2.09, 3.88] |
| 4 50% reduction in seizure frequency ‐ Sensitivity analysis by intervention (dosage) | 9 | 1052 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.32 [2.51, 4.38] |
| 5 50% reduction in seizure frequency ‐ Best‐case analysis | 12 | 1650 | Risk Ratio (IV, Random, 95% CI) | 3.54 [2.67, 4.69] |
| 6 50% reduction in seizure frequency ‐ Worst‐case analysis | 12 | 1650 | Risk Ratio (IV, Random, 95% CI) | 1.96 [1.54, 2.50] |
| 7 Seizure freedom | 8 | 1177 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.67 [1.79, 7.54] |
| 8 Treatment withdrawal | 12 | 1650 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.37 [1.66, 3.37] |
| 9 Adverse effects | 12 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 9.1 Ataxia | 4 | 757 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.29 [1.10, 4.77] |
| 9.2 Difficulty with concentration/concentration impaired/concentration‐attention difficulties | 6 | 702 | Risk Ratio (M‐H, Fixed, 99% CI) | 7.81 [2.08, 29.29] |
| 9.3 Dizziness | 8 | 1385 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.52 [1.07, 2.16] |
| 9.4 Fatigue | 10 | 1427 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.08 [1.37, 3.15] |
| 9.5 Headache | 9 | 1092 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.96 [0.67, 1.39] |
| 9.6 Nausea/vomiting | 4 | 492 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.50 [0.71, 3.15] |
| 9.7 Paraesthesia | 7 | 1071 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.65 [1.58, 8.39] |
| 9.8 Somnolence | 9 | 1462 | Risk Ratio (M‐H, Fixed, 99% CI) | 2.44 [1.61, 3.68] |
| 9.9 Speech difficulties/language problems | 3 | 310 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.37 [0.80, 14.13] |
| 9.10 Thinking abnormally | 4 | 640 | Risk Ratio (M‐H, Fixed, 99% CI) | 5.70 [2.26, 14.38] |
| 9.11 Weight loss/decrease | 9 | 1070 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.99 [1.82, 8.72] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ben‐Menachem 1996.
| Methods | Double‐blind, placebo‐controlled, parallel‐group study 2 treatment arms: 1 placebo, 1 topiramate Pre‐randomisation baseline period: 8 weeks Treatment period: 13 weeks | |
| Participants | A multicentre study (Sweden, Norway, Denmark, Germany)
56 people randomised (all with drug‐resistant focal epilepsy): 28 to placebo and 28 to 800 mg topiramate Age range: 18 to 65 years Mean age: 37.2 years 84% males Other AEDs: 2 or fewer Median baseline monthly seizure frequency: 11.4 for placebo group, 14.2 for topiramate group |
|
| Interventions | 800 mg topiramate per day or placebo | |
| Outcomes |
|
|
| Notes | Trial sponsored by Johnson & Johnson | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "computer‐generated randomization schedule" Comment: random permuted blocks |
| Allocation concealment (selection bias) | Low risk | Comment: sealed, numbered packages allocated sequentially |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: identical tablets and packaging |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: it is likely that blinding was maintained due to the methods used |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: specific details of outcome assessment blinding not provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: all participants were included in the analysis Quote: "intention‐to‐treat analysis" |
| Selective reporting (reporting bias) | Low risk | Comment: protocol unavailable, but appears all expected and prespecified outcomes are reported |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias |
Chung 2014.
| Methods | Double‐blind, placebo‐controlled, parallel‐group study 2 treatment arms: 1 placebo, 1 topiramate Pre‐randomisation baseline period: 8 weeks Treatment period: 11 weeks | |
| Participants | A multicentre study (Argentina, Australia, Belgium, Canada, Chile, Germany, Greece, Hungary, India, Israel, New Zealand, Poland, Russia, South Africa, Spain, and USA)
249 people randomised (all with drug‐resistant focal epilepsy): 125 to placebo and 124 mg to 200 mg per day topiramate Age range: 18 to 75 years Mean age: 37.6 years 53% males Other AEDs: 1 or more Median baseline seizure frequency/week: 2.7 for placebo group, 2.3 for topiramate group |
|
| Interventions | 200 mg topiramate per day or placebo | |
| Outcomes | Primary outcome: median percentage reduction in weekly partial onset seizure frequency Secondary outcomes:
|
|
| Notes | Trial sponsored by Upsher‐Smith Laboratories, Inc | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization by an independent statistician was generated using permuted blocks with a block size of 4 without stratification" |
| Allocation concealment (selection bias) | Low risk | Quote: "The interactive voice response group programmed the randomization schedule for investigators to dispense study drug." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "matching placebo" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: it is likely that blinding was maintained |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: specific details of outcome assessment blinding not provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no concerns about missing data Quote: "Efficacy analyses were performed using the intent‐to‐treat (ITT) population" |
| Selective reporting (reporting bias) | Low risk | Comment: protocol unavailable, but appears all expected and prespecified outcomes are reported |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias |
Elterman 1999.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study 2 treatment arms: 1 placebo, 1 topiramate Pre‐randomisation baseline period: 8 weeks Treatment period: 16 weeks | |
| Participants | A multicentre study (USA, Costa Rica)
86 people were randomised (all with drug‐resistant focal epilepsy): 45 to placebo and 41 to topiramate
Age range: 2 to 16 years Mean age: 9.0 years for placebo group, 8.8 years for topiramate group 56% males Other AEDs: 2 or fewer, except for person who was on more than 2 AEDs Median baseline monthly seizure frequency: 19 for placebo group, 22 for topiramate group (focal‐onset seizures) |
|
| Interventions | 6 mg/kg/day topiramate or placebo | |
| Outcomes | Primary outcome: median percentage reduction in average monthly focal‐onset seizure rate Secondary outcomes:
|
|
| Notes | Trial sponsored by Johnson & Johnson | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: random permuted blocks |
| Allocation concealment (selection bias) | Low risk | Comment: sealed, numbered packages allocated sequentially |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "tablets that were similar in shape, size and color" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: it is likely that blinding was maintained |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: specific details of outcome assessment blinding not provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: all participants were included in the analysis Quote: "intention‐to‐treat basis" |
| Selective reporting (reporting bias) | Low risk | Comment: protocol unavailable, but appears all expected and prespecified outcomes are reported |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias |
Faught 1996.
| Methods | Double‐blind, placebo‐controlled, parallel‐group study 4 treatment arms: 1 placebo, 3 topiramate Pre‐randomisation baseline period: 12 weeks Treatment period: 16 weeks | |
| Participants | A multicentre study (USA)
181 people were randomised (all with drug‐resistant focal epilepsy): 45 to placebo, 45 mg to 200 mg per day topiramate, 45 mg to 400 mg per day topiramate, 46 mg to 600 mg per day topiramate
Age range: 19 to 68 years Mean age: 36.9 years Other AEDs: 2 or fewer 70% males Median baseline monthly seizure frequency: 10.8 (10.0 for placebo group, 11.5 for 200 mg/d topiramate group, 11.0 for 400 mg/d topiramate group, 11.2 for 600 mg/d topiramate group) |
|
| Interventions | 200 mg topiramate per day or 400 mg topiramate per day or 600 mg topiramate per day or placebo | |
| Outcomes |
|
|
| Notes | Trial sponsored by Johnson & Johnson | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: random permuted blocks |
| Allocation concealment (selection bias) | Low risk | Comment: sealed, numbered packages allocated sequentially |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: identical tablets and packaging |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: it is likely that blinding was maintained |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: specific details of outcome assessment blinding not provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no concerns about missing data Quote: "intention‐to‐treat efficacy analysis" |
| Selective reporting (reporting bias) | Low risk | Comment: protocol unavailable, but appears all expected and prespecified outcomes are reported |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias |
Guberman 2002.
| Methods | Randomised, double‐blind, placebo‐controlled study 3 treatment arms: 1 placebo, 2 topiramate Pre‐randomisation baseline period: 4 weeks Treatment period: 12 weeks |
|
| Participants | A multicentre study (Hungary, Poland, Israel, Canada, Russia, Czech Republic)
263 people were randomised (all with drug‐resistant focal epilepsy): 91 to placebo, 85 to topiramate escalated weekly by 25 mg/day to 200 mg/day, 83 to topiramate escalated weekly by 50 mg/day to 200 mg/day
Age range: 18 to 67 years for placebo group, 18 to 64 years for topiramate groups
Mean age: 36 years for placebo group, 37 years for topiramate groups 50% males for placebo group, 46% males for topiramate groups Other AEDs: 2 or fewer Median baseline monthly seizure frequency: 7 for placebo group, 7 for topiramate group |
|
| Interventions | 200 mg topiramate per day escalated over 8 weeks by 25 mg/day increases weekly or 200 mg topiramate per day escalated over 4 weeks by 50 mg/day increases weekly or placebo | |
| Outcomes | Primary outcome: medial % reduction from baseline in monthly focal‐onset seizure frequency for the combined topiramate groups versus placebo group Secondary outcomes:
|
|
| Notes | Trial sponsored by Johnson & Johnson | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized in equal proportions" Comment: insufficient information provided to determine judgement for sequence generation |
| Allocation concealment (selection bias) | Unclear risk | Comment: details regarding allocation concealment were not provided |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: identical tablets and packaging |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: it is likely that blinding was maintained |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: specific details of outcome assessment blinding not provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no concerns about missing data Quote: "intention‐to‐treat" |
| Selective reporting (reporting bias) | Low risk | Comment: protocol unavailable, but appears all expected and prespecified outcomes are reported |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias |
Korean Topiramate Study Group 1999.
| Methods | Randomised, double ‐blind, placebo ‐controlled study 2 treatment arms: 1 placebo, 1 topiramate Pre‐randomisation baseline period: 12 weeks Treatment period: 18 weeks | |
| Participants | A multicentre study (Korea)
177 people were randomised (all with drug‐resistant focal epilepsy): 86 to placebo, 91 to topiramate
Age range: 16 to 65 years Mean age: 29.8 years for placebo group, 29.6 years for topiramate group 54% males Other AEDs: 2 or fewer Median baseline monthly seizure frequency: 5.6 for placebo group, 5.6 for topiramate group |
|
| Interventions | 600 mg topiramate per day or placebo | |
| Outcomes |
|
|
| Notes | Trial sponsored by Janssen Korea Ltd | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: random number tables with permuted blocks of 4 |
| Allocation concealment (selection bias) | Low risk | Comment: sealed, numbered packages allocated sequentially |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: identical tablets and packaging |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: it is likely that blinding was maintained |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: specific details of outcome assessment blinding not provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: 2 patients in the topiramate group and 1 in the placebo group were excluded from the analysis. It was determined that this did not represent a potentially clinically important impact Quote: "intention‐to‐treat analysis (ITTA) was performed for efficacy and safety measures" |
| Selective reporting (reporting bias) | Low risk | Comment: protocol unavailable, but appears all expected and prespecified outcomes are reported |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias |
Privitera 1996.
| Methods | Double‐blind, parallel‐group study 4 treatment arms: 1 placebo, 3 topiramate Pre‐randomisation baseline period: 12 weeks Treatment period: 18 weeks | |
| Participants | A multicentre study (USA)
190 people were randomised (all with drug‐resistant focal epilepsy): 47 to placebo, 48 to 600 mg topiramate, 48 to 800 mg topiramate, 47 to 1000 mg topiramate
Age range: 18 to 68 years Mean age: 35.5 years 80% males Other AEDs: 2 or fewer Median baseline monthly seizure frequency: 11 (9.3 for placebo group, 10.0 for 600 mg/d topiramate group, 16.2 for 800 mg/d topiramate group, 11.7 for 1000 mg/d topiramate group) |
|
| Interventions | 600 mg topiramate per day or 800 mg topiramate per day or 1000 mg topiramate per day or placebo | |
| Outcomes |
|
|
| Notes | Trial sponsored by Johnson & Johnson | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: random permuted blocks |
| Allocation concealment (selection bias) | Low risk | Comment: sealed, numbered packages allocated sequentially |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "topiramate and placebo tablets were identical in appearance and packaging" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: it is likely that blinding was maintained |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: specific details of outcome assessment blinding not provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "all 209 subjects who enrolled in the double‐blind phase of the trial were included in the efficacy analysis" |
| Selective reporting (reporting bias) | Low risk | Comment: data published in full according to the protocol |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias |
Rosenfeld 1996.
| Methods | Double‐blind, placebo‐controlled, parallel‐group study 2 treatment arms: 1 placebo, 1 topiramate Prospective pre‐randomisation baseline period: 8 weeks Treatment period: 19 weeks | |
| Participants | A multicentre study (USA)
209 people were randomised (all with drug‐resistant focal epilepsy): 42 to placebo, 167 to 1000 mg topiramate
Age range 18 to 65 years Mean age: Unknown 49% male Other AEDs: 1 Baseline seizure frequency (unknown). Patients had to have a minimum of 6 focal seizures during the 8‐week baseline phase |
|
| Interventions | 1000 mg topiramate per day or placebo | |
| Outcomes | Proportion with a 50% reduction in seizure frequency | |
| Notes | Limited information regarding trial. All information was provided by a single poster abstract. Trial sponsored by Johnson & Johnson |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: random permuted blocks |
| Allocation concealment (selection bias) | Low risk | Comment: sealed, numbered packages allocated sequentially |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "tablets were identical in appearance and packaging" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: only states "double‐blinded", and so blinding is likely though not confirmed |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: specific details of outcome assessment blinding not provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no concerns about missing data Quote: "intention‐to‐treat" |
| Selective reporting (reporting bias) | Low risk | Comment: appears all expected and prespecified outcomes are reported |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias |
Sharief 1996.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study 2 treatment arms: 1 placebo, 1 topiramate Pre‐randomisation baseline period: 8 weeks Treatment period: 11 weeks | |
| Participants | A multicentre study (Sweden, Spain, UK and France)
47 people were randomised (all with drug‐resistant focal epilepsy): 24 to placebo, 23 to 400 mg topiramate
Age range: 18 to 65 years Mean age: 34 years 85% males Other AEDs: 2 or fewer Median baseline monthly seizure frequency: 12.5 (10 for placebo group, 18 for topiramate group) |
|
| Interventions | 400 mg topiramate per day or placebo | |
| Outcomes |
|
|
| Notes | Trial sponsored by Johnson & Johnson | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: random permuted blocks Quote: "computer‐generated randomization schedule" |
| Allocation concealment (selection bias) | Low risk | Comment: sealed, numbered packages allocated sequentially |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: identical tablets and packaging |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: patients and clinical staff blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: investigators blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no participants were excluded from the analysis and intention‐to‐treat was followed |
| Selective reporting (reporting bias) | Low risk | Comment: protocol unavailable, but appears all expected and prespecified outcomes are reported |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias |
Tassinari 1996.
| Methods | Double‐blind, placebo‐controlled, parallel‐group study 2 treatment arms: 1 placebo, 1 topiramate Pre‐randomisation baseline period: 8 weeks Treatment period: 12 weeks | |
| Participants | A multicentre study (UK, Italy, France, Norway and Denmark)
60 people were randomised (all with drug‐resistant focal epilepsy): 30 to placebo 30 to 600 mg topiramate
Age range: 18 to 65 years Mean age: 32.9 years 68% males Other AEDs: 2 or fewer Median baseline monthly seizure frequency: 15.0 for placebo group, 16.8 for topiramate group |
|
| Interventions | 600 mg topiramate per day or placebo | |
| Outcomes |
|
|
| Notes | Trial sponsored by Johnson & Johnson | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: random permuted blocks |
| Allocation concealment (selection bias) | Low risk | Comment: sealed, numbered packages allocated sequentially |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "topiramate and placebo tablets were identical" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: it is likely that blinding was maintained |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: specific details of outcome assessment blinding not provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no concerns about missing data Quote: "intent‐to‐treat" |
| Selective reporting (reporting bias) | Low risk | Comment: protocol unavailable, but appears all expected and prespecified outcomes are reported |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias |
Yen 2000.
| Methods | Double‐blind, placebo‐controlled, parallel‐group 2 treatment arms: 1 placebo, 1 topiramate Pre‐randomisation baseline period: 8 weeks Treatment period: 14 weeks | |
| Participants | A single‐centre study (China)
46 people were randomised (all with drug‐resistant focal epilepsy): 23 to placebo, 23 to topiramate
Age range: 18 to 54 years Mean age: 32.0 years for placebo group, 31.4 years for topiramate group 41% males Other AEDs: up to 4 or more Median baseline monthly seizure frequency: 5 to 10 for placebo group, < 5 for topiramate group |
|
| Interventions | 300 mg topiramate per day or placebo | |
| Outcomes |
|
|
| Notes | Trial sponsored in part by grants from Taipei Veterans General Hospital (88‐V229) and the Yen Tjing Ling Medical Foundation | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: random permuted blocks Quote: "computer‐generated randomization schedule" |
| Allocation concealment (selection bias) | Low risk | Comment: sealed, numbered packages allocated sequentially |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "matching placebo tablets" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "investigators, patients, study monitors and observers remained blinded to codes" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "investigators, patients, study monitors and observers remained blinded to codes" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "intention‐to‐treat efficacy analysis" Comment: no concerns about missing data |
| Selective reporting (reporting bias) | Low risk | Comment: protocol unavailable, but appears all expected and prespecified outcomes are reported |
| Other bias | Low risk | Comment: the study appeared to be free of other sources of bias |
Zhang 2011.
| Methods | Randomised, placebo‐controlled trial 2 treatment arms: 1 placebo, 1 topiramate Pre‐randomisation baseline period: 8 weeks Treatment period: 12 weeks |
|
| Participants | A single‐centre study (Shanghai, China) 86 participants were randomised (documented drug‐resistant focal epilepsy) Age range: > 65 years old eligible Mean age: 73.4 years 57% males 8 patients on 1 AED, 37 on 2 AEDs, 41 on 3 AEDs Median baseline monthly seizure frequency: 17.3 for placebo group, 16.9 for topiramate group |
|
| Interventions | 200 mg per day topiramate or placebo | |
| Outcomes |
|
|
| Notes | None No trial sponsorship information provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "were randomly assigned, according to a computer‐generated randomization schedule" |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Comment: no information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: no information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no dropouts (data available for all patients) |
| Selective reporting (reporting bias) | Low risk | Comment: protocol unavailable, but appears all expected and prespecified outcomes are reported |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias |
AED: antiepileptic drug
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Christensen 2003 | Alternative dose‐controlled study, no placebo control group |
| Chung 2009 | Active comparator‐controlled study, no placebo control group |
| Coles 1999 | Study did not investigate outcomes of interest: 50% or greater reduction in seizure frequency, treatment withdrawal or adverse effects. Studied seizure severity as outcome using seizure severity scales |
| Novotny 2010 | Baseline period too short (48 hours) |
| Ramsay 2008 | Alternative dose‐controlled study, no placebo control group |
Characteristics of studies awaiting assessment [ordered by study ID]
Aranguiz 1991.
| Methods | A double‐blind, parallel, placebo‐controlled study |
| Participants | Patients with drug‐resistant focal seizures |
| Interventions | Investigated oral doses of topiramate 100 mg, 200 mg and 300 mg twice daily |
| Outcomes | Not known |
| Notes | None |
Kazibutowska 2000.
| Methods | RCT |
| Participants | 45 patients aged 12 to 51 with simple focal seizures or complex focal seizures, with or without secondary generalisation receiving 1 to 4 AEDs |
| Interventions | Added tiagabine in 19 patients, gabapentin in 10 patients and topiramate in 16 patients to the previous treatment |
| Outcomes | Efficacy was evaluated as: seizure‐free, improvement of at least 50%, no improvement, worsening. Results of treatment were as follows.
|
| Notes | None |
AED: antiepileptic drug RCT: randomised controlled trial
Differences between protocol and review
In keeping with the previous review update, we used Mantel‐Haenszel risk ratio (RR) as the preferred estimator of treatment effect. This is in contrast to the published protocol which stated that Peto's odds ratio (OR) would be the preferred estimator. Peto's OR is recommended for use when the event rate is very low and therefore it was not necessary to use this estimator in this review.
In accordance with the latest classification of epilepsies published by the International League Against Epilepsy (ILAE) (Scheffer 2017), the title of this review has been changed from 'Topiramate add‐on for drug‐resistant partial epilepsy' (registered for the original review and protocol) to 'Topiramate add‐on therapy for drug‐resistant focal epilepsy' (for the current update). Any reference to "partial epilepsy" or "refractory epilepsy" throughout this review has been changed to "focal epilepsy" and "drug‐resistant epilepsy", respectively.
Contributions of authors
Rebecca Bresnahan was responsible for the current update of this review and assessed studies for eligibility, updated the methods and assessed risk of bias.
Juliet Hounsome extracted data and assessed risk of bias for the current update of this review.
Tony Marson acted as third review author and resolved any disagreements during the review process.
Jane L Hutton and Nathalie Jette contributed to the previous versions of this review and/or to the original protocol.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK.
This review update was supported by the National Institute for Health Research, via Cochrane Programme Grant funding to the Epilepsy Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service (NHS), or the Department of Health and Social Care.
Declarations of interest
RB: none known JH: none known NJ: receives grant funding paid to her institution for grants unrelated to this work from NINDS (NIH U24NS107201, NIH IU54NS100064), PCORI and Alberta Health. She also receives an honorarium for her work as an Associate Editor of Epilepsia. JLH: none known AGM: a consortium of pharmaceutical companies (GSK, EISAI, UCB Pharma) funded the National Audit of Seizure Management in Hospitals (NASH) through grants paid to the University of Liverpool. Professor Tony Marson is part funded by National Institute for Health Research Collaboration for Leadership in Applied Health Research and Care North West Coast (NIHR CLAHRC NWC).
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Ben‐Menachem 1996 {published data only}
- Ben‐Menachem E, Dam M, Henriksen O, Schmidt D. Double‐blind, placebo‐controlled trial of 800 mg/day topiramate as add‐on therapy in patients with refractory partial epilepsy. Epilepsia 1995;36(Suppl 3):S150. [DOI] [PubMed] [Google Scholar]
- Ben‐Menachem E, Dam M, Mikkelsen M, Engelskjon T, Henriksen O, Johannessen S, et al. Topiramate add‐on treatment in patients with intractable partial epilepsy: a multicenter study. Epilepsia 1993;34(Suppl 2):109. [Google Scholar]
- Ben‐Menachem E, Henriksen O, Dam M, Mikkelsen M, Schmidt D, Reid S, et al. Double‐blind, placebo‐controlled trial of topiramate as add‐on therapy in patients with refractory partial seizures. Epilepsia 1996;37(6):539‐43. [PUBMED: 8641230] [DOI] [PubMed] [Google Scholar]
- Martinez‐Lage J, Ben‐Menachem E, Shorvon SD, Weber M. Double‐blind, placebo‐controlled trial of 400 mg/day topiramate as add‐on therapy in patients with refractory partial epilepsy. Epilepsia 1995;36(Suppl 3):S149‐50. [Google Scholar]
- Ostergard L, Dam M, Mikkelsen M. Topiramate as add‐on therapy in refractory partial epilepsy. Epilepsia 1992;33(Suppl 3):105. [Google Scholar]
Chung 2014 {published data only}
- Arnold S, Blatt I, Clark AM, Halvorsen MB, Nagaraddi VN. USL255, a once‐daily, extended‐release topiramate, has positive effects on clinical outcomes and quality of life: results from the phase 3 PREVAIL clinical trial. Epilepsy Currents 2014;14(Suppl 1):105, Abstract no: 1.223. [Google Scholar]
- Blatt I, Chung SS, Hogan RE, Clark A, Anders B, Halvorsen M. Efficacy and safety of USL255, once‐daily extended‐release topiramate, in adults with partial onset seizures: the PREVAIL study. Epilepsia 2014;55(Suppl 2):42‐43, Abstract no: p112. [Google Scholar]
- Blatt I, Nagaraddi V, Hogan R, Arnold S, Lawson B, Anders B, et al. Efficacy of USL255 across partial‐onset seizure types and refractory patient status: subgroup analyses from the PREVAIL study. Neurology 2014;82(10 Suppl):Abstract no: P3.263. [Google Scholar]
- Blatt I, Nagaraddi VN, Anders B, Clark AM, Halvorsen MB, Hogan RE. USL255 is efficacious across all partial‐onset seizure types and with a variety of concomitant antiepileptic drugs: Results from subgroup analyses of the phase 3 PREVAIL clinical trial. Epilepsy Currents 2014;14(Suppl 1):204, Abstract no: 2.116. [Google Scholar]
- Chung S. USL255 extended‐release topiramate for the treatment of epilepsy. Expert Review of Neurotherapeutics 2014;14(10):1127‐37. [PUBMED: 25220748] [DOI] [PubMed] [Google Scholar]
- Chung SS. A review of the efficacy and safety of extended‐release topiramate in the adjunctive treatment for refractory partial‐onset seizures. Therapeutic Advances in Neurological Disorders 2015;8(3):131‐6. [PUBMED: 25941540] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SS, Fakhoury TA, Hogan RE, Nagaraddi VN, Blatt I, Lawson B, et al. Once‐daily USL255 as adjunctive treatment of partial‐onset seizures: randomized phase III study. Epilepsia 2014;55(7):1077‐87. [PUBMED: 24902983] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SS, Hogan R, Blatt I, Lawson PB, Nguyen H, Clark AM, et al. Long‐term safety and sustained efficacy of USL255 (topiramate extended‐release capsules) in patients with refractory partial‐onset seizures. Epilepsy & Behavior 2016;59:13‐20. [PUBMED: 27084978] [DOI] [PubMed] [Google Scholar]
- Clark AM, Chung SS, Blatt I, Anders B, Halvorsen MB, Hogan RE. Efficacy of USL255 (qudexyTM XR; Extended‐release topiramate) in patients with refractory partial‐onset seizures: PREVAIL and PREVAIL OLE. Epilepsy Currents 2015;15(Suppl 1):142‐3, Abstract no: 1.307. [Google Scholar]
- Hogan R, Arnold S, Fakhoury T, Anders B. Adverse event profile of USL255 in patients with refractory partial‐onset seizures: the PREVAIL study. Neurology 2014;82(10 Suppl):Abstract no: P3.262. [Google Scholar]
- Hogan RE, Arnold S, Fakhoury TA, Anders B, Laine D, Todd WM, et al. Safety and tolerability of USL255 in subjects with refractory partial‐onset seizures: results from the randomized, phase 3 PREVAIL clinical trial. Epilepsy Currents 2014;14(Suppl 1):103‐4, Abstract no: 1.219. [Google Scholar]
- Hogan RE, Blatt I, Lawson B, Nagaraddi V, Fakhoury TA, Anders B, et al. Efficacy of once‐daily extended‐release topiramate (USL255): a subgroup analysis based on the level of treatment resistance. Epilepsy & Behavior 2014;41:136‐9. [PUBMED: 25461205] [DOI] [PubMed] [Google Scholar]
- Lawson B, Chung SS, Clark AM, Halvorsen MB, Blatt I. Time to onset of efficacy and sustained treatment effects of USL255: results from the phase 3 PREVAIL clinical trial. Epilepsy Currents 2014;14(Suppl 1):104, Abstract no: 1.222. [Google Scholar]
- Nagaraddi V, Chung S, Arnold S, Clark A, Anders B, Fakhoury T. Early onset of efficacy and safety outcomes with USL255 treatment: The PREVAIL study. Neurology 2014;82(10 Suppl):Abstract no: P3.264. [Google Scholar]
Elterman 1999 {published data only}
- Elterman RD, Glauser TA, Wyllie E, Reife R, Wu SC, Pledger G, et al. A double‐blind, randomized trial of topiramate as adjunctive therapy for partial‐onset seizures in children. Neurology 1999;52(7):1338‐44. [PUBMED: 10227615] [DOI] [PubMed] [Google Scholar]
Faught 1996 {published data only}
- Faught E, Wilder BJ, Ramsay RE. Topiramate dose‐ranging trial in refractory partial epilepsy. Epilepsia 1995;36(Suppl 4):33, Abstract no: D.9. [Google Scholar]
- Faught E, Wilder BJ, Ramsay RE, Reife RA, Kramer LD, Pledger GW, et al. Topiramate placebo‐controlled dose‐ranging trial in refractory partial epilepsy using 200‐, 400‐, and 600‐mg daily dosages. Neurology 1996;46(6):1684‐90. [PUBMED: 8649570] [DOI] [PubMed] [Google Scholar]
Guberman 2002 {published data only}
- Guberman A, Neto W, Gassmann‐Mayer C, EPAJ‐119 Study Group. Low‐dose topiramate in adults with treatment‐resistant partial‐onset seizures. Acta Neurologica Scandinavica 2002;106(4):183‐9. [PUBMED: 12225311] [DOI] [PubMed] [Google Scholar]
- Guberman A, Neto W, Gassmann‐Mayer C, Topiramate EPAJ‐119 Study Group. Efficacy of 200 mg/day topiramate in treatment‐resistant partial seizures when added to an enzyme‐inducing anti‐epileptic drug (AED). Epilepsia 2001;42(Suppl 7):179‐80, Abstract no: 2.244. [Google Scholar]
- Neto W, Gassmann‐Mayer C, Topiramate EPAJ‐119 study group. Efficacy and tolerability of 200 mg/day topiramate in a placebo‐controlled comparison of titration rates. Epilepsia 2000;41:35. [Google Scholar]
- Oene J, Neto W, Nilsson J, Hamilton G, Baeten B, Schreiner A, et al. Weight reduction during topiramate treatment of partial seizures. Epilepsia 2002;43(Suppl 8):152, Abstract no: P482. [Google Scholar]
Korean Topiramate Study Group 1999 {published data only}
- Korean Topiramate Study Group. Topiramate in medically intractable partial epilepsies: double‐blind placebo‐controlled randomized parallel group trial. Epilepsia 1999;40(12):1767‐74. [PUBMED: 10612342] [DOI] [PubMed] [Google Scholar]
- Lee BI. Double‐blind placebo‐controlled randomized clinical trial of topiramate add‐on therapy in medically intractable partial epilepsies. Journal of the Korean Neurology Association 1998;16(6):809‐19. [Google Scholar]
Privitera 1996 {published data only}
- Privitera M, Fincham R, Penry J, Reife R, Kramer L, Pledger G, et al. Topiramate placebo‐controlled dose‐ranging trial in refractory partial epilepsy using 600‐, 800‐, and 1,000‐mg daily dosages. Neurology 1996;46(6):1678‐83. [PUBMED: 8649569] [DOI] [PubMed] [Google Scholar]
- Privitera M, Fincham R, Penry JK. Dose‐ranging trial with higher doses of topiramate in patients with resistant partial seizures. Epilepsia 1995;36(Suppl 4):33, Abstract no: D.7. [Google Scholar]
Rosenfeld 1996 {unpublished data only}
- Rosenfeld W, Abou‐Khalil B, Reife R, Hegadus R, Pledger G, Topiramate YF/YG Study Group. Placebo‐controlled trial of topiramate as adjunctive therapy to carbamazepine or phenytoin for partial‐onset seizures. Epilepsia 1996;37(Suppl 5):153, Abstract no: 6.7. [Google Scholar]
Sharief 1996 {published data only}
- Sharief M, Viteri C, Ben‐Menachem E, Weber M, Reife R, Pledger G, et al. Double‐blind, placebo‐controlled study of topiramate in patients with refractory partial epilepsy. Epilepsy Research 1996;25(3):217‐24. [PUBMED: 8956919] [DOI] [PubMed] [Google Scholar]
- Sharief MK, Sander JW, Patsalos PN, Shorvon SD. Adjuvant topiramate treatment in intractable partial epilepsy. Epilepsia 1993;6:41. [Google Scholar]
Tassinari 1996 {published data only}
- Tassinari C, Chauvel P, Chodkiewicz J, Shorvon SD, Henriksen O, Dam M, et al. Double‐blind, placebo‐controlled trial of 600 mg/day topiramate as add‐on therapy in patients with refractory partial epilepsy. Epilepsia 1995;36(Suppl 3):S150. [Google Scholar]
- Tassinari CA, Michelucci R, Chauvel P, Chodkiewicz J, Shorvon S, Henriksen O, et al. Double‐blind, placebo‐controlled trial of topiramate (600 mg daily) for the treatment of refractory partial epilepsy. Epilepsia 1996;37(8):763‐8. [PUBMED: 8764816] [DOI] [PubMed] [Google Scholar]
Yen 2000 {published data only}
- Yen DJ, Yu HY, Guo YC, Chen C, Yiu CH, Su MS. A double‐blind, placebo‐controlled study of topiramate in adult patients with refractory partial epilepsy. Epilepsia 2000;41(9):1162‐6. [PUBMED: 10999555] [DOI] [PubMed] [Google Scholar]
Zhang 2011 {published data only}
- Zhang L, Huang J, Zhuang JH, Huang LQ, Zhao ZX. Topiramate as an adjunctive treatment for refractory partial epilepsy in the elderly. Journal of International Medical Research 2011;39(2):408‐15. [PUBMED: 21672344] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Christensen 2003 {published data only}
- Christensen J, Andreasen F, Poulsen JH, Dam M. Randomized, concentration‐controlled trail of topiramate in refractory focal epilepsy. Neurology 2003;61(9):1210‐8. [PUBMED: 14610122] [DOI] [PubMed] [Google Scholar]
Chung 2009 {published data only}
- Chung SS, Kerls S, Hammer A, Kustra R. Cognitive effects of lamotrigine versus topiramate as adjunctive therapy in older adults with epilepsy. Neurology international 2009;1(1):e6. [PUBMED: 21577364] [DOI] [PMC free article] [PubMed] [Google Scholar]
Coles 1999 {published data only}
- Coles H, Baker G, O'Donoghue M. Seizure severity in patients with partial onset (POS) or primary generalised tonic‐clonic (PGTC) seizures following treatment with topiramate. A comparison of two different methodologies in a randomised controlled trial. Epilepsia 1999;40(Suppl 2):285. [Google Scholar]
Novotny 2010 {published data only}
- Novotny E, Renfroe B, Yardi N, Nordli D, Ness S, Wang S, et al. Randomized trial of adjunctive topiramate therapy in infants with refractory partial seizures. Neurology 2010;74(9):714‐20. [PUBMED: 20089937] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfroe JB, Novotny EJ, Yardi N, Nordli D, Ness S, Wang S, et al. A randomized, double‐blind, placebo‐controlled study of topiramate as an adjunct to anticonvulsant therapy in infants with refractory partial onset seizures. Epilepsia 2008;49(Suppl 7):233, Abstract no: 2.140. [Google Scholar]
Ramsay 2008 {published data only}
- Ramsay R, Rowan A, Spitz M. Topiramate (TPM) in older adults with partial‐onset seizures: a double‐blind, dose‐comparison study. Epilepsia 2005;46(Suppl 6):114, Abstract no: p235. [DOI] [PubMed] [Google Scholar]
- Ramsay RE, Uthman B, Pryor FM, Rowan AJ, Bainbridge J, Spitz M, et al. Topiramate in older patients with partial‐onset seizures: a pilot double‐blind, dose‐comparison study. Epilepsia 2008;49(7):1180‐5. [DOI: 10.1111/j.1528-1167.2008.01584.x; PUBMED: 18494791] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Aranguiz 1991 {unpublished data only}
- Aranguiz C, McJilton J, Vega M, Ramsay R. Safety and effectiveness of three oral doses of topiramate in the treatment of patients with refractory partial epilepsy. Epilepsia 1991;32(Suppl 3):11. [Google Scholar]
Kazibutowska 2000 {unpublished data only}
- Kazibutowska Z, Stelmach‐Wawrzyczek M. Efficacy of therapy with new antiepileptic drugs (tiagabine, gabapentin, topiramate) in drug‐resistant epilepsy. Epilepsia 2000;41:38. [Google Scholar]
Additional references
Brown 1993
- Brown SD, Wolf HH, Swinyard EA, Twyman RE, White HS. The novel anticonvulsant topiramate enhances GABA‐mediated chloride flux. Epilepsia 1993; Vol. 34, issue Suppl 2:122‐3.
Cockerell 1995
- Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the national general practice study of epilepsy. Lancet 1995;346(8968):140‐4. [PUBMED: 7603228] [DOI] [PubMed] [Google Scholar]
Coulter 1993
- Coulter DA, Sombati S, DeLorenzo RJ. Selective effects of topiramate on sustained repetitive firing and spontaneous bursting in cultured hippocampal neurons. Epilepsia 1993;34(Suppl 2):123. [DOI] [PubMed] [Google Scholar]
Fiest 2017
- Fiest KM, Sauro KM, Wiebe S, Patten SB, Kwon CS, Dykeman J, et al. Prevalence and incidence of epilepsy: A systematic review and meta‐analysis of international studies. Neurology 2017;88(3):296‐303. [PUBMED: 27986877] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Version accessed 17 January 2019. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Gryder 2003
- Gryder DS, Rogawski MA. Selective antagonism of GluR5 kainate‐receptor‐mediated synaptic currents by topiramate in rat basolateral amygdala neurons. The Journal of Neuroscience 2003;23(18):7069‐74. [PUBMED: 12904467] [DOI] [PMC free article] [PubMed] [Google Scholar]
Herrero 2002
- Herrero AI, Olmo N, Gonzalez‐Escalada JR, Solis JM. Two new actions of topiramate: inhibition of depolarizing GABA(A)‐mediated responses and activation of a potassium conductance. Neuropharmacology 2002;42(2):210‐20. [PUBMED: 11804617] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Kirkham 2010
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:c365. [DOI: 10.1136/bmj.c365; PUBMED: 20156912] [DOI] [PubMed] [Google Scholar]
Kwan 2000
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. New England Journal of Medicine 2000;342(5):314‐9. [DOI: 10.1056/NEJM200002033420503; PUBMED: 10660394] [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Murad 2018
- Murad MH, Chu H, Lin L, Wang Z. The effect of publication bias magnitude and direction on the certainty in evidence. BMJ Evidence‐Based Medicine 2018;23(3):84‐6. [DOI: 10.1136/bmjebm-2018-110891; PUBMED: 29650725] [DOI] [PMC free article] [PubMed] [Google Scholar]
Perucca 1997
- Perucca E. A pharmacological and clinical review on topiramate, a new antiepileptic drug. Pharmacological Research 1997;35(4):241‐56. [PUBMED: 9264038] [DOI] [PubMed] [Google Scholar]
Privitera 1997
- Privitera MD. Topiramate: a new antiepileptic drug. The Annals of Pharmacotherapy 1997;31(10):1164‐73. [PUBMED: 9337443] [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Reynolds 1981
- Reynolds EH, Shorvon SD, Galbraith AW, Chadwick D, Dellaportas CI, Vydelingum L. Phenytoin for epilepsy: a long term prospective study, assisted by serum level monitoring, in previously untreated patients. Epilepsia 1981;22(4):475‐88. [PUBMED: 6790274] [DOI] [PubMed] [Google Scholar]
Scheffer 2017
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s). Handbook for grading the quality of evidence and the strength of recommendations using the GRADE approach (updated October 2013). GRADE Working Group, 2013. Available from gdt.guidelinedevelopment.org/app/handbook/handbook.html.
Turner 2000
- Turner RM, Omar RZ, Yang M, Goldstein H, Thompson SG. A multilevel model framework for meta‐analysis of clinical trials with binary outcomes. Statistics in Medicine 2000;19(24):3417‐32. [PUBMED: 11122505] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Jette 1999a
- Jette N, Marson AG, Kadir ZA, Hutton JL, Chadwick DW. TOPIRAMATE FOR DRUG RESISTANT PARTIAL EPILEPSY. Cochrane Database of Systematic Reviews 1999, Issue 1. [DOI: 10.1002/14651858.CD001417] [DOI] [PubMed] [Google Scholar]
Jette 1999b
- Jette NJ, Marson AG, Kadir ZA, Hutton JL. TOPIRAMATE IN DRUG‐RESISTANT PARTIAL EPILEPSY. Cochrane Database of Systematic Reviews 1999, Issue 3. [DOI: 10.1002/14651858.CD001417] [DOI] [PubMed] [Google Scholar]
Jette 2000
- Jette NJ, Marson AG, Kadir ZA, Hutton JL. Topiramate for drug‐resistant partial epilepsy. Cochrane Database of Systematic Reviews 2000, Issue 1. [DOI: 10.1002/14651858.CD001417] [DOI] [PubMed] [Google Scholar]
Jette 2002
- Jette NJ, Marson AG, Hutton JL. Topiramate add‐on for drug‐resistant partial epilepsy. Cochrane Database of Systematic Reviews 2002, Issue 3. [DOI: 10.1002/14651858.CD001417] [DOI] [PubMed] [Google Scholar]
Jette 2008
- Jette N, Hemming K, Hutton J, Marson A. Topiramate add‐on for drug‐resistant partial epilepsy. Cochrane Database of Systematic Reviews 2008, Issue 3. [DOI: 10.1002/14651858.CD001417.pub2] [DOI] [PubMed] [Google Scholar]
Marson 1996
- Marson AG, Kadir ZA, Chadwick DW. New antiepileptic drugs; a systematic review of their efficacy and tolerability. BMJ 1996;313:1169‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marson 1997
- Marson AG, Kadir ZA, Hutton JL, Chadwick DW. The new antiepileptic drugs; a systematic review of their efficacy and tolerability. Epilepsia 1997;38:859‐80. [DOI] [PubMed] [Google Scholar]
Pulman 2014
- Pulman J, Jette N, Dykeman J, Hemming K, Hutton JL, Marson AG. Topiramate add‐on for drug‐resistant partial epilepsy. Cochrane Database of Systematic Reviews 2014, Issue 2. [DOI: 10.1002/14651858.CD001417.pub3] [DOI] [PubMed] [Google Scholar]