

Abstract
Induction of lysosomal exocytosis alleviates lysosomal storage of undigested metabolites in cell models of lysosomal disorders (LDs). However, whether this strategy affects other vesicular compartments, e.g., those involved in endocytosis, is unknown. This is important both to predict side effects and to use this strategy in combination with therapies that require endocytosis for intracellular delivery, such as lysosomal enzyme replacement therapy (ERT). We investigated this using δ-tocopherol as a model previously shown to induce lysosomal exocytosis and cell models of type A Niemann-Pick disease, a LD characterized by acid sphingomyelinase (ASM) deficiency and sphingomyelin storage. δ-Tocopherol and derivative CF3-T reduced net accumulation of fluid phase, ligands, and polymer particles via phagocytic, caveolae-, clathrin-, and cell adhesion molecule (CAM)-mediated pathways, yet the latter route was less affected due to receptor overexpression. In agreement, δ-tocopherol lowered uptake of recombinant ASM by deficient cells (known to occur via the clathrin pathway) and via targeting intercellular adhesion molecule-1 (associated to the CAM pathway). However, the net enzyme activity delivered and lysosomal storage attenuation were greater via the latter route. Data suggest stimulation of exocytosis by tocopherols is not specific of lysosomes and affects endocytic cargo. However, this effect was transient and became unnoticeable several hours after tocopherol removal. Therefore, induction of exocytosis in combination with therapies requiring endocytic uptake, such as ERT, may represent a new type of drug interaction, yet this strategy could be valuable if properly timed for minimal interference.
Introduction
Lysosomal diseases (LDs) are a group of 50 to 60 different monogenic disorders characterized by deficient lysosomal function (Futerman and van Meer, 2004). This is often caused by a defective lysosomal hydrolase, and, as a consequence, lysosomes accumulate respective undigested metabolites (Ballabio and Gieselmann, 2009). Because lysosomes are vital to many cellular functions, this aberrant storage leads to alterations in signaling pathways, cytoskeletal arrangement, autophagy, lipid trafficking, etc., manifesting into a wide array of life-threatening symptoms, including premature death (Simons and Gruenberg, 2000; Fukuda et al., 2006; Parkinson-Lawrence et al., 2010).
Currently, there are a number of clinical and experimental treatments for LDs, including therapies based on small molecules that act as chemical chaperones or inhibit metabolite synthesis, gene therapy to produce functional enzyme activity, and i.v. infusion of recombinant enzymes to replace those that are deficient, known as enzyme replacement therapy (ERT) (Solomon and Muro, 2017). These approaches are highly valuable for individual diseases; however, they are specific to the genetic defect and/or lysosomal storage of each particular disease, for which they do not represent universal treatment options for LDs. An alternative or complementary approach recently proposed, which would be applicable to most LDs, is the induction of lysosomal exocytosis. This would enable cellular secretion of the metabolites stored in diseased lysosomes to the extracellular milieu, where they can be removed by the lymphatics, the immune system, etc. (Klein et al., 2005; Chen et al., 2010; Strauss et al., 2010; Medina et al., 2011; Xu et al., 2012; Spampanato et al., 2013; Cao et al., 2015; Long et al., 2016; Zhong et al., 2016).
Lysosomal exocytosis is a natural event (Schultz et al., 2011; Samie and Xu, 2014), and, in fact, secreted lysosomal substrates have been found in the urine of LD patients, validating this clearance mechanism as a potential treatment strategy (Whitley et al., 1989a,b; Wisniewski et al., 1994). Hence, this process can be capitalized for therapeutic purposes. Approaches to stimulate lysosomal exocytosis include the use of 2-hydroxypropyl-β-cyclodextrin in mouse-derived, type C1 Niemann-Pick (NPC1) cell models (Chen et al., 2010) and agonists of lysosomal big conductance calcium-activated Big Potassium (BK) channels in NPC1, mucolipidosis type IV, NPD-A, and Fabry disease (Cao et al., 2015; Zhong et al., 2016). Also, overexpression of transcription factor EB, a key regulator of lysosomal biogenesis and function, or that of one of its target proteins (TRPML1), enhances lysosomal exocytosis and alleviates storage in several LDs (Medina et al., 2011; Spampanato et al., 2013; Samie and Xu, 2014). Although less relevant translationally, this paradigm can be modeled in cell culture using the vitamin E species, δ-tocopherol (Xu et al., 2012; Long et al., 2016). Incubation of LD cells with this compound and some of its derivatives results in reduction in lysosome size, lysosome distribution closer to the plasma membrane, enhanced lysosomal enzyme activity in the extracellular medium, and significant attenuation of lysosomal storage, as shown for cell models of type C Niemann-Pick disease, type A Niemann-Pick disease (NPD-A), Wolman disease, and other LDs (Xu et al., 2012; Long et al., 2016).
Although lysosomal exocytosis holds interest as a potential treatment option for LDs, it is unclear whether induction of lysosomal exocytosis may also cause exocytosis of related vesicles, such as endocytic ones. This is critical for many cellular functions, such as uptake of nutrients, signal transduction, plasmalemma recycling, pathogen defense, etc. (Mellman, 1996; Conner and Schmid, 2003), and also for delivery of other therapeutics that require endocytic uptake for intracellular delivery (Muro, 2012a; Rappaport et al., 2016). This is particularly relevant for LDs because many of these diseases have been reported to associate with diminished endocytic activity via clathrin, caveolae, cell adhesion molecule (CAM), macropinocytosis, etc. (Liscum and Faust, 1987; Marks and Pagano, 2002; Dhami and Schuchman, 2004; Hortsch et al., 2010; Teixeira et al., 2014; Kuech et al., 2016; Rappaport et al., 2016). This affects uptake of bulk fluid phase, ligands of receptor-mediated pathways, recycling of membrane proteins and lipids, etc., as found for NPC1, NPD-A, Pompe, Fabry, and Gaucher diseases (Liscum and Faust, 1987; Marks and Pagano, 2002; Dhami and Schuchman, 2004; Hortsch et al., 2010; Kuech et al., 2016; Rappaport et al., 2016). Induction of exocytosis could affect endocytic vesicles that bring cargo into diseased cells, further lowering their uptake capacity. Because most of these pathways have been explored for drug delivery and lysosomal ERT (Muro et al., 2003, 2006b; Nichols, 2003; Hillaireau and Couvreur, 2009; Boado et al., 2011; Chrastina et al., 2011; McMahon and Boucrot, 2011; Duncan and Richardson, 2012; Muro, 2012b; Rappaport et al., 2016; Garnacho et al., 2017), collateral exocytosis of endocytic vesicles could also hinder treatment by means that require endocytic uptake, such as gene therapy or ERT.
In this work, we explored this question using the δ-tocopherol model of lysosomal exocytosis and cellular models of acid sphingomyelinase (ASM)-deficient NPD-A because of the following reasons: 1) this LD associates with endocytic alterations in various routes (Dhami and Schuchman, 2004; Rappaport et al., 2016); 2) several of these routes are being investigated for ERT delivery (Solomon and Muro, 2017); and 3) lysosomal exocytosis can be induced in NPD-A cells by δ-tocopherol (Xu et al., 2012). Our results described below shed light into the potential effects of inducing lysosomal exocytosis on endocytic uptake and implications for combination therapy with recombinant enzymes.
Materials and Methods
Antibodies and Reagents.
δ-Tocopherol was from Sigma-Aldrich (St. Louis, MO). CF3-T was synthesized internally, as published (Marugan et al., 2014). Alexa Fluor 594-transferrin (Tf) and cholera toxin subunit B (CTB), Texas Red dextran (10,000 MW), BODIPY-FL-C12-sphingomyelin, and Texas Red goat anti-mouse IgG were from Invitrogen (Carlsbad, CA). Lysenin and rabbit anti-lysenin serum were from Wako Chemicals (Richmond, VA). Nile Red, filipin, imipramine hydrochloride, and 4-methylumbelliferyl-N-acetyl-β-D-glucosaminide were from Sigma-Aldrich. Mouse monoclonal anti-human intercellular adhesion molecule 1 (ICAM-1) clone R6.5 (anti-ICAM) was purified from HB-9580 hybridoma (American Type Culture Collection, Manassas, VA). Rabbit polyclonal anti-early endosome antigen 1 (anti-EEA-1) was from Cell Signaling Technologies (Danvers, MA). Mouse IgG, fluorescein isothiocyanate (FITC) anti-rabbit IgG, and Alexa Fluor 488 mouse anti-rabbit IgG were from Jackson ImmunoResearch (West Grove, PA). Polystyrene beads were from Polysciences (Warrington, PA). Recombinant human ASM (He et al., 1999) was provided by E. Schuchman (Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, NY) or commercially obtained from Abcam (Cambridge, UK). 6-Hexadecanoylamino-4-methylumbelliferyl-phosphorylcholine was from Moscerdam Substrates (Oegstgeest, The Netherlands). 125Iodine (125I) was from Perkin-Elmer (Waltham, MA), and Pierce iodination tubes were from Thermo Fisher Scientific (Waltham, MA). Unless otherwise noted, all other reagents were from Sigma-Aldrich.
Cell Cultures.
Pharmacological models consisted of human umbilical vein endothelial cells (Lonza, Walkersville, MD) seeded on gelatin-coated coverslips and incubated at 37°C, 5% CO2, and 95% relative humidity in M-199 medium (Invitrogen) supplemented with 15% FBS, 15 µg/ml endothelial cell growth supplement, 2 mM L-glutamine, 100 µg/ml heparin, and antibiotics. To mimic lysosomal storage characteristic of NPD-A, human umbilical vein endothelial cells were incubated for 4 days at 37°C with 20 µM imipramine, which degrades endogenous ASM (Hurwitz et al., 1994). Genetic models were primary, human skin fibroblasts from wild-type or NPD-A (GM13205 with deletion of a cytosine in codon 330) subjects from the Coriell Cell Repository (Camden, NJ) that were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY) supplemented with 10% FBS, 2 mM L-glutamine, and antibiotics. Where indicated, cells were treated for 48 hours prior to assays with 40 µM δ-tocopherol or 20 µM CF3-T.
Preparation of Nano- and Microparticles.
For studies on the CAM pathway and enzyme delivery, respectively, anti-ICAM alone (4.8 µM) or a 50:50 mass ratio of anti-ICAM:ASM (1.36 µM:2.72 µM) was coated by surface adsorption onto 100-nm–diameter, green Fluoresbrite polystyrene particles (anti-ICAM and anti-ICAM/ASM nanocarriers (NCs), respectively), as described (Serrano et al., 2016; Garnacho et al., 2017). Anti-ICAM NCs loaded with ASM at this ratio efficiently target the CAM route and deliver active ASM to lysosomes, leading to effective degradation of accumulated substrates in cells and mice (Muro et al., 2006b; Garnacho et al., 2008, 2017). The hydrodynamic diameter and polydispersity index of coated particles were measured by dynamic light scattering (Malvern Instruments, Westborough, MA) and found to be, respectively, 269 ± 4 nm and 0.17 ± 0.01 for anti-ICAM NCs, and 243 ± 2 nm and 0.15 ± 0.01 for anti-ICAM/ASM NCs. To characterize coating, anti-ICAM or ASM was conjugated to 125I, and the amount of antibody or enzyme per particle was quantified using a γ counter (2470 Wizard2; Perkin Elmer) and their respective specific activities (cpm/mass), as reported (Serrano et al., 2016; Garnacho et al., 2017). Anti-ICAM NCs contained 257 ± 9 anti-ICAM molecules per NC, and anti-ICAM/ASM NCs had 135 ± 5 anti-ICAM molecules and 259 ± 1 ASM molecules per NC, in accord with previous reports (Garnacho et al., 2008, 2017). In saline or serum-containing medium, these formulations exhibit no aggregation, minimal antibody, or enzyme release (≤10% release), and minimal coating with serum albumin (≤5%) (Hsu et al., 2011, 2012; Serrano et al., 2016). As demonstrated (Muro et al., 2006a; Garnacho et al., 2008, 2017), these model carriers are comparable to clinically-relevant biodegradable poly(lactic-co-glycolic acid) ones in regard to coating efficiency, in vivo targeting, and intracellular trafficking, validating this model.
For studies on phagocytosis, nonfluorescent 1-µm–diameter polystyrene particles were coated by adsorption with mouse IgG (1 µm IgG-coated microparticles), rendering final size of 1.15 ± 0.04 µm, polydispersity index of 0.38 ± 0.06, and total coating of 47,522 ± 6007 IgG molecules per particle.
Intracellular Lipid Storage.
Lipid levels and the number of vesicular storage compartments were measured in control versus diseased cells, including pharmacological and genetic NPD-A cell models, prior or after 1- to 24-hour treatment with δ-tocopherol or derivative CF3-T. For sphingomyelin staining, cells were fixed with cold 2% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked with 2% bovine serum albumin–PBS, and immunostained with 0.5 µg/ml lysenin, followed by rabbit antilysenin serum and 4 µg/ml (green) FITC-labeled secondary antibody. Also, fixed cells were stained with 50 µg/ml (blue) filipin to label cholesterol, or with 100 ng/ml (red) Nile Red to generically label lipids. Samples were visualized by fluorescence microscopy using an Olympus IX81 microscope (Olympus, Center Valley, PA); 40× or 60× UPlanApo oil immersion objectives (Olympus); and blue, green, and red fluorescence filters (1160A-OMD, 3540B-OMF, 4040B-OMF; Semrock, Rochester, NY). Images were obtained with an ORCA-ER camera (Hamamatsu, Bridgewater, NJ) and SlideBook 4.2 software (Intelligent Imaging Innovations, Denver, CO) and analyzed using Image-Pro 6.3 (Media Cybernetics, Bethesda, MD). To quantify lysosomal sphingomyelin, cholesterol, and generic lipids, the mean fluorescence intensity in the perinuclear region (3 µm around the nucleus) was quantified with subtraction of the mean fluorescence intensity of the cell periphery, which was considered nonlysosomal (likely plasmalemma) background. Cells were also imaged by phase contrast, from which the number of dark-refringent intracellular (storage) compartments was quantified. These analyses were performed for individual cells to capture the intercellular variability of the population.
Sphingomyelin levels were also addressed in bulk for the entire cell population, either prior or after 4-hour treatment using 5 µg/ml naked recombinant ASM. For this, cells were preincubated overnight with 0.2 µg/ml BODIPY-FL-C12-sphingomyelin, which fluoresces green (513 nm) at high concentrations (Luzio et al., 2007a,b). Cells were washed and fixed, and the nuclei stained with Hoechst 33342. Sphingomyelin fluorescence was analyzed using an InCell2200 imaging system (GE Healthcare Bio-Sciences, Pittsburgh, PA).
ASM Activity.
Endogenous ASM activity was measured by incubating cells for 2 hours at 37°C with 20 µM 6-hexadecanoylamino-4-methylumbelliferyl-phosphorylcholine, a profluorescence ASM substrate in buffer containing 1% Triton X-100. The supernatant was measured in a fluorescence plate reader with excitation and emission wavelengths of 385 and 450 nm. ASM activity was assessed in a similar manner after 48-hour treatment with 40 µM δ-tocopherol and/or after 4-hour treatment with 2.3 µg/ml recombinant ASM.
Bulk Fluid-Phase Uptake.
Control versus diseased cells were treated for 48 hours at 37°C with 40 µM δ-tocopherol or 20 µM CF3-T. Between 0 and 8 hours after removal of these agents, cells were incubated in fresh medium containing 1 mg/ml Texas Red dextran (a fluid-phase marker of endocytosis) for 1 hour at 37°C. Cells were then washed and fixed, and the number of fluorescent dextran-filled compartments per cell was analyzed by microscopy with an algorithm that quantifies fluorescent objects of 100- to 200-nm diameter with intensity above a background threshold level (Serrano et al., 2016).
Ligand and Particle Uptake.
To evaluate the effect of δ-tocopherol on ligand uptake, healthy versus imipramine-induced diseased cells were treated for 48 hours at 37°C with 40 µM δ-tocopherol. Diseased cells that did not receive δ-tocopherol were used as control. Cells were then incubated between 1 and 3 hours at 37°C with one of the following agents: 20 µg/ml red Alexa Fluor594-labeled CTB (caveolae-mediated uptake), 50 µg/ml red Alexa Fluor594–labeled Tf (clathrin-mediated uptake), 7 × 1010 (green) Fluoresbrite anti-ICAM NCs/ml (CAM-mediated uptake), or 1 × 108 nonfluorescent 1 µm IgG-coated microparticles per milliliter (phagocytosis). Samples were then washed, fixed without permeabilization, and immunostained with secondary antibodies conjugated to green FITC or red Texas Red, opposite to the ligand or particle color. As previously demonstrated (Muro et al., 2003), this provides differential labeling of surface-located materials that are accessible for secondary staining versus internalized materials that are not accessible to said secondary staining (Muro et al., 2003). Thus, internalized materials appear as single-colored objects versus surface-bound materials, which appear as dual-colored objects (Muro et al., 2003). In the case of 1 µm IgG-coated microparticles, internalized microparticles lacked a fluorescent label, but these microparticles are visible and quantifiable from phase-contrast images, whereas surface-bound microparticles were immunolabeled in red. Where indicated, cells were treated with 10 ng/ml tumor necrosis factor α (TNFα) overnight prior to assay to mimic the inflammatory state found in NPD-A (Schuchman and Desnick, 2017).
Quantification of Early Endosomes.
To discern whether δ-tocopherol may induce exocytosis of endosomal compartments, diseased cells were incubated at 37°C with 40 µM δ-tocopherol for either 15 minutes, 1 hour, or 3 hours. Thereafter, cells were fixed and permeabilized, and early endosomes were immunostained using anti–EEA-1 antibody, followed by washing and incubation with a secondary antibody labeled with green Alexa Fluor 488. Fluorescence microscopy images were taken and quantified using the settings and algorithm described above, to discern the number of ≥100-nm–diameter fluorescent objects with intensity level above the background level.
Lysosomal Secretion.
To verify lysosome secretion, the release of the lysosomal enzyme β-hexosaminidase A and B (HEX) from the cell culture into the cell medium was examined. Control or diseased cells were treated for 24 hours at 37°C with 40 μM δ-tocopherol, and then this compound was removed and cells were incubated for up to 3 hours at 37°C with 10 μM ionomycin, an inducer of lysosome release, or 40 μM δ-tocopherol. The medium fraction was collected, and cells were washed, trypsinized, suspended in acidic buffer (10 mM monosodium phosphate, 5% glycerol buffer, pH 6.0), subjected to five freeze–thaw cycles, and centrifuged at 16,000g for 20 minutes to pellet the insoluble membrane fraction, so that the soluble intracellular supernatant (cell fraction) was collected. Both the medium and cell fractions were incubated for 1 hour at 37°C in HEX substrate solution (2 mM 4-methylumbelliferyl-N-acetyl-β−D-glucosaminide, dissolved in 0.1 M citric acid/0.2 M disodium phosphate buffer, pH 4.5). Then the reaction was stopped with 0.4 M glycine–sodium hydroxide buffer (pH 10.4), and the fluorescent product was quantified in a plate reader (Spectramax M2; Molecular Devices) at λex = 360 nm and λem = 450 nm. The relative fluorescence units (RFU) were used to determine the percentage of HEX in each fraction using the following formula:
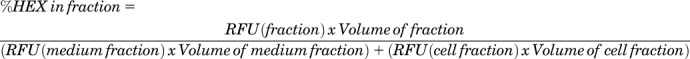 |
Uptake of Recombinant ASM.
Diseased cells treated for 48 hours with 40 µM δ-tocopherol were incubated overnight with 10 ng/ml TNFα to mimic inflammation. The cells were then washed and incubated for 2 hours at 37°C with 2.1 µg/ml naked 125I-ASM or a similar concentration of 125I-ASM coupled to anti-ICAM NCs. Cells were washed to remove nonbound counterparts and incubated with an acid glycine solution (50 mM glycine and 100 mM NaCl at pH 2.5) to remove noninternalized ASM from the cell surface (Rappaport et al., 2015). The cells were then lysed in 2% Triton X-100, and the total 125I-ASM present in the cell lysates was measured in a γ counter and corrected by subtraction of free 125I, as determined by trichloroacetic acid precipitation.
Statistics.
Data were calculated as the mean ± standard error of the mean (mean ± S.E.M.), where statistical significance for two-way comparisons was determined by Student’s t test with a threshold of P < 0.05. Microscopy assays involved two to four experiments, each with at least two wells, from which 5 to 10 regions located throughout each sample, representative of the entire population, were selected for quantification. Each region contained a range of cells (4–10), all of which were individually analyzed. Fluorescence plate reader, enzyme activity, and radiotracing tests involved n ≥ 2 independent experiment and n ≥ 3 repeats per experiment.
Results
Effect of Tocopherols on Lysosomal Storage in NPD-A Cells.
We first verified the effect of tocopherols on lysosomal exocytosis using vascular endothelial cells because they constitute one of the first cell linings encountered by i.v. injected therapies. As such, exocytosis induced in these cells could affect transport of recombinant enzymes to tissues. Because endothelial cells from NPD-A patients are unavailable, we first used a pharmacological model consisting of endothelial cells treated with imipramine (Muro et al., 2006b), whereas experiments described hereafter verify data in NPD-A patient fibroblasts. Imipramine enhances the degradation of endogenous ASM and mimics the intracellular lipid storage (sphingomyelin and cholesterol) characteristic of ASM-deficient NPD-A (Schuchman and Desnick, 2017). As expected, imipramine highly reduced endogenous ASM activity (87% reduction from control), similar to NPD-A patient fibroblasts (79% reduction from wild-type; Supplemental Fig. 1). Imipramine-induced diseased cells had enhanced intracellular storage of sphingomyelin (2.0-fold), cholesterol (3.5-fold), and overall lipids (2.3-fold) in perinuclear compartments, a location consistent with lysosomes, which drastically increased the total number of dark-refringent storage vesicles (22.4-fold) compared with control cells (Supplemental Fig. 2). This level of storage was comparable to NPD-A patient fibroblasts, which had 2.8-fold greater sphingomyelin and 3.8-fold greater cholesterol accumulation versus wild-type fibroblasts (Supplemental Fig. 3), validating this model.
Next, we examined whether the effect of tocopherols on lysosomal storage, which has been reported for patient fibroblasts and neural stem cells (Xu et al., 2012; Long et al., 2016), also applies to vascular endothelial cells. For this, imipramine-diseased endothelial cells were incubated for 48 hours with 40 µM δ-tocopherol or 20 µM analog CF3-T (Fig. 1A; Supplemental Fig. 4). As expected, both agents significantly decreased sphingomyelin storage by 33% and 48%, respectively, validating their effect on endothelial cells. In addition, treatment of imipramine-diseased endothelial cells with δ-tocopherol decreased the intracellular activity level of a control lysosomal enzyme HEX (20% reduction), whereas it increased the extracellular activity of this enzyme (30% increase; Fig. 1B), validating secretion of lysosomal content. This effect was almost equal to that of ionomycin, an agent known to induce lysosomal secretion, on control cells (Fig. 1B).
Fig. 1.

Effect of tocopherols on sphingomyelin storage and lysosomal exocytosis in diseased endothelial cells. (A) Micrograph quantification of the perinuclear lysosomal staining of sphingomyelin using fluorescent lysenin, in control (Ctrl) vs. imipramine-diseased endothelial cells (Dis), prior or after 48-hour incubation with 40 µM δ-tocopherol (δ-Toc) or 20 µM CF3-T. Data are normalized to untreated control cells (horizontal dashed line). (B) Fluorimetric quantification of the enzymatic activity of lysosomal β-hexosaminidase in the cell medium (exocytosed) or the cell fraction (lysosomal) upon treatment of imipramine-diseased endothelial cells with δ-Toc, as in (A). Control endothelial cells treated or not with 10 µM ionomycin to cause lysosomal secretion are shown for a comparison (A and B). Data are mean ± S.E.M. (n ≥ 4 independent wells). *Comparison with untreated control cells; †comparison with untreated diseased cells (P < 0.05 by Student’s t test).
Effect of Tocopherols on Bulk Fluid-Phase Endocytosis.
Having validated the imipramine-induced diseased cell model and tocopherol-induced reduction of storage, we focused on the effect of tocopherols on endocytic uptake. First, we tested nonspecific pinocytosis (bulk fluid-phase uptake), a process by which cells internalize extracellular fluid and solutes into endocytic vesicles. We used fluorescence microscopy to measure pinocytic uptake of the fluid-phase marker Texas Red dextran in imipramine-induced diseased endothelial cells treated with δ-tocopherol or CF3-T for 48 hours, then removed for 1 hour, versus diseased cells that lacked this treatment (Fig. 2A). As compared with control cells (Fig. 2B), dextran uptake was significantly reduced in diseased cells (53% reduction), which is consistent with published findings (Rappaport et al., 2015) on NPD-A patient fibroblasts (∼40% reduction compared with wild-type), further validating this model. Importantly, bulk dextran endocytosis was further diminished in cells treated with δ-tocopherol or CF3-T (Fig. 2B), with CF3-T being a somewhat more potent inhibitor (61% and 78% reduction, respectively), in accord with its more potent effect on exocytosis (Fig. 1A). Reduced sphingomyelin storage was verified (26% and 41% reduction from untreated diseased cells, respectively; Supplemental Figs. 4 and 5), suggesting that induction of exocytosis by tocopherols alleviates lysosomal storage, but also lowers fluid-phase endocytosis.
Fig. 2.

Effect of tocopherols on bulk fluid-phase uptake in diseased endothelial cells. (A) Fluorescence microscopy of control (Ctrl) and imipramine-diseased endothelial cells (Dis) incubated for 1-hour pulse with Texas Red dextran, 1 hour after treating cells for 48 hours with 40 µM δ-tocopherol or 20 µM CF3-T. Dotted lines mark the cell borders, observed by phase-contrast microscopy. Scale bar, 10 µm. (B) Dextran uptake was quantified per cell and normalized to untreated diseased cells (horizontal solid line). Data are mean ± S.E.M. (n ≥ 4 independent wells). *Comparison with untreated control cells; †comparison with untreated diseased cells (P < 0.05 by Student’s t test).
Effect of δ-Tocopherol on Receptor-Mediated Endocytosis.
Next, using fluorescently-labeled ligands, we examined the effect of δ-tocopherol on receptor-mediated endocytosis via caveoli (CTB), clathrin-coated pits (Tf), the CAM pathway [100 nm polymer nanocarriers targeted to ICAM-1; hereafter called anti-ICAM NCs (see Materials and Methods for characterization)], and phagocytosis (1 µm IgG-coated microparticles). Specificity of the uptake pathways for these ligands has been validated and can be found in our previous studies (Rappaport et al., 2014, 2015, 2016). Because the effects observed were similar for δ-tocopherol and CF3-T, we focused on the first compound as an example.
We recently reported decreased caveolar endocytosis of CTB, which binds to ganglioside GM1, and clathrin-mediated uptake of Tf, which binds to the Tf receptor, in NPD-A patient fibroblasts (Rappaport et al., 2014, 2015). Uptake of anti-ICAM NCs via the CAM pathway was also decreased in this disease (Serrano et al., 2012), although this route appeared more efficient than others (Rappaport et al., 2015, 2016). In this work, we found that imipramine-induced diseased endothelial cells also showed a similar pattern (14%, 58%, and 80% uptake of control cells for clathrin, caveolae, and CAM pathways, and 80% of control for phagocytosis; Fig. 3A), validating again this model. Importantly, δ-tocopherol treatment further reduced CTB, Tf, and microparticle uptake versus untreated diseased cells (35%, 39%, and 55% reduction, respectively; Fig. 3B). In contrast, anti-ICAM NC uptake via the CAM pathway was unaffected (92% of untreated diseased cells; Fig. 3B). Treatment of control endothelial cells with δ-tocopherol showed a similar trend, with clathrin-, caveoae-, and phagocytosis-, but not CAM-mediated uptake being decreased (Supplemental Fig. 6), suggesting that this effect is specific of δ-tocopherol, not imipramine, used to generate the disease phenotype. Similar experiments were repeated upon treatment of diseased cells with TNFα, a cytokine that stimulates an inflammatory phenotype, as observed in NPD-A and other LDs (Muro et al., 2006b; Schuchman and Desnick, 2017). Under TNFα activation, δ-tocopherol caused even more pronounced reduction of caveolar CTB uptake and phagocytic uptake of IgG-coated microparticles (58% and 71% reduction from untreated TNFα-activated diseased cells), whereas it had less of an effect (20% reduction) on clathrin-mediated Tf uptake, and it did not impact CAM uptake of anti-ICAM NCs (97% of untreated TNFα-activated diseased cells; Fig. 3C).
Fig. 3.

Effect of δ-tocopherol on receptor-mediated uptake in diseased endothelial cells. (A) Microscopy quantification of the uptake (2-hour) of fluorescent ligands of individual endocytic pathways in imipramine-diseased endothelial cells (Dis) vs. control (Ctrl) cells. (B) Uptake of fluorescent ligands (3-hour) in imiprimine-diseased cells treated for 48 hours with 40 µM δ-tocopherol under noninflammatory or (C) inflammatory-like conditions (overnight incubation with TNFα). Ligands were CTB (caveolae-mediated endocytosis), Tf (clathrin-mediated endocytosis), 200 nm polymer nanocarriers targeted to ICAM-1 (anti-ICAM NCs; CAM-mediated endocytosis), and 1 µm IgG-coated microparticles (phagocytosis; Phag.). (A–C) After ligand incubation, cells were washed and fixed, and cell surface–bound ligands were immunostained with antibodies fluorescently labeled in a different color to distinguish internalized vs. surface-bound localization (see Materials and Methods). Data are mean ± S.E.M. (n ≥ 4 independent wells), normalized to conditions shown in the horizontal solid lines. *Comparison with untreated diseased cells; †comparison with untreated diseased cells; #comparison with the CAM pathway; $comparison with the clathrin pathway (P < 0.05 by Student’s t test).
Based on this, it appeared δ-tocopherol lowered ligand uptake by all pathways except for CAM-mediated endocytosis. This was surprising because if tocopherol had caused this effect by inducing exocytosis of endocytic vesicles, the CAM pathway should also be affected. Hence, we investigated this in more detail. As expected, given that inflammatory conditions cause ICAM-1 overexpression (Muro et al., 2006a), TNFα increased binding of anti-ICAM NCs to diseased cells (2.44-fold increase; Supplemental Fig. 7). Unexpectedly, δ-tocopherol treatment further enhanced binding in TNFα-activated diseased cells (1.37-fold increase over untreated TNFα-activated diseased cells; Fig. 4A). However, the rate of NC uptake in TNFα-activated cells treated with δ-tocopherol, measured as the internalized fraction of all cell-associated NCs, was indeed reduced by 31% (Fig. 4A). Therefore, as for the other pathways, δ-tocopherol does diminish uptake via CAM-mediated endocytosis, but, because it also enhances binding to ICAM-1, absolute uptake via this pathway does not change. Increased anti-ICAM NC binding by δ-tocopherol treatment was not found for nonactivated diseased cells (104% of binding in untreated nonactivated diseased cells; Fig. 4A); hence, this effect may result from the combination of TNFα activation and δ-tocopherol.
Fig. 4.

δ-Tocopherol modulation of the TNFα effect on endocytosis in diseased endothelial cells. Imipramine-diseased endothelial cells (Dis) treated for 48 hours with 40 µM δ-tocopherol were left quiescent or activated overnight with TNFα to mimic inflammation. Cells were then incubated for 3 hours with (A) fluorescent anti-ICAM NCs or (B) fluorescent ligands of all individual endocytic pathways described in Fig. 3. Cells were washed and fixed, and cell surface ligands were fluorescently immunostained in a different color to distinguish internalized vs. surface-bound counterparts. (A) Binding was quantified as the total number of cell-associated fluorescent NCs, of which also the percentage of NCs internalized was measured. Data were normalized to untreated diseased cells (horizontal solid line). (B) Uptake of fluorescent ligands in TNFα-activated cells was normalized to nonactivated cells (horizontal solid line). (A and B) Data are mean ± S.E.M. (n ≥ 4 independent wells). *Comparison with untreated diseased cells; †comparison with nonactivated cells (P < 0.05 by Student’s t test).
Similarly, the effect of TNFα on the activity of each endocytic pathway was also assessed (Fig. 4B). As expected, absolute uptake via the CAM pathway was significantly increased under inflammation regardless of δ-tocopherol treatment (∼3-fold). Caveolae-mediated uptake of CTB and phagocytosis of IgG-coated microparticles by diseased cells were also increased by TNFα (1.4-fold and 1.9-fold, respectively), but δ-tocopherol inhibited this effect (0.9-fold and 1.2-fold of nonactivated diseased cells). Finally, clathrin-mediated uptake of Tf, which was lowered by δ-tocopherol (Fig. 3B), was not affected by TNFα (Fig. 4B).
Effect of δ-Tocopherol on Endosomal Compartments.
Altogether, the data described to date indicate a generic effect of δ-tocopherol on the endocytic events. Because this vitamin has been shown to induce exocytosis of lysosomes (Xu et al., 2012; Long et al., 2016), which was verified in this study in Fig. 1B, we tested whether this was also the case for early endosomal compartments. As shown in Fig. 5A, treatment with δ-tocopherol decreased over time the visualization by fluorescence microscopy of EEA-1–positive vesicles, indicative of early endosomes, within the cell body of both imipramine-diseased endothelial cells and NPD-A fibroblasts. Image quantification (Fig. 5B) verified this: δ-tocopherol significantly reduced the number of EEA-1–positive compartments compared with controls (e.g., 29% and 34%, respectively, by 15-minute treatment).
Fig. 5.

δ-Tocopherol reduction of the number of EEA-1–positive compartments in diseased cells. Imipramine-diseased endothelial cells and NPD-A patient fibroblasts were left untreated (Ctr) or were treated with 40 µM δ-tocopherol for 15 minutes, 1 hour, or 3 hours. Cells were washed, fixed, and permeabilized, and early endosomes were labeled using anti–EEA-1, followed by secondary antibody conjugated to green Alexa Fluor 488. (A) Fluorescence microscopy images of endothelial cells, shown as an example. Scale bar, 10 µm. (B) Number of EEA-1–positive compartments per cell compared with absence of δ-tocopherol (Ctr), expressed as a percentage. Data are mean ± S.E.M. (n ≥ 4 independent wells). *Comparison with Ctrl cells (P < 0.05 by Student’s t test).
Effect of δ-Tocopherol on Cell Uptake of Recombinant Enzymes Used for ERT.
Given that tocopherols lowered endocytic content in diseased cells and the fact that ERT requires endocytic uptake of recombinant enzymes, we examined whether δ-tocopherol would affect delivery of recombinant ASM, the enzyme deficient in NPD-A (Schuchman and Desnick, 2017). As other enzymes used for lysosomal ERT, ASM undergoes clathrin-mediated endocytosis upon binding to M6P receptors (Dhami and Schuchman, 2004), which is blocked in the presence of competing M6P (Muro et al., 2006b). We first tested NPD-A patient fibroblasts to verify their lower level of endogenous ASM activity versus healthy cells (21% of control wild-type activity; Fig. 6A), and to verify that incubation with recombinant ASM increased their ASM activity, as expected for delivery of an active enzyme (Fig. 6A). Recombinant ASM increased ASM activity of wild-type fibroblasts by 25% (not statistically significant due to high activity level in these cells) and to a greater extent (63% increase) in diseased cells, consistent with their lower endogenous activity. In contrast, treatment of cells (either endothelial cells or fibroblasts; Fig. 6B) with δ-tocopherol inhibited the acquisition of recombinant ASM activity by diseased cells (∼22% reduction; Fig. 6B) and also lowered acquired ASM activity in healthy cells (9%–30% reduction; Fig. 6B). However, in the absence of recombinant ASM, δ-tocopherol did not affect endogenous ASM activity: 107% and 89% activity of untreated cells were found in imipramine-induced diseased endothelial cells or NPD-A fibroblasts, respectively (Supplemental Fig. 8). Thus, δ-tocopherol does not impact ASM activity per se, but diminishes acquisition of exogenous ASM activity, which is consistent with its effect in lowering the clathrin pathway, as was observed for Tf uptake (Fig. 3). As such, treatment with recombinant ASM was 45% less effective in reducing sphingomyelin storage in diseased cells treated with δ-tocopherol compared with those that did not receive this vitamin (Fig. 6C).
Fig. 6.

Effect of δ-tocopherol on the activity provided by recombinant ASM to diseased cells. (A) ASM activity in wild-type (Ctrl) or NPD-A patient fibroblasts (Dis) incubated for 4 hours in the absence vs. presence of 2.3 µg/ml recombinant ASM. (B) ASM activity in healthy (Ctrl) vs. imipramine-diseased endothelial cells or NPD-A fibroblasts (Dis) after incubation for 4 hours with 2.3 µg/ml recombinant ASM, which was added after treatment with 40 µM δ-tocopherol. Data are normalized to (A) wild-type fibroblasts without ASM addition and to (B) untreated cells (respective horizontal dashed lines). (C) NPD-A fibroblasts treated for 48 hours with 40 µM δ-tocopherol and labeled with fluorescent sphingomyelin were washed and incubated with 5 µg/ml recombinant ASM for 4 hours, after which the sphingomyelin levels were measured in a plate reader. Data show sphingomyelin levels as a percentage of that found in cells not incubated with ASM. All data are mean ± S.E.M. (n ≥ 4 independent wells). (A and B) *Comparison with wild-type fibroblasts without ASM addition; †comparison with untreated cells; (C) *comparison with untreated cells (all P < 0.05 by Student’s t test).
Then, because absolute uptake via the CAM pathway was relatively unaffected by δ-tocopherol, we tested uptake of recombinant ASM coupled to anti-ICAM NCs as an alternative approach (see Materials and Methods for NC characterization). Using ligand competition assays, this formulation has been verified to bind to ICAM-1, not M6PR, unlike naked ASM (Muro et al., 2006b). After 2 hours of incubation (Fig. 7A), total NC-mediated uptake of ASM (CAM pathway) was significantly enhanced versus uptake of naked ASM (clathrin route), both in the case of untreated diseased cells (48-fold enhancement by NCs) and δ-tocopherol–treated diseased cells (84-fold enhancement; Fig. 7A). As well, δ-tocopherol significantly reduced naked ASM uptake via the clathrin route (60% of untreated diseased cells; Fig. 7B), whereas the CAM pathway was unaffected (105% of untreated diseased cells), as expected based on previous experiments (Fig. 3).
Fig. 7.

Effect of δ-tocopherol on uptake of recombinant ASM via the clathrin vs. CAM pathways. Imipramine-diseased endothelial cells (Dis), activated overnight with TNFα and treated for 48 hours with 40 µM δ-tocopherol, were incubated for 2 hours with 2.1 µg/ml naked 125I-ASM or 125I-ASM coupled to anti-ICAM NCs (anti-ICAM/ASM NCs). After washing cells, an acid glycine solution was used to elute noninternalized ASM from the cell surface. ASM delivered into cells was then measured in the cell lysates. (A) Internalized ASM in cell lysates. (B) ASM uptake after treatment with δ-tocopherol as a percentage of untreated diseased cells (horizontal solid line). Data are mean ± S.E.M. (n ≥ 4 independent wells). *Comparison with untreated diseased cells; †comparison with naked ASM (P < 0.05 by Student’s t test).
Kinetics of the Tocopherol Effect on Lowering Endocytosis.
Although we identified a pathway (CAM) less affected by tocopherols, the fact that δ-tocopherol lowered uptake of recombinant enzymes used for ERT suggests that combination treatment using both strategies may need optimal timing. To evaluate whether this effect is reversible, we examined the ability of cells to restore dextran pinocytosis upon removal of tocopherols because this marker showed good indication of uptake effects (Fig. 2). As shown in Fig. 8A, dextran uptake remained affected 3 hours after removal of δ-tocopherol or CF3-T from the cell medium (15% and 28% reduction vs. untreated diseased cells, respectively). Yet, 8 hours after removal, dextran uptake returned to the vitamin-untreated level in diseased cells (0.98-fold and 0.95-fold), suggesting that this effect is transient. During this 8-hour period, sphingomyelin storage in cells treated with δ-tocopherol or CF3-T remained below that in vitamin-untreated diseased cells (26% to 41% reduction; Fig. 8B). Hence, restoration of endocytosis seems due to cessation of tocopherol-induced exocytosis, not an increase in the sphingomyelin storage levels after tocopherol removal. As a control, sphingomyelin storage did not increase to diseased levels until 24 hours postremoval of δ-tocopherol or CF3-T (0.92-fold and 0.83-fold storage vs. vitamin-untreated diseased cells; Fig. 8B). Similar findings were obtained by quantifying the number of dark-refringent storage compartments by phase-contrast microscopy (Supplemental Fig. 9). Finally, a similar trend was observed for uptake of anti-ICAM NCs, whose binding and endocytosis recovered 5 hours after removal of δ-tocopherol (Supplemental Fig. 10), indicating this effect is reversible and combination therapy may be an option if properly timed.
Fig. 8.

Kinetics of tocopherol effect on bulk fluid-phase uptake and sphingomyelin levels. (A) Control (Ctrl) and imipramine-diseased endothelial cells (Dis) were incubated with Texas Red dextran (1 hour pulse) 1 to 8 hours after a 48-hour treatment with 40 µM δ-tocopherol or 20 µM CF3-T. Dextran uptake was normalized to untreated diseased cells (horizontal solid line). Dextran uptake by untreated control cells is shown by the horizontal dashed line. (B) Sphingomyelin was stained with fluorescent lysenin 1 to 24 hours after a 48-hour treatment with δ-tocopherol or CF3-T, quantified as described in Fig. 1, and normalized to untreated control cells (horizontal dashed line). Sphingomyelin levels in untreated diseased cells are shown by the horizontal solid line. Data are mean ± S.E.M. (n ≥ 4 independent wells). *Comparison with untreated control cells; †comparison with untreated diseased cells (P < 0.05 by Student’s t test).
Discussion
Lysosomal exocytosis is a constitutive event (Schultz et al., 2011; Samie and Xu, 2014) whose induction offers an avenue for LD treatment (Chen et al., 2010; Medina et al., 2011; Xu et al., 2012; Spampanato et al., 2013; Samie and Xu, 2014; Cao et al., 2015; Long et al., 2016; Zhong et al., 2016). Yet, the effect of this strategy on vesicles derived from endocytic pathways is unknown. Endosome–lysosome (like autophagosome–lysosome) fusion is a transient event, followed by lysosome reformation once substrate processing is complete (Luzio et al., 2007a,b; Fraldi et al., 2010). However, several LDs exhibit defects in intracellular trafficking, membrane fusion, and lysosome reformation, resulting in accumulation of early endosomes (Holroyd et al., 1999; Simons and Gruenberg, 2000; Fukuda et al., 2006; Yu et al., 2010; Andersen and Moestrup, 2014; Teixeira et al., 2014). Therefore, materials contained within endocytic vesicles may be expelled from the cell upon exocytosis induction, reducing endocytic delivery of therapeutics, such as recombinant enzymes used for ERT. We evaluated this using tocopherols to induce lysosomal exocytosis. As suspected, this agent reduced endocytic uptake in NPD-A, the LD model studied, including dextran pinocytosis (Fig. 2) and receptor-mediated endocytosis of CTB, Tf, anti-ICAM NCs, and IgG-coated microparticles via caveolae-, clathrin-, CAM-, and phagocytosis-mediated mechanisms (Figs. 3 and 4), leading to decreased delivery and activity of a recombinant enzyme (Figs. 6 and 7).
This suggests that tocopherol-induced exocytosis affects endocytic vesicles and endosomes where separation of the ligand–receptor complex typically occurs prior to lysosomal trafficking (cargo) or recycling to the plasmalemma (receptor) (Mellman, 1996), lowering both cargo accumulation (Figs. 3 and 4) and number of EEA-1–positive early endosomes (Fig. 5). This would be possible if tocopherol-induced exocytosis operates through a mechanism common to different types of vesicles, which is supported by literature. For instance, endocytic recycling and vesicular trafficking along the endolysosomal route are calcium-dependent, just like lysosomal exocytosis (Knight, 2002; Luzio et al., 2007a,b; Lloyd-Evans et al., 2010; Yu et al., 2010; Cao et al., 2015). Additionally, tocopherols can undergo insertion in the membrane and alter its physical status (Ohyashiki et al., 1986; Bradford et al., 2003; Lemaire-Ewing et al., 2010). Because the expression and distribution of endocytic receptors, their adaptors, and/or signal transduction partners depend on the membrane physical properties, this could explain a generic effect of tocopherol on endocytosis regardless of the uptake route (Wells et al., 2010; Zingg, 2015). In fact, tocopherols can disrupt plasmalemma recruitment of protein kinase C, which is involved in all of the endocytic routes tested (Caron and Hall, 2001; Wells et al., 2010; Zingg, 2015).
Despite this generic effect, endocytosis was differently affected by δ-tocopherol for the caveolar-, clathrin-, phagocytic-, and CAM-mediated routes (Fig. 3). For the CAM pathway, the rate of anti-ICAM NC uptake was diminished by δ-tocopherol (Fig. 4A); yet, tocopherol enhanced NC binding to TNFα-activated cells (Fig. 4A), so that one effect balanced the other out (Fig. 3). Enhanced anti-ICAM NC binding by δ-tocopherol could be due to greater ICAM-1 expression or accessibility. If the first premise was true, binding to ICAM-1 should be greater upon δ-tocopherol treatment regardless whether or not cells were treated with TNFα; but this was not the case, ruling out this explanation. If tocopherol would have improved receptor access (Lemaire-Ewing et al., 2010; Wells et al., 2010), increased binding would also be expected regardless of TNFα, ruling out the second explanation. However, this could be explained if tocopherols induced generic exocytosis of intracellular vesicles: cells possess an intracellular pool of ICAM-1 in subplasmalemma vesicles that recycle back and forth the cell surface (Jo et al., 2010; Ghaffarian and Muro, 2014). The amount of ICAM-1 in these vesicles and the cell surface depends upon activation by cytokines such as TNFα (Descamps et al., 1997; Muro et al., 2003). Although tocopherols would not increase ICAM-1 expression (TNFα does), by enhancing exocytosis of intracellular ICAM-1, tocopherols would result in a greater membrane display of ICAM-1, supporting enhanced binding.
Similarly, δ-tocopherol reduced Tf uptake in TNFα-activated and nonactivated cells, although this effect was less acute in the former scenario (Fig. 3). Exocytosis and surface display of clathrin-associated receptors often sequestered in the Golgi–lysosome route in LDs (Dhami and Schuchman, 2004; Futerman and van Meer, 2004; Rappaport et al., 2014) would explain this. For the caveolar route, reduction of endocytic cargo by tocopherols could be due to several reasons. For instance, some tocopherol species (α-tocopherol) undergo membrane insertion predominantly into lipid rafts (Jiang et al., 2004; Royer et al., 2009), and treatment with γ- and δ-tocopherols interferes with sphingolipid synthesis (Jiang et al., 2004; Royer et al., 2009). Therefore, tocopherols may alter lipid raft formation and caveolae-mediated endocytosis, as observed (Fig. 3). Interestingly, TNFα increased caveolae-mediated uptake, in tune with previous reports (Wang et al., 2008; Chidlow and Sessa, 2010); yet, δ-tocopherol attenuated this effect (Fig. 4B).
For phagocytosis, impaired uptake of IgG-coated microparticles by δ-tocopherol seems counterintuitive because phagocytosis depends on endosome/lysosome exocytosis to recruit membrane at the cell surface for pseudopod extension (Champion and Mitragotri, 2006; Samie et al., 2013). Alterations in the membrane composition, structure, fluidity, etc., by δ-tocopherol may hinder large plasmalemma deformations. Although phagocytic uptake was improved by TNFα, consistent with literature (Nikolova and Russell, 1995), δ-tocopherol also reduced phagocytosis in the presence of this cytokine (Fig. 3).
Therefore, induction of lysosomal exocytosis as treatment of chronic diseases, such as LDs, may impact intracellular accumulation of endocytic cargo. Whether other exocytosis inductors lead to similar effects remains undetermined. This is important for the use of lysosomal exocytosis along with therapies needing intracellular delivery via endocytosis, such as ERT. For example, we observed reduced ASM activity delivered in cells treated with δ-tocopherol (Figs. 6 and 7), despite lack of an effect on endogenous ASM (Supplemental Fig. 8), indicating that tocopherols impact uptake of therapeutic enzymes. However, using lysosomal exocytosis to clear lysosomal storage first, followed by ERT, is an interesting alternative given that storage lowers uptake of recombinant enzymes (Dhami and Schuchman, 2004; Cardone et al., 2008; Rappaport et al., 2014). This is supported by the fact that reduced lysosomal storage was still observed 8 hours after δ-tocopherol removal (Fig. 8B; Supplemental Fig. 9), whereas endocytosis recovered between 5 and 8 hours (Fig. 8A; Supplemental Fig. 10). This duration is in relative agreement with observations on exocytosis induced by vitamin E derivatives (Xu et al., 2012) or by 2-hydroxypropyl-β-cyclodextrin (Chen et al., 2010; Liu et al., 2010). Said duration may depend on the cell type, lysosomal disease, agent employed to induce exocytosis, its concentration, in vivo pharmacokinetics, etc. As such, the timing for the use of agents inducing exocytosis along with agents requiring endocytosis must be investigated for each scenario.
Similarly, the effects of agents inducing exocytosis on the uptake of drug delivery systems may vary depending on the nanocarrier type, surface characteristics, targeting moieties, and drug cargo. For instance, the carrier composition, surface charge, stiffness, etc., can modulate cell interaction, uptake, and trafficking (Howard et al., 2014; Merkel et al., 2012; Muro, 2012a; Farokhirad et al., 2019). Yet, this varies for different targets: e.g., polystyrene, poly(lactic-co-glycolic acid), and DNA-built nanocarriers targeting ICAM-1 showed relatively similar binding and uptake in cell culture and biodistribution in mice (Muro et al., 2006a; Muro, 2014; Garnacho et al., 2017). Also, the size of a drug carrier impacts its endocytic uptake, yet this effect depends on the mechanism of endocytosis: clathrin- and caveolae-mediated pathways are more severely affected by increasing the size of drug vehicles compared with CAM-mediated endocytosis (Champion and Mitragotri, 2006; Muro et al., 2008; Merkel et al., 2012; Howard et al., 2014). Carrier surface-to-volume ratio and angle of interaction with a cell also modulate uptake (Champion and Mitragotri, 2006), as well as the number of targeting moieties displayed on a nanocarrier (Serrano et al., 2016). Furthermore, the drug carried by a drug delivery vehicle can influence intracellular uptake and trafficking. For instance, ASM used in this work reduces NPD-A lysosomal storage (Garnacho et al., 2017). Because said lysosomal storage decreases uptake and endolysosomal trafficking in a “traffic jam” type of effect, by decreasing lysosomal storage, ASM also helps improve uptake and endolysosomal trafficking (Rappaport et al., 2016). Yet, other drugs without this property may be more severely affected by agents inducing exocytosis. In fact, many pharmaceutical agents impact negatively endocytosis and intracellular sorting, including drugs that alter the cytoskeleton, lysosomotropic compounds, and aminoglycoside antibiotics, which are used to fight certain cancers, infections, parasitoses, etc. (Muro, 2018). Therefore, it is expected that drug and nanocarrier parameters will further influence uptake efficacy in cells treated with δ-tocopherol or other agents causing exocytosis. Nevertheless, the fact that δ-tocopherol impacted uptake via all pathways examined even though very different cargoes were tested (a sugar polymer, proteins, and submicrometer- or micrometer-sized particles) suggests that any drug vehicle using endocytosis to access cells may be affected by agents that induce exocytosis.
Altogether, the results reported in this work will help guide future in vivo work to evaluate combination therapies for LDs using induction of lysosomal exocytosis and ERT, to minimize potential side effects and enhance treatment outcomes.
Acknowledgments
We thank Dr. Edward Schuchman (Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, NY) for providing recombinant human ASM.
Abbreviations
- ASM
acid sphingomyelinase
- CAM
cell adhesion molecule
- CTB
cholera toxin subunit B
- ERT
enzyme replacement therapy
- FITC
fluorescein isothiocyanate
- HEX
hexosaminidase
- ICAM-1
intercellular adhesion molecule 1
- LD
lysosomal disorder
- NPC1
type C1 Niemann-Pick disease
- NPD-A
type A Niemann-Pick disease
- Tf
transferrin
- TNFα
tumor necrosis factor α
Authorship Contributions
Participated in research design: Muro, Marugan, Zheng.
Conducted experiments: Rappaport, Manthe, Long, Solomon, Hildreth, Veluvolu, Gugutkov.
Contributed new reagents or analytic tools: Marugan, Zheng.
Performed data analysis: Rappaport, Manthe, Long, Solomon, Hildreth, Veluvolu, Gugutkov, Zheng, Muro.
Wrote or contributed to the writing of the manuscript: Manthe, Rappaport, Solomon, Marugan, Zheng, Muro.
Footnotes
This work was supported by the National Institutes of Health [Grant R01 HL98416]; Spanish Ministry of Economy and Competitiveness—MINECO/FEDER Project SEV-2014-0425, Spanish Ministry of Science, Innovation and University—MINECO/EXPLORA Project SAF2017-91909-EXP and MINECO/RETOS Project RTI2018-101034-B-I00, and CERCA Program of the Generalitat de Catalunya (awarded to S.M.); National Science Foundation Graduate Research Fellowship [DGE-0750616], University of Maryland Flagship Fellowship, and the National Institutes of Health Ruth L. Kirschstein Predoctoral Fellowship [F31-HL128121 (awarded to R.L.M.)]; Howard Hughes Medical Institute Fellowship Program under the University of Maryland Undergraduate Science Education Program (awarded to J.A.R.); and Intramural Research Program of the National Institutes of Health National Center for Advancing Translational Sciences (awarded to W.Z.).
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Andersen CB, Moestrup SK. (2014) How calcium makes endocytic receptors attractive. Trends Biochem Sci 39:82–90. [DOI] [PubMed] [Google Scholar]
- Ballabio A, Gieselmann V. (2009) Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta 1793:684–696. [DOI] [PubMed] [Google Scholar]
- Boado RJ, Hui EK, Lu JZ, Zhou QH, Pardridge WM. (2011) Reversal of lysosomal storage in brain of adult MPS-I mice with intravenous Trojan horse-iduronidase fusion protein. Mol Pharm 8:1342–1350. [DOI] [PubMed] [Google Scholar]
- Bradford A, Atkinson J, Fuller N, Rand RP. (2003) The effect of vitamin E on the structure of membrane lipid assemblies. J Lipid Res 44:1940–1945. [DOI] [PubMed] [Google Scholar]
- Cao Q, Zhong XZ, Zou Y, Zhang Z, Toro L, Dong XP. (2015) BK channels alleviate lysosomal storage diseases by providing positive feedback regulation of lysosomal Ca2+ release. Dev Cell 33:427–441. [DOI] [PubMed] [Google Scholar]
- Cardone M, Porto C, Tarallo A, Vicinanza M, Rossi B, Polishchuk E, Donaudy F, Andria G, De Matteis MA, Parenti G. (2008) Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics 1:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron E, Hall A. (2001) Phagocytosis, in Endocytosis (Marsh M. ed) pp 58–77, Oxford University Press, Oxford, UK. [Google Scholar]
- Champion JA, Mitragotri S. (2006) Role of target geometry in phagocytosis. Proc Natl Acad Sci USA 103:4930–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FW, Li C, Ioannou YA. (2010) Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS One 5:e15054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidlow JH, Jr, Sessa WC. (2010) Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc Res 86:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrastina A, Massey KA, Schnitzer JE. (2011) Overcoming in vivo barriers to targeted nanodelivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol 3:421–437. [DOI] [PubMed] [Google Scholar]
- Conner SD, Schmid SL. (2003) Regulated portals of entry into the cell. Nature 422:37–44. [DOI] [PubMed] [Google Scholar]
- Descamps L, Cecchelli R, Torpier G. (1997) Effects of tumor necrosis factor on receptor-mediated endocytosis and barrier functions of bovine brain capillary endothelial cell monolayers. J Neuroimmunol 74:173–184. [DOI] [PubMed] [Google Scholar]
- Dhami R, Schuchman EH. (2004) Mannose 6-phosphate receptor-mediated uptake is defective in acid sphingomyelinase-deficient macrophages: implications for Niemann-Pick disease enzyme replacement therapy. J Biol Chem 279:1526–1532. [DOI] [PubMed] [Google Scholar]
- Duncan R, Richardson SC. (2012) Endocytosis and intracellular trafficking as gateways for nanomedicine delivery: opportunities and challenges. Mol Pharm 9:2380–2402. [DOI] [PubMed] [Google Scholar]
- Farokhirad S, Ranganathan A, Myerson J, Muzykantov VR, Ayyaswamy PS, Eckmann DM, Radhakrishnan R. (2019) Stiffness can mediate balance between hydrodynamic forces and avidity to impact the targeting of flexible polymeric nanoparticles in flow. Nanoscale 11:6916–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraldi A, Annunziata F, Lombardi A, Kaiser HJ, Medina DL, Spampanato C, Fedele AO, Polishchuk R, Sorrentino NC, Simons K, et al. (2010) Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J 29:3607–3620. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Fukuda T, Ewan L, Bauer M, Mattaliano RJ, Zaal K, Ralston E, Plotz PH, Raben N. (2006) Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol 59:700–708. [DOI] [PubMed] [Google Scholar]
- Futerman AH, van Meer G. (2004) The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol 5:554–565. [DOI] [PubMed] [Google Scholar]
- Garnacho C, Dhami R, Simone E, Dziubla T, Leferovich J, Schuchman EH, Muzykantov V, Muro S. (2008) Delivery of acid sphingomyelinase in normal and Niemann-Pick disease mice using intercellular adhesion molecule-1-targeted polymer nanocarriers. J Pharmacol Exp Ther 325:400–408. [DOI] [PubMed] [Google Scholar]
- Garnacho C, Dhami R, Solomon M, Schuchman EH, Muro S. (2017) Enhanced delivery and effects of acid sphingomyelinase by ICAM-1-targeted nanocarriers in type B Niemann-Pick disease mice. Mol Ther 25:1686–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaffarian R, Muro S. (2014) Distinct subcellular trafficking resulting from monomeric vs multimeric targeting to endothelial ICAM-1: implications for drug delivery. Mol Pharm 11:4350–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Miranda SR, Xiong X, Dagan A, Gatt S, Schuchman EH. (1999) Characterization of human acid sphingomyelinase purified from the media of overexpressing Chinese hamster ovary cells. Biochim Biophys Acta 1432:251–264. [DOI] [PubMed] [Google Scholar]
- Hillaireau H, Couvreur P. (2009) Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol Life Sci 66:2873–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holroyd C, Kistner U, Annaert W, Jahn R. (1999) Fusion of endosomes involved in synaptic vesicle recycling. Mol Biol Cell 10:3035–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortsch R, Lee E, Erathodiyil N, Hebbar S, Steinert S, Lee JY, Chua DS, Kraut R. (2010) Glycolipid trafficking in Drosophila undergoes pathway switching in response to aberrant cholesterol levels. Mol Biol Cell 21:778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard M, Zern BJ, Anselmo AC, Shuvaev VV, Mitragotri S, Muzykantov V. (2014) Vascular targeting of nanocarriers: perplexing aspects of the seemingly straightforward paradigm. ACS Nano 8:4100–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J, Northrup L, Bhowmick T, Muro S. (2012) Enhanced delivery of α-glucosidase for Pompe disease by ICAM-1-targeted nanocarriers: comparative performance of a strategy for three distinct lysosomal storage disorders. Nanomedicine (Lond) 8:731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J, Serrano D, Bhowmick T, Kumar K, Shen Y, Kuo YC, Garnacho C, Muro S. (2011) Enhanced endothelial delivery and biochemical effects of α-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J Control Release 149:323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz R, Ferlinz K, Sandhoff K. (1994) The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol Chem Hoppe Seyler 375:447–450. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Wong J, Fyrst H, Saba JD, Ames BN. (2004) gamma-Tocopherol or combinations of vitamin E forms induce cell death in human prostate cancer cells by interrupting sphingolipid synthesis. Proc Natl Acad Sci USA 101:17825–17830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo JH, Kwon MS, Choi HO, Oh HM, Kim HJ, Jun CD. (2010) Recycling and LFA-1-dependent trafficking of ICAM-1 to the immunological synapse. J Cell Biochem 111:1125–1137. [DOI] [PubMed] [Google Scholar]
- Klein D, Büssow H, Fewou SN, Gieselmann V. (2005) Exocytosis of storage material in a lysosomal disorder. Biochem Biophys Res Commun 327:663–667. [DOI] [PubMed] [Google Scholar]
- Knight DE. (2002) Calcium-dependent transferrin receptor recycling in bovine chromaffin cells. Traffic 3:298–307. [DOI] [PubMed] [Google Scholar]
- Kuech EM, Brogden G, Naim HY. (2016) Alterations in membrane trafficking and pathophysiological implications in lysosomal storage disorders. Biochimie 130:152–162. [DOI] [PubMed] [Google Scholar]
- Lemaire-Ewing S, Desrumaux C, Néel D, Lagrost L. (2010) Vitamin E transport, membrane incorporation and cell metabolism: is alpha-tocopherol in lipid rafts an oar in the lifeboat? Mol Nutr Food Res 54:631–640. [DOI] [PubMed] [Google Scholar]
- Liscum L, Faust JR. (1987) Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J Biol Chem 262:17002–17008. [PubMed] [Google Scholar]
- Liu B, Ramirez CM, Miller AM, Repa JJ, Turley SD, Dietschy JM. (2010) Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J Lipid Res 51:933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Evans E, Waller-Evans H, Peterneva K, Platt FM. (2010) Endolysosomal calcium regulation and disease. Biochem Soc Trans 38:1458–1464. [DOI] [PubMed] [Google Scholar]
- Long Y, Xu M, Li R, Dai S, Beers J, Chen G, Soheilian F, Baxa U, Wang M, Marugan JJ, et al. (2016) Induced pluripotent stem cells for disease modeling and evaluation of therapeutics for Niemann-Pick disease type A. Stem Cells Transl Med 5:1644–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Bright NA, Pryor PR. (2007a) The role of calcium and other ions in sorting and delivery in the late endocytic pathway. Biochem Soc Trans 35:1088–1091. [DOI] [PubMed] [Google Scholar]
- Luzio JP, Pryor PR, Bright NA. (2007b) Lysosomes: fusion and function. Nat Rev Mol Cell Biol 8:622–632. [DOI] [PubMed] [Google Scholar]
- Marks DL, Pagano RE. (2002) Endocytosis and sorting of glycosphingolipids in sphingolipid storage disease. Trends Cell Biol 12:605–613. [DOI] [PubMed] [Google Scholar]
- Marugan JJ, Zheng W, Xiao J, and McKew J (2014) inventors, National Institutes of Health assignee. Tocopherol and tocopheryl quinone derivatives as correctors of lysosomal storage disorders. U.S. patent 2014 WO/2014/078573.
- McMahon HT, Boucrot E. (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12:517–533. [DOI] [PubMed] [Google Scholar]
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, et al. (2011) Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell 21:421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I. (1996) Endocytosis and molecular sorting. Annu Rev Cell Dev Biol 12:575–625. [DOI] [PubMed] [Google Scholar]
- Merkel TJ, Chen K, Jones SW, Pandya AA, Tian S, Napier ME, Zamboni WE, DeSimone JM. (2012) The effect of particle size on the biodistribution of low-modulus hydrogel PRINT particles. J Control Release 162:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro S. (2012a) Challenges in design and characterization of ligand-targeted drug delivery systems. J Control Release 164:125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro S. (2012b) Strategies for delivery of therapeutics into the central nervous system for treatment of lysosomal storage disorders. Drug Deliv Transl Res 2:169–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro S. (2014) A DNA-device that mediates selective endosomal escape and intracellular delivery of drugs and biological. Adv Funct Mater 24:2899–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro S. (2018) Alterations in cellular processes involving vesicular trafficking and implications in drug delivery. Biomimetics 3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro S, Dziubla T, Qiu W, Leferovich J, Cui X, Berk E, Muzykantov VR. (2006a) Endothelial targeting of high-affinity multivalent polymer nanocarriers directed to intercellular adhesion molecule 1. J Pharmacol Exp Ther 317:1161–1169. [DOI] [PubMed] [Google Scholar]
- Muro S, Garnacho C, Champion JA, Leferovich J, Gajewski C, Schuchman EH, Mitragotri S, Muzykantov VR. (2008) Control of endothelial targeting and intracellular delivery of therapeutic enzymes by modulating the size and shape of ICAM-1-targeted carriers. Mol Ther 16:1450–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro S, Schuchman EH, Muzykantov VR. (2006b) Lysosomal enzyme delivery by ICAM-1-targeted nanocarriers bypassing glycosylation- and clathrin-dependent endocytosis. Mol Ther 13:135–141. [DOI] [PubMed] [Google Scholar]
- Muro S, Wiewrodt R, Thomas A, Koniaris L, Albelda SM, Muzykantov VR, Koval M. (2003) A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J Cell Sci 116:1599–1609. [DOI] [PubMed] [Google Scholar]
- Nichols B. (2003) Caveosomes and endocytosis of lipid rafts. J Cell Sci 116:4707–4714. [DOI] [PubMed] [Google Scholar]
- Nikolova EB, Russell MW. (1995) Dual function of human IgA antibodies: inhibition of phagocytosis in circulating neutrophils and enhancement of responses in IL-8-stimulated cells. J Leukoc Biol 57:875–882. [DOI] [PubMed] [Google Scholar]
- Ohyashiki T, Ushiro H, Mohri T. (1986) Effects of alpha-tocopherol on the lipid peroxidation and fluidity of porcine intestinal brush-border membranes. Biochim Biophys Acta 858:294–300. [DOI] [PubMed] [Google Scholar]
- Parkinson-Lawrence EJ, Shandala T, Prodoehl M, Plew R, Borlace GN, Brooks DA. (2010) Lysosomal storage disease: revealing lysosomal function and physiology. Physiology (Bethesda) 25:102–115. [DOI] [PubMed] [Google Scholar]
- Rappaport J, Garnacho C, Muro S. (2014) Clathrin-mediated endocytosis is impaired in type A-B Niemann-Pick disease model cells and can be restored by ICAM-1-mediated enzyme replacement. Mol Pharm 11:2887–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappaport J, Manthe RL, Garnacho C, Muro S. (2015) Altered clathrin-independent endocytosis in type A Niemann-Pick disease cells and rescue by ICAM-1-targeted enzyme delivery. Mol Pharm 12:1366–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappaport J, Manthe RL, Solomon M, Garnacho C, Muro S. (2016) A comparative study on the alterations of endocytic pathways in multiple lysosomal storage disorders. Mol Pharm 13:357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer MC, Lemaire-Ewing S, Desrumaux C, Monier S, Pais de Barros JP, Athias A, Néel D, Lagrost L. (2009) 7-Ketocholesterol incorporation into sphingolipid/cholesterol-enriched (lipid raft) domains is impaired by vitamin E: a specific role for alpha-tocopherol with consequences on cell death. J Biol Chem 284:15826–15834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samie M, Wang X, Zhang X, Goschka A, Li X, Cheng X, Gregg E, Azar M, Zhuo Y, Garrity AG, et al. (2013) A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell 26:511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samie MA, Xu H. (2014) Lysosomal exocytosis and lipid storage disorders. J Lipid Res 55:995–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchman EH, Desnick RJ. (2017) Types A and B Niemann-Pick disease. Mol Genet Metab 120:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz ML, Tecedor L, Chang M, Davidson BL. (2011) Clarifying lysosomal storage diseases. Trends Neurosci 34:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano D, Bhowmick T, Chadha R, Garnacho C, Muro S. (2012) Intercellular adhesion molecule 1 engagement modulates sphingomyelinase and ceramide, supporting uptake of drug carriers by the vascular endothelium. Arterioscler Thromb Vasc Biol 32:1178–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano D, Manthe RL, Paul E, Chadha R, Muro S. (2016) How carrier size and valency modulate receptor-mediated signaling: understanding the link between binding and endocytosis of ICAM-1-targeted carriers. Biomacromolecules 17:3127–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Gruenberg J. (2000) Jamming the endosomal system: lipid rafts and lysosomal storage diseases. Trends Cell Biol 10:459–462. [DOI] [PubMed] [Google Scholar]
- Solomon M, Muro S. (2017) Lysosomal enzyme replacement therapies: historical development, clinical outcomes, and future perspectives. Adv Drug Deliv Rev 118:109–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato C, Feeney E, Li L, Cardone M, Lim JA, Annunziata F, Zare H, Polishchuk R, Puertollano R, Parenti G, et al. (2013) Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med 5:691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss K, Goebel C, Runz H, Möbius W, Weiss S, Feussner I, Simons M, Schneider A. (2010) Exosome secretion ameliorates lysosomal storage of cholesterol in Niemann-Pick type C disease. J Biol Chem 285:26279–26288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira CA, Miranda CO, Sousa VF, Santos TE, Malheiro AR, Solomon M, Maegawa GH, Brites P, Sousa MM. (2014) Early axonal loss accompanied by impaired endocytosis, abnormal axonal transport, and decreased microtubule stability occur in the model of Krabbe’s disease. Neurobiol Dis 66:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lim EJ, Toborek M, Hennig B. (2008) The role of fatty acids and caveolin-1 in tumor necrosis factor alpha-induced endothelial cell activation. Metabolism 57:1328–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SR, Jennings MH, Rome C, Hadjivassiliou V, Papas KA, Alexander JS. (2010) Alpha-, gamma- and delta-tocopherols reduce inflammatory angiogenesis in human microvascular endothelial cells. J Nutr Biochem 21:589–597. [DOI] [PubMed] [Google Scholar]
- Whitley CB, Draper KA, Dutton CM, Brown PA, Severson SL, France LA. (1989a) Diagnostic test for mucopolysaccharidosis. II. Rapid quantification of glycosaminoglycan in urine samples collected on a paper matrix. Clin Chem 35:2074–2081. [PubMed] [Google Scholar]
- Whitley CB, Ridnour MD, Draper KA, Dutton CM, Neglia JP. (1989b) Diagnostic test for mucopolysaccharidosis. I. Direct method for quantifying excessive urinary glycosaminoglycan excretion. Clin Chem 35:374–379. [PubMed] [Google Scholar]
- Wisniewski KE, Golabek AA, Kida E. (1994) Increased urine concentration of subunit c of mitochondrial ATP synthase in neuronal ceroid lipofuscinoses patients. J Inherit Metab Dis 17:205–210. [DOI] [PubMed] [Google Scholar]
- Xu M, Liu K, Swaroop M, Porter FD, Sidhu R, Firnkes S, Ory DS, Marugan JJ, Xiao J, Southall N, et al. (2012) δ-Tocopherol reduces lipid accumulation in Niemann-Pick type C1 and Wolman cholesterol storage disorders [published correction appears in J Biol Chem (2013) 288:296]. J Biol Chem 287:39349–39360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465:942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong XZ, Sun X, Cao Q, Dong G, Schiffmann R, Dong XP. (2016) BK channel agonist represents a potential therapeutic approach for lysosomal storage diseases. Sci Rep 6:33684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingg JM. (2015) Vitamin E: a role in signal transduction. Annu Rev Nutr 35:135–173. [DOI] [PubMed] [Google Scholar]