

Abstract
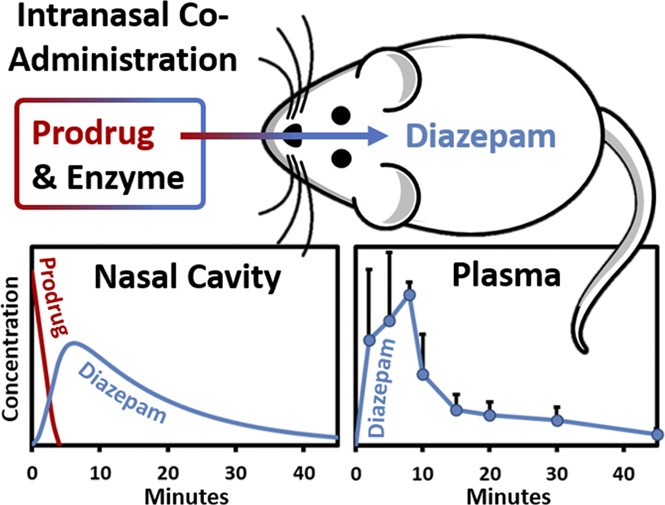
Intranasal administration is an attractive route for systemic delivery of small, lipophilic drugs because they are rapidly absorbed through the nasal mucosa into systemic circulation. However, the low solubility of lipophilic drugs often precludes aqueous nasal spray formulations. A unique approach to circumvent solubility issues involves coadministration of a hydrophilic prodrug with an exogenous converting enzyme. This strategy not only addresses poor solubility but also leads to an increase in the chemical activity gradient driving drug absorption. Herein, we report plasma and brain concentrations in rats following coadministration of a hydrophilic diazepam prodrug, avizafone, with the converting enzyme human aminopeptidase B. Single doses of avizafone equivalent to diazepam at 0.500, 1.00, and 1.50 mg/kg were administered intranasally, resulting in 77.8% ± 6.0%, 112% ± 10%, and 114% ± 7% bioavailability; maximum plasma concentrations 71.5 ± 9.3, 388 ± 31, and 355 ± 187 ng/ml; and times to peak plasma concentration 5, 8, and 5 minutes for each dose level, respectively. Both diazepam and a transient intermediate were absorbed. Enzyme kinetics incorporated into a physiologically based pharmacokinetic model enabled estimation of the first-order absorption rate constants: 0.0689 ± 0.0080 minutes−1 for diazepam and 0.122 ± 0.022 minutes−1 for the intermediate. Our results demonstrate that diazepam, which is practically insoluble, can be delivered intranasally with rapid and complete absorption by coadministering avizafone with aminopeptidase B. Furthermore, even faster rates of absorption might be attained simply by increasing the enzyme concentration, potentially supplanting intravenous diazepam or lorazepam or intramuscular midazolam in the treatment of seizure emergencies.
Introduction
Status epilepticus and acute repetitive seizures are serious medical emergencies that can cause lasting neurologic damage and other life-threatening complications. Prolonged seizure activity associated with these conditions may lead to hypoxia, metabolic acidosis, hyperthermia, aspiration, pulmonary edema, brain injury, and an increase in the likelihood of experiencing additional seizures in the future (Mirski and Varelas, 2008). The risk of injury, medical complications, or death can be mitigated by prompt medical treatment (Pellock, 2007; Sutter et al., 2013). Benzodiazepines are the drugs of choice for treating seizure emergencies. Intravenous diazepam (DZP), intravenous lorazepam, or intramuscular midazolam (MDZ) are the preferred first-line rescue therapies (Glauser et al., 2016). However, drug delivery by injection can be problematic, especially when administered by nonmedical personnel. Injection is difficult to carry out on obese or emaciated individuals, in the presence of multiple layers of clothing, or in public places. Furthermore, treatment of seizure emergencies can be significantly delayed if the patient must wait for medical personnel or transportation to the emergency room. Therefore, there is a need for outpatient treatments that can be administered immediately at the onset of a seizure by nonmedical personnel.
Currently, the only outpatient treatment of a seizure emergency approved by the United States Food and Drug Administration (FDA) is rectally administered DZP (Diastat). Patients often object to this route of administration despite its effectiveness (Haut et al., 2016). Off-label alternatives to rectal diazepam including intranasal (IN) and buccal MDZ, using, respectively, injectable and oral solution formulations (Maglalang et al., 2018). Other routes to deliver benzodiazepines for the treatment of seizure emergencies currently under development include buccal DZP films (Heller et al., 2018) and IN sprays for DZP or MDZ (Agarwal et al., 2013; Bancke et al., 2013).
Intranasal DZP is an attractive treatment option because administration is easily performed by nonmedical personnel, and it has an extended duration of action with low risk of adverse events (Maglalang et al., 2018). However, the poor aqueous solubility of benzodiazepines presents a challenge in formulating them as nasal sprays because they require cosolvents or other additives to improve solubility (Kälviäinen, 2015). These solubilizing excipients can be highly irritating to nasal mucosa. Transient epistaxis, nasal discomfort, nasal congestion, and rhinitis have been reported as side effects with some of these formulations, including those with DZP (Agarwal et al., 2013; Buck, 2013; Sperling et al., 2014). Further, the time to reach the maximum plasma concentration (tmax) for IN DZP formulations utilizing solubilizing excipients is relatively slow, ranging from 0.75 to 1.5 hours (Agarwal et al., 2013; Ivaturi et al., 2013; Maggio and Pillion, 2013).
One strategy to overcome these challenges is to formulate a nasal DZP spray in an aqueous vehicle employing a water-soluble lysine prodrug of DZP, avizafone (AVF). Although prodrug formulations typically rely on endogenous enzymes for in vivo biotransformation to the active drug (Jornada et al., 2015), unpublished results from a pilot canine study performed by our group suggested that exogenous enzyme would need to be included in the formulation to achieve the desired prodrug conversion rate and bioavailability. A series of in vitro experiments combining water soluble benzodiazepine prodrugs with the fungal converting enzyme Aspergillus oryzae protease (AOP) demonstrated that the formulations were capable of producing supersaturated solutions of the active drug, leading to an enhanced permeation rate of drug across cultured cell monolayers (Kapoor and Siegel, 2013; Kapoor et al., 2014, 2016; Siegel et al., 2015). Further in vitro studies characterized the conversion kinetics of AVF to DZP (Rautiola et al., 2018), which first involves enzymatically catalyzed release of the lysine and formation of an open-ring intermediate (ORI), followed by spontaneous closure of ORI to form DZP (Fig. 1). These studies also showed that supersaturated DZP undergoes liquid/liquid phase separation at a critical degree of supersaturation, and resists crystallization over periods of days.
Fig. 1.

Prodrug/enzyme reaction scheme. Formation of DZP by enzymatic hydrolysis of AVF with APB progresses through an intermediate, ORI.
On the basis of these in vitro results, we hypothesized that intranasal administration of prodrug/enzyme formulations should result in rapid and complete in vivo absorption of DZP, with rapid distribution into the brain. In the present study, we tested this hypothesis in a rat model, in which we replaced AOP with a human enzyme, aminopeptidase B (APB), and compared the plasma and brain pharmacokinetics of IN AVF/APB with IN AVF and intravenous (IV) DZP. In addition to standard noncompartmental pharmacokinetic analyses, we augmented an established physiologically based pharmacokinetic (PKPB) model to incorporate mechanistic components, including enzyme reaction kinetics, absorption of both ORI and DZP across the nasal mucosa, supersaturation of DZP, and liquid-liquid phase separation of DZP. Finally, we carried out preliminary histologic studies to determine whether acute damage would result from the AVF/APB formulations.
Materials and Methods
Experimental Design.
The primary aim of this study was to determine whether IN administration of an AVF/APB combination to rats would result in rapid absorption of DZP without damaging the nasal mucosa. In brief, single doses of admixed solutions of AVF and APB equivalent to DZP at either 1.50, 1.00, or 0.500 mg/kg were deposited in the nasal cavities of rats. Plasma samples and brains were collected at time points ranging from 2 to 90 minutes. DZP concentrations measured in these tissues were compared against concentrations measured in samples collected from rats that were administered a 1.00 mg/kg dose of DZP i.v. Nasal tissue samples collected from rats sacrificed at the 90-minute time point were examined for lesions. Details about the dosing and sample collection schedule are given in the sections that follow and summarized in the supplemental data (Supplemental Table 1S).
Animals.
All animal experiments were approved by the Institutional Animal Care and Use Committees of the University of Minnesota (protocol 1508-32889A, principal investigator: J.C.C.) and HealthPartners (protocol 14-073, principal investigator: L.R.H.). The experiments were conducted at the HealthPartners Animal Care Facility and in accordance with the Guide for the Care and Use of Laboratory Animals (U.S. National Institutes of Health). Sixty-six male Sprague Dawley rats 8 to 9 weeks old (0.271 ± 0.023 kg) from Envigo RMS, Inc. (Indianapolis, IN) were used for this study. Rats were housed under controlled environmental conditions with a standard 12-hour light/dark cycle and free access to food and water.
Formulation Components.
S-avizafone dihydrochloride (AVF•2HCl) was synthesized with >99% purity, 99% enantiomeric excess from 5-chloro-2-methylaminobenzophenone according to the procedure described previously (Kapoor et al., 2016). Recombinant human aminopeptidase B (APB) enzyme was produced by inserting the aminopeptidase B gene (RNPEP) DNA sequence encoding for APB and a His tag into a pQE-30 Xa vector, transforming the JM109 strain of Escherichia coli with the resultant plasmid, inducing APB expression with IPTG, and purifying the expressed APB by affinity capture chromatography (Ogawa et al., 2014; Ohnishi et al., 2015). Purity of APB was >80% by gel electrophoresis. Phosphate-buffered saline (PBS) IN delivery vehicle was prepared at pH 7.4 and concentration 20 mM using sodium phosphate dibasic and sodium phosphate monobasic purchased from Fisher Scientific, Inc. (cat. nos. BP332 and BP329; Hampton, NH), made isotonic with sodium chloride purchased from EMD Millipore (cat. no. 567440; Burlington, MA), and sterile-filtered during formulation. For the intravenous control arm of the study, a commercial formulation of injectable diazepam USP (5 mg/ml DZP, 40% propylene glycol, 10% alcohol, 5% benzoate-benzoic acid, and 1.5% benzyl alcohol) was purchased from Hospira, Inc. (NDC 0409-3213-12, lot 58-257-EV; Rocky Mount, NC).
Enzyme Kinetics.
The Michaelis constant (KM) and maximum reaction velocity (Vmax) for the hydrolysis of AVF by APB in pH 7.4 PBS at 32°C were determined by fitting the Michaelis-Menten equation to the initial rates of substrate consumption for a series of AVF concentrations. Reactions with 62.5–4000 μM AVF and 15 μg/ml (0.203 μM) APB were carried out in an Eppendorf Thermomixer 5436 at 500 rpm. After 1 minute, APB was denatured by addition of methanol. Cyclization of ORI to DZP was allowed to progress to completion before measurement of UV spectra of the quenched reaction mixtures in a Cary 100 Bio UV/Vis spectrophotometer (Agilent Technologies, Inc., Santa Clara, CA). The second derivative of the spectrum at 338 nm was used to quantify DZP, and this quantity was taken to be the molar equivalent to the amount of AVF consumed. A curve was fit to the AVF consumption versus initial AVF concentration with KM and Vmax as fitting parameters using the nlinfit function in MATLAB version R2017b by The MathWorks, Inc. (Natick, MA).
Dosing.
For IN dosing, rats were anesthetized, placed in the supine position, and cannulated. Rats designated for nasal tissue histology were not cannulated. Solutions of AVF and APB prepared in PBS were admixed to the appropriate concentration for each animal immediately prior to administration. See Table 1 for dose levels. After mixing, the formulation was quickly instilled into the nasal cavity using an Eppendorf pipettor with a gel loading pipette tip inserted to a depth of 14 mm past the nares. A total volume of 30 μl was delivered, 15 μl into the right nostril followed by 15 μl into the left nostril within 0.5 minutes. There were four IN dose groups: AVF/APB at low, medium, and high doses, and an AVF-only group at the medium dose. These doses were chosen because DZP near 1 mg/kg is commonly used in rat studies and results in plasma concentrations in rats that are clinically relevant.
TABLE 1.
IN doses
| Dose Level | Equivalent DZP | AVF•2HCl | APB |
|---|---|---|---|
| mg/kg | mg/kg | μg/kg | |
| High | 1.50 | 2.66 | 91.4 |
| Medium | 1.00 | 1.77 | 60.7 |
| Low | 0.500 | 0.885 | 30.4 |
For intravenous dosing, a commercial formulation of DZP was injected directly into the femoral vein. A small incision was made in the skin along the midline of the left hind leg of anesthetized rats that were already cannulated in the right femoral vein. The incision was enlarged by blunt dissection to expose the left femoral vein. A 30-gauge needle was inserted into the vein to deliver a bolus dose of 1 mg/kg (medium-dose level, ∼54 μl) of DZP using a 100-μl syringe. Upon withdrawal of the needle, moderate pressure was applied to the site of injection with a cotton swab for 5–10 seconds to facilitate coagulation and seal the puncture.
Sample Collection.
Prior to cannulation for serial blood draws, rats were anesthetized with a 1.0-ml/kg subcutaneous ketamine cocktail containing 43 mg/ml ketamine HCl, 8.6 mg/ml of xylazine HCl, and 1.4 mg/ml acepromazine. Anesthesia was maintained throughout surgery, dosing, and sample collection by administration of subcutaneous boosters of 50 mg/kg of ketamine alternating with boosters of ketamine cocktail. Blood was drawn from the right femoral vein through a PU 3Fr 25-cm catheter (cat. no. C30PU-RFV1308; Instech Laboratories Inc., Plymouth Meeting, PA). Leapfrog sampling was performed, with one cohort of three rats sampled at 2, 10, 20, 30, and 60 minutes and another cohort of three rats sampled at 5, 15, 45, 75, and 90 minutes for each dosing group. Each blood draw was 500 μl, with a 500-μl saline replacement volume pushed following every other draw. Whole blood was centrifuged at 2000g for 3 minutes in BD Microtainer Tubes (cat. no. 365974; Becton Dickinson Co., Franklin Lakes, NJ) containing EDTA to obtain plasma. Additional cohorts of three rats each were designated for destructive sampling to obtain 2-, 5-, 8-, and 10-minute brain samples for the medium-dose IV DZP and medium-dose IN AVF/APB groups. At the final time points, blood samples were collected by cardiac puncture. The rats were then euthanized by transcardial perfusion with 120 ml saline. Brains, including the olfactory bulbs, were removed by gross dissection and hemisected sagittally. Brain and plasma samples were snap-frozen in liquid nitrogen and stored at −80°C until analysis.
Liquid Chromatography–Tandem Mass Spectrometry Method.
DZP and AVF were extracted from plasma and brain samples and quantified by LC-MS/MS. For extraction, brain tissue was first combined with two volumes (w/v) of lysis buffer and homogenized. The lysis buffer was composed of 25% methanol in PBS. An internal standard, tolbutamide, was spiked into 50-μl aliquots of plasma and 200-μl aliquots of brain lysates. The aliquots were then extracted with 2.5 ml of ethyl acetate. The supernatant was removed by centrifugation at 2500 rpm for 10 minutes and evaporated to dryness under nitrogen at 37°C. The dried extract was resuspended in 150 μl of mobile phase for LC-MS/MS analysis.
A Dionex UltiMate 3000 LC and TSQ Quantum Access MAX triple quadrupole mass spectrometer (MS/MS) with electrospray ionization (ESI) manufactured by Thermo Fisher Scientific, Inc. (Waltham, MA) was used for the analysis. Analyte separation was performed at a flow rate of 0.2 ml/min through a Zorbax Eclipse XDB C18 (3.0 × 150 mm × 3.5 μm) column (cat. no. 963954-302; Agilent Technologies, Inc.) with a step-gradient mobile phase. Retention times for AVF, DZP, and internal standard were 3.4, 9.3, and 7.6 minutes, respectively (Supplemental Figs 3S and 4S). The mobile phase consisted of acetonitrile (solvent A) and 0.1% acetic acid in water (solvent B). The composition of the mobile phase was held for 2 minutes at 5:95 A/B, increased to 60:40 A/B for 8 minutes, and finally decreased to 5:95 A/B for 3 minutes to condition the column before the next injection. The mass spectrometer was operated in multiple reaction monitoring mode with a capillary voltage of 3200 V and capillary temperature of 2500°C. Precursor and product ions for the analytes were detected in positive ion mode with m/z 431 [MH]+ → 246 for AVF and 286 [MH]+ → 193 for DZP; the internal standard was detected in negative mode with m/z 269 [M-H]− → 170. The concentration ranges for DZP were 25.0–1600 ng/ml in plasma and 6.25–400 ng/ml in brain lysate (Supplemental Table 3S).
Nasal Tissue Histology.
A separate cohort of rats was used to assess potential trauma to the nasal mucosa from the prodrug/enzyme formulation. This cohort did not receive catheters and no blood or brain samples were collected, but the animals did receive anesthesia and were dosed intranasally in the same manner as the others. Three rats at each dose level (vehicle-only, low, medium, and high, n = 12) were euthanized 90 minutes postdose by perfusion with 120 ml of saline followed by 120 ml of 10% neutral buffered formalin (NBF). The heads were removed, fixed in 10% NBF, and submitted to Colorado Histo-Prep (Fort Collins, CO) for histopathological evaluation by a board-certified veterinary pathologist. The samples were decalcified, trimmed, processed, embedded, and sectioned. Five sections of the nasal turbinates from each of the rats were stained with hematoxylin and eosin for microscopic imaging. Slides of the sections were read without knowledge of the dose group. The observed tissue changes were categorized using standardized toxicological pathology criteria (Boorman, 1990; Banks, 1993).
Noncompartmental Analysis.
Naïve pooled noncompartmental analysis (NCA) of plasma and brain concentration-time profiles was performed using WinNonlin software version 8.0.0.3176 by Certara USA, Inc. (Princeton, NJ). Maximum concentration (Cmax) and the tmax were determined from the average concentrations at nominal time points. The apparent terminal rate constant (λz) was estimated by regression of the log-linear portion of the concentration-time profiles. The apparent terminal half-life (t1/2,z) was calculated as ln(2)/λz. Concentrations measured from animals subjected to destructive and repeated sampling were pooled. Data were treated as sparse and the linear trapezoidal method was used for calculating the area under the curve (AUC) from the time of dosing to 90 minutes. Intranasal bioavailability (F) was calculated by
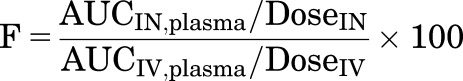 |
(1) |
Drug-targeting efficiency (DTE) was used to assess direct nose-to-brain transport via the olfactory or trigeminal pathways (Mittal et al., 2014). DTE represents the time-average partition ratio between brain and plasma for IN administration versus intravenous administration. Data from the medium-dose level from time of dosing to 90 minutes was used to calculate DTE from the following equation
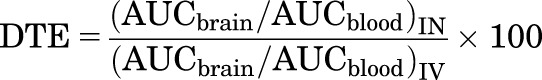 |
(2) |
Physiologically Based Pharmacokinetic Model.
A physiologically based pharmacokinetic (PBPK) model was used for further analysis. The model’s structure and parameters for DZP disposition in rats were originally reported by Igari et al. (1983). Variants of the model have been used by other researchers, such as Gueorguieva et al. (2004) and Thompson et al. (2012), to explore aspects of predicting DZP concentration in tissues. We have appended to the model a nasal compartment for IN delivery of AVF and APB as illustrated in Fig. 2.
Fig. 2.

PBPK model schematic. An established PBPK model for DZP disposition in rats was modified to include a nasal cavity compartment. ORI and DZP absorb from the nasal cavity into circulation after IN administration of AVF and APB.
The conversion rate for AVF to DZP in the nasal cavity is governed by enzyme kinetics and the rate of ORI cyclization. To characterize the enzyme reaction, KM and Vmax were measured in vitro in the formulation vehicle (pH 7.4 PBS) at 32°C. This temperature is approximately the average temperature of rodent nasal passages (Jackson and Schmidt-Nielsen, 1964). The catalytic rate constant (kcat) for enzymatic hydrolysis was then calculated from Vmax, i.e., kcat = Vmax/Cnasal,APB, where Cnasal,APB is the concentration of enzyme in the nasal cavity. Thus, the rate of AVF hydrolysis in vivo can be predicted by
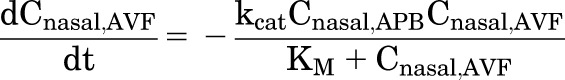 |
(3) |
where Cnasal,AVF is the concentration of AVF in the nasal cavity. ORI, the immediate product of AVF hydrolysis, can accumulate in the nasal cavity and has physicochemical properties such that ORI has the potential to passively diffuse across nasal epithelium. Transport of ORI was modeled as unidirectional owing to the relatively small volume of the nasal cavity. The volume of the nasal cavity (Vnasal) was assumed to be the volume of the deposited formulation, 30 μl. Thus, the rate of change in the concentration of ORI (Cnasal,ORI) in the nasal cavity can be expressed as
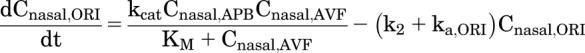 |
(4) |
where k2 is the first-order intramolecular cyclization rate constant of ORI, and ka,ORI is the nasal absorption rate constant of ORI. Likewise, ka,DZP is the nasal absorption rate constant of DSP. However, DZP generated in excess of the solution concentration limit for diazepam (CDZP,lim) forms an amorphous phase separate from the solution phase. Assuming drug absorption only occurs from the solution phase, the rate of change in the concentration of DZP (Cnasal,DZP) in the nasal cavity can be expressed as
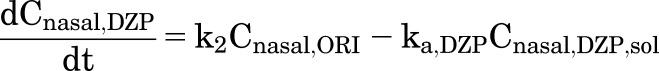 |
(5) |
where Cnasal,DZP,sol is the concentration of DZP in the solution phase and Cnasal,DZP,sol ≤ CDZP,lim.
This PBPK model assumes perfusion rate–limited distribution and tissue-compartment concentrations that are in instantaneous equilibrium with venous blood flows. A general expression for the rate of change in concentration of DZP in non-eliminating organ or tissue compartments can be written as
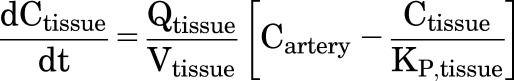 |
(6) |
where Ctissue is the concentration in the specified tissue, Vtissue is the volume of that tissue, Qtissue is the blood flow rate, Cartery is the concentration in the arterial blood, and KP,tissue is the partition coefficient.
DZP is extensively metabolized in the liver and minimally excreted unchanged, so elimination was assumed to be solely by hepatic metabolism. The hepatic extraction ratio for DZP in rats is approximately unity, even for doses as high as 4 mg/kg (Diaz-Garcia et al., 1991). Therefore, the concentration of DZP in the liver was assumed to stay well below KM for metabolism by liver enzymes. An expression for the rate of change in the concentration of DZP in the liver can be written as
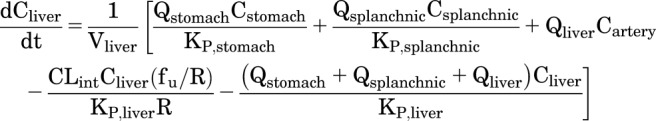 |
(7) |
where CLint is intrinsic clearance, fu is the fraction unbound in plasma, and R is the blood-to-plasma ratio. In rats, the blood-to-plasma DZP concentration ratio has been reported to be R = 1.037 ± 0.007 with the fraction unbound in plasma fu = 0.14 ± 0.003 (Igari et al., 1983, 1984).
Expressions for the rate of change in the concentration of DZP in the venous and arterial blood compartments are
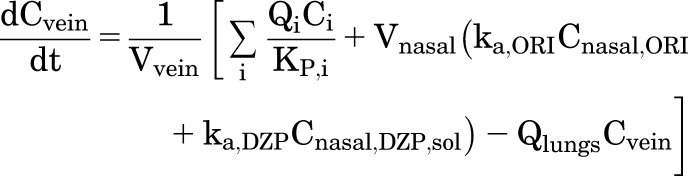 |
(8) |
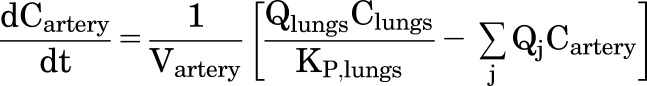 |
(9) |
where i represents each tissue compartment that has an outflow to the venous pool and j represents each tissue compartment that has an inflow from the arterial pool. At pH 7.4, the half-life for cyclization of ORI is 1.47 minutes (Rautiola et al., 2018), so we assume most of the conversion to DZP occurs in the nasal cavity. Absorbed ORI would continue to transform into DZP in the mucosa and in the blood (Upshall et al., 1990). Therefore, systemic exposure to ORI was limited, and any ORI that did reach the circulation was regarded as DZP for the purposes of pharmacokinetic analysis.
The PBPK model, represented by eqs. 3–9, was constructed in MATLAB. Values for the parameters of interest (CLint, ka,ORI, ka,DZP, and CDZP,lim) were determined by fitting numerically solved solutions of the differential equations to experimental data using the ode45 function in conjunction with the lsqcurvefit function. Experimental data were input as averages of the DZP concentrations measured at the nominal time points, with data from animals subjected to destructive and repeated sampling pooled. Fitting was performed in two stages. First, CLint was determined by fitting the model to DZP concentrations measured in plasma from rats administered IV DZP. Tissue compartment volumes, blood flow rates, and partition coefficients were fixed at the values reported by Gueorguieva et al. (2004) and Thompson et al. (2012). These constants are listed in the supplemental data (Supplemental Table 2S). Then, CLint was fixed and in vivo values for ka,ORI, ka,DZP, and CDZP,lim were determined by fitting the model to a matrix of DZP concentrations measured in plasma from rats administered IN AVF/APB at the low-, medium-, and high-dose levels.
Statistical Methods.
Naïve pooled noncompartmental analysis was performed using sparse sampling computations (Carter et al., 1995). Concentrations of DZP measured below the limit of quantification were retained in the data set (Jusko, 2012). Squared correlation coefficients adjusted (R2adj) for the number of points used in the estimation of the terminal rate constant λz were >0.7 for all treatment groups. AUC, S.E. of the mean estimate was calculated using WinNonlin, which employs equations described by Nedelman and Jia (1998) and unbiased sample covariance estimation described by Holder (2001). The S.E. associated with any F estimate was calculated by propagating the S.E. from the AUC. Likewise, the S.E. of any DTE was calculated by propagating the S.E. from the AUC. Descriptive statistics for the variability in tmax are not reported because leapfrog serial and destructive sampling data were pooled. The S.D. of measured concentrations at tmax was used to describe the dispersion of Cmax. Any S.E. reported for a determined PBPK parameter estimate was obtained from MATLAB by back-calculating from a 95% confidence interval computed using the nlparci function with the Jacobian from the lsqcurvefit function. Root mean square error (RMSE) for the model fits was calculated from the average concentration at the nominal time points.
Results
Noncompartmental Analysis.
As shown in Fig. 3, DZP generated from IN administration of the AVF/APB formulation appeared rapidly in plasma. Plasma and brain tissue samples were also analyzed for AVF, but concentrations were below the limit of quantification. The short half-life of ORI did not allow for its quantification. The average plasma concentration of DZP at the first measured time point, 2 minutes, was greater for IN AVF/APB than for IV DZP at an equivalent dose owing to rapid distribution following intravenous dosing (Fig. 3A). Plasma concentrations from IN administration continued to rise, reaching Cmax = 388 ± 31 ng/ml at tmax = 8 minutes for the medium-dose level. Similar tmax values were observed at each dose level, but Cmax was not proportional to the dose. In fact, Cmax for the high dose was not statistically different from that of the medium dose (Fig. 3B). When AVF was administered without APB, there was a prominent decrease in the rate of DZP absorption. As shown in Table 2, Cmax was nearly 4-fold lower without coadministered enzyme.
Fig. 3.

DZP concentration-time profiles measured in plasma. Error bars are S.D. For IV DZP and IN AVF/APB at the medium-dose level (equivalent to DZP at 1.00 mg/kg), n = 6 at 2-, 5-, and 10-minute time points; n = 3 for all other data points. (A) IN-administered AVF/APB compared with intravenously administered DZP indicated high bioavailability of the IN formulation, F = 112% ± 10% (± S.E.). The IN-administered AVF/APB formulation has a pharmacokinetic advantage with regard to Cmax and AUC0–15min compared with the AVF-only control. (B) Saturation of the absorption process resulted in nearly identical initial rates of absorption for IN-administered AVF/APB at the medium- and high-dose levels.
TABLE 2.
Noncompartmental analysis of plasma DZP concentrations
| Formulation | DZP | AVF | AVF/APB | AVF/APB | AVF/APB |
|---|---|---|---|---|---|
| Route | Intravenous | Intranasal | Intranasal | Intranasal | Intranasal |
| Equiv. DZP dose (mg/kg) [dose level] | 1.00 [medium] | 1.00 [medium] | 0.500 [low] | 1.00 [medium] | 1.50 [high] |
| Tmax (min) | 0a | 10 | 5 | 8 | 5 |
| Cmax ± S.D. (ng/ml) | 441a | 108 ± 30 | 71.5 ± 9.3 | 388 ± 31 | 355 ± 187 |
| t1/2,z (min) | 32.9 | 33.4 | 136b | 26.7 | 37.8 |
| AUC0–15 min (min⋅ng/ml) | 1910 ± 70 | 837 ± 28 | 793 ± 54 | 3250 ± 190 | 3850 ± 270 |
| AUC0–90 min ± S.E. (min⋅ng/ml) | 5430 ± 190 | 4230 ± 120 | 2110 ± 150 | 6080 ± 510 | 9280 ± 470 |
| F % ± S.E. | n/a | 77.8 ± 3.5 | 77.8 ± 6.0 | 112 ± 10 | 114 ± 7 |
Cmax reported for IV DZP is the concentration extrapolated to t = 0 min (i.e., the initial concentration C0) as calculated by WinNonlin noncompartmental analysis.
Overestimation of terminal half-life t1/2,z owing to inherent variability of data below limit of quantification in terminal phase of low-dose group.
There was complete absorption of DZP from IN AVF/APB at the medium- and high-dose levels, as was evident by bioavailabilities of 112% ± 10% and 114% ± 7%, respectively. The AVF-only control group and low-dose APB/AVF group had nearly identical bioavailability (F = 77.8%) on the basis of exposure from 0 to 90 minutes. However, there was a notable difference between these groups at early time points (Fig. 3B). Dose normalized AUC0–15 min was twice as large for the AVF/APB low-dose group compared with the AVF-only control (Table 2). AUC0–15 min for the IN AVF/APB medium-and high-dose groups were similar, suggesting saturation of the DZP absorption rate.
Noncompartmental analysis results for DZP measured in brain tissue are listed in Table 3. For the IN AVF/APB medium-dose group, tmax was reached at 8 minutes in both plasma and brain tissue. However, the concentration of DZP measured in plasma at the first time point (Cplasma,DZP = 270 ± 181 ng/ml ± S.D. at t = 2 minutes) was already greater than the therapeutic concentration that is expected to lead to seizure threshold elevation in rats, Cplasma,DZP ≥70 ng/ml (Dhir and Rogawski, 2018). Brain concentrations were not measured at time points between 10 and 60 minutes, but comparable concentrations were observed at 8 and 10 minutes (415 ± 32 and 404 ± 35 ng/g, respectively), suggesting the true maxima were in this region. Brain Cmax for IV DZP was recorded at the first time point, 2 minutes. Rapid transport from blood into brain tissue is normal for small, lipophilic drugs. DTE, a metric commonly used to assess nose-to-brain transport, was less than unity, negating significant transport of DZP through direct nose-to-brain pathways.
TABLE 3.
Noncompartmental analysis of brain DZP concentrations
| Formulation | Route | Equiv. DZP Dose | tmax | Cmax ± S.D. | AUC0–90 min ± S.E. | DTE % ± S.E. |
|---|---|---|---|---|---|---|
| mg/kg | min | ng/ml | min⋅ng/ml | |||
| DZP | IV | 1.00 | 2 | 938 ± 142 | 18,600 ± 200 | N/A |
| AVF/APB | IN | 1.00 | 8 | 415 ± 33 | 15,400 ± 500 | 74.0 ± 7.3 |
PBPK Model.
Concentration-time profiles were well described by a PBPK model with the rate of drug input controlled by enzyme kinetics. At 32°C in pH 7.4 PBS, the Michaelis-Menten enzyme kinetic parameters for APB measured in vitro were KM = 370 ± 63 μM and kcat = 1250 ± 60 minutes−1 ± S.E. (see Supplemental Fig. 1S). The rate constant, k2 ± S.D., for cyclization of ORI, previously measured in vitro under the same conditions (Rautiola et al., 2018), was assumed to remain constant at 0.470 ± 0.012 minutes−1 in vivo. As listed in Table 1, the APB concentrations were scaled with the AVF concentration so that hydrolysis of AVF would be complete at t ≈ 4 minutes, regardless of the dose level. This also means that the predicted mole fraction of the total dose that had been converted to DZP was the same for each dose level at a given time point. For example, approximately 75% of the dose was expected to be converted to DZP at t = 5 minutes regardless of the dose level.
Fitting the PBPK model to DZP concentrations measured in plasma samples from the IV DZP group resulted in CLint = 15.4 l/min, which was expected since DZP is well known to have a hepatic extraction ratio of approximately one in rats (Rowland et al., 1984) and confirms the assumption that DZP clearance from the liver was flow-limited in this PBPK model. Subsequent fitting of the model to DZP concentrations measured in plasma samples from the IN APB/AVF groups allowed estimation of the parameters governing in vivo absorption from the nasal cavity: ka,ORI = 0.122 ± 0.022 minutes−1, ka,DZP = 0.0689 ± 0.0080 minutes−1, and CDZP,lim = 20.8 ± 3.1 mM (± S.E.). Conversion of AVF to DZP was faster than the combined absorption of ORI and DZP, leading to an accumulation of DZP in the nasal cavity. Since the aqueous equilibrium solubility of DZP is only 130 μM, concentrations entered the supersaturated region. The apparent concentration at which supersaturated DZP began to phase separate from solution in the nasal cavity, CDZP,lim, was a useful parameter to model the nonlinear absorption kinetics observed at early time points. However, the value of CDZP,lim should be interpreted within the context of the assumptions that Vnasal was equal to the dose volume and only DZP in the solution phase could be absorbed.
Well-stirred PBPK models predict high initial plasma concentrations with intravenous administration because the initial volume of distribution is assumed to be the volume of the venous blood compartment. This is illustrated in Fig. 4A where the predicted plasma concentration for the IV DZP group decreases dramatically from the time of dosing as it becomes distributed out of the venous compartment to reach the first measured plasma concentration at t = 2 minutes. The high concentrations of DZP measured in the brain at the first time point were predicted more accurately with this PBPK distribution behavior compared with a modeling approach that relied on a standard two-compartment model function to drive brain concentrations. In general, concentrations of DZP in the brain were predicted reasonably well by the PBPK model. For the IN AVF/APB medium-dose group, brain concentrations lagged behind plasma concentrations during the first few minutes. This observation provided additional evidence that transport of DZP through direct nose-to-brain pathways was not significant.
Fig. 4.

DZP concentration-time profiles in plasma and brain tissue. Experimental data (points) and model predictions (lines) at the medium-dose level (equivalent to DZP at 1.00 mg/kg). For plasma concentrations, n = 6 at 2-, 5-, and 10-minute time points; n = 3 at all other time points for plasma and brain. Error bars are S.D. (A) Intravenously administered DZP. RMSE of the predicted curve was 44 ng/ml for plasma and 246 ng/g for brain. The open purple triangle at the origin is a marker to highlight the steep initial increase in brain DZP concentration; it is not a measured data point. (B) IN-administered AVF/APB. Predicted curve RMSE = 66 ng/ml for plasma and 67 ng/g for Bain.
Although there was significant variablity in the data, similar average plasma concentrations were observed for the medium and high IN AVF/APB dose groups at early time points (Fig. 5A). This indicated the absorption process was saturable. The nonlinearity was probably caused by phase separation of DZP from solution when CDZP,lim was reached in the nasal cavity, a phenomenon that has been observed in vitro (Rautiola et al., 2018). Phase separation attenuated DZP absorption and produced a shoulder at t = 10 minutes in the predicted concentration-time profile for the high-dose group (Fig. 5A, green curve). The peak preceding the shoulder comes from the absorption of ORI, which contributes to the concentration of DZP observed in plasma. In the first few minutes after administration, the amount of DZP produced by the reaction is relatively low compared with ORI. Predictions for the relative amount of each reaction species in the nasal cavity are shown in Fig. 5B. Decomposition of the plasma concentration-time profile for the medium IN AVF/APB dose group, shown in Fig. 5C, illustrates how absorption of ORI contributes to the rate at which DZP enters the circulation.
Fig. 5.

DZP concentration-time profiles in plasma from IN administration of AVF/APB at each dose level. Experimental data (points) and model predictions (lines). For medium-dose level (equivalent to DZP at 1.00 mg/kg), n = 6 at 2-, 5-, and 10-minute time points; n = 3 for all other data points. Error bars are S.D. (A) Saturation of absorption rate observed with the high dose. Predicted curve RMSE = 43 ng/ml for low dose, 66 ng/ml for medium dose, and 61 ng/ml for the high dose. (B) Predicted amounts of each reaction species in the nasal cavity. Solid lines represent the low dose, dashed lines medium dose, and dotted lines high-dose level for a 0.250 kg rat. Inset shows expanded view of the first 10 minutes. (C) Absorption of ORI contributes to the rate of DZP delivery into the systemic circulation.
Nasal Tissue Histology.
No inflammatory response was observed in the nasal tissue samples. Lesions (changes) found in the samples were few, with severities graded as either minimal or mild. These were located in the anterior portion of the nasal cavity and were consistent with mechanical trauma from insertion of the pipette tip used for dosing. There were some small, focal areas of missing epithelium on the wall of the nasal septum and/or nasoturbinate, and occasionally the maxilloturbinate. Cellular debris, consisting of small clusters of sloughed epithelial cells and cellular fragments, were found on the wall of the nasal septum or the nasoturbinate and anterior meatus. Other than two instances of minimal focal hemorrhage, the blank vehicle control group was unremarkable. There were no lesions observed in the ethmoid turbinates, olfactory nerves, and olfactory bulbs. The low-, medium-, and high-dose groups (IN AVF/APB equivalent to 0.500, 1.00, and 1.50 mg/kg DZP, respectively) did not display a dose response with regard to location, frequency, or severity of the lesions. Some examples of the tissue sections are shown in Fig. 6. A detailed enumeration of the locations and severity of the observations can be found in the supplemental data (Supplemental Table 4S).
Fig. 6.

Representative examples of nasal tissue section images. Minor mechanical trauma from insertion of the dosing pipette tip was observed in some animals. There were no lesions attributed to the drug formulation. (A) Normal tissue, (B) hemorrhage, and (C) necrosis.
Discussion
Intranasal coadministration of AVF and APB resulted in rapid absorption of DZP with excellent bioavailability, thus demonstrating that a prodrug/enzyme combination in an aqueous vehicle efficiently delivers a hydrophobic drug. PBPK analysis indicated that both DZP and transient ORI were readily absorbed through the nasal mucosa into the systemic circulation. There was no evidence of transport through direct nose-to-brain pathways. Histologic analysis of the nasal tissues postdose did not reveal significant differences between the vehicle-only control group and treatment groups, suggesting that the formulation was well tolerated.
Although aminopeptidases capable of hydrolyzing Lys from AVF to produce DZP exist in the nasal mucosa and blood of both rats and humans (Sarkar, 1992; Hussain et al., 1995; Agu et al., 2009), exogenous APB is necessary to ensure complete and rapid conversion of AVF. In Fig. 3B, the appearance of DZP in plasma at early time points for the IN AVF-only control group suggests that some AVF conversion occurred in the nasal cavity by endogenous enzymes. However, the therapeutic concentration of DZP that is expected to lead to seizure threshold elevation in rats (Cplasma,DZP ≥ 70 ng/ml, Dhir and Rogawski, 2018) was not reached until t = 10 minutes and was not sustained thereafter. AVF also converts to DZP when absorbed into the circulation. Upshall et al. (1990) found the half-life for conversion of AVF in blood was 0.49 ± 0.06, 3.17 ± 0.09, and 3.85 ± 0.26 minutes for guinea pig, rhesus monkey, and human, respectively (blood and plasma were not significantly different). However, substantial transport of AVF across nasal epithelium is improbable since it does not readily partition into membranes. Using wild-type Madin-Darby canine kidney epithelial-cell (MDCKII-wt) monolayers as an in vitro model for nasal epithelium to estimate apparent permeability coefficients (Papp), we observed negligible permeation of AVF (Papp = 1.0–1.5 × 10−6 cm/s) compared with DZP (Papp = 2.2 × 10−5 cm/s) (Kapoor et al., 2014).
IN-administered drugs tend to exhibit variable absorption rates and bioavailabilities when mucociliary clearance to the digestive tract competes with absorption though the nasal mucosa. The high bioavailability calculated for the medium and high IN AVF/APB dose groups suggests that those doses were fully converted and absorbed through the nasal mucosa of the rats (Table 2). The lower bioavailability observed for the low-dose IN AVF/APB and AVF-only groups may have been the result of a portion of the dose being cleared from the nasal cavity before complete conversion could occur. The first-order rate constant for mucociliary clearance is roughly 0.035 minutes−1 in healthy adult humans (Gizurarson, 2015) and 0.14 minutes−1 in Sprague-Dawley rats (Donovan and Zhou, 1995). However, the rate of clearance depends on many factors, including dose volume, viscosity, pH, tonicity, component concentrations, and pharmacodynamic effects on the cilia. A separate study to assess the effects of dose level on the beat frequency of cilia may be warranted to reconcile the discrepant bioavailabilities.
From the plasma DZP concentration-time profiles measured at each dose level (Fig. 5A), absorption during the first 15 minutes displayed nonlinear pharmacokinetics, whereas concentrations at later time points were approximately dose proportional. This behavior was not entirely unexpected since it was known that phase separation of DZP would occur if the accumulation of DZP in the nasal cavity exceeded CDZP,lim. However, it was surprising that the in vivo estimate of CDZP,lim (20.8 ± 3.1 mM ± S.E.), determined by fitting the PBPK model, was nearly 17 times greater than CDZP,lim measured in vitro (1.22 ± 0.03 mM ± S.D.) (Rautiola et al., 2018). The in vivo estimation is conspicuously high because Vnasal was assumed to be equivalent to the dosed formulation volume. However, the total volume would increase when the deposited dose mixes with nasal mucus.
Absorption of DZP directly from phase-separated DZP could also cause overestimation of CDZP,lim. Transcytosis of the precipitated DZP colloidal particles presents one possible mechanism of transport across the nasal epithelium. Brooking et al. (2001) reported size-dependent transcytosis of latex nanoparticles through the nasal mucosa and into the blood of rats. Another potential mechanism invokes an increase in driving force for absorption arising from higher local concentrations of DZP in the diffuse layer of particles that are proximal to cell membranes. Both mechanisms require that DZP-precipitate particles be close to cell membranes. Confounding of DZP absorption from solution with absorption from the precipitated phase is exacerbated by the small volume-to-surface–area ratio that results when the formulation spreads across the surface of the mucosa, minimizing the distance particles need to travel to reach the cell membranes.
The hydrophobic reaction intermediate, ORI, can accumulate in the nasal cavity as well, so there is potential for phase separation of this species. Since we did not observe precipitate formation at time points where there was expected to be high concentrations of ORI during our in vitro experiments (Rautiola et al., 2018), phase separation of ORI was not considered in this PBPK model. However, it was necessary to incorporate absorption of ORI into the model to account for the rapid appearance of DZP in plasma and the shape of the concentration-time profiles. Satisfying Lipinski’s rule of five, with cLogD = 1.69 at pH 7.4 (calculated octanol/water partition coefficient at specified pH; https://chemicalize.com/), ORI can partition into membranes and permeate epithelium. Papp for ORI across MDCKII-wt monolayers was predicted to be 1.77 × 10−5 cm/s using structure/activity relationship calculations (Chen et al., 2005; https://preadmet.bmdrc.kr). Since ORI quickly transforms into DZP, absorption of ORI fortuitously contributes to the rate at which DZP appears in plasma. This contribution is illustrated by the dotted curve in Fig. 5C, but the area under the ORI curve is not representative of exposure. Exposure to ORI was miniscule owing to its short half-life.
Exposure to the other reaction species, AVF and APB, was likewise short and most probably only topical. On the basis of enzyme kinetic calculations, AVF was completely hydrolyzed in the nasal cavity in less than 4 minutes, and increasing the amount of APB could easily speed up its consumption. The residence time of APB in the nasal cavity is controlled by mucociliary clearance and possibly by proteases in the mucus. Once APB or its degradation products reach the digestive tract, they would be digested like any other protein. Histologic analysis of the posterior, absorptive nasal tissue sections suggested that the formulation was innocuous. However, the histologic findings are incomplete. Collection of tissue samples 90 minutes postdose does not provide sufficient time for an inflammatory response to develop. Future in vivo studies will incorporate later time points to more fully assess acute nasal toxicology.
Approximately 50% of the nasal surface in a rat is olfactory epithelium (Dhuria et al., 2010). Although dosing did not specifically target the olfactory epithelium, rats were in the supine position during treatment. Presumably the formulation spread across the olfactory epithelium, providing ample opportunity for direct nose-to-brain transport via the olfactory bulb. However, there was no evidence that direct nose-to-brain transport was an important pathway for delivery of DZP to the brain, in agreement with the findings of Kaur and Kim (2008). DTE was only 74% (Table 3) and DZP concentrations in plasma preceded brain concentrations (Fig. 4B), indicating absorption into the blood and subsequent advection to the brain was faster than diffusive transport along olfactory or trigeminal nerves.
In conclusion, the pharmacokinetic results presented here demonstrate that IN coadministration of AVF with APB is a viable method to rapidly deliver DZP into the systemic circulation and, subsequently, the brain. Since the highly concentrated formulation does not contain organic solvents, it is expected to be better tolerated and absorb faster than IN formulations of DZP that use solubilizing excipients. Administered as a noninvasive nasal spray, IN AVF/APB could be used to quickly terminate seizure emergencies in humans, resulting in reduced emergency department visits and improved quality of life for patients who experience seizure emergencies. Further progress necessitates development of a device that can store the prodrug and enzyme separately, and then combine them into sprayed solution at the time of administration.
Acknowledgments
We thank Dr. William F. Elmquist for his insightful tutelage in pharmacokinetic modeling and Dr. Michael A. Walters for providing the resources necessary to produce the APB enzyme used in this study. Histopathology was conducted by Paul K. Hildebrandt, Diplomate (ACVP), Matthew Stone, and Rajan Bawa at Colorado Histo-Prep, 702 W. Drake, G101, Fort Collins, CO 80526.
Abbreviations
- APB
aminopeptidase B
- AUC
area under the curve
- AVF
avizafone
- CDZP,lim
solution concentration limit for diazepam
- CLint
intrinsic clearance
- Cmax
maximum concentration
- DTE
drug-targeting efficiency
- DZP
diazepam
- IN
intranasal, intranasally
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- IV
intravenous, intravenously
- k2
cyclization rate constant
- ka,DZP
nasal absorption rate constant of diazepam
- ka,ORI
nasal absorption rate constant of open-ring intermediate
- kcat
catalytic rate constant
- KM
Michaelis constant
- ORI
open-ring intermediate
- Papp
permeability coefficient
- PBPK
physiologically based pharmacokinetic
- PBS
phosphate-buffered saline
- RMSE
root mean square error
- tmax
time to peak concentration
- Vmax
maximum velocity
Authorship Contributions
Participated in research design: Rautiola, Maglalang, Cheryala, Nelson, Georg, Fine, Svitak, Faltesek, Hanson, Mishra, Coles, Cloyd, Siegel.
Conducted experiments: Rautiola, Maglalang, Svitak, Faltesek, Mishra.
Contributed new reagents or analytic tools: Cheryala, Nelson, Georg, Mishra.
Performed data analysis: Rautiola, Maglalang, Mishra, Coles, Cloyd, Siegel.
Wrote or contributed to the writing of the manuscript: Rautiola, Maglalang, Cheryala, Nelson, Georg, Fine, Svitak, Faltesek, Hanson, Mishra, Coles, Cloyd, Siegel.
Footnotes
This study was supported by a grant [Grant U01HL127479] from the University of Minnesota’s NIH Research Evaluation and Commercialization Hub (MN-REACH). Financial support was also provided by the 3M Science and Technology Fellowship in Drug Delivery, American Foundation for Pharmaceutical Education (AFPE), and the Pharmaceutical Research and Manufacturers of America (PhRMA) Foundation.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Agarwal SK, Kriel RL, Brundage RC, Ivaturi VD, Cloyd JC. (2013) A pilot study assessing the bioavailability and pharmacokinetics of diazepam after intranasal and intravenous administration in healthy volunteers. Epilepsy Res 105:362–367. [DOI] [PubMed] [Google Scholar]
- Agu RU, Obimah DU, Lyzenga WJ, Jorissen M, Massoud E, Verbeke N. (2009) Specific aminopeptidases of excised human nasal epithelium and primary culture: a comparison of functional characteristics and gene transcripts expression. J Pharm Pharmacol 61:599–606. [DOI] [PubMed] [Google Scholar]
- Bancke L, Dworak H, Halvorsen M. (2013) Pharmacokinetics, pharmacodynamics, and safety of three doses of Usl261, a midazolam formulation optimized for intranasal administration: p178. Epilepsia 54:62. [DOI] [PubMed] [Google Scholar]
- Banks WJ. (1993) Applied Veterinary Histology, Mosby-Year Book, Inc., St. Louis, MO. [Google Scholar]
- Boorman GA, Eutis SL, Elwell MR, Montgomery CA, MacKenzie WF. (1990) Pathology of the Fischer Rat: Reference and Atlas. Academic Press, San Diego, CA. [Google Scholar]
- Brooking J, Davis SS, Illum L. (2001) Transport of nanoparticles across the rat nasal mucosa. J Drug Target 9:267–279. [DOI] [PubMed] [Google Scholar]
- Buck ML. (2013) Intranasal administration of benzodiazepines for the treatment of acute repetitive seizures in children. Pediatr Pharmacol 19. [Google Scholar]
- Carter AA, Rosenbaum SE, Dudley MN. (1995) Review of methods in population pharmacokinetics. Clin Res Regul Aff 12:1–21. [Google Scholar]
- Chen LL, Yao J, Yang JB, Yang J. (2005) Predicting MDCK cell permeation coefficients of organic molecules using membrane-interaction QSAR analysis. Acta Pharmacol Sin 26:1322–1333. [DOI] [PubMed] [Google Scholar]
- Dhir A, Rogawski MA. (2018) Determination of minimal steady-state plasma level of diazepam causing seizure threshold elevation in rats. Epilepsia 59:935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhuria SV, Hanson LR, Frey WH., II (2010) Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci 99:1654–1673. [DOI] [PubMed] [Google Scholar]
- Diaz-Garcia JM, Oliver-Botana J, Fos Galve D. (1991) Pharmacokinetics of diazepam in the rat: influence of an experimentally induced hepatic injury. Eur J Drug Metab Pharmacokinet Spec No 3:94–101. [PubMed] [Google Scholar]
- Donovan MD, Zhou M. (1995) Drug effects on in vivo nasal clearance in rats. Int J Pharm 116:77–86. [Google Scholar]
- Gizurarson S. (2015) The effect of cilia and the mucociliary clearance on successful drug delivery. Biol Pharm Bull 38:497–506. [DOI] [PubMed] [Google Scholar]
- Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, Bare M, Bleck T, Dodson WE, Garrity L, et al. (2016) Evidence-based guideline: Treatment of convulsive status epileptius in children and adults: Report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr 16:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueorguieva II, Nestorov IA, Rowland M. (2004) Fuzzy simulation of pharmacokinetic models: case study of whole body physiologically based model of diazepam. J Pharmacokinet Pharmacodyn 31:185–213. [DOI] [PubMed] [Google Scholar]
- Haut SR, Seinfeld S, Pellock J. (2016) Benzodiazepine use in seizure emergencies: a systematic review. Epilepsy Behav 63:109–117. [DOI] [PubMed] [Google Scholar]
- Heller AH, Wargacki S, Jung C, Wyatt DJ, Bonnefois G, Barriere O, Tremblay P-O, Schobel AM. (2018) Population pharmacokinetic modeling of diazepam buccal soluble film. Neurotherapeutics 15:819–835. [Google Scholar]
- Holder DJ. (2001) Comments on Nedelman and Jia’s extension of Satterthwaite’s approximation applied to pharmacokinetics. J Biopharm Stat 11:75–79. [DOI] [PubMed] [Google Scholar]
- Hussain MA, Seetharam R, Wilk RR, Aungst BJ, Kettner CA. (1995) Nasal mucosal metabolism and absorption of pentapeptide enkephalin analogs having varying N-terminal amino acids. J Pharm Sci 84:62–64. [DOI] [PubMed] [Google Scholar]
- Igari Y, Sugiyama Y, Sawada Y, Iga T, Hanano M. (1983) Prediction of diazepam disposition in the rat and man by a physiologically based pharmacokinetic model. J Pharmacokinet Biopharm 11:577–593. [DOI] [PubMed] [Google Scholar]
- Igari Y, Sugiyama Y, Sawada Y, Iga T, Hanano M. (1984) In vitro and in vivo assessment of hepatic and extrahepatic metabolism of diazepam in the rat. J Pharm Sci 73:826–828. [DOI] [PubMed] [Google Scholar]
- Ivaturi V, Kriel R, Brundage R, Loewen G, Mansbach H, Cloyd J. (2013) Bioavailability of intranasal vs. rectal diazepam. Epilepsy Res 103:254–261. [DOI] [PubMed] [Google Scholar]
- Jackson DC, Schmidt-Nielsen K. (1964) Countercurrent heat exchange in the respiratory passages. Proc Natl Acad Sci USA 51:1192–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jornada DH, dos Santos Fernandes GF, Chiba DE, de Melo TRF, dos Santos JL, Chung MC. (2015) The prodrug approach: a successful tool for improving drug solubility. Molecules 21:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jusko WJ. (2012) Use of pharmacokinetic data below lower limit of quantitation values. Pharm Res 29:2628–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kälviäinen R. (2015) Intranasal therapies for acute seizures. Epilepsy Behav 49:303–306. [DOI] [PubMed] [Google Scholar]
- Kapoor M, Cheryala N, Rautiola D, Georg GI, Cloyd JC, Siegel RA. (2016) Chirally pure prodrugs and their converting enzymes lead to high supersaturation and rapid transcellular permeation of benzodiazepines. J Pharm Sci 105:2365–2371. [DOI] [PubMed] [Google Scholar]
- Kapoor M, Siegel RA. (2013) Prodrug/Enzyme based acceleration of absorption of hydrophobic drugs: an in vitro study. Mol Pharm 10:3519–3524. [DOI] [PubMed] [Google Scholar]
- Kapoor M, Winter T, Lis L, Georg GI, Siegel RA. (2014) Rapid delivery of diazepam from supersaturated solutions prepared using prodrug/enzyme mixtures: toward intranasal treatment of seizure emergencies. AAPS J 16:577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P, Kim K. (2008) Pharmacokinetics and brain uptake of diazepam after intravenous and intranasal administration in rats and rabbits. Int J Pharm 364:27–35. [DOI] [PubMed] [Google Scholar]
- Maggio ET, Pillion DJ. (2013) High efficiency intranasal drug delivery using Intravail® alkylsaccharide absorption enhancers. Drug Deliv Transl Res 3:16–25. [DOI] [PubMed] [Google Scholar]
- Maglalang PD, Rautiola D, Siegel RA, Fine JM, Hanson LR, Coles LD, Cloyd JC. (2018) Rescue therapies for seizure emergencies: new modes of administration. Epilepsia 59 (Suppl 2):207–215. [DOI] [PubMed] [Google Scholar]
- Mirski MA, Varelas PN. (2008) Seizures and status epilepticus in the critically ill. Crit Care Clin 24:115–147, ix. [DOI] [PubMed] [Google Scholar]
- Mittal D, Ali A, Md S, Baboota S, Sahni JK, Ali J. (2014) Insights into direct nose to brain delivery: current status and future perspective. Drug Deliv 21:75–86. [DOI] [PubMed] [Google Scholar]
- Nedelman JR, Jia X. (1998) An extension of Satterthwaite’s approximation applied to pharmacokinetics. J Biopharm Stat 8:317–328. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Ohnishi A, Goto Y, Sakuma Y, Watanabe J, Hattori A, Tsujimoto M. (2014) Role of glutamine-169 in the substrate recognition of human aminopeptidase B. Biochim Biophys Acta 1840:1872–1881. [DOI] [PubMed] [Google Scholar]
- Ohnishi A, Watanabe J, Ogawa Y, Goto Y, Hattori A, Tsujimoto M. (2015) Involvement of phenylalanine 297 in the construction of the substrate pocket of human aminopeptidase B. Biochemistry 54:6062–6070. [DOI] [PubMed] [Google Scholar]
- Pellock JM. (2007) Overview: definitions and classifications of seizure emergencies. J Child Neurol 22 (5 Suppl):9S–13S. [DOI] [PubMed] [Google Scholar]
- Rautiola D, Cloyd JC, Siegel RA. (2018) Conversion of a soluble diazepam prodrug to supersaturated diazepam for rapid intranasal delivery: kinetics and stability. J Control Release 289:1–9. [DOI] [PubMed] [Google Scholar]
- Rowland M, Leitch D, Fleming G, Smith B. (1984) Protein binding and hepatic clearance: discrimination between models of hepatic clearance with diazepam, a drug of high intrinsic clearance, in the isolated perfused rat liver preparation. J Pharmacokinet Biopharm 12:129–147. [DOI] [PubMed] [Google Scholar]
- Sarkar MA. (1992) Drug metabolism in the nasal mucosa. Pharm Res 9:1–9. [DOI] [PubMed] [Google Scholar]
- Siegel RA, Kapoor M, Cheryala N, Georg GI, Cloyd JC. (2015) Water-soluble benzodiazepine prodrug/enzyme combinations for intranasal rescue therapies. Epilepsy Behav 49:347–350. [DOI] [PubMed] [Google Scholar]
- Sperling MR, Haas KF, Krauss G, Seif Eddeine H, Henney HR, III, Rabinowicz AL, Bream G, Squillacote D, Carrazana EJ. (2014) Dosing feasibility and tolerability of intranasal diazepam in adults with epilepsy. Epilepsia 55:1544–1550. [DOI] [PubMed] [Google Scholar]
- Sutter R, Kaplan PW, Rüegg S. (2013) Outcome predictors for status epilepticus--what really counts. Nat Rev Neurol 9:525–534. [DOI] [PubMed] [Google Scholar]
- Thompson MD, Beard DA, Wu F. (2012) Use of partition coefficients in flow-limited physiologically-based pharmacokinetic modeling. J Pharmacokinet Pharmacodyn 39:313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upshall D, Gouldstone S, Macey N, Maidment M, Wast S, Yeadon M. (1990) Conversion of a peptidoaminobenzophenone prodrug to diazepam in vitro. Enzyme isolation and characterisation. J Biopharm Sci 1:111–126. [Google Scholar]