

Abstract
Influenza is a long-standing health problem. For treatment of seasonal flu and possible pandemic infections, there is a need to develop new anti-influenza drugs that have good bioavailability against a broad spectrum of influenza viruses, including the resistant strains. Relenza™ (zanamivir), Tamiflu™ (the phosphate salt of oseltamivir), Inavir™ (laninamivir octanoate) and Rapivab™ (peramivir) are four anti-influenza drugs targeting the viral neuraminidases (NAs). However, some problems of these drugs should be resolved, such as oral availability, drug resistance and the induced cytokine storm. Two possible strategies have been applied to tackle these problems by devising congeners and conjugates. In this review, congeners are the related compounds having comparable chemical structures and biological functions, whereas conjugate refers to a compound having two bioactive entities joined by a covalent bond. The rational design of NA inhibitors is based on the mechanism of the enzymatic hydrolysis of the sialic acid (Neu5Ac)-terminated glycoprotein. To improve binding affinity and lipophilicity of the existing NA inhibitors, several methods are utilized, including conversion of carboxylic acid to ester prodrug, conversion of guanidine to acylguanidine, substitution of carboxylic acid with bioisostere, and modification of glycerol side chain. Alternatively, conjugating NA inhibitors with other therapeutic entity provides a synergistic anti-influenza activity; for example, to kill the existing viruses and suppress the cytokines caused by cross-species infection.
Keywords: Influenza, Neuraminidase, Inhibitor, Drug, Congener, Conjugate
Background
Influenza is a serious and long-standing health problem
Influenza virus is one of major human pathogens responsible for respiratory diseases, causing high morbidity and mortality through seasonal flu and global pandemics. Vaccines and antiviral drugs can be applied to prevent and treat influenza infection, respectively [1, 2]. Unfortunately, the RNA genome of influenza virus constantly mutates and the genomic segments may undergo reassortment to form new virus subtypes. Although vaccine is the most effective way for prophylaxis of influenza, vaccine formulations must be updated annually due to changes in circulating influenza viruses [3], and the production of influenza vaccine takes several months. If prediction of the incoming influenza strains is incorrect, the vaccines may just give limited efficacy in protection.
Several influenza pandemics have occurred in the past, such as Spanish flu caused by H1N1 virus in 1918, Asian flu by H2N2 virus in 1957, Hong Kong flu by H3N2 virus in 1968, bird flu by H5N1 and H7N9 viruses in 2003 and 2013, respectively, as well as swine flu by H1N1 virus in 2009 (Fig. 1) [4–6]. The influenza pandemics have claimed a large number of human lives and caused enormous economic loss in many countries. A universal vaccine for flu remains elusive.
Fig. 1.

Timeline showing influenza pandemics caused by influenza A viruses
Genome organization of influenza A virus
Influenza viruses are negative-sense RNA viruses of the Orthomyxoviridae family [7]. The viral genome is divided into multiple segments and differs in host range and pathogenicity. There are A, B and C types of influenza viruses, and influenza A viruses are the most virulent. Influenza A viruses infect a wide range of avian and mammalian hosts, whereas influenza B viruses infect almost exclusively humans. Much attention has been paid to influenza A viruses because they have brought about pandemic outbreaks. The structure of influenza virus contains three parts: core, envelope and matrix proteins. These proteins are hemagglutinin (HA), neuraminidase (NA), matrix protein 1 (M1), proton channel protein (M2), nucleoprotein (NP), RNA polymerase (PA, PB1 and PB2), non-structural protein 1 (NS1) and nuclear export protein (NEP, NS2). In addition, some proteins (e.g. PB1-F2, PB1-N40 and PA-X) were found in particular strains [8, 9]. Influenza A viruses are further classified by HA and NA subtypes [10]. There are 18 subtypes of HA and 11 subtypes of NA; for example, H1N1 and H3N2 are human influenza viruses, while H5N1 and H7N9 are avian influenza viruses. HA and NA constantly undergo point mutations (antigenic drift) in seasonal flu. Genetic reassortment (antigenic shift) between human and avian viruses may occur to cause pandemics [11, 12].
Infection and propagation route of influenza virus
The life cycle of influenza virus is a complex biological process that can be divided into the following steps (Fig. 2): (i) virion attachment to the cell surface (receptor binding); (ii) internalization of the virus into the cell (endocytosis); (iii) viral ribonucleoprotein (vRNP) decapsidation, cytoplasmic transport and nuclear import; (iv) viral RNA transcription and replication; (v) nuclear exportation and protein synthesis; (vi) viral progeny assembly, budding and release from the cell membrane. All of these steps in the life cycle of influenza virus are essential for its virulence, replication and transmission. Developing a small molecule inhibitor that blocks any of these steps can produce a potentially efficient strategy to control and prevent influenza infection [13].
Fig. 2.

Schematic representation of the life cycle of influenza virus
The influenza HA exists as a trimer and mediates the attachment to host cell via interactions with the cell surface glycoproteins that contain a terminal sialic acid (N-acetylneuraminic acid, Neu5Ac, compound 1 in Fig. 3) linked to galactose in α2,3 or α2,6 glycosidic bond [14]. Influenza viruses from avian recognize the 2,3-linked Neu5Ac receptor on host cell, whereas the human-derived viruses recognize 2,6-linked Neu5Ac receptor. The viruses from swine recognize both α2,3 and α2,6 receptors (Fig. 3a). After endocytosis and fusion of the viral envelope membrane into the host endosomal membrane, the viral ribonucleoprotein (RNP) complexes will enter the host cell, and proceed with replication by the machinery of host cell. The newly generated virus will bud on the plasma membrane, and its NA will break the connection between HA and host cell, thereby releasing the progeny virus to infect surrounding cells. NA is a tetrameric transmembrane glycoprotein that catalyzes the hydrolytic reaction to cleave the terminal Neu5Ac residue from the sialo-receptor on the surface of host cell. Thus, HA and NA play the central roles in influenza virus infection [15].
Fig. 3.

Actions of hemagglutinin and neuraminidase. a Binding of HA to the surface Neu5Ac-linked glycoproteins on host cell. b NA catalyzes the hydrolytic reaction to cleave the terminal Neu5Ac residue from the sialo-receptor
Development of anti-influenza drugs
Drugs are needed for treatment of patients infected by influenza viruses, especially during influenza pandemics without effective vaccine. Even broadly protective flu vaccines were available, anti-influenza drugs are still needed, especially important for treating the patients with poor responses to vaccination. The currently available anti-influenza drugs directly target the virus at various stages of the viral life cycle, while therapeutics targeting the host are under development [16, 17].
Approved anti-influenza drugs
Figure 4 shows the approved anti-influenza drugs [18], including M2 ion channel blockers, neuraminidase inhibitors, and a nucleoprotein inhibitor [19]. However, the emerging drug-resistant influenza viruses have posed problems in treatment [20]. Two M2 ion channel inhibitors Fig. 4a (a in black), amantadine (2) [21] and rimantadine (3) [22], were widely used against influenza. However, the efficacy of M2 ion channel inhibitors is limited to influenza A because influenza B viruses lack M2 protein. In addition, almost all of influenza strains have developed high resistance against both amantadine and rimantadine [23]. The M2 ion channel inhibitors are now largely discontinued and replaced by NA inhibitors [24, 25].
Fig. 4.

Chemical structures of currently available licensed anti-flu drugs. a M2 ion-channel inhibitors, b neuraminidase inhibitors, and c nucleoprotein inhibitor
Baloxavir marboxil (Xofluza™, Shionogi/Hoffmann-La Roche, 2018) is used as single-dose oral drug for treatment of influenza [19]. Baloxavir acid, the active form of baloxavir marboxil, is a cap-dependent endonuclease inhibitor targeting the viral PA polymerase and interferes with the transcription of viral mRNA [19]. Moreover, the combination treatment with baloxavir marboxil and oseltamivir, a neuraminidase inhibitor, showed synergistic effect against influenza virus infections in mice experiments [26]. It is possible to develop the combination therapy using sub-optimal dose of baloxavir marboxil and NA inhibitor.
The current medical treatment of influenza patients is based on the administration of neuraminidase inhibitors [27]. NA catalyzes the hydrolytic cleavage of the glycosidic bond of sialic acid, so that the progeny virion can be released from the host cell, and spread to infect the surrounding cells. Thus, an effective way to control influenza is to block the function of NA with specific inhibitors [28]. Currently, four NA inhibitors Fig. 4b are used in clinical practice: zanamivir (4) (Relenza™; GlaxoSmithKline, 1999) [29, 30], oseltamivir phosphate salt (5) (Tamiflu™; Hoffmann-La Roche, 1999) [31, 32], laninamivir octanoate (6) (Inavir™; Biota/Daiichi-Sankyo, 2010) [33] and peramivir (7) (Rapivab™; BioCryst Pharm, 2014) [34, 35].
Zanamivir (ZA) is more effective than oseltamivir, but the oral bioavailability of ZA in humans is poor (< 5%) [36], presumably because ZA is a hydrophilic compound that is water soluble and readily eliminated through renal system. ZA is usually delivered by intranasal or dry powder inhalation [29, 30, 37]. After inhaling dry powder, about 7–21% is deposited in the lower respiratory tract, and the rest is deposited in the oropharynx [36]. To prevent influenza, the recommended dose of ZA is 20 mg/50 kg/day for adults by inhalation twice daily (half dose at each inhalation). Adverse drug reactions of zanamivir are rarer than oseltamivir because zanamivir carries a glycerol side chain similar to the chemical structure of sialic acid, the natural NA substrate.
Tamiflu, the phosphate salt of oseltamivir (OS), is a popular orally available anti-flu drug, which is well absorbed and rapidly cleaved by endogenous esterases in the gastrointestinal tract, liver and blood to give OS carboxylate (OC). To treat influenza, the recommended dose of OS for adults is 75 mg, twice a day, for 5 days. Tamiflu is less effective if used after 48 h of influenza infection. The preventive dose is usually 75 mg, once a day for at least 10 days or up to 6 weeks during a community outbreak. In comparison with ZA, oseltamivir has more adverse effects and tends to induce resistant viral strains. The cause of drug resistance is related to the change of binding mode that will be discussed in section 2.3.2.
Laninamivir octanoate is a long-acting anti-flu prodrug that is converted by endogenous esterases in the airway to give laninamivir, the C7-methoxy analog of ZA as a potent NA inhibitor [38]. Currently, laninamivir octanoate is only approved for use in Japan to treat and prevent influenza A and B infection. A single inhalation of the drug powder at a dose of 20 mg daily for 2 days is recommended for prophylaxis, and at 40 mg dosage for treatment of individuals greater than or equal to 10 years of age.
Peramivir (PE) has low oral bioavailability and is administered by a single intravenous drip infusion at a dose of 300 mg in 15 min during influenza treatment. PE is a highly effective inhibitor against influenza A and B viruses with good safety. PE can be used to treat the patients who cannot use oral drugs or insensitive to OS and ZA [39].
Why do we need new anti-influenza drugs?
Anti-influenza drugs are needed to treat seasonal flu and particularly unexpected global influenza infection. Our recent challenge is to deal with new influenza strains, cross-species transmission, and drug resistance. The pandemic influenza A/H1N1 virus in 2009 is currently circulating as a seasonal virus and resistant to M2 inhibitors [40]. Since 2009, only NA inhibitors have been able to provide protection against the circulating human influenza A and B viruses. Small molecular NA inhibitors are powerful tools to fight against influenza viruses. Like other antiviral therapeutics, influenza NA inhibitor is not an exception to encounter the problem of drug-resistant mutations in the target enzyme. Since the drug-resistant H1N1 influenza virus became popular in 2007 and quickly dominated in the 2008–2009 season, the emergence of OS resistance is of particular concern [41, 42]. The resistant phenotype is associated with an H275Y mutation in NA. In comparison with other permissive mutations, H275Y-mutant viruses do not display any fitness deficits, and thus remain in circulation [43, 44]. The clinically relevant H5N1 avian influenza virus from a patient even shows an increasing resistance against OS. Fortunately, the H275Y mutant is still sensitive to ZA.
In this review, we highlight the latest advances in structural modification of oseltamivir, zanamivir and peramivir for the development of effective anti-influenza drugs, especially focusing on using congeners and conjugates of the existing NA inhibitors. Congeners are the related compounds having comparable chemical structures and biological functions, whereas conjugate refers to a compound having two bioactive entities joined by a covalent bond.
Rational design of neuraminidase inhibitor congeners
Mechanism and assay of neuraminidase catalyzed reaction
Influenza virus NA is an ideal drug target because NA is an essential enzyme that located on virus membrane for easy access of drugs. Moreover, all subtypes of influenza NAs have a similar conserved active site. On NA-catalyzed hydrolysis of sialo-glycoprotein, the scaffold of Neu5Ac is flipped to a pseudo-boat conformation, so that cleavage of the glycoside bond is facilitated by anomeric effect, giving an oxocarbenium intermediate (Fig. 3b). Based on this reaction mechanism, a fluorometric assay using 2-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid (MUNANA) as NA substrate is designed (Fig. 5a). On hydrolysis of MUNANA, the anion of 4-methylumbelliferone will be released to show strong fluorescence at 460 nm (excitation at 365 nm). The fluorescence dims in the presence of NA inhibitor to suppress the enzymatic hydrolysis. A sialic acid 1,2-dioxetane derivative (NA-Star™, Applied Biosystems) can be used as the luminescence substrate to assess the NA inhibitory activity when the test compound contains a fluorescent moiety to interfere with the fluorescence assay (Fig. 5b).
Fig. 5.
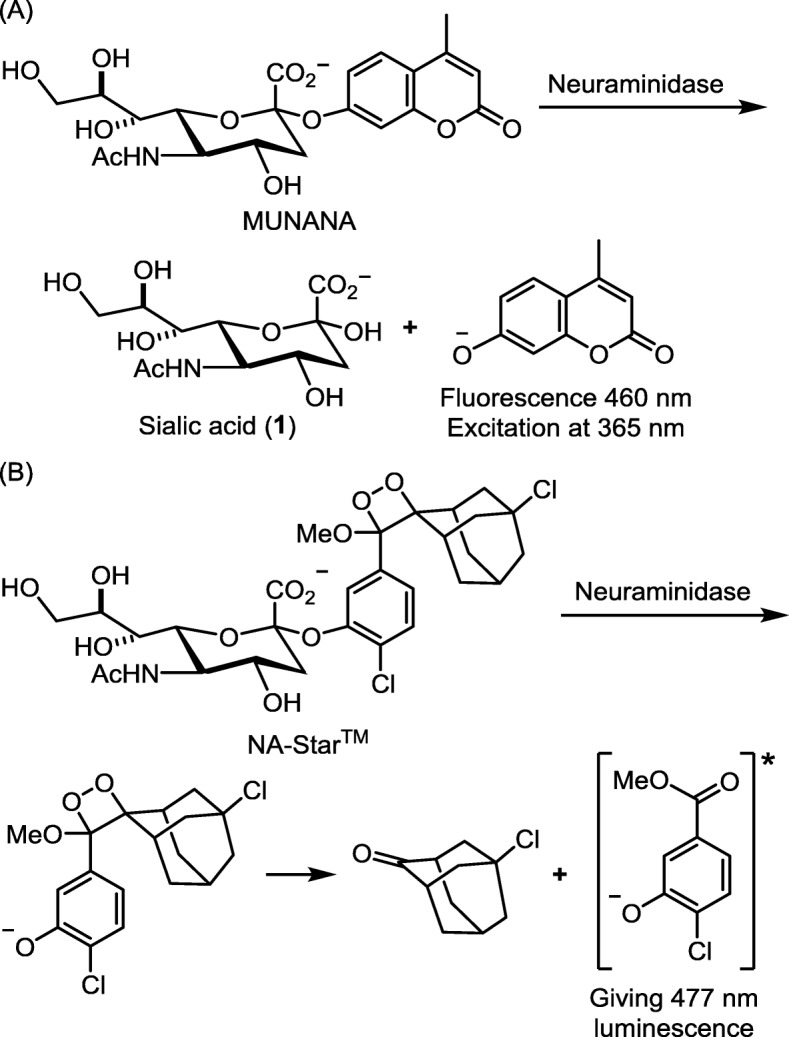
Substrates for assays of influenza NA inhibitors. a fluorescent substrate 2-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid (MUNANA), and b luminescent substrate NA-Star™
Neuraminidase inhibitors and binding modes
Didehydro-2-deoxy-N-acetylneuraminic acid (Neu5Ac2en, DANA, 8) is the first reported influenza NA inhibitor [45]. The crystal structure of NA–DANA complex (Fig. 6a) has been used as a template for the discovery of more potent NA inhibitors. ZA and OS are two NA inhibitors having (oxa)cyclohexene ring to mimic the oxocarbenium intermediate (Fig. 3). ZA is a DANA guanidino derivative designed by von Itzstein and coworkers [46, 47]; the key interactions of ZA in NA active site are depicted in Fig. 6b. The carboxylate group shows electrostatic interactions with the three arginine residues (Arg118, Arg292 and Arg371 as a tri-arginine motif) in the S1 site of influenza NA [48, 49], whereas the basic guanidino group exhibits strong electrostatic interactions with the acidic residues of Glu119, Asp151 and Glu227 in the S2 site. In addition, the glycerol side chain provides hydrogen bonds with Glu276 in the S5 site.
Fig. 6.
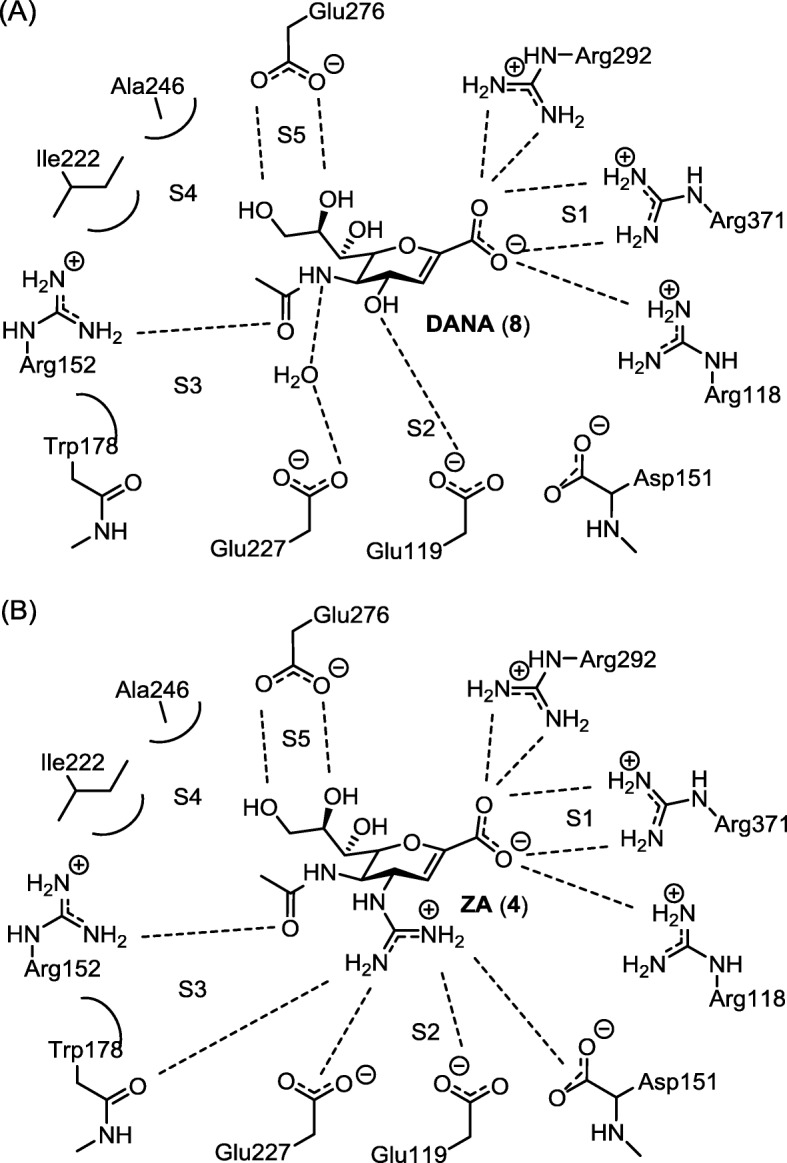
Key interactions of NA inhibitors in the active site based on the crystal structures of the NA–inhibitor complexes. a NA–DANA complex; b NA–ZA complex
Oseltamivir carboxylate (OC) contains an amine group at C5-position to interact with the acidic residues (Glu119, Asp151 and Glu227). Instead of glycerol side chain, OC has a 3-pentoxy group at the C-3 position. Upon binding to OC, NA redirects the Glu276 residue to Arg224 to form a larger hydrophobic pocket for incorporation of the 3-pentoxy group [50, 51]. However, the salt bridge between Glu276 and Arg224 in H275Y mutant will collapse by substitution of the histidine with a bulkier tyrosine residue, thus altering the hydrophobic pocket of NA and causing decreased affinity with OC [51, 52]. In contrast, ZA rarely induces resistant viruses because it is structurally similar to the natural substrate Neu5Ac.
Conversion of carboxylic acid to ester prodrug for better bioavailability
Lipophilicity is an important factor in the pharmacokinetics behavior of drugs. The partition coefficient (log P) of a compound between octanol and water can be taken as a measure of lipophilicity. Compounds with log P values between − 1 and 5 are likely developed as orally available drugs [53]. In lieu of log P, the distribution coefficient (log D) between octanol and PBS buffer is used to predict the lipophilicity of ionic compounds.
OC has low lipophilicity and oral bioavailability (< 5%). To solve this problem, the ethyl ester OS was prepared as prodrug with improved oral bioavailability (35%) [54]. The phosphate salt of OS was formulated with appropriate filler materials to make tamiflu capsule with good bioavailability (79%).
A similar strategy has been applied to modify ZA molecule to develop better anti-influenza drugs with improved pharmacokinetic properties and oral bioavailability. Li and coworkers have shown that (heptadecyloxy)ethyl ester of ZA is an effective drug for mice by oral or intraperitoneal administration [55]. Similar to oseltamivir, the ZA ester can undergo enzymatic hydrolysis to release ZA as an active anti-influenza agent. Compared to the rapid elimination of ZA in body, the ZA ester appears to sustain by oral administration. However, no pharmacokinetics studies were performed to determine the value of bioavailability. Amidon and coworkers have synthesized several acyloxy ester prodrugs of zanamivir with conjugation of amino acids [56]. For example, [(L-valyl)oxy] ethyl ester of ZA improved the cell permeability by targeting hPepT1, an oligopeptide transporter present in gastrointestinal tract with broad substrate specificity. This ZA ester is a carrier-linked prodrug with a bioreversible covalent bond, and may be developed as an oral drug.
Besides the carboxylate group, the highly hydrophilic guanidinium group also accounts for the low oral bioavailability of ZA and guanidino-oseltamivir carboxylate (GOC). In one approach to improve bioavailability, Amidon and coworkers [57] prepared ZA heptyl ester and used 1-hydroxy-2-naphthoic acid (HNAP) as a counterion of the guanidinium group (Fig. 7a) [58, 59]. This intact ion-pair prodrug (9) showed an enhanced permeability across Caco-2 and rat jejunum cell membranes. Moreover, Fang and coworkers have synthesized an intramolecular ion-pair ZA ester prodrug 10 by annexing an HNAP moiety [60]. Compound 10 has improved lipophilicity (log D = 0.75 at pH 7.4) by incorporating an aromatic moiety of HNAP and forming the guanidinium–phenoxide ion-pair. The ZA–HNAP prodrug resumes high anti-influenza activity, EC50 = 48 nM in cell-based anti-influenza assays, by enzymatic hydrolysis to release zanamivir along with nontoxic HNAP.
Fig. 7.

Tackling the hydrophilic guanidinium group in zanamivir and guanidine-oseltamivir carboxylate. a Using 1-hydroxy-2-naphthoic acid to form ion-pair. b Forming acylguanidine as prodrug
Conversion of guanidine to acylguanidine for better bioavailability
Though the guanidinium moiety in ZA and GOC plays an important role in NA binding, its polar cationic nature is detrimental to oral administration. Modification of the guanidine group to acylguanidine by attachment of lipophilic acyl substituent improves bioavailability (Fig. 7b) [61]. Moreover, appropriate acyl substituents at the external N-position of the guanidine group in ZA are proposed to attain extra bindings in the 150-cavity [47, 62] and 430-cavity [63] of H1N1 virus [61, 64, 65]. Some GOC acylguanidines also possess higher activities than OC against wild-type H1N1 and OS-resistant H259Y viruses [66]. The ZA and GOC acylguanidine derivatives 11 and 12 are stable in acidic media, but slowly hydrolyzed in neural phosphate buffer, and the hydrolytic degradation is accelerated in basic conditions [61]. The hydrolysis of ZA and GOC acylguanidines in animal plasma at physiological condition liberates the parental anti-influenza agents ZA and GOC. Thus, influenza infected mice receiving the octanoylguanidine derivative 11 (or 12) by intranasal instillation have better or equal survival rate than those treated with parental ZA or GOC [61].
Substitution of carboxylic acid with bioisosteres
Bioisosteres are the surrogates mimicking the structure of an active compound while keep similar chemical, physical, electronic, conformational and biological properties [67, 68]. There are two types of bioisosteres, mimicking the enzyme substrate or the reaction transition state. For example, hydroxamic acid, sulfinic acid and boronic acid can mimic the planar structure of carboxylic acid, whereas phosphonic acid, sulfonic acid, sulfonamide, and trifluoroborate can mimic the transition state in enzymatic hydrolysis of peptide bond.
Sialic acid (Neu5Ac, 1), the product of NA-catalyzed hydrolysis, exists as a mixture of two anomers. The affinity of Neu5Ac to influenza NA was weak (Ki = 5 mM to A/H2N2 virus) [69], presumably due to low proportion (~ 5%) of appropriate anomer in the solution [70]. By substitution of the C2-OH group in Neu5Ac with hydrogen atom, the configurations at C-1 position are fixed [71]. Compounds 13a and 13b (Fig. 8) have the carboxylate group axially and equatorially located on the chair conformation of pyranose ring, respectively. The inhibition constant of 13b against V. cholera NA is 2.6 mM, but 13a is inactive.
Fig. 8.
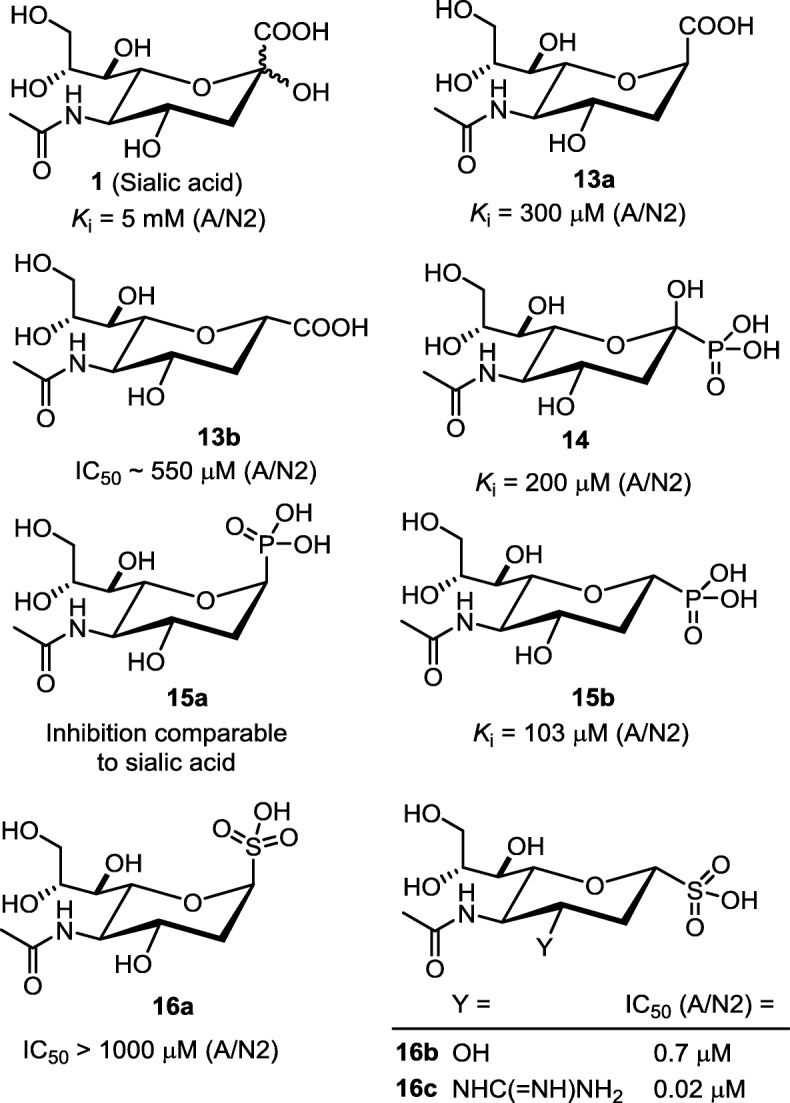
Influenza virus NA inhibitors based on bioisostere-substituted surrogates of sialic acid
Considering phosphonic acid and sulfonic acid are more acidic than carboxylic acid, the phosphonate and sulfonate congeners are predicted to have higher affinity toward NA by enhancing the binding strength with the tri-arginine cluster in NA. The phosphonate congener 14 (equatorial PO3H2) was found to inhibit the NAs of influenza A/N2 and V. cholera viruses with IC50 values of 0.2 and 0.5 mM, better than the natural carboxylate substrate Neu5Ac [72]. The 2-deoxy phosphonate congeners 15a (axial PO3H) and 15b (equatorial PO3H) were synthesized [71], and shown to bind V. cholera NA with Ki values of 0.23 and 0.055 mM, respectively. In a related study [73], 15b shows inhibitory activity against H2N2 virus with Ki and IC50 values of 103 and 368 μM, respectively. However, the binding affinity of epimer 15a is too low to be detected.
The sulfonate derivative 16b (equatorial SO3H) is a more potent inhibitor (Ki = 2.47 μM against H2N2 virus NA) than the epimer 16a (axial SO3H) and the phosphonate congener 15b (equatorial PO3H) by 14 and 42 fold, respectively. Sulfonate 16b also inhibits the NAs of H5N1 and the drug-resistant H275Y mutant at the same level with Ki values of 1.62 and 2.07 μM. In another report [74], the sulfonate derivatives 16a and 16b were evaluated for their inhibitory ability against H3N2 (A/Perth/16/2009) virus by fluorometric enzymatic assay. The experiments indicate that 16b is a much stronger NA inhibitor than the axially substituted sulfonate 16a (IC50 > 1000 μM). The cell-based assay confirms that 16b has good ability to block H3N2 virus infection of MDCK cells in vitro (IC50 = 0.7 μM).
Furthermore, the C4-OH group in 16b is replaced by basic guanidino group to give the derivative 16c to engage strong bindings with the negatively charged residues (Glu119 and Asp151) in the active site of influenza NA [75]. Thus, the inhibitory activity of 16c (IC50 = 19.9 nM) against H3N2 virus NA is greatly enhanced. The C3-guanidino sulfonate 16c is a very potent inhibitor against influenza NAs of various strains, including H1N1, pandemic California/2009 H1N1 and H5N1-H274Y viruses, with potencies of 7.9 to 65.2 nM. Importantly, 16c at 1 mM is still inactive to human sialidase Neu2. As 16c inhibits in vitro infection of influenza H3N2 virus to MDCK-II cells with a high potency of 5 nM, it provides good opportunity for lead optimization.
Zanamivir phosphonate congener
Phosphonate group is commonly used as a bioisostere of carboxylate in drug design [76]. Compared with carboxylic acid (pKa = 4.74), phosphonic acid (pKa1 = 2.38) has higher acidity and stronger electrostatic interactions with guanidinium group. In a helical protein, the formation of phosphonate–guanidinium complex (ΔG0 = − 2.38 kJ/mol) is more stable than the carboxylate–guanidinium ion-pair (ΔG0 = + 2.51 kJ/mol) [77, 78]. A phosphonate ion in tetrahedral structure is also topologically complementary to bind with Arg118, Arg292 and Arg371 in influenza NAs. The molecular docking experiment [79] shows that zanaphosphor (ZP, compound 21 in Fig. 9), the phosphonate bioisostere of ZA, has higher affinity to NA. Compared the bonding mode of ZA in NA, ZP attains two more hydrogen bonds with the tri-arginine motif while other functional groups (C4-guanidinium, C5-acetamide and glycerol side chain) maintain comparable interactions. ZP possesses high affinity to influenza NAs with IC50 values in nanomolar range. Though the phosphonate analogs (e.g. 14 and 15b) of sialic acid are weak NA inhibitors with IC50 values in sub-millimolar range [72, 80], ZP mimicking the transition state of oxonium-like geometry in the enzymatic hydrolysis is a very effective NA inhibitor. ZP also showed higher activity than ZA in protecting the canine MDCK cells challenged by various influenza viruses including the resistant H275Y strain [79].
Fig. 9.
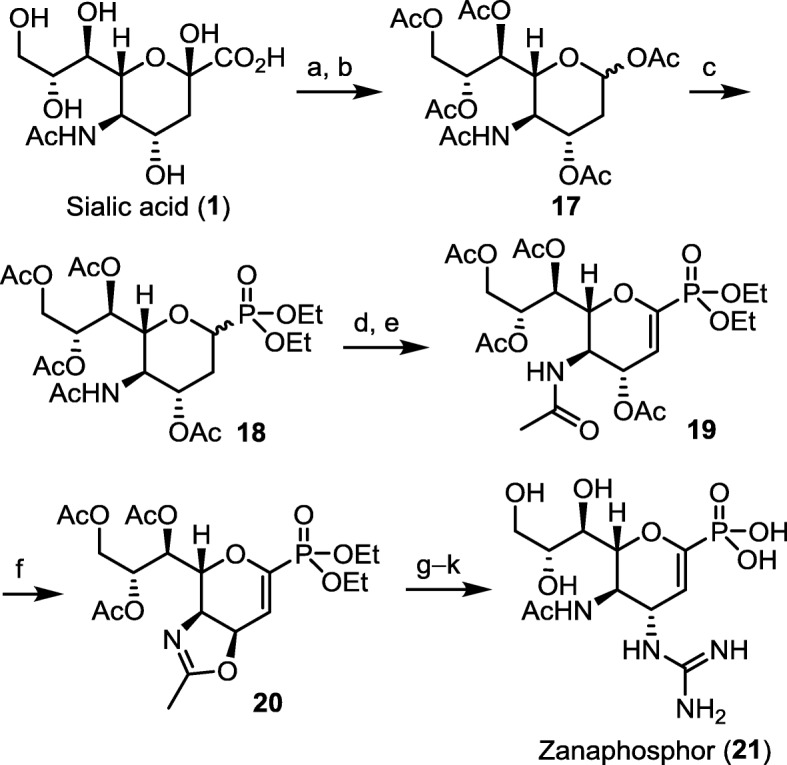
A practical synthesis of zanaphosphor. (a) Ac2O, py, rt., 12 h; (b) 100 °C, 5 h, 50% yield for two steps; (c) TMSOTf, P(OEt)2OTMS, 0 °C to rt., 24 h, 62% yield; (d) NBS, CH2Cl2, hv; (e) py, 50 °C, 1 h, 75% yield for two steps; (f) conc. H2SO4, Ac2O, AcOH, rt., 48 h; 80% yield; (g) TMSN3; (h) H2, Lindlar cat.; (i) MeS-C(=NBoc)NHBoc, HgCl2, Et3N, CH2Cl2; (j) TMSBr, CH2Cl2; (k) MeONa, MeOH, 55% yield for 5 steps. Boc = tert-butoxycarbonyl, NBS = N-bromosuccinimide, py = pyridine, TMS = trimethylsilyl, TMSOTf = trimethylsilyl trifluoromethanesulfonate
The first practical synthesis of ZP was achieved by Fang and coworkers using sialic acid as a viable starting material (Fig. 9) [79]. Sialic acid is firstly protected as a peracetate derivative, which undergoes a concomitant decarboxylation at 100 °C to give the acetyl glycoside 17. The anomeric acetate was replaced with phosphonate group by using diethyl (trimethylsilyl)phosphite as the nucleophile in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) as a promoter. After photochemical bromination, the intermediate is treated with a base to eliminate an HBr molecule for construction of the oxacyclohexene core structure. Following the previously reported procedure [81], the guanidine substituent is introduced to the C-4 position to furnish ZP. Another synthetic route to ZP is also explored by using inexpensive d-glucono-δ-lactone as the starting material, which proceeds through an asymmetric aza-Henry reaction as a key step [82].
Oseltamivir phosphonate congener
In the related study, tamiphosphor (TP, 22) was synthesized as the phosphonate congener of oseltamivir carboxylate by several methods (Fig. 10). The first synthesis [83] begins with introduction of a (diphosphoryl)methyl substituent to the C-5 position of d-xylose, and the subsequent intramolecular Horner−Wadsworth−Emmons (HWE) reaction constructs the cyclohexene-phosphonate core structure. Intramolecular HWE reaction was also applied to build up the scaffold of the polysubstituted cyclohexene ring in another TP synthesis starting with N-acetyl-d-glucosamine (d-GlcNAc) [84]. d-GlcNAc contains a preset acetamido group to manipulate the required absolute configuration in the TP synthesis. In the three-component one-pot approach [85], a chiral amine-promoted Michael reaction of 2-ethylbutanal with nitroenamide, a second Michael addition to 1,1-diphosphorylethene and an intramolecular HWE reaction are sequentially performed in one flask to construct the cyclohexene-phosphonate core structure. TP is thus synthesized by subsequent reduction of the nitro group and hydrolysis of the phosphonate ester. In another synthetic strategy of TP, palladium-catalyzed phosphonylation of 1-halocyclohexene is effectively applied as a key reaction [86–88].
Fig. 10.

Strategies for synthesis of oseltamivir (OS, 5), tamiphosphor (TP, 22), tamiphosphor monoethyl ester (TP1Et, 23), guanidino tamiphosphor (TPG, 24) and guanidino tamiphosphor monoethyl ester (TPG1Et, 25)
In addition to TP having C5-amino substituent, the TPG analog (24) having C5-guanidino group is also synthesized for evaluation its NA inhibitory activity. It is noted that treatment of phosphonate diethyl esters with bromotrimethylsilane (TMSBr) gives the phosphonic acids TP and TPG, whereas treatment with sodium ethoxide gives the corresponding phosphonate monoesters 23 and 25.
TP containing a phosphonate group is a potent inhibitor against human and avian influenza viruses, including A/H1N1 (wild-type and H275Y mutant), A/H5N1, A/H3N2 and type B viruses. TPG is even a stronger NA inhibitor because the guanidine group is more basic for stronger interactions with Glu119, Asp151 and Glu227 [18–20, 89].
Though TP (log D = − 1.04) has double negative charges on the phosphonate group, it is more lipophilic than OC (log D = − 1.69) carrying a single negative charge. The improved lipophilicity of TP is attributable to higher acidity of phosphonic acid to enhance the intramolecular zwitterionic structure or the intermolecular ion-pair structures [57, 60, 90, 91]. The guanidino compounds are also more lipophilic than their corresponding amino compounds because guanidine is more basic and preferable to form zwitterionic/ion-pair structures with the phosphonate group.
Though oseltamivir as a carboxylate ester is inactive to NA, the phosphonate monoester 23 exhibits high NA inhibitory activity because it retains a negative charge in the monoalkyl phosphonate moiety to exert adequate electrostatic interactions with the tri-arginine motif. The phosphonate diester is inactive to NA, while both phosphonate monoesters 23 and 25 show the anti-influenza activity comparable to phosphonic acids 22 and 24. This result may be attributed to better lipophilicity of monoesters to enhance intracellular uptake. The alkyl substituent in phosphonate monoester can be tuned to improve pharmacokinetic properties including bioavailability. For example, TP and TP monoethyl ester have 7 and 12% oral availability in mice, respectively. It is worth noting that TPG and its monoester 25 also possess significant inhibitory activity against the H275Y oseltamivir-resistant strain with IC50 values of 0.4 and 25 nM, respectively. In another study [92], TP monoester molecules are immobilized on gold nanoparticles, which bind strongly and selectively to all seasonal and pandemic influenza viruses through the NAs.
The mice experiments are conducted by oral administration of TP or its derivative after challenge with a lethal dose (10 LD50) of influenza virus [93]. When administered at doses of 1 mg/kg/day or higher, TP, TPG and their phosphonate monoesters (22–25) all render significant protection of mice infected with influenza viruses. Despite the low bioavailability (≤ 12%), all four phosphonates maintain the plasma concentrations in mice above the concentration required to inhibit influenza viruses. The metabolism studies indicate that almost no phosphonate monoesters 23 and 25 were transformed into their parental phosphonic acids 22 and 24. Therefore, these phosphonate monoesters are active drugs, unlike OS prodrug that releases the active carboxylic acid by endogenous hydrolysis.
Peramivir phosphonate congener
Peraphosphor (PP, 33) is the phosphonate congener of peramivir (PE). An efficient synthetic method of peraphosphor [94] comprises a [3 + 2] cycloaddition of 2-ethylbuanenitrile oxide (27) with a cyclopentene dipolarophile 26 (Fig. 11). After reduction with NiCl2 − NaBH4 to give multiple substituted cyclopentane-1-carboxylic acid 29, Barton–Crich iododecarboxylation successfully provides the iodo compound 30 with retention of the S-configuration as confirmed by X-ray diffraction analysis. The ring-opening reaction of epoxide 31 is performed at a low temperature (− 78 °C) by using diethyl phosphite and boron trifluoride etherate to afford the phosphonate diester 32, which is further transformed into PP (33) and the phosphonate monoester (34).
Fig. 11.

Synthesis of peraphosphor (PP, 33) and the monoethyl ester (PP1Et, 34) via a key step of [3 + 2] cycloaddition of 2-ethylbutanenitrile oxide with a cyclopentene dipolarophile
Although PP is a good NA inhibitor (IC50 = 5.2 nM against A/WSN/33 H1N1), its inhibitory activity is unexpectedly 74 times lower than that of PE, contrary to the previous computational study [95] that predicted PP to be a stronger binder for N1 neuraminidase. Due to the flexible cyclopentane core structure, the phosphonate congener (PP) can display different conformation than the carboxylate compound (PE). Therefore, the NA inhibitory activity of PP series is less predictable. The phosphonate compounds 33 and 34 show reduced binding affinity to the H275Y mutant with IC50 of 86 and 187 nM, respectively, presumably because less hydrophobic interactions are acquired by the 3-pentyl group in the active site of the mutant NA [96, 97]. However, the phosphonate monoalkyl ester 34 exhibits the anti-influenza activity superior to that of parental phosphonic acid 33 in the cell-based assay. Inferred from the calculated partition and distribution coefficients, the phosphonate monoalkyl ester can increase lipophilicity to enhance intracellular uptake.
Since the crystal structure of PE–NA complex (PDB code: 1L7F) [96] reveals that the C2-OH group of peramivir has no direct interaction with influenza NA, a dehydration analog of PP is prepared for bioactivity evaluation. By forming a more rigid cyclopentene ring, the PP dehydration analog regains extensive electrostatic interactions with the tri-arginine cluster in NA, thus exhibiting high NA inhibitory activity (IC50 = 0.3 nM) against influenza H1N1 virus.
Oseltamivir boronate, trifluoroborate, sulfinate, sulfonate and sulfone congeners
Compared to carboxylic acid (pKa ≈ 4.5), boronic acid is a weaker acid (pKa ≈ 10.0) while sulfinic acid (pKa ≈ 2.0) and sulfonic acid (pKa ≈ − 0.5) are stronger acids. Figure 12 outlines the synthetic methods for the oseltamivir boronate, trifluoroborate, sulfinate, sulfonate and sulfone congeners [98]. Oseltamivir carboxylic acid (OC) is converted to a Barton ester, which undergoes photolysis in the presence of CF3CH2I to give the iodocyclohexene derivative 35. This pivotal intermediate is subjected to palladium-catalyzed coupling reactions with appropriate diboron and thiol reagents to afford OS boronate (36a), trifluoroborate (37a), sulfinate (39a), sulfonate (40a) and sulfone (42a) congeners. The corresponding guanidino analogs (GOC congeners) are also synthesized. The GOC congeners (b series) consistently display better NA inhibition and anti-influenza activity than the corresponding OC congeners (a series). The GOC sulfonate congener (40b) is the most potent anti-influenza agent in this series and shows EC50 of 2.2 nM against the wild-type H1N1 virus. Since sulfonic acid is a stronger acid than carboxylic acid, it can exert stronger electrostatic interactions than GOC on the three arginine residues (R118, R292 and R371) in the NA active site. The sulfonate compound 40b may exist in zwitterionic structure and form the sulfonate−guanidinium ion-pair more effectively than GOC to attain higher lipophilicity as predicted by the distribution coefficients (cLog D) values. Interestingly, the congeners with trifluoroborate, sulfone or sulfonate ester still exhibit significant NA inhibitory activity, indicating that the polarized B−F and S → O bonds still provide sufficient interactions with the tri-arginine motif.
Fig. 12.

Synthesis of oseltamivir boronates (36a/36b), trifluoroborates (37a/37b), sulfinates (39a/39b), sulfonates (40a/40b) and sulfones (42a/42b) from oseltamivir carboxylic acid (OC)
Modification of zanamivir at the glycerol side chain
Replacing the glycerol chain in ZA with tertiary amides (e.g. 43b, in Fig. 13) still keeps good NA inhibitory activity with the IC50 values similar to that of ZA [99, 100]. Compared to the function of 3-pentoxy group in oseltamivir, the dialkylamide moiety in 43b may render similar hydrophobic interactions in the S5 site of NA. To support this hypothesis, the crystallographic and molecular dynamics studies of compound 43a with influenza NA were carried out to show that the Glu276 and Arg224 residues form a salt bridge to produce a lipophilic pocket, and an extended lipophilic cleft is formed between Ile222 and Ala246 near the S4 site. The N-isopropyl and phenylethyl substituents of 43a can properly reside in the lipophilic pocket and cleft, respectively [101, 102].
Fig. 13.

Modification of zanamivir at the glycerol side chain. The C7-OH group points away from the NA active site according to the crystallographic analysis of the ZA–NA complex [103]
The three-dimensional structure of ZA–NA complex [103] shows that the C7-OH group exposes to water without direct interaction with NA. Therefore, the C7-OH is an ideal site for structural modification. Laninamivir (compound 44) derives from ZA by changing the C7-OH group to a methoxy group without reduction of NA inhibitory activity. Laninamivir is developed to Inavir (6) as a long-acting drug by further converting the C9-OH group to an octanoate ester. The lipophilic octanoyl group is proposed to make compound 6 more permeable to cells. Compound 6 is rapidly hydrolyzed by esterases to give laninamivir, which is hydrophilic and may be captured in endoplasmic reticulum and Golgi. When the influenza NA matures in endoplasmic reticulum and Golgi apparatus, laninamivir can firmly retain the NA, thereby preventing the formation of progeny virus particles [104]. The half-life of prodrug 6 was about 2 h in man, and the active ingredient 44 appeared at 4 h after inhalation administration. Compound 44 was slowly eliminated over 144 h [38, 105, 106]. Inavir only needs one inhalation with 40 mg dose to last 5 days for influenza treatment, compared to Relenza and Tamiflu which require twice daily administration at 10 mg and 75 mg doses. Moreover, ZA analogs having the C7-OH derived to carbamates (e.g. compound 45) do not cause significant reduction in anti-influenza activity [107].
Conjugating neuraminidase inhibitors with enhanced anti-influenza activity
Using NA inhibitor is a good therapy by preventing the spread of progeny viral particles. However, there are related problems in quest of solutions. For example, how to kill the existing viruses in severely infected patients? Is it possible to develop anti-influenza drugs that also suppress the complication of inflammation, especially the cytokine storm caused by cross-species infection? To address these issues, one may consider conjugating NA inhibitors with other therapeutic entity to provide better anti-influenza activity.
Multi-component drug-cocktails may have complex pharmacokinetics and unpredictable drug−drug interactions [108], whereas conjugate inhibitors are designed to incorporate multiple therapeutic entities into a single drug by covalent bond [109, 110].
Conjugating zanamivir with porphyrin to kill influenza viruses
Porphyrins and the related compounds have been used as photosensitizers to activate molecular oxygen [111–113]. Activated singlet oxygen (1O2) is a highly reactive oxidant that can be utilized to kill adjacent cells in photodynamic therapy (PDT), which has been successfully applied to cancer treatment, and occasionally for treatments of bacterial and viral infections [114–116].
Because ZA has strong affinity to influenza NA, it is an excellent payload to deliver porphyrins to influenza virus in a specific way. Using the C7-OH group as connection hinge, four ZA molecules are linked to a porphyrin core structure to furnish the dual functional ZA conjugate 46 (Fig. 14) [117]. The ZA–porphyrin conjugate inhibits human and avian influenza NAs with the IC50 values in nanomolar range. By plaque yield reduction assay, conjugate 46 shows 100-fold potency than monomeric ZA in inactivation of influenza viruses. Influenza H1N1 viruses are reduced to less than 5% on treatment with conjugate 46 at 200 nM for 1 h under illumination of room light, whereas 60% titer of viruses remain on treatment with ZA alone or combination of ZA and porphyrin at micromolar concentrations. The viral inactivation by 46 is associated with the high local concentration of the ZA–porphyrin conjugate brought to the viral surface by the high affinity of the ZA moiety for NA. Under irradiation of room light, the porphyrin component of conjugate 46 brings about reactive singlet oxygen to kill the attached viruses without damaging other healthy host cells. In contrast, a similar concentration of free porphyrin alone or in combination with zanamivir cannot accumulate to a high local concentration on the viral surface, and thus the destruction of influenza virus by light irradiation is ineffective.
Fig. 14.

A strategy to kill influenza virus by ZA–porphyrin conjugate. ZA carries the conjugate 46 to viral surface through binding with neuraminidase, and porphyrin is light sensitized (λmax = 420 nm) to generate singlet oxygen in close proximity, causing inactivation of influenza virus
In another aspect, the tetrameric ZA conjugate 46 can also take advantage of multivalent effect [118–121] to enhance the binding with influenza NA, which exists as a homotetramer on the surface of the virus, thus inducing aggregation of viral particles for physical reduction of the infectivity. Di-, tri-, tetra- and polyvalent ZA conjugates are also designed to increase the binding affinity with NA [122–128]. Klibanov and coworkers [129] implanted ZA and sialic acid molecules on the poly(isobutylene-alt-maleic anhydride) backbone for concurrent bindings with viral NAs and HAs, thus greatly increasing the anti-influenza activity by more than 1000 fold.
Conjugating zanamivir with caffeic acid to alleviate inflammation
Influenza infection may induce uncontrolled cytokine storms as that happened in 2003 avian flu, resulting in the cross-species transmission of H5N1 avian virus to humans to claim a large number of lives. Since extension from the C7-OH would not interfere with NA binding, the dual functional ZA–caffeate conjugates 47a and 47b (Fig. 15) are prepared by connection of caffeic acid to ZA with ester or amide linkage [130]. The cell-based assays indicate that conjugates 47a and 47b effectively inactivate H1N1 and H5N1 influenza viruses with EC50 in nanomolar range. These conjugates also significantly inhibit proinflammatory cytokines, such as interleukin-6 (IL-6) and interferon-gamma (INF-γ), compared to ZA alone or in the presence of caffeic acid (CA).
Fig. 15.
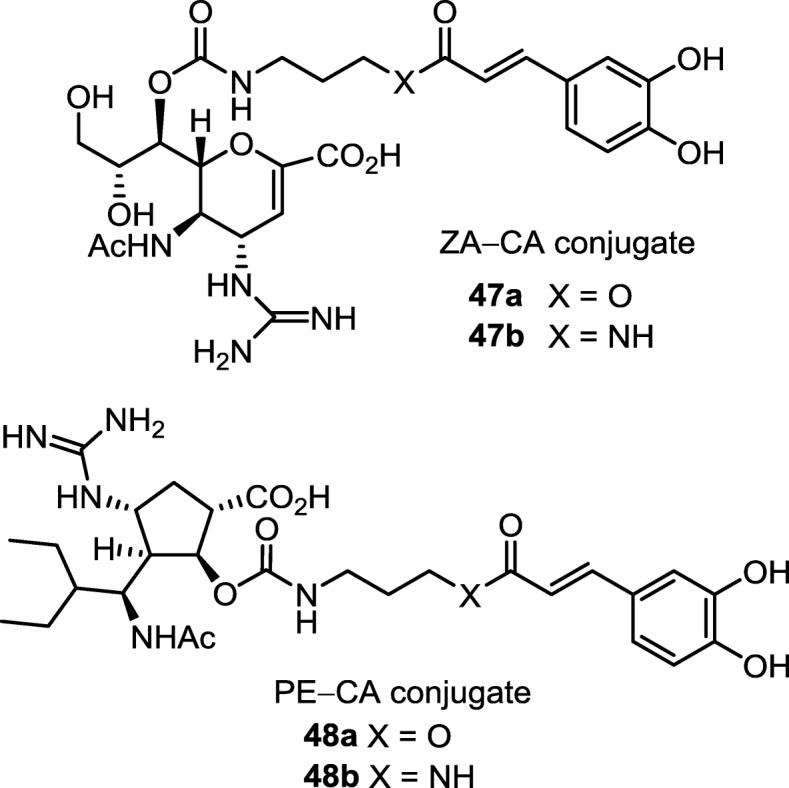
Enhanced anti-influenza activity of ZA−caffeate and PE−caffeate conjugates by synergistic inhibition of neuraminidase and suppression of the virus-induced cytokines
Treatment with the ZA conjugates 47a and 47b improves the survival of mice infected with influenza virus. For example, treatment of conjugates 47a and 47b at 1.2 μmol/kg/day, i.e. the human equivalent dose, provides 100% protection of mice from lethal-dose challenge of influenza H1N1 or H5N1 viruses in the 14-day experimental period. Even at a low dose of 0.12 μmol/kg/day, conjugates 47a and 47b still significantly protect the H1N1 virus-infected mice, showing greater than 50% survival on day 14. ZA alone or anti-inflammatory agent alone cannot reach such high efficacy for influenza therapy [131, 132]. Although the combination of an NA inhibitor with anti-inflammatory agents is effective in treating influenza-infected mice [133, 134], the drug development may encounter problems with complex pharmacokinetics behavior. On the other hand, conjugates 47a and 47b bear ZA component for specific binding to influenza virus, thus delivering the anti-inflammatory component for in situ action to suppress the virus-induced cytokines. By incorporating a caffeate component, conjugates 47a and 47b also have higher lipophilicity to improve the pharmacokinetic properties.
Conjugating peramivir with caffeic acid as enhanced oral anti-influenza drug
The C2-OH group, which does not directly interact with NA protein [135, 136], is used for conjugation of peramivir with caffeic acid. The PE–caffeate conjugates 48a and 48b (Fig. 15) are nanomolar inhibitors against wild-type and mutated H1N1 viruses [137]. The molecular modeling of conjugate 48b reveals that the caffeate moiety is preferably located in the 295-cavity of H275Y neuraminidase, thus providing additional interactions to compensate for the peramivir moiety, which has reduced binding affinity to H275Y mutant caused by Glu276 dislocation. By incorporating a caffeate moiety, conjugates 48a and 48b also have higher lipophilicity than PE. Thus, conjugates 48a and 48b provide better effect in protecting MDCK cells from infection of H275Y virus at low EC50 (~ 17 nM). Administration of conjugates 48a or 48b by oral gavage is effective in treating mice infected by a lethal dose of wild-type or H275Y influenza virus. In view of drug metabolism, since the ester bond in the conjugate 48a is easily hydrolyzed in plasma, the conjugate 48b having a robust amide bond may be a better candidate for development into oral drug that is also active against mutant viruses.
Conclusion
In this review, the anti-influenza drugs are discussed with an emphasis on those targeting the NA glycoprotein. In order to generate more potent NA inhibitors and counter the surge of resistance caused by natural mutations, the structures of on-market anti-influenza drugs are used as templates for design of new NA inhibitors. In particular, we highlight the modifications of these anti-influenza drugs by replacing the carboxylate group in oseltamivir, zanamivir and peramivir with bioisosteres (e.g. phosphonate and sulfonate) to attain higher binding strength with influenza NA. The carboxylic acid can also be converted to ester prodrugs for better lipophilicity and bioavailability. Using lipophilic acyl derivatives of guanidine as prodrug of zanamivir and guanidino-oseltamivir can mitigate the problem of low bioavailability. The C7-OH in zanamivir and C2-OH in peramivir, which point outward from the active site of influenza NA, are suitable for derivatization. Conjugating zanamivir molecules to porphyrin not only enhances the NA inhibitory activity, but also effectively activates molecular oxygen to kill influenza viruses. The ZA–caffeate and PE–caffeate conjugates render higher efficacy than their parental compounds (ZA or PE) in treatments of the mice infected with human or avian influenza viruses. Using congeners and conjugates is a viable strategy to develop orally available anti-influenza drug that is also active to mutant viruses. Interdisciplinary collaboration is essential in development of new anti-influenza drugs, and synthetic chemists play an important role to reach the goal.
Acknowledgments
Not applicable.
Abbreviations
- Boc
tert-butoxycarbonyl
- CA
caffeic acid
- DANA
didehydro-2-deoxy-N-acetylneuraminic acid
- d-GlcNAc
N-acetyl-d-glucosamine
- GOC
guanidino-oseltamivir carboxylate
- HA
hemagglutinin
- HNAP
1-hydroxy-2-naphthoic acid
- HWE
Horner−Wadsworth−Emmons
- log D
distribution coefficient
- log P
partition coefficient
- MUNANA
2-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid
- NA
neuraminidase
- NBS
N-bromosuccinimide
- Neu5Ac
sialic acid
- OC
oseltamivir carboxylate
- OS
oseltamivir
- PDT
photodynamic therapy
- PE
peramivir
- PP
peraphosphor
- PP1Et
peraphosphor monoethyl ester
- py
pyridine
- RNP
ribonucleoprotein
- TMS
trimethylsilyl
- TMSBr
bromotrimethylsilane
- TMSOTf
trimethylsilyl trifluoromethanesulfonate
- TP
tamiphosphor
- TP1Et
tamiphosphor monoethyl ester
- TPG
guanidino tamiphosphor
- TPG1Et
guanidino tamiphosphor monoethyl ester
- ZA
zanamivir
- ZP
zanaphosphor
Authors’ contributions
Two authors jointly prepared this manuscript. JM provided outlines for JJ to draft manuscript, and JM then made substantial modifications. Both authors read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Das K. Antivirals targeting influenza a virus. J Med Chem. 2012;55(14):6263–6277. doi: 10.1021/jm300455c. [DOI] [PubMed] [Google Scholar]
- 2.Syrjänen RK, Jokinen J, Ziegler T, Sundman J, Lahdenkari M, Julkunen I, Kilpi TM. Effectiveness of pandemic and seasonal influenza vaccines in preventing laboratory-confirmed influenza in adults: a clinical cohort study during epidemic seasons 2009-2010 and 2010-2011 in Finland. PLoS One. 2014;9(9):e108538. doi: 10.1371/journal.pone.0108538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajão DS, Pérez DR. Universal vaccines and vaccine platforms to protect against influenza viruses in humans and agriculture. Front Microbiol. 2018;9:123. doi: 10.3389/fmicb.2018.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taubenberger JK, Morens DM. 1918 influenza: the mother of all pandemics. Emerg Infect Dis. 2006;12(1):15–22. doi: 10.3201/eid1209.05-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, et al. Antigenic and genetic characteristics of swine-origin 2009 a(H1N1) influenza viruses circulating in humans. Science. 2009;325(5937):197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan JF, To KK. Tse H, Jin DY, Yuen KY. Interspecies transmission and emergence of novel viruses: lessons from bats and birds. Trends Microbiol. 2013;21(10):544–555. doi: 10.1016/j.tim.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palese P. Influenza: old and new threats. Nat Med. 2004;10(12 Suppl):S82–S87. doi: 10.1038/nm1141. [DOI] [PubMed] [Google Scholar]
- 8.Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, et al. An overlapping protein-coding region in influenza a virus segment 3 modulates the host response. Science. 2012;337(6091):199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wise HM, Foeglein A, Sun J, Dalton RM, Patel S, Howard W, et al. A complicated message: identification of a novel PB1-related protein translated from influenza a virus segment 2 mRNA. J Virol. 2009;83(16):8021–8031. doi: 10.1128/JVI.00826-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kreijtz JH, Fouchier RA, Rimmelzwaan GF. Immune responses to influenza virus infection. Virus Res. 2011;162(1–2):19–30. doi: 10.1016/j.virusres.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 11.Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, et al. New world bats harbor diverse influenza a viruses. PLoS Pathog. 2013;9(10):e1003657. doi: 10.1371/journal.ppat.1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chambers BS, Parkhouse K, Ross TM, Alby K, Hensley SE. Identification of Hemagglutinin residues responsible for H3N2 antigenic drift during the 2014–2015 influenza season. Cell Rep. 2015;12(1):1–6. doi: 10.1016/j.celrep.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu X, Wu X, Sun Q, Zhang C, Yang S, Li L, Jia Z. Progress of small molecular inhibitors in the development of anti-influenza virus agents. Theranostics. 2017;7(4):826–845. doi: 10.7150/thno.17071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossman JS, Lamb RA. Influenza virus assembly and budding. Virology. 2011;411(2):229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaymard A, Le Briand N, Frobert E, Lina B, Escuret V. Functional balance between neuraminidase and haemagglutinin in influenza viruses. Clin Microbiol Infect. 2016;22(12):975–983. doi: 10.1016/j.cmi.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 16.Das K. Antivirals targeting influenza a virus. J Med Chem. 2012;55(14):6263–6277. doi: 10.1021/jm300455c. [DOI] [PubMed] [Google Scholar]
- 17.Das K, Aramini JM, Ma LC, Krug RM, Arnold E. Structures of influenza a proteins and insights into antiviral drug targets. Nat Struct Mol Biol. 2010;17(5):530–538. doi: 10.1038/nsmb.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Clercq E. Antiviral agents active against influenza a viruses. Nat Rev Drug Discov. 2006;5(12):1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayden FG, Sugaya N, Hirotsu N, Lee N, de Jong MD, Hurt AC, et al. Baloxavir marboxil for uncomplicated influenza in adults and adolescents. N Engl J Med. 2018;379(10):913–923. doi: 10.1056/NEJMoa1716197. [DOI] [PubMed] [Google Scholar]
- 20.von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Discov. 2007;6(12):967–974. doi: 10.1038/nrd2400. [DOI] [PubMed] [Google Scholar]
- 21.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451(7178):596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza a virus. Nature. 2008;451(7178):591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barik S. New treatments for influenza. BMC Med. 2012;10:104. doi: 10.1186/1741-7015-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheu TG, Fry AM, Garten RJ, Deyde VM, Shwe T, Bullion L, Peebles PJ, Li Y, Klimov AI, Gubareva LV. Dual resistance to adamantanes and oseltamivir among seasonal influenza a(H1N1) viruses: 2008-2010. J Infect Dis. 2011;203(1):13–17. doi: 10.1093/infdis/jiq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chamni S, De-Eknamkul W. Recent progress and challenges in the discovery of new neuraminidase inhibitors. Expert Opin Ther Pat. 2013;23(4):409–423. doi: 10.1517/13543776.2013.765861. [DOI] [PubMed] [Google Scholar]
- 26.Fukao K, Noshi T, Yamamoto A, Kitano M, Ando Y, Noda T, Baba K, Matsumoto K, Higuchi N, Ikeda M, Shishido T, Naito A. Combination treatment with the cap-dependent endonuclease inhibitor baloxavir marboxil and a neuraminidase inhibitor in a mouse model of influenza a virus infection. J Antimicrob Chemother. 2019;74(3):654–662. doi: 10.1093/jac/dky462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laborda P, Wang SY, Voglmeir J. Influenza neuraminidase inhibitors: synthetic approaches, derivatives and biological activity. Molecules. 2016;21(11):1513–1553. doi: 10.3390/molecules21111513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moscona A. Neuraminidase inhibitors for influenza. N Engl J Med. 2005;353(13):1363–1373. doi: 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]
- 29.Dunn CJ, Goa KL. Zanamivir, a review of its use in influenza. Drugs. 1999;58(4):761–784. doi: 10.2165/00003495-199958040-00016. [DOI] [PubMed] [Google Scholar]
- 30.Cheer SM, Wagstaff AJ. Zanamivir, an update of its use in influenza. Drugs. 2002;62(1):71–106. doi: 10.2165/00003495-200262010-00004. [DOI] [PubMed] [Google Scholar]
- 31.Kim CU, Lew W, Williams MA, Wu H, Zhang L, Chen X, Escarpe PA, Mendel DB, Laver WG, Stevens RC. Structure-activity relationship studies of novel carbocyclic influenza neuraminidase inhibitors. J Med Chem. 1998;41(12):2451–2460. doi: 10.1021/jm980162u. [DOI] [PubMed] [Google Scholar]
- 32.McClellan K, Perry CM. Oseltamivir, a review of its use in influenza. Drugs. 2001;61(2):263–283. doi: 10.2165/00003495-200161020-00011. [DOI] [PubMed] [Google Scholar]
- 33.Kubo S, Tomozawa T, Kakuta M, Tokumitsu A, Yamashita M. Laninamivir prodrug CS-8958, a long-acting neuraminidase inhibitor, shows superior anti-influenza virus activity after a single administration. Antimicrob Agents Chemother. 2010;54(3):1256–1264. doi: 10.1128/AAC.01311-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smee DF, Sidwell RW. Peramivir (BCX-1812, RWJ-270201): potential new therapy for influenza. Expert Opin Investig Drugs. 2002;11(6):859–869. doi: 10.1517/13543784.11.6.859. [DOI] [PubMed] [Google Scholar]
- 35.Jain S, Fry AM. Peramivir: another tool for influenza treatment? Clin Infect Dis. 2011;52(6):707–709. doi: 10.1093/cid/cir010. [DOI] [PubMed] [Google Scholar]
- 36.Cass LMR, Efthymiopoulos C, Bye A. Pharmacokinetics of zanamivir after intravenous, oral, inhaled or intranasal administration to healthy volunteers. Clin Pharmacokinet. 1999;36(Suppl 1):1–11. doi: 10.2165/00003088-199936001-00001. [DOI] [PubMed] [Google Scholar]
- 37.Burch J, Corbett M, Stock C, Nicholson K, Elliot AJ, Duffy S, Westwood M, Palmer S, Stewart L. Prescription of anti-influenza drugs for healthy adults: a systematic review and meta-analysis. Lancet Infect Dis. 2009;9(9):537–545. doi: 10.1016/S1473-3099(09)70199-9. [DOI] [PubMed] [Google Scholar]
- 38.Ikematsu H, Kawai N. Laninamivir octanoate: a new long-acting neuraminidase inhibitor for the treatment of influenza. Expert Rev Anti-Infect Ther. 2011;9(10):851–857. doi: 10.1586/eri.11.112. [DOI] [PubMed] [Google Scholar]
- 39.Birnkrant D, Cox E. The emergency use authorization of peramivir for treatment of 2009 H1N1 influenza. N Engl J Med. 2009;361(23):2204–2207. doi: 10.1056/NEJMp0910479. [DOI] [PubMed] [Google Scholar]
- 40.Deyde VM, Xu X, Bright RA, Shaw M, Smith CB, Zhang Y, Shu Y, Gubareva LV, Cox NJ, Klimov AI. Surveillance of resistance to adamantanes among influenza a(H3N2) and a(H1N1) viruses isolated worldwide. J Infect Dis. 2007;196(2):249–257. doi: 10.1086/518936. [DOI] [PubMed] [Google Scholar]
- 41.Hayden F. Developing new antiviral agents for influenza treatment: what does the future hold? Clin Infect Dis. 2009;48(Suppl 1):S3–S13. doi: 10.1086/591851. [DOI] [PubMed] [Google Scholar]
- 42.Meijer A, Lackenby A, Hungnes O, Lina B, van-der Werf S, Schweiger B, Opp M, Paget J, van-de Kassteele J, Hay A, Zambon M. Oseltamivir-resistant influenza virus A (H1N1), Europe, 2007–2008 season. Emerg Infect Dis. 2009;15(4):552–560. doi: 10.3201/eid1504.181280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bloom JD, Gong LI, Baltimore D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science. 2010;328(5983):1272–1275. doi: 10.1126/science.1187816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abed Y, Pizzorno A, Bouhy X, Boivin G. Role of permissive neuraminidase mutations in influenza a/Brisbane/59/2007-like (H1N1) viruses. PLoS Pathog. 2011;7(12):e1002431. doi: 10.1371/journal.ppat.1002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burmeister WP, Henrissat B, Bosso C, Cusack S, Ruigrok RWH. Influenza-B virus neuraminidase can synthetize its own inhibitor. Structure. 1993;1(1):19–26. doi: 10.1016/0969-2126(93)90005-2. [DOI] [PubMed] [Google Scholar]
- 46.von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363(6428):418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 47.Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, Blackburn GM, Hay AJ, Gamblin SJ, Skehel JJ. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443(7107):45–49. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
- 48.Taylor NR, von Itzstein M. Molecular modeling studies on ligand binding to sialidase from influenza virus and the mechanism of catalysis. J Med Chem. 1994;37(5):616–624. doi: 10.1021/jm00031a011. [DOI] [PubMed] [Google Scholar]
- 49.Janakiraman MN, White CL, Laver WG, Air GM, Luo M. Structure of influenza virus neuraminidase B/Lee/40 complexed with sialic acid and a dehydro analog at 1.8-Å resolution: implications for the catalytic mechanism. Biochemistry. 1994;33(27):8172–8179. doi: 10.1021/bi00193a002. [DOI] [PubMed] [Google Scholar]
- 50.Wang MZ, Tai CY, Mendel DB. Mechanism by which mutations at His274 alter sensitivity of influenza a virus N1 neuraminidase to oseltamivir carboxylate and zanamivir. Antimicrob Agents Chemother. 2002;46(12):3809–3816. doi: 10.1128/AAC.46.12.3809-3816.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Collins PJ, Haire LF, Lin YP, Liu J, Russell RJ, Walker PA, Skehel JJ, Martin SR, Hay AJ, Gamblin SJ. Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature. 2008;453(7199):1258–1262. doi: 10.1038/nature06956. [DOI] [PubMed] [Google Scholar]
- 52.McKimm-Breschkin JL. Resistance of influenza viruses to neuraminidase inhibitors – a review. Antivir Res. 2000;47(1):1–17. doi: 10.1016/S0166-3542(00)00103-0. [DOI] [PubMed] [Google Scholar]
- 53.Proudfoot JR. The evolution of synthetic oral drug properties. Bioorg Med Chem Lett. 2005;15(4):1087–1090. doi: 10.1016/j.bmcl.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 54.Widmer N, Meylan P, Ivanyuk A, Aouri M, Decosterd LA, Buclin T. Oseltamivir in seasonal, avian H5N1 and pandemic 2009 a/H1N1 influenza. Clin Pharmacokinet. 2010;49(11):741–765. doi: 10.2165/11534730-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 55.Liu ZY, Wang B, Zhao LX, Li YH, Shao HY, Yi H, You XF, Li ZR. Synthesis and anti-influenza activities of carboxyl alkoxyalkyl esters of 4-guanidino-Neu5Ac2en (zanamivir) Bioorg Med Chem Lett. 2007;17(17):4851–4854. doi: 10.1016/j.bmcl.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 56.Gupta SV, Gupta D, Sun J, Dahan A, Tsume Y, Hilfinger J, Lee KD, Amidon GL. Enhancing the intestinal membrane permeability of zanamivir: a carrier mediated prodrug approach. Mol Pharm. 2011;8(6):2358–2367. doi: 10.1021/mp200291x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller JM, Dahan A, Gupta D, Varghese S, Amidon GL. Enabling the intestinal absorption of highly polar antiviral agents: ion-pair facilitated membrane permeation of zanamivir heptyl ester and guanidino oseltamivir. Mol Pharm. 2010;7(4):1223–1234. doi: 10.1021/mp100050d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gould PL. Salt selection for basic drugs. Int J Pharm. 1986;33(1–3):201–217. doi: 10.1016/0378-5173(86)90055-4. [DOI] [Google Scholar]
- 59.Cazzola M, Testi R, Matera MG. Clinical pharmacokinetics of salmeterol. Clin Pharmacokinet. 2002;41(1):19–30. doi: 10.2165/00003088-200241010-00003. [DOI] [PubMed] [Google Scholar]
- 60.Liu KC, Lee PS, Wang SY, Cheng YSE, Fang JM, Wong CH. Intramolecular ion-pair prodrugs of zanamivir and guanidino-oseltamivir. Bioorg Med Chem. 2011;19(16):4796–4802. doi: 10.1016/j.bmc.2011.06.080. [DOI] [PubMed] [Google Scholar]
- 61.Hsu PH, Chiu DC, Wu KL, Lee PS, Jan JT, Cheng YSE, Tsai KC, Cheng TJ, Fang JM. Acylguanidine derivatives of Zanamivir and Oseltamivir: potential orally available Prodrugs against influenza viruses. Eur J Med Chem. 2018;154:314–323. doi: 10.1016/j.ejmech.2018.05.030. [DOI] [PubMed] [Google Scholar]
- 62.Rudrawar S, Dyason JC, Rameix-Welti MA, Rose FJ, Kerry PS, Russell RJ, van der Werf S, Thomson RJ, Naffakh N, von Itzstein M. Novel sialic acid derivatives lock open the 150-loop of an influenza a virus group-1 sialidase. Nat Commun. 2010;1:113. doi: 10.1038/ncomms1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amaro RE, Minh DDL, Cheng LS, Lindstrom WM, Jr, Olson AJ, Lin JH, Li WW, McCammon JA. Remarkable loop flexibility in avian influenza N1 and its implications for antiviral drug design. J Am Chem Soc. 2007;129(25):7764–7765. doi: 10.1021/ja0723535. [DOI] [PubMed] [Google Scholar]
- 64.Lin CH, Chang TC, Das A, Fang MY, Hung HC, Hsu KC, Yang JM, von Itzstein M, Mong KK, Hsu TA, Lin CC. Synthesis of acylguanidine zanamivir derivatives as neuraminidase inhibitors and the evaluation of their bio-activities. Org Biomol Chem. 2013;11(24):3943–3948. doi: 10.1039/c3ob40624e. [DOI] [PubMed] [Google Scholar]
- 65.Das A, Adak AK, Ponnapalli K, Lin CH, Hsu KC, Yang JM, Hsu TA, Lin CC. Design and synthesis of 1,2,3-triazole-containing N-acyl zanamivir analogs as potent neuraminidase inhibitors. Eur J Med Chem. 2016;123:397–406. doi: 10.1016/j.ejmech.2016.07.064. [DOI] [PubMed] [Google Scholar]
- 66.Li Z, Meng Y, Xu S, Shen W, Meng Z, Wang Z, Ding G, Huang W, Xiao W, Xu J. Discovery of acylguanidine oseltamivir carboxylate derivatives as potent neuraminidase inhibitors. Bioorg Med Chem. 2017;25(10):2772–2781. doi: 10.1016/j.bmc.2017.03.052. [DOI] [PubMed] [Google Scholar]
- 67.Patani GA, LaVoie EJ. Bioisosterism: a rational approach in drug design. Chem Rev. 1996;96(8):3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- 68.Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54(8):2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 69.Walop JN, Boschman TAC, Jacobs J. Affinity of N-acetylneuraminic acid for influenza virus neuraminidase. Biochim Biophys Acta. 1960;44:185–186. doi: 10.1016/0006-3002(60)91544-4. [DOI] [PubMed] [Google Scholar]
- 70.Friebolin H, Supp M, Brossmer R, Keilich G, Ziegler D. 1H-NMR investigations on the mutarotation of N-acetyl-D-neuraminic acid. Angew Chem Int Ed Engl. 1980;19(3):208–209. doi: 10.1002/anie.198002081. [DOI] [Google Scholar]
- 71.Wallimann K, Vasella A. Phosphonic-acid analogues of the N-acetyl-2-deoxyneuraminic acids: synthesis and inhibition of Vibrio cholerae sialidase. Helv Chim Acta. 1990;73(5):1359–1372. doi: 10.1002/hlca.19900730523. [DOI] [Google Scholar]
- 72.Chan TH, Xin YC, von Itzstein M. Synthesis of phosphonic acid analogues of sialic acids (Neu5Ac and KDN) as potential sialidase inhibitors. J Org Chem. 1997;62(11):3500–3504. doi: 10.1021/jo961891p. [DOI] [Google Scholar]
- 73.Vavricka CJ, Muto C, Hasunuma T, Kimura Y, Araki M, Wu Y, Gao GF, Ohrui H, Izumi M, Kiyota H. Synthesis of sulfo-sialic acid analogues: potent neuraminidase inhibitors in regards to anomeric functionality. Sci Rep. 2017;7(1):8239. doi: 10.1038/s41598-017-07836-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hadházi Á, Pascolutti M, Bailly B, Dyason JC, Borbás A, Thomson RJ, von Itzstein M. A sialosyl sulfonate as a potent inhibitor of influenza virus replication. Org Biomol Chem. 2017;15(25):5249–5253. doi: 10.1039/C7OB00947J. [DOI] [PubMed] [Google Scholar]
- 75.Hadházi Á, Li L, Bailly B, Maggioni A, Martin G, Dirr L, Dyason JC, Thomson RJ, Gao GF, Borbás A, Ve T, Pascolutti M, von Itzstein M. A sulfonozanamivir analogue has potent anti-influenza virus activity. ChemMedChem. 2018;13(8):785–789. doi: 10.1002/cmdc.201800092. [DOI] [PubMed] [Google Scholar]
- 76.Ballatore C, Huryn DM, Smith AB., 3rd Carboxylic acid (bio) isosteres in drug design. ChemMedChem. 2013;8(3):385–395. doi: 10.1002/cmdc.201200585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schug KA, Lindner W. Noncovalent binding between guanidinium and anionic groups: focus on biological- and synthetic-based arginine/guanidinium interactions with phosph[on]ate and sulf[on]ate residues. Chem Rev. 2005;105(1):67–114. doi: 10.1021/cr040603j. [DOI] [PubMed] [Google Scholar]
- 78.Klenchin VA, Czyz A, Goryshin IY, Gradman R, Lovell S, Rayment I, Reznikoff WS. Phosphate coordination and movement of DNA in the Tn5 synaptic complex: role of the (R)YREK motif. Nucleic Acids Res. 2008;36(18):5855–5862. doi: 10.1093/nar/gkn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shie JJ, Fang JM, Lai PT, Wen WH, Wang SY, Cheng YSE, Tsai KC, Yang AS, Wong CH. A practical synthesis of zanamivir phosphonate congeners with potent anti-influenza activity. J Am Chem Soc. 2011;133(44):17959–17965. doi: 10.1021/ja207892q. [DOI] [PubMed] [Google Scholar]
- 80.Vasella A, Wyler R. Synthesis of a phosphonic acid analogue of N-acetyl-2,3-didehydro-2-deoxyneuraminic acid, an inhibitor of Vibrio cholerae sialidase. Helv Chim Acta. 1991;74(2):451–463. doi: 10.1002/hlca.19910740223. [DOI] [Google Scholar]
- 81.von Itzstein M, Wu WY, Jin B. The synthesis of 2,3-dehydro-2,4-dideoxy-4-guanidinyl-N-acetylneuraminic acid: a potent influenza virus sialidase inhibitor. Carbohydr Res. 1994;259(2):301–305. doi: 10.1016/0008-6215(94)84065-2. [DOI] [PubMed] [Google Scholar]
- 82.Lin LZ, Fang JM. Total synthesis of anti-influenza agents zanamivir and zanaphosphor via asymmetric aza-Henry reaction. Org Lett. 2016;18(17):4400–4403. doi: 10.1021/acs.orglett.6b02131. [DOI] [PubMed] [Google Scholar]
- 83.Shie JJ, Fang JM, Wang SY, Tsai KC, Cheng YSE, Yang AS, Hsiao SC, Su CY, Wong CH. Synthesis of tamiflu and its phosphonate congeners possessing potent anti-influenza activity. J Am Chem Soc. 2007;129(39):11892–11893. doi: 10.1021/ja073992i. [DOI] [PubMed] [Google Scholar]
- 84.Chen CA, Fang JM. Synthesis of oseltamivir and tamiphosphor from N-acetyl-D-glucosamine. Org Biomol Chem. 2013;11(44):7687–7699. doi: 10.1039/c3ob41622d. [DOI] [PubMed] [Google Scholar]
- 85.Lo YW, Fang JM. A short synthetic pathway via three-component coupling reaction to tamiphosphor possessing anti-influenza activity. Tetrahedron. 2015;71(2):266–270. doi: 10.1016/j.tet.2014.11.062. [DOI] [Google Scholar]
- 86.Shie JJ, Fang JM, Wong CH. A concise and flexible synthesis of the potent anti-influenza agents-tamiflu and tamiphosphor. Angew Chem Int Ed. 2008;47(31):5788–5791. doi: 10.1002/anie.200801959. [DOI] [PubMed] [Google Scholar]
- 87.Carbain B, Collins PJ, Callum L, Martin SR, Hay AJ, McCauley J, Streicher H. Efficient synthesis of highly active phospha-isosteres of the influenza neuraminidase inhibitor oseltamivir. ChemMedChem. 2009;4(3):335–337. doi: 10.1002/cmdc.200800379. [DOI] [PubMed] [Google Scholar]
- 88.Gunasekera DS. Formal synthesis of tamiflu: conversion of tamiflu into tamiphosphor. Synlett. 2012;23(4):573–576. doi: 10.1055/s-0031-1290356. [DOI] [Google Scholar]
- 89.Schmidt AC. Antiviral therapy for influenza : a clinical and economic comparative review. Drugs. 2004;64(18):2031–2046. doi: 10.2165/00003495-200464180-00003. [DOI] [PubMed] [Google Scholar]
- 90.Woods AS, Ferré S. Amazing stability of the arginine−phosphate electrostatic interaction. J Proteome Res. 2005;4(4):1397–1402. doi: 10.1021/pr050077s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pantos A, Tsogas I, Paleos CM. Guanidinium group: a versatile moiety inducing transport and multicompartmentalization in complementary membranes. Biochim Biophys Acta. 2008;1778(4):811–823. doi: 10.1016/j.bbamem.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 92.Stanley M, Cattle N, McCauley J, Rashid M, Field AR, Carbain B, Streicher H. 'TamiGold': phospha-oseltamivir-stabilised gold nanoparticles as the basis for influenza therapeutics and diagnostics targeting the neuraminidase (instead of the hemagglutinin) Med Chem Commun. 2012;3(11):1373–1376. doi: 10.1039/c2md20034a. [DOI] [Google Scholar]
- 93.Cheng TJ, Weinheimer S, Tarbet EB, Jan JT, Cheng YS, Shie JJ, Chen CL, Chen CA, Hsieh WC, Huang PW, Lin WH, Wang SY, Fang JM, Hu OY, Wong CH. Development of oseltamivir phosphonate congeners as anti-influenza agents. J Med Chem. 2012;55(20):8657–8670. doi: 10.1021/jm3008486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang PC, Fang JM, Tsai KC, Wang SY, Huang WI, Tseng YC, Cheng YSE, Cheng TJR, Wong CH. Peramivir phosphonate derivatives as influenza neuraminidase inhibitors. J Med Chem. 2016;59(11):5297–5310. doi: 10.1021/acs.jmedchem.6b00029. [DOI] [PubMed] [Google Scholar]
- 95.Udommaneethanakit T, Rungrotmongkol T, Bren U, Frecer V, Stanislav M. Dynamic behavior of avian influenza a virus neuraminidase subtype H5N1 in complex with oseltamivir, zanamivir, peramivir, and their phosphonate analogues. J Chem Inf Model. 2009;49(10):2323–2332. doi: 10.1021/ci900277r. [DOI] [PubMed] [Google Scholar]
- 96.Smith BJ, McKimm-Breshkin JL, McDonald M, Fernley RT, Varghese JN, Colman PM. Structural studies of the resistance of influenza virus neuraminidase to inhibitors. J Med Chem. 2002;45(11):2207–2212. doi: 10.1021/jm010528u. [DOI] [PubMed] [Google Scholar]
- 97.Hurt AC, Holien JK, Parker MW, Barr IG. Oseltamivir resistance and the H274Y neuraminidase mutation in seasonal, pandemic and highly pathogenic influenza viruses. Drugs. 2009;69(18):2523–2531. doi: 10.2165/11531450-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 98.Hong BT, Cheng YSE, Cheng TJ, Fang JM. Boronate, trifluoroborate, sulfone, sulfinate and sulfonate congeners of oseltamivir carboxylic acid: synthesis and anti-influenza activity. Eur J Med Chem. 2019;163:710–721. doi: 10.1016/j.ejmech.2018.12.027. [DOI] [PubMed] [Google Scholar]
- 99.Sollis SL, Smith PW, Howes PD, Cherry PC, Bethell RC. Novel inhibitors of influenza sialidase related to GG167. Synthesis of 4-amino and guanidino-4H-pyran-2-carboxylic acid-6-propylamides; selective inhibitors of influenza a virus sialidase. Bioorg Med Chem Lett. 1996;6(15):1805–1808. doi: 10.1016/0960-894X(96)00318-6. [DOI] [Google Scholar]
- 100.Smith PW, Sollis SL, Howes PD, Cherry PC, Starkey ID, Cobley KN, et al. Dihydropyrancarboxamides related to zanamivir: a new series of inhibitors of influenza virus sialidases. 1. Discovery, synthesis, biological activity, and structure-activity relationships of 4-guanidino- and 4-amino-4H-pyran-6-carboxamides. J Med Chem. 1998;41(6):787–797. doi: 10.1021/jm970374b. [DOI] [PubMed] [Google Scholar]
- 101.Smith PW, Sollis SL, Howes PD, Cherry PC, Bethell RC. Novel inhibitors of influenza sialidases related to GG167 structure–activity, crystallographic and molecular dynamics studies with 4H-pyran-2-carboxylic acid 6-carboxamides. Bioorg Med Chem Lett. 1996;6(24):2931–2936. doi: 10.1016/S0960-894X(96)00542-2. [DOI] [Google Scholar]
- 102.Taylor NR, Cleasby A, Singh O, Skarzynski T, Wonacott AJ, Smith PW, Sollis SL, Howes PD, Cherry PC, Bethell R, Colman P, Varghese J. Dihydropyrancarboxamides related to zanamivir: a new series of inhibitors of influenza virus sialidases. 2. Crystallographic and molecular modeling study of complexes of 4-amino-4H-pyran-6-carboxamides and sialidase from influenza virus types a and B. J Med Chem. 1998;41(6):798–807. doi: 10.1021/jm9703754. [DOI] [PubMed] [Google Scholar]
- 103.Varghese JN, Epa VC, Colman PM. Three-dimensional structure of the complex of 4-guanidino-NeuSAc2en and influenza virus neuraminidase. Protein Sci. 1995;4(6):1081–1087. doi: 10.1002/pro.5560040606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yamashita M, Tomozawa T, Kakuta M, Tokumitsu A, Nau H, Kubo S. CS-8958, a prodrug of the new neuraminidase inhibitor R-125489, shows long-acting anti-influenza virus activity. Antimicrob Agents Chemother. 2009;53(1):186–192. doi: 10.1128/AAC.00333-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yamashita M. Laninamivir and its prodrug, CS-8958: long-acting neuraminidase inhibitors for the treatment of influenza. Antivir Chem Chemother. 2010;21(2):71–84. doi: 10.3851/IMP1688. [DOI] [PubMed] [Google Scholar]
- 106.Ishizuka H, Yoshiba S, Okabe H, Yoshihara K. Clinical pharmacokinetics of laninamivir, a novel long-acting neuraminidase inhibitor, after single and multiple inhaled doses of its prodrug, CS-8958, in healthy male volunteers. J Clin Pharmacol. 2010;50(11):1319–1329. doi: 10.1177/0091270009356297. [DOI] [PubMed] [Google Scholar]
- 107.Andrews DM, Cherry PC, Humber DC, Jones PS, Keeling SP, Martin PF, Shaw CD, Swanson S. Synthesis and influenza virus sialidase inhibitory activity of analogues of 4-guanidino-Neu5Ac2en (zanamivir) modified in the glycerol side-chain. Eur J Med Chem. 1999;34(3–4):563–574. doi: 10.1016/s0223-5234(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 108.Frantz S. The trouble with making combination drugs. Nat Rev Drug Discov. 2006;5(11):881–882. doi: 10.1038/nrd2189. [DOI] [PubMed] [Google Scholar]
- 109.Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48(21):6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 110.Medina-Franco JL, Giulianotti MA, Welmaker GS, Houghten RA. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov Today. 2013;18(9–10):495–501. doi: 10.1016/j.drudis.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bonnett R. Photosensitizers of the porphyrin and phthalocyanine series for photodynamic therapy. Chem Rev Soc. 1995;24(1):19–33. doi: 10.1039/cs9952400019. [DOI] [Google Scholar]
- 112.Macdonal IJ, Dougherty TJ. Basic principles of photodynamic therapy. J Porphyrins Phthalocyanines. 2001;5(2):105–129. doi: 10.1002/jpp.328. [DOI] [Google Scholar]
- 113.Castano AP, Demidova TN, Hambline MR. Mechanism in photodynamic therapy: part one-photosensitizers, photochemistry and cellular localization. Photodiagn Photodyn Ther. 2004;1(4):279–293. doi: 10.1016/S1572-1000(05)00007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Berthiaume F, Reiken SR, Toner M, Tompkins RG, Yarmush ML. Antibody-targeted photolysis of bacteria in vivo. Nat Biotechnology. 1994;12(7):703–706. doi: 10.1038/nbt0794-703. [DOI] [PubMed] [Google Scholar]
- 115.Wainwright M. Photoinactivation of viruses. Photochem Photobiol Sci. 2004;3(5):406–411. doi: 10.1039/b311903n. [DOI] [PubMed] [Google Scholar]
- 116.Hamblin MR, Hasan T. Photodynamic therapy a new antimicrobial approach to infectious disease? Photochem Photobiol Sci. 2004;3(5):436–450. doi: 10.1039/b311900a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wen WH, Lin M, Su CY, Wang SY, Cheng YS, Fang JM, Wong CH. Synergistic effect of zanamivir–porphyrin conjugates on inhibition of neuraminidase and inactivation of influenza virus. J Med Chem. 2009;52(15):4903–4910. doi: 10.1021/jm900515g. [DOI] [PubMed] [Google Scholar]
- 118.Lee YC, Lee RT. Carbohydrate–protein interactions: basis of glycobiology. Acc Chem Res. 1995;28(8):321–327. doi: 10.1021/ar00056a001. [DOI] [Google Scholar]
- 119.Mammen M, Dahmann G, Whitesides GM. Effective inhibitors of hemagglutination by influenza virus synthesized from polymers having active ester groups. Insight into mechanism of inhibition. J Med Chem. 1995;38(21):4179–4190. doi: 10.1021/jm00021a007. [DOI] [PubMed] [Google Scholar]
- 120.Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed Engl. 1998;37(20):2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 121.Lundquist JJ, Toone EJ. The cluster glycoside effect. Chem Rev. 2002;102(2):555–578. doi: 10.1021/cr000418f. [DOI] [PubMed] [Google Scholar]
- 122.Macdonald SJ, Watson KG, Cameron R, Chalmers DK, Demaine DA, Fenton RJ, et al. Potent and long-acting dimeric inhibitors of influenza virus neuraminidase are effective at a once-weekly dosing regimen. Antimicrob Agents Chemother. 2004;48(12):4542–4549. doi: 10.1128/AAC.48.12.4542-4549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Watson KG, Cameron R, Fenton RJ, Gower D, Hamilton S, Jin B, et al. Highly potent and long-acting trimeric and tetrameric inhibitors of influenza virus neuraminidase. Bioorg Med Chem Lett. 2004;14(6):1589–1592. doi: 10.1016/j.bmcl.2003.09.102. [DOI] [PubMed] [Google Scholar]
- 124.Macdonald SJ, Cameron R, Demaine DA, Fenton RJ, Foster G, Gower D, et al. Dimeric zanamivir conjugates with various linking groups are potent, long-lasting inhibitors of influenza neuraminidase including H5N1 avian influenza. J Med Chem. 2005;48(8):2964–2971. doi: 10.1021/jm040891b. [DOI] [PubMed] [Google Scholar]
- 125.Fraser BH, Hamilton S, Krause-Heuer AM, Wright PJ, Greguric I, Tucke SP, et al. Synthesis of 1,4-triazole linked zanamivir dimers as highly potent inhibitors of influenza a and B. Med Chem Commun. 2013;4:383–386. doi: 10.1039/C2MD20300F. [DOI] [Google Scholar]
- 126.Honda T, Masuda T, Yoshida S, Arai M, Kobayashi Y, Yamashita M. Synthesis and anti-influenza virus activity of 4-guanidino-7-substituted Neu5Ac2en derivatives. Bioorg Med Chem Lett. 2002;12(15):1921–1924. doi: 10.1016/S0960-894X(02)00328-1. [DOI] [PubMed] [Google Scholar]
- 127.Weight AK, Haldar J, Álvarez de Cienfuegos L, Gubareva LV, Tumpey TM, Chen J, Klibanov AM. Attaching zanamivir to a polymer markedly enhances its activity against drug-resistant strains of influenza a virus. J Pharm Sci. 2011;100(3):831–835. doi: 10.1002/jps.22338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee CM, Weight AK, Haldar J, Wang L, Klibanov AM, Chen J. Polymer-attached zanamivir inhibits synergistically both early and late stages of influenza virus infection. Proc Natl Acad Sci U S A. 2012;109(50):20385–20390. doi: 10.1073/pnas.1219155109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Haldar J. Álvarez de Cienfuegos L, Tumpey TM, Gubareva LV, Chen J, Klibanov AM. Bifunctional polymeric inhibitors of human influenza a viruses. Pharm Res. 2010;27(2):259–263. doi: 10.1007/s11095-009-0013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu KC, Fang JM, Jan JT, Cheng TJ, Wang SY, Yang ST, Cheng YSE, Wong CH. Enhanced anti-influenza agents conjugated with anti-inflammatory activity. J Med Chem. 2012;55(19):8493–8501. doi: 10.1021/jm3009844. [DOI] [PubMed] [Google Scholar]
- 131.Salomon R, Hoffmann E, Webster RG. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc Natl Acad Sci U S A. 2007;104(30):12479–12481. doi: 10.1073/pnas.0705289104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fedson DS. Confronting the next influenza pandemic with anti-inflammatory and immunomodulatory agents: why they are needed and how they might work. Influenza Other Respir Viruses. 2009;3(4):129–142. doi: 10.1111/j.1750-2659.2009.00090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ottolini M, Blanco J, Porter D, Peterson L, Curtis S, Prince G. Combination anti-inflammatory and antiviral therapy of influenza in a cotton rat model. Pediatr Pulmonol. 2003;36(4):290–294. doi: 10.1002/ppul.10320. [DOI] [PubMed] [Google Scholar]
- 134.Zheng BJ, Chan KW, Lin YP, Zhao GY, Chan C, Zhang HJ, Chen HL, Wong SS, Lau SK, Woo PC, Chan KH, Jin DY, Yuen KY. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza a/H5N1 virus. Proc Natl Acad Sci U S A. 2008;105(23):8091–8096. doi: 10.1073/pnas.0711942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Babu YS, Chand P, Bantia S, Kotian P, Dehghani A, El-Kattan Y, Lin TH, Hutchison TL, Elliott AJ, Parker CD, Ananth SL, Horn LL, Laver GW, Montgomery JA. BCX-1812 (RWJ-270201): discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. J Med Chem. 2000;43(19):3482–3486. doi: 10.1021/jm0002679. [DOI] [PubMed] [Google Scholar]
- 136.Alame MM, Massaad E, Zaraket H. Peramivir: a novel intravenous neuraminidase inhibitor for treatment of acute influenza infections. Front Microbiol. 2016;7:450. doi: 10.3389/fmicb.2016.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang PC, Chiu DC, Jan JT, Huang WI, Tseng YC, Li TT, Cheng TJ, Tsai KC, Fang JM. Peramivir conjugates as orally available agents against influenza H275Y mutant. Eur J Med Chem. 2018;145:224–234. doi: 10.1016/j.ejmech.2017.12.072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.