

Abstract
Background:
Molecular hydrogen (H2) is now recognized as a therapeutic gas for the treatment of numerous diseases including neurodegenerative diseases, metabolic disorders, and inflammatory diseases. Non-polar, neutral H2 is assumed to have health benefits facilitated by its passive diffusion across the human body immediately after administration and is considered a safe therapeutic inert gas that does not interfere with physiological enzymatic reactions. The effects of H2 on mammalian cells are assumed to be based on non-enzymatic reactions with Reactive Oxygen Species (ROS) exhibiting extremely high reactivity. However, many reports on therapeutic applications of H2 have the limitation to regard H2 only as a scavenger for the hydroxyl radical and peroxynitrite.
Methods:
Apart from this proposed principle, a new possible mechanism of H2 activation and consumption in mammalian cells is considered in this review, which is specifically focused on the mitochondrial complex I that has a close evolutionary relationship with energy-converting, membrane-bound [NiFe]-hydrogenases (MBH). Notably, the possibility that H2 may function as both electron and proton donor in the ubiquinone-binding chamber of complex I is discussed.
Results:
H2 is proposed to act as the rectifier of the mitochondrial electron flow in the disordered or pathological state when the accumulation of electrons leads to ROS production, specifically during the re-supply of O2 after hypoxia in the mitochondria.
Conclusion:
Furthermore, H2 is proposed to convert the quinone intermediates to the fully reduced ubiquinol, thereby increasing the antioxidant capacity of the quinone pool as well as preventing the generation of ROS.
Keywords: Molecular hydrogen, semiquinone, hydrogenase, rectifier of electron flow, mitochondrial respiratory complex, electron and proton donor
1. INTRODUCTION
Molecular hydrogen (H2) is a non-polar molecule with a stable covalent bond. Many microorganisms are equipped with hydrogenase, an enzyme that reversibly splits H2 into protons and electrons. Most hydrogenases have a catalytic center containing two metal atoms, [NiFe] or [FeFe], along with iron-sulfur (Fe-S) clusters. Interestingly, there are energy-converting, membrane-bound [NiFe]-hydrogenases (MBH) with O2 tolerance that function in H2 uptake by oxidizing it in the presence of certain electron acceptors including ubiquinone or menaquinone [1, 2]. Due to these characteristics, MBH-type enzymes are considered to be close to the ancestor of respiratory complex I. Despite the lack of hydrogenases in mammalian cells, H2 is now recognized as a therapeutic gas, with therapeutic effects on numerous diseases including neurodegenerative diseases, metabolic disorders, and inflammatory diseases. Therapeutic H2 possesses neutral properties that are assumed to facilitate its passive diffusion across the human body immediately after administration and to ensure a beneficial safety profile as a relatively inert gas that does not affect physiological enzymatic reactions. To date, the effects of H2 on mammalian cells are assumed to be mostly related to non-enzymatic interactions with extremely reactive oxygen species (ROS) by random collisions (named “scavenger theory” here and explained below). Apart from this insight, a possible mechanism of H2 activation and consumption in mammalian cells is discussed in this review, specifically focusing on the mitochondrial complex I with its close evolutionary relationship with MBH.
In reports since 1975, the therapeutic potential of H2 has been demonstrated in some diseases using hydrogen gas containing helium or alkaline water containing H2 produced by electrolysis [3, 4]. The scavenger theory is based on a report from 2007 by Ohsawa and colleagues. They explained the biological effects of H2 in vivo as liquid-phase chemical reactions of H2, which can act as an electron donor for ROS molecules such as the extremely reactive hydroxyl radical or peroxynitrite [5]. The amphipathic properties of H2 are thought to contribute to these scavenging effects in the mitochondrial inner membrane composed of a lipid bilayer [6]. The authors demonstrated the protective potential of H2 against ischemia-reperfusion (I/R) injury in a mouse model where H2 reduced oxidative stress and scavenged hydroxyl radicals. Following this publication, many studies demonstrated the efficacy of hydrogen in rodents with induced oxidative stress due to physiological treatments impairing circulation or the administration of oxidative stress-inducing chemical compounds. However, most of these animal experiments were designed to investigate the prophylactic efficacy of H2 by administering H2 prior to, simultaneously, or immediately after the oxidative stress-inducing treatment to develop a pathologic disorder rather than to evaluate the therapeutic efficacy of H2 [6], in animals that had developed diseases prior to the experiments.
In contrast, in human trials, the enrolled patients suffered from the disease for a certain period and the pathological conditions and/or the adaptation of the body to compensate for the disorders were established when they were diagnosed and enrolled in the clinical trials. Because of this critical difference in the experimental strategy between efficacy testing in animals/cultured cells and clinical trials, the therapeutic application or precise strategy for using H2 in human disease should be developed further, specifically when H2 is used in combination with pharmaceutical drugs. Therefore, insights into the mechanisms of action of H2 are critical for assessing the benefit of H2 therapy even after the disorders have become irreversible because treatment decisions should not be solely based on the scavenging reactions for the prophylactic application. This review discusses a new possible mechanism of action of H2, apart from the scavenger properties of H2 against ROS.
2. LIMITATIONS OF THE SCAVENGER THEORY OF H2
In the most recent clinical report by Yoritaka A. and colleagues, the efficacy of H2 on Parkinson’s disease (PD) could not be confirmed, indicating the need for further investigations [7]. However, in an earlier clinical trial on PD, a daily dose of 0.8 mM H2 (1.6 ppm) dissolved in 1 L of water was ingested over 48 weeks by one treatment group. As a potential therapeutic effect, the results suggested that the neurodegenerative symptoms of PD could be improved by this daily dose of H2 even in patients with modified Hoen and Yahr stage 1-4 (approximate average, 2.0) [8]. The onset of PD occurred prior to study enrollment, indicating that the duration of the disease was more than 4 years on average in the study. This fact is an indication of improvement rather than prevention. It is assumed that ROS promote the PD pathogenesis by causing the cell death of dopamine-producing cells, but the observed improvement of PD by H2 therapy was not inferior to the nonergot dopamine therapy; however, the significance of this finding is limited due to the small number of participants (placebo and H2 therapy group size: n = 9 each). Even if H2 only blocks oxidative damage, the improvement of established PD should require the regeneration of the dopaminergic cells in the neuron network including the restoration of mitochondrial function in damaged cells. Therefore, the efficacy of H2 for human PD could not be fully explained by the reduction of ROS, if this early trial by Yoritaka A. and colleagues [8] generated representative data. There remains the possibility that H2 does not specifically affect established PD but may compensate for the effects of neurodegenerative disorders by another collateral pathway associated with the repair of the injured or disordered tissues by the regeneration of mitochondria in the affected cells. At the same time, the improvement of circulation appears to be required for the reconstruction of tissues, and the consumption of H2 could improve endothelial function [9].
Moreover, in the field of anti-inflammation therapy, to improve the established disorder of the immune system caused by rheumatoid arthritis (RA), it seems that it is not sufficient to reduce the oxidative stress that promotes the deleterious effects amplified by the NF-κB and pro-inflammatory cytokines including TNF-α and IL-6. In our recent review, to explain the mechanistic insight into the anti-rheumatoid effect of H2, it was discussed that continuous stimulation by ROS appears to be required to maintain the rheumatic immune reactions; therefore, H2 could improve RA by shutting down the vicious cycle solely as a ROS scavenger [10]. The efficacy of H2 on RA was explained partially by the reduction in the content of hydroxyl radicals.
In the field of PD and RA, improvements of certain aspects of these diseases by H2 therapy cannot be fully explained by the scavenging properties of H2 against ROS, indicating a limitation of the scavenger theory [3]. From a medical point of view, it appears to be a logical conclusion to consider the regeneration of tissues or biological networks accompanied by mitochondrial regeneration as a requirement for improvement in these established diseases. In addition, the rate constant for the hydroxyl radical (k value, L mol-1 s-1) in pure water is 4.2 x 107 against H2, which is much lower than the k values for two precursors of the hydroxyl radical, H2O2 and O•2- (1.2 x 1010 and 8 x 109, respectively), which are expected to exist in proximity [11]. Furthermore, there remains the basic assumption that the probability for a collision between H2 and the hydroxyl radical may be limited to a half-life in the range of nanoseconds before this radical encounters the surrounding biomolecules. Once the hydroxyl radical is formed, it reacts with the closest molecules including lipids, proteins, or nucleic acids packed and concentrated in a cell [12]. The k values for the hydroxyl radical against other molecules including the amino acids proline, glutamic acid, and pyroglutamic acid are 3.1 x 108, 2.3 x 108, and 1.05 x 109, respectively, which are much higher than the values against H2 [13]. Thus, as it appears, the higher the oxidative potential of the ROS, the lower is its chance to collide specifically with H2.
In addition to the clinical and biochemical limitations of the scavenger theory, H2 administration caused an improvement of the metabolic system in db/db mice lacking the functional leptin receptor [14]. Oxidative stress reduction and energy metabolism enhancement associated with the increased expression of FGF21 are shown and discussed as the mechanisms of H2 for the improvement of obesity and diabetes in those mice. It is interesting that H2 could improve the congenitally established disorder of energy metabolism. Presumably, there appears to be a big gap between the reduction of the hydroxyl radical and the improvement of energy metabolism. The pathology of chronic human diseases, including that of RA and PD assessed in the clinical trials, had been established several years before testing the administration of H2. To improve the prognosis of these congenital or established diseases associated with chronic disorders, it appears that the functional regeneration of mitochondria from the existing cells in the affected tissues is required. Consequently, these cells and tissues could play an important role in the amelioration of long-term pathological states; hence, the reported efficacy of H2 should be related to these processes. To fill the gap between the scavenging property of H2 and the improvements in the metabolism and the cellular regeneration, researchers are investigating the signal transduction triggered by H2 [3]. However, to restore the established disorder and/or partial degeneration of the neuronal or immune systems, it seems insufficient or too late that only some altered or non-specific signal transductions can restore the normal function after the abnormal tissue had been formed. The improvement by H2 administration was rapid, i.e., within 4 weeks in RA [15] and within 1 hour for the endothelial function of the brachial artery in our pilot studies [9]. It seems to be necessary to improve the cellular dysfunction towards a radical switch of cellular energetics, in addition to the indirect metabolic alterations in response to the signals from outside the cell. The key events linking the actual properties of H2 to the energy metabolism should occur somewhere upstream of the generation of ROS, as opposed to the function as a scavenger molecule downstream of the oxidative stress. These mechanisms directly related to the energy metabolism are predicted to be critical for using the H2 molecule to establish treatment strategies for long-term adjuvant administrations because the energy metabolism processes are located upstream in relation to most of the pathological events. The center of energy metabolism is the mitochondrion. Accordingly, the possible behavior of H2 in the catalytic center of energy-converting respiratory complexes is discussed further.
3. MITOCHONDRIAL RESPIRATORY CHAIN AS A POSSIBLE TARGET FOR H2
In reviewing potential targets for H2, the processes upstream of the production of superoxide were focused, apart from the reduction of the hydroxyl radical. Here, the possibility is discussed that H2 can suppress the electron leakage in the electron transport chain (ETC), thereby preventing superoxide overproduction, which is the first step during mitochondrial oxidative stress. Mammalian cells contain approximately 1000 mitochondria (with 100–10000 genome copies per organelle) [16]. A mechanism that can regenerate mitochondrial dysfunction by improving the uncontrolled electron flow or by preventing the deleterious electron leakage from the ETC is predicted to have the potential to regenerate the dysfunction of the cells by rescuing the disordered population of mitochondria. Improvement of mitochondrial dysfunction is also expected to improve the disordered signal transduction that depends on or is related to cellular redox balance.
Mitochondrial respiratory complexes I, III, and IV are the energy-converting enzymes that are key to establishing the ETC, which converts redox energy into the electrochemical potential between the inner mitochondrial membrane and mitochondrial matrix [17]. The electron transfer, which begins with NADH (nicotinamide adenine dinucleotide) and ends with the reduction of O2 to H2O, generates the proton-motive force across the mitochondrial inner membrane, thereby driving complex V (mitochondrial ATPase) to synthesize almost 90% of the ATP from ADP [18]. Specifically in the mitochondrial complex I that converts approximately 40% of the energy of the ETC from redox energy to membrane potential, the elaborate catalytic mechanisms employed by hydrogenases are conserved through evolution [19]. Although living organisms have selected and established this elaborate machine through evolution, it is not perfect at least in higher organisms in which the inevitable electron leakage from the ETC is often the origin of dysfunction of the energy-producing organelle, the mitochondrion. About 2% of O2 is assumed to be reduced by leaked electrons [20]. This level appears to be within the tolerance level, and some ROS are utilized in redox signaling, especially in the endothelium [21]. However, higher electron leakage levels increase the production of deleterious ROS and induce the vicious circle of mitochondrial dysfunction that causes inflammatory diseases, neurodegenerative diseases, endothelial dysfunction, and metabolic disorders [10, 22]. Optimal efficiency of electron transfer along the ETC is critical for preventing excess electron leakage that interferes with the appropriate energy conversion in mitochondria.
In the mammalian ETC, ubiquinone (abbreviated as quinone or Q) is the most important electron and proton carrier. In complex I, the intermediate quinone species including semiquinones (SQ) between ubiquinone and ubiquinol (QH2) play a pivotal role in converting the energy of the electrons into the proton-motive force by still unidentified coupling mechanisms linking the peripheral arm with the catalytic center including the iron-sulfur cluster, N2 and the membrane domain with the proton pumps [23, 24]. The electron and proton transfers via SQ intermediates are also indispensable for the Q-cycle in complex III, whereas SQ generated excessively or inappropriately is believed to be one of the major sources of superoxide [21, 25]. Much of the electron leakage and subsequent production of superoxide by a single electron reduction of O2 is assumed to relate to the semiquinone intermediates in the catalytic center of the complexes as well as in the Q pools in the mitochondrial membrane. The unstable SQ itself with an unpaired electron can directly donate one electron to the oxygen molecule to produce the superoxide according to the following reaction [26, 27].
Q•- + O2 → Q + O•2-
To prevent the electron flow in ETC from generating excessive superoxide and causing oxidative damage to the respiratory complexes, it is critical to suppress the production of the deleterious or uncontrolled SQ. If an appropriate target can be identified, it will become one of the ideal procedures to improve or prevent the numerous diseases induced by the mitochondrial dysfunctions. In this review, I focus on the quinone intermediate species including semiquinones as a possible target of H2.
The highly reactive and unstable semiquinone species have been observed by electron paramagnetic resonance (EPR) spectroscopy both in complex I and III [25, 28]. The stability constant of SQ (Q•H) at physiological temperature is approximately 10-10 [29]. Therefore, in complex I, at least 105 times of the binding stability is required to keep the highly reactive semiquinone form (termed SQNf) in the Q-binding site during the energy conversion [30]. There appears to be the possibility that these quinone intermediate species, including the unstable SQNf in complex I and the unstable semiquinone (Q•-) at the Q(o)-site in complex III, may react non-enzymatically with H2 because of their high reactivity. It should be noted, however, that the leading enzyme in the ETC, the complex I, is known to have evolved from the energy-converting MBH [2, 19]. To consider the interaction of H2 with the quinone species, it is important to focus on the possibility that H2 is activated in the quinone-binding space (Q-chamber) of the catalytic center of complex I, where quinone intermediates species play a pivotal role for the energy conversion.
4. THE STRUCTURE OF THE BINDING SITES OF QUINONE SPECIES IN COMPLEX I
Dysfunction of complex I is involved in numerous neurodegenerative diseases including PD as well as congenital diseases that are caused by the mutation of genes encoding the amino acids forming the Q-chamber [31, 32]. Many of the reports concerning the efficacy of H2 are on neurodegenerative diseases, and therefore, it is important to discuss the site in complex I, where H2 could interact to regenerate the disordered ETC. In the mammalian complex I, two electrons are transferred from NADH to the 8 Fe-S clusters via the non-covalently bound FMN (flavin mononucleotide) as a hydride ion in the hydrophilic matrix arm of the complex [23, 33]. The electrons from the array of Fe-S clusters ending at N2 are transferred one by one to the quinone from the N2 cluster, thereby generating quinone intermediates species including SQ radicals. The N2 cluster is positioned at the deepest tip of the Q-chamber, formed by the 49-kDa and PSST subunits that have a pH-dependent redox midpoint potential of approximately -150 mV (60 mV per 1 pH), which is higher than the arrays upstream of the Fe-S clusters (approximately -250 mV), which function as electron reservoir in complex I [34, 35].
To consider the possibility that H2 is activated in the catalytic center of complex I, the binding site of quinone species including the reactive intermediates should be the pivot of the discussion. If the activation of H2 is possible, it likely could occur during the electron and proton transfer in the presence of unstable quinone intermediates, preferentially when the electron leakage is induced in the Q-chamber at the catalytic center of complex I. Based on this model, I discuss the possible site where H2 could be activated as electron and proton donor according to the structural information and the redox mechanisms recently proposed.
In the first report on the entire structure of the crystallized bacterial complex I (from Thermus thermophilus), the binding site of the quinone head is hydrogen-bonded to the hydroxyl group of Tyr87, which is located in proximity to the N2 cluster (9 Å) and is essential for the electron transfer with N2 [24, 36]. Although an endogenous quinone was not bound to the complex in the crystal structure, the folding of the complex I catalytic center is assumed to be similar to that of the resolved structure co-crystalized with piericidin and decylubiquinone in this study. It should be noted that the quinone analog decylubiquinone acted on complex I and was hydrogen-bonded to His38 as well as Tyr87. In the structure, the quinone binding chamber has a length of 30 Å and a diameter of 2-3 x 4-5 Å [36].
Recently, the mitochondrial complex I structures of yeast (Yarrowia lipolytica) [37], ovine (Ovis aries) [38], and bovine (Bos taurus) [39, 40] complexes have been reported. The 14 core subunits conserved from bacteria are surrounded by multiple subunits specific for mitochondria with a total mass of nearly 1 MDa. An additional feature specific for the mitochondrial complex I is the conformational flexibility linked to the movement associated with the activated (A-form)-deactivated (D-form) transition that plays an important role in controlling the generation of ROS as well as in the catalysis of the energy-converting redox reactions via the quinone species during the enzyme turnover [37, 41]. In the D-form of mammalian complex I (Ovis aries), the distance between the Q head and the N2 cluster is not less than 20 Å due to the extension of the β1-β2 loop of the 49-kDa subunit into the catalytic center [38]. Because only the structure of the D-form has been obtained, the A-form is assumed to be unstable. Although the structural data have not been verified, the distance between the quinone head groups and N2 cluster in the A-form is assumed to be similar to the findings in bacteria; 12 Å was identified as the fast relaxing signal of the semiquinone which was analyzed using EPR and was sensitive to the proton-motive force (designated SQNf) [42]. During the catalytic reactions in the A-from, the mammalian Q head is also assumed to enter between His 59 and Tyr108 (corresponding to His95 and Tyr144 in Y. lipolytica, and His38 and Tyr87 in T. thermophilus), whereas these two residues are predicted to be located closer to each other in the absence of the Q head in the D-form [40]. Because the binding of NADH and quinone is assumed to reactivate the Q-site to the A-form, the structural requirement for this conformational change to the A-form appears to reflect the flexibility that allows the accessibility and binding of different types of quinone analogs including inhibitors with bulky extensions (such as rotenone). This flexible structure of the Q-chamber near the N2 cluster also facilitates the mobility of authentic quinone head groups, and it may relate to the absence of endogenous ubiquinone in the purified complex I. The dynamics of all the possible interactions between the quinone species and the catalytic center with His59 prompted me to consider the possibility of the usage of H2 as the electron and proton donor during the transition from the D- to the A-form of complex I, thereby preventing the deleterious electron leakage that generates the superoxide, especially in cases of I/R injury. Considering the complex I conformations in the A-form (including bacterial complex I) and D-form, I propose that H2 is present in the quinone/semiquinone reaction chamber, when it is administered. In this scenario, the acquired H2 could be positioned between His 59 and the quinone head groups, forming a triangle with Tyr108. This possible configuration including H2, quinone, and basic residues resembles the situation during the cleavage reaction of H2 in the [NiFe] hydrogenase, supporting the assumption that the structure of the catalytic center and the energy-converting mechanisms are conserved through an evolution including that in complex I [43]. Although complex I lacks a metal catalyst, it should be noted that activation and cleavage of H2 do not necessarily require metals and the mechanistic basis for the metal-free activation of H2 has emerged in the recent decade [44].
5. POSSIBLE MECHANISMS TO ACTIVATE H2 IN THE Q-CHAMBER OF COMPLEX I
Complex I-related oxidoreductases are thought to have evolved from MBH without acquiring new essential subunits or components from energy-converting hydrogenases-related (Ehr) complexes [19]. The structures forming the 49-kDa/PSST subunits of complex I and the large/small subunits in the hydrogenase are highly conserved, including the N2 cluster, the proton delivery pathways, and the similar folding around the catalytic center, which is assumed to be important for the energy-coupling conformational change in both proteins [43]. The structure composing the catalytic metals in [NiFe] hydrogenase is conserved in complex I except for the space occupying the metal center in the hydrogenase of Themus thermophilus with the amino acids 85-89 including Tyr87 (Tyr108 in the mammalian protein) that functions as the quinone binding site in complex I [43]. It is suggested that the ancestor of complex I-related oxidoreductases, proposed as the missing link in the evolution from the hydrogenase to complex I, may have utilized both H2 and ubiquinone as the electron/proton carrying substrate [45]. In this review, the possibility that even mitochondrial complex I may have the ancestral properties to convert the energy of electron by using both quinone and H2 is considered. Among the conserved and essential arrays of basic residues close to the catalytic center in complex I, His59 in the mammalian Q-chamber (His38 in bacteria) and should be examined first as a possible candidate that may activate H2.
The precise mechanisms to cleave the covalent bond of H2 in [Ni-Fe] hydrogenase have not been fully understood. However, in a recent study, H2 bound to the metal catalyst increased polarity and acidity by abstraction of a proton to the adjacent basic amino acid in a reaction that involves an arginine located close to the Ni atom [46]. This mechanism is based on and akin to the metal-free hydrogen activation theory, termed a frustrated Lewis pair (FLP) mechanism [47]. In a model of this mechanism, H2 is activated by being sandwiched between Lewis acid and Lewis base, which are sterically prevented from reacting with each other [44]. The interpretation and involvement of the basic residue in the metal-free FLP mechanism even in [NiFe] hydrogenases suggest the possibility that the cleavage reaction of H2 in complex I occurs without the Ni atom available as the acid catalyst but with a corresponding electrophilic reactant. In the mammalian complex I, the location of the proton-abstracting bases with His59, the oxidized N2 cluster, and the electrophilic/protophilic quinone species may act as accelerant for the reaction where two electrons and two protons are supplied from the activated H2 to the ETC similar to the typical reaction from Q to QH2 in complex I. Here, it is hypothesized that the cleavage reaction of H2 in complex I proceeds via a stepwise electron transfer; it replaces the formation of the hydride ion (H-) bound to the Ni-Fe cluster in the catalytic center of hydrogenase. In the presence of the H2 molecule that entered the catalytic center of complex I, there is the possibility that the electrophilic/protophilic form of the quinone species, including the unstable semiquinone intermediates, could affect the polarity of H2 and increase the activity of H2 by pulling the electron/proton to the quinone head groups, thereby facilitating the cleavage of H2 according to the partially similar FLP mechanism by the concerted reaction with the nearby base and acid, respectively. The de-protonated bases located in the proximity of the N2 cluster are assumed to activate H2 in a scenario when the electrophilic quinone species increase the acidity of H2. The oxidized N2 with Tyr108 may also induce the polarity in H2 and abstract the electron from the H2 molecule while the protophilic quinone species could absorb the proton from H2. However, it is still unknown whether the N2 cluster is oxidized (N2ox) or reduced (N2red) even in the presence of semiquinone species [24]. Because the quinone species are not metal and not fixed to the catalytic center, the mobility of the quinone head groups is associated with a certain velocity that appears to provide the collision energy typically required in these chemical reactions. Moreover, the structural flexibility of the catalytic center and mobile quinone species may create the ideal intermolecular distance required for the FLP-like reaction involving H2, quinone head groups, and nearby bases, as well as the N2 cluster to activate H2. Among the possible combinations of electrophilic (acid) and protophilic (base) factors described here, in the scenario with quinone or semiquinone species as an electrophilic acid, the His59 residue could function as the corresponding base in the mechanism akin to FLP. Histidine acts as the elemental base in the catalytic center of the amine oxidase or alcohol oxidase where the proton is abstracted from a hydroxyl group [48, 49]. Although Histidine is not involved in the FLP-related mechanisms, the deprotonated imidazole appears to have the potential to absorb the proton and polarize H2 by co-operative interaction with the electrophilic quinone species and/or oxidized N2 cluster.
In [NiFe] hydrogenases, the distance between the guanidine base of the Arginine and the Ni atom, which are both involved in an FLP-like mechanism, is within 4.5 Å and is occupied by the bound H2 molecule [50]. In the case of the catalytic center of the A-form of mammalian complex I, the predicted distance between the semiquinone species and the hydroxyl of Tyr108 is predicted to be ~3 Å, and the His59 residue is assumed to be positioned in proximity at the opposite side of the tyrosine [24, 36]. Because the quinone head groups are mobile, it is assumed that the position of the H2 between the quinone head groups and the His59 base could have the ideal distance for the H2 activation by an FLP-like interaction. His55 is another conserved residue in the proximity of His59 that also may be involved in the oxidation of H2; although, this possibility depends on whether the C1 or C4 side groups of the Q-head species in the electrophilic state are accessible to H2 during the transition from the D- to the A-form. Furthermore, other bases close to the catalytic center could be considered as hypothetical candidates, which abstract the protons from the H2 molecule by the FLP-like mechanism that polarizes this molecule when it is trapped between the quinone head groups. The residues Arg63 and Arg53 (residues Arg99 and Arg89 in Y. lipolytica, respectively [37]) are potential candidates that could function as proton-abstracting bases, although it is not clear how close they come to the Q-head during the conformational change to the A-form [37]. In addition, the distance between the N2 cluster and the His190 residue, which is known as the redox-Bohr group (His226 in Y. lipolytica, and His169 in T. thermophilus), is within 4 Å [51-53]; thus, this pair may have a mechanism similar to the FLP-like mechanism, although the probability is low without the association of a quinone species. Many complex I inhibitors are analogs or antagonists of quinone. Investigations using such inhibitors may demonstrate the involvement of the quinone species in the activation of H2 in complex I.
Although the accessibility of H2 close to His59 and Tyr108-N2 during the D-form cannot be currently confirmed by the mammalian structure, it is very likely, because the complex I evolved from [NiFe] hydrogenases and shares highly conserved amino acid residues with these hydrogenases that are critical for the conformation of the catalytic chamber [43, 54]. Importantly, the efficacy of H2 in the I/R injury is consistent with the fact that the D-form prevented the generation of superoxide during I/R injury and decreased the risk of electron leakage during the D-A transition [41]. Notably, besides the structure of the quinone-binding site, which is highly conserved with [NiFe] hydrogenases, there are also many polar and hydrophilic amino acids on the surface of the Q-chamber that the two enzyme complexes share [43]. The surface in proximity to the N2 cluster of the Q-chamber is positively charged due to conserved basic residues, and the distal part is negatively charged where the bound hydrophobic Q tail is positioned to block the entry of solvent molecules [36]. The separation of the Q-chamber into the acidic and basic parts (by shifting the charges from negative to positive toward the tip of the chamber) may electrostatically affect the polarity of the quinone molecule occupying the entire length of the Q-chamber, which may also affect the redox reactivity of the quinone head groups as well as the velocity of the quinone species entering the catalytic center. And even in the presence of the isoprenoid Q tail at the entry of the Q-chamber, there still is the possibility that H2 could move to the tip of the chamber due to the amphipathicity of H2. Any mutation of these strictly conserved and charged residues on the surface of the Q-cavity impairs the function of complex I and is related to human diseases [36, 55].
6. MODELS FOR THE ELECTRON FLOW FROM THE ACTIVATED H2 IN THE Q-CHAMBER
The midpoint potential (Em) is estimated to be approximately -420 mV for H2/2H+, +90 mV for Q/QH2, and (-150)-(-250) mV for the N2 cluster (pH dependency: -59 mV/pH) [2, 43]. The negatively charged semiquinone anion and quinol anion provide the energy that is converted by proton pumping, by being stabilized with a shift of the midpoint potential of approximately 200 mV according to the two-state model for proton pumping [34]. The activated H2 has the thermodynamic potential to provide the redox energy to these stabilized anionic intermediates. In this scenario, the potential convertible for proton pumping will be charged again. In addition, the H2 molecules have the thermodynamic potential to donate the electrons to the unstable quinone intermediates including the negatively charged semiquinones with low Em values (~ (-200)-(-300) mV) [34]. The dissociation constant for the deprotonation of the quinol anion is estimated to be 2 x 10-13 M. The quinol anion appears to have the potential to activate H2 as a proton drawing base, whereas the semiquinone anion could work either as an electrophilic or protophilic molecule. Furthermore, the basic residues (including His59) could absorb protons as described in the former section; alternatively, the electrophilic N2 cluster associated with Thy108 may also polarize and activate H2 for the collision with the quinone species. The possible electron and proton transfer from H2 to the quinone species (Q: quinone, Q•-: semiquinone anion, Q•H: protonated neutral semiquinone, QH-: quinol anion) are presented below. H+-base indicates the proton-absorbing bases (including His59) and N2ox-e- indicates the oxidized N2 cluster that accepts the electron.
Electron transfer from H2 to Q
Q + H2 → Q•-+ H+-base + H•→ Q•H + H+-base + e-→ QH- + H+-base→ QH2 (1)
Electron transfer from H2 to Q•-
Q•- + H2 → Q2- + H+-base + N2ox -e-+ H+ → QH- + H+-base + N2 red → QH2 + N2 red (2)
Proton transfer from H2 to Q•-
Q•- + H2 → Q•H + N2ox -e- + H+-base + e- → QH-+ N2 red + H+-base → QH2 + N2 red (3)
Electron transfer from H2 to Q•H
Q•H + H2 → QH- + H+-base + N2ox -e- + H+ → QH2 + N2 red + H+-base (4)
Proton transfer from H2 to QH-
QH- + H2 → QH2 + N2ox -e- + e- + H+-base → QH2 + N2 red + e- + H+-base (5)
Hypothetical reaction (1) would require an earlier activation of H2 by the neighboring bases and the electrophilic N2 cluster, whereas reactions (2) to (5) would depend on the bifurcated electron flow from N2ox to NAD+ via N2red (reverse flow) and from quinone intermediate species to QH2 (forward flow). It should be noted that during the first step, H2 would act as a reductant in reactions (1), (2), and (4), but as an oxidant in reactions (3) and (5) against the anionic quinone species. The mechanism of multistep reactions (3) and (5) could be named “oxidant (or oxidation)-induced reduction,” which is traditionally used for the oxidoreduction mechanism in complex III (described later). In both reactions, H2 would initially behave as proton-donating oxidant against the anionic form of the quinone species. Then, the oxidized N2 cluster would be reduced by accepting the electrons from H2 as a reverse step; moreover, in reaction (3), protonated neutral semiquinone would also be reduced subsequently. In the hypothetical description here, H2 would rectify the electron flow in both directions as reductant and oxidant by donating electrons and protons, respectively, to the ubiquinone species including the reactive intermediates. This reaction would prevent progression to the pathological dead end of the ETC, which is the stage prior to the I/R injury.
The midpoint potential of H2 has the thermodynamic potential to enable the reverse flow to the reduced NADH with the Em of 320 mV. The two-way flow of electrons is expected to decrease the risk of oxidative burst during I/R injury by reducing the quinone intermediates to quinol and donating electrons to the oxidized N2, which could, in turn, transfer the electrons to the Fe-S cluster array to reduce oxidized FMN with free radicals and/or to reduce NAD+ to NADH. In addition, the quinone intermediate species are fully reduced to ubiquinol by the forward electron transfer from H2, which decreases the risk for the superoxide formation related to the semiquinone radicals in the Q-pool with the benefit of increasing the antioxidant potential of the Q-pool. Further, the hypothetical reverse electron transfer from H2 to the oxidized N2 would be facilitated by the pH-dependent Em of the N2 cluster. In this model, the redox energy of H2 is supposed to be separately consumed to reduce the unstable quinone intermediates with the activation energy of H2; in this case, the remaining redox energy will be favorable if the Em of N2 is higher than the other Fe-S clusters with ~250 mV [34, 43] according to the redox-Bohr effect. Downhill reverse electron transfer from the cleaved H2 will proceed more easily to the N2 cluster with the higher Em of -150 mV [42]. In contrast, in the absence of quinone species and the membrane potential providing the proton motive force, the lower Em of N2 cluster might contribute to the simple reactions with H2, N2, and the nearby bases because of the closer Em of N2 to H2.
In addition to the catalytic center of complex I, another hypothetical candidate of the site where H2 could be activated in the Q-chamber is the bottleneck of the channel where the basic residues Arg71, Arg274, and hydroxy-Arg77 are surrounding the neck [38, 40]. Hydroxyl-Arginine is known as the oxidized intermediate of arginine that is produced during the biosynthesis of nitric oxide due to the oxidation of arginine, following the generation of nitrite and citrulline [56]. The narrowest diameter is predicted to be 1.9-2.2 Å in the D-form, whereas the quinone head with 6 Å width will block the deeper access to the N2 catalytic tip. Even in the D-form or in the state when the channel is not fully open, the quinone head has to go through the bottleneck, and a collision with H2 during the passage might induce an activation by the FLP-like mechanism, which would involve the catalytic quinone head groups, the proton-abstracting arginine bases (Arg71 or Arg274), and the proton-donating oxidized arginine bases (hydroxy-Arg77). This bottleneck appears to be near the position of the SQNs, which was detected by EPR as the slow relaxing signal with a distance of 30 Å between the SQNs and the N2 site [25]. Thus, there is the possibility of a reaction between SQNs and the surrounding bases. The hypothetical model for the electron flow from the activated H2 in the Q-chamber is schematically presented in Fig. 1.
Fig. (1).
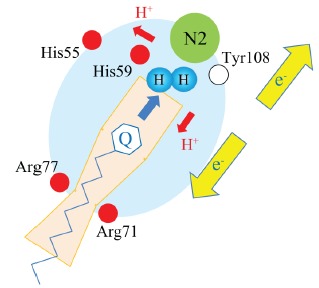
A hypothetical schematic for the activation of H2 in the Q-chamber.
The yellow arrows pointing in opposite directions indicate the bifurcate flow of electrons donated by the activated H2. The red arrows indicate the donation of protons from H2. The pale orange box represents the Q-chamber, and the “Q” head with the isoprenoid tail represents the quinone species including the semiquinone intermediates. Red circles represent the basic amino acids assumed to be indispensable for the catalytic function of complex I. The open circle indicates the conserved tyrosine residue required for the electron transfer of the N2 cluster. The large blue background circle depicts the part of complex I involved in the H2 activation of this hypothetical interaction. The pair of blue circles symbolizes H2. The blue arrow in the Q-chamber indicates the movement of quinone species with a certain velocity to collide with H2.
7. SEMIQUINONE RADICALS IN COMPLEX III
The production of superoxide in complex III (cytochrome bc1 complex) is well defined. The mechanism of the so-called Q-cycle, which couples the electron transfer from ubiquinol to cytochrome c by the proton pumping at the stoichiometry of 2e-/2H+, was originally proposed by Michell and later modified [57, 58]. During the cycle, unstable semiquinone is generated at the Q(o) site (also called the positively charged P-side). Although complex III does not have an evolutionary relationship with hydrogenases, and there is no rationale to explain the potential that H2 could intervene with the Q-cycle as electron and/or proton donor, there appears to be the possibility that the unstable semiquinone radical at the Q(o) site, where most of the superoxide is supposed to be generated, may react with H2 and suppress superoxide production. The superoxide at the Q(o) site is assumed to be generated in the presence of the semiquinone intermediate during forward electron transfer from ubiquinol to the b-type heme (cytochrome bL), especially under pathological conditions, such as I/R injury, when the so-called “oxidant-induced reduction” is observed due to the high electrochemical membrane potential with the reduced Q-pool (1) [21, 59]. Alternatively, the reverse electron transfer from the reduced cytochrome bL to the oxidized quinone appears to be a more plausible mechanism for the generation of superoxide at the Q(o) site (2) [27]. In this scenario, the highly oxidized Q-pool is a critical factor for the superoxide burst. Therefore, at the Q(o) site, it would be important to forwardly reduce the semiquinones of the Q-pool upstream of complex III to prevent the reverse reduction of oxidized quinone under physiological conditions and/or to re-reduce the semiquinone, which was forwardly reduced from quinol before the semiquinone donated the electron to the O2 which would be oversupplied under the reperfusion after inducing the hypoxic condition. The hypothetical bifurcate electron donation from H2 to the unstable semiquinone and oxidized Rieske [2Fe2S] cluster at the Q(o) site appears to make it possible to prevent the two mechanisms for superoxide production (1 and 2, above) and may rescue the Q-cycle in both directions. The semiquinone at the Q(i) site is stabilized by the enzyme and the midpoint potential of cytochrome bH is relatively high. If H2 could be reacted with semiquinone at this site, the reduction itself appears energetically favorable, but the activation of H2 seems to be difficult.
8. SUPERCOMPLEX AND DYNAMICS OF QUINONE SPECIES IN ETC
The appropriate amount and the distribution of quinone species in the mitochondrial inner membrane are not completely defined. Recently, studies on mice with genetically blocked or partially reduced quinone biosynthesis provided new insights on the general requirement of quinone and the appropriate quinone content in the ETC, which has become a focus of recent research. These genetic approaches suggested that intricate mechanisms are controlling the quinone content in the ETC. Interestingly, Mclk1+/- heterozygous mice with a partially decreased quinone phenotype had an extended lifespan associated with an approximately 20% lower content of quinone in the inner mitochondrial membrane [60]. Interestingly, the experiments with Mclk1 conditionally knockout mice indicated that the lowest required level of quinone in the ETC is unexpectedly lower than the normal level [61]. Although a 90% reduction in quinone severely affected the heart, kidneys, and skeletal muscles as indicated by the dysfunction of mitochondrial respiration (50% of state 3 respiration), only 10% of the normal quinone content could supply sufficient energy to the heart to ensure survival [62]. The least functional ETC with the low content of quinone may relate to the formation of the supercomplex in the mitochondrial respiratory system. Recently, the supercomplex containing all components (complex I-III-IV) required for mitochondrial respiration, starting from NADH to O2 as the terminal electron acceptor, has been isolated [63]. It is suggested that the relevant conformations of the supercomplex may relate to the efficient electron transfer and also suppress the excess generation of ROS [33]. It should be noted that the distance between the Q-binding site of complex I and III in the bovine assembly is approximately 13 nm that allows quantum tunneling [64, 65]. It suggests that quinone is not necessarily required to move out from the Q-chamber of complex I to carry the electrons into complex III. In the hypothetical utilization of the H2 molecule in the Q-binding site in complex I or III as described in the previous section, it is possible that the FLP-like activation of H2 includes this non-diffusive interaction where the quinone species may act like a catalyst rather than a simple electron carrier. It is suggested that the Q-content in the supercomplex correlates with the size of the Q-pool [66] and the rate of oxidized quinone in the Q-pool affects the generation of ROS in complex III [27]. Either in the ETC carried out by the mobile quinone species with random collision to complex I-III-IV or in the ETC completed by the stalled quinone species in the supercomplex assembly, the unfavorable level of quinone intermediates stuck in both forward and reverse directions should be completely reduced to suppress the generation of ROS. The hypothetical possibility where the activated H2 donates electron and proton with the catalytic quinone species may contribute to rectifying the electron and proton flow in the disordered ETC described above. Although the isolated mammalian supercomplex is equipped with one or a few quinones [64], the precise ratio of the Q-content to complex I assembled in the mammalian supercomplex is not clear in vivo. The EPR-detectable SQ-signals are assumed to be less than one quinone per complex I in bovine mitochondria and also Q-content in the fully active complex I of yeast was substoichiometric (0.2-0.4 per complex I) [53, 67]. These observations may suggest the properties of quinone species as a catalyst.
9. EFFECTIVE CONCENTRATION OF H2
Animal experiments in rats showed that the inhalation of H2 could suppress the I/R injury of the brain at an effective H2 concentration in the artery and vein 1 hour after inhalation of 2% H2 gas at a concentration of nearly 20 μM and 10 μM, respectively [5]. In a clinical report, the concentration of H2 in blood 30 min after inhaling 3% H2 gas was close to the corresponding values in the study above [68]. In cases where H2 was dissolved in water for administration (15 mL/kg of 0.8 mM H2 water was administered to the rats), molecular H2 did effectively alleviate nephrotoxicity caused by cisplatin at a concentration of 5–6 μM of H2 in blood [69]. The measured blood concentrations of H2 were in the range of 5–20 μM, which typically means that a similar concentration is achieved throughout the body due to the passive diffusion property of H2, which appeared to be enough for the enzymatic activity of [NiFe] hydrogenase to oxidize H2. The Km value for [NiFe] hydrogenase is in the range of 1-10 μM and the enzyme can oxidize H2 even at a partial pressure of 0.01 bar (corresponding to 8 μM dissolved H2) [70, 71]. Although the catalytic activity of complex I for the oxidation of H2 proposed here is hypothetical, the accessibility and presence of H2 in the quinone catalytic site is very instructive, considering the structural similarity around the conserved catalytic centers of complex I-related enzymes and [NiFe] hydrogenases [43]. Apart from the physiological activity of complex I, the possibility to utilize H2 seems to be restricted even if it exists. The hypothesis here is based on a pathological complex I with leaky electrons due to the disorders affecting the ETC and/or an increase of highly reactive semiquinone intermediates. Recently, the beneficial effects of H2 were discussed by Ostojic from the view of bioenergetics [72]. In the commentary, the author suggested that H2 affects energy metabolism via activation of the metabolism-related factors including ghrelin, GLUT1, GLUT4, and FGF21. Although it is not known how H2 triggers these signal cascades, the hypothetical insight presented here may prompt future studies to consider the relationship between the mitochondrial energy metabolism and H2, which might be involved in the activation of the cascades. In Japan, water containing extremely high concentrations of H2 (recently reaching 8 ppm in the water) is available, and we have observed over the past 7 years that many people have adapted the habit of drinking H2-enriched water daily. It is important to identify and describe the proper mechanisms, which should explain the benefits of H2 intake. Furthermore, future studies should focus on the effectiveness of H2 to determine the optimal administration method and dose needed for having health benefits and therapeutic activity in various diseases.
CONCLUSION
Many reports examining the therapeutic efficacy of H2 are limited in their suggestion that H2 exerts its effects as the scavenger molecule for the hydroxyl radical and peroxynitrite (“scavenger theory”). However, it is critical to consider other potential mechanisms that could explain the efficacy associated with the consumption of H2, which has been observed in clinical trials. Although the experimental evidence for the activation of H2 in the Q-chamber is lacking, the recently developed FLP theory and the conserved structural features shared between the energy-converting complex I and the hydrogenases provide a strong argument for considering the significance of a new possible mechanism discussed here. H2 is proposed to function as the rectifier for the mitochondrial electron flow in the disordered or pathological state. Notably, the possibility exists that H2 may work as both reductant and oxidant by donating electrons and protons, respectively, to the ubiquinone species including the reactive intermediates, thereby preventing the premature leakage of electrons from the ETC and, consequently, the generation of ROS. Our group currently investigates this possible mechanism of action for H2 treatment. It appears to be important that researchers interested in H2 therapy are introduced to this possible mechanism in addition to the conventional scavenger theory, which will expand our knowledge about the potential health benefits associated with the consumption of H2.
ACKNOWLEDGEMENTS
I thank G. Ishihara, K. Kawamoto, and N. Komori from the Anicom Speciality Medical Institute for their kind advise.
LIST OF ABBREVIATIONS
- H2
Molecular hydrogen
- ROS
Reactive oxygen species
- Fe-S
Iron-sulfur
- MBH
Energy-converting, membrane-bound [NiFe]-hydrogenases
- I/R
Ischemia-reperfusion
- PD
Parkinson’s disease
- RA
Rheumatoid arthritis
- ETC
Electron transport chain
- NADH
Nicotinamide adenine dinucleotide
- Q
Ubiquinone
- SQ
Semiquinones
- QH2
Ubiquinol
- EPR
Electron paramagnetic resonance
- FMN
Flavin mononucleotide
- A-form
Activated form
- D-form
Deactivated form
- Ehr
Energy-converting hydrogenases-related
- FLP
A frustrated Lewis pair
- H-
Hydride ion
- N2ox
Oxidized N2 cluster
- N2red
Reduced N2 cluster
- Em
Midpoint potential
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Vignais P.M., Colbeau A. Molecular biology of microbial hydrogenases. Curr. Issues Mol. Biol. 2004;6(2):159–188. [PubMed] [Google Scholar]
- 2.Schut G.J., Zadvornyy O., Wu C.H., Peters J.W., Boyd E.S., Adams M.W. The role of geochemistry and energetics in the evolution of modern respiratory complexes from a proton-reducing ancestor. Biochim. Biophys. Acta. 2016;1857(7):958–970. doi: 10.1016/j.bbabio.2016.01.010. [DOI] [PubMed] [Google Scholar]
- 3.Ichihara M., Sobue S., Ito M., Ito M., Hirayama M., Ohno K. Beneficial biological effects and the underlying mechanisms of molecular hydrogen - comprehensive review of 321 original articles. Med. Gas Res. 2015;5:12. doi: 10.1186/s13618-015-0035-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shirahata S., Kabayama S., Nakano M., et al. Electrolyzed-reduced water scavenges active oxygen species and protects DNA from oxidative damage. Biochem. Biophys. Res. Commun. 1997;234(1):269–274. doi: 10.1006/bbrc.1997.6622. [DOI] [PubMed] [Google Scholar]
- 5.Ohsawa I., Ishikawa M., Takahashi K., et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 2007;13(6):688–694. doi: 10.1038/nm1577. [DOI] [PubMed] [Google Scholar]
- 6.Ohta S. Molecular hydrogen as a preventive and therapeutic medical gas: initiation, development and potential of hydrogen medicine. Pharmacol. Ther. 2014;144(1):1–11. doi: 10.1016/j.pharmthera.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Yoritaka A., Ohtsuka C., Maeda T., et al. Randomized, double-blind, multicenter trial of hydrogen water for Parkinson’s disease. Mov. Disord. 2018;33(9):1505–1507. doi: 10.1002/mds.27472. [DOI] [PubMed] [Google Scholar]
- 8.Yoritaka A., Takanashi M., Hirayama M., Nakahara T., Ohta S., Hattori N. Pilot study of H2 therapy in Parkinson’s disease: a randomized double-blind placebo-controlled trial. Mov. Disord. 2013;28(6):836–839. doi: 10.1002/mds.25375. [DOI] [PubMed] [Google Scholar]
- 9.Sakai T., Sato B., Hara K., et al. Consumption of water containing over 3.5 mg of dissolved hydrogen could improve vascular endothelial function. Vasc. Health Risk Manag. 2014;10:591–597. doi: 10.2147/VHRM.S68844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishibashi T. Molecular hydrogen: new antioxidant and anti-inflammatory therapy for rheumatoid arthritis and related diseases. Curr. Pharm. Des. 2013;19(35):6375–6381. doi: 10.2174/13816128113199990507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buxton G.V., Greenstock C.L., Helman W.P., Ross A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (・OH/・OH-) in aqueous solution. J. Phys. Chem. Ref. Data. 1988;17:513–886. doi: 10.1063/1.555805. [DOI] [Google Scholar]
- 12.Filippin L.I., Vercelino R., Marroni N.P., Xavier R.M. Redox signalling and the inflammatory response in rheumatoid arthritis. Clin. Exp. Immunol. 2008;152(3):415–422. doi: 10.1111/j.1365-2249.2008.03634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones K.G., Cooper W.J., Mezykk S.P. Bimolecular rate constant determination for the reaction of hydroxyl radicals with domoic and kainic acid in aqueous solution. Environ. Sci. Technol. 2009;43(17):6764–6768. doi: 10.1021/es901128c. [DOI] [PubMed] [Google Scholar]
- 14.Kamimura N., Nishimaki K., Ohsawa I., Ohta S. Molecular hydrogen improves obesity and diabetes by inducing hepatic FGF21 and stimulating energy metabolism in db/db mice. Obesity (Silver Spring) 2011;19(7):1396–1403. doi: 10.1038/oby.2011.6. [DOI] [PubMed] [Google Scholar]
- 15.Ishibashi T., Sato B., Rikitake M., et al. Consumption of water containing a high concentration of molecular hydrogen reduces oxidative stress and disease activity in patients with rheumatoid arthritis: an open-label pilot study. Med. Gas Res. 2012;2(1):27. doi: 10.1186/2045-9912-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feagin J.E. The 6-kb element of Plasmodium falciparum encodes mitochondrial cytochrome genes. Mol. Biochem. Parasitol. 1992;52(1):145–148. doi: 10.1016/0166-6851(92)90046-M. [DOI] [PubMed] [Google Scholar]
- 17.Moser C.C., Farid T.A., Chobot S.E., Dutton P.L. Electron tunneling chains of mitochondria. Biochim. Biophys. Acta. 2006;1757(9-10):1096–1109. doi: 10.1016/j.bbabio.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 18.Murphy E., Steenbergen C. Preconditioning: the mitochondrial connection. Annu. Rev. Physiol. 2007;69:51–67. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- 19.Marreiros B.C., Batista A.P., Duarte A.M., Pereira M.M. A missing link between complex I and group 4 membrane-bound [NiFe] hydrogenases. Biochim. Biophys. Acta. 2013;1827(2):198–209. doi: 10.1016/j.bbabio.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 20.Boveris A., Oshino N., Chance B. The cellular production of hydrogen peroxide. Biochem. J. 1972;128(3):617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y.R., Zweier J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014;114(3):524–537. doi: 10.1161/CIRCRESAHA.114.300559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reuter S., Gupta S.C., Chaturvedi M.M., Aggarwal B.B. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic. Biol. Med. 2010;49(11):1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parey K., Brandt U., Xie H., et al. Cryo-EM structure of respiratory complex I at work. eLife. 2018;7:e39213. doi: 10.7554/eLife.39213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirst J., Roessler M.M. Energy conversion, redox catalysis and generation of reactive oxygen species by respiratory complex I. Biochim. Biophys. Acta. 2016;1857(7):872–883. doi: 10.1016/j.bbabio.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohnishi T., Ohnishi S.T., Salerno J.C. Five decades of research on mitochondrial NADH-quinone oxidoreductase (complex I). Biol. Chem. 2018;399(11):1249–1264. doi: 10.1515/hsz-2018-0164. [DOI] [PubMed] [Google Scholar]
- 26.Song Y., Buettner G.R. Thermodynamic and kinetic considerations for the reaction of semiquinone radicals to form superoxide and hydrogen peroxide. Free Radic. Biol. Med. 2010;49(6):919–962. doi: 10.1016/j.freeradbiomed.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dröse S., Brandt U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J. Biol. Chem. 2008;283(31):21649–21654. doi: 10.1074/jbc.M803236200. [DOI] [PubMed] [Google Scholar]
- 28.Sarewicz M., Borek A., Daldal F., Froncisz W., Osyczka A. Demonstration of short-lived complexes of cytochrome c with cytochrome bc1 by EPR spectroscopy: implications for the mechanism of interprotein electron transfer. J. Biol. Chem. 2008;283(36):24826–24836. doi: 10.1074/jbc.M802174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitchell P. Possible molecular mechanisms of the protonmotive function of cytochrome systems. J. Theor. Biol. 1976;62(2):327–367. doi: 10.1016/0022-5193(76)90124-7. [DOI] [PubMed] [Google Scholar]
- 30.Ohnishi T., Ohnishi S.T., Shinzawa-Itoh K., Yoshikawa S., Weber R.T. EPR detection of two protein-associated ubiquinone components (SQ(Nf) and SQ(Ns)) in the membrane in situ and in proteoliposomes of isolated bovine heart complex I. Biochim. Biophys. Acta. 2012;1817(10):1803–1809. doi: 10.1016/j.bbabio.2012.03.032. [DOI] [PubMed] [Google Scholar]
- 31.Breuer M.E., Koopman W.J., Koene S., et al. The role of mitochondrial OXPHOS dysfunction in the development of neurologic diseases. Neurobiol. Dis. 2013;51:27–34. doi: 10.1016/j.nbd.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Holper L., Ben-Shachar D., Mann J.J. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology. 2018;2018:1. doi: 10.1038/s41386-018-0090-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Letts J.A., Sazanov L.A. Clarifying the supercomplex: the higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 2017;24(10):800–808. doi: 10.1038/nsmb.3460. [DOI] [PubMed] [Google Scholar]
- 34.Brandt U. A two-state stabilization-change mechanism for proton-pumping complex I. Biochim. Biophys. Acta. 2011;1807(10):1364–1369. doi: 10.1016/j.bbabio.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 35.Ingledew W.J., Ohnishi T. An analysis of some thermodynamic properties of iron-sulphur centres in site I of mitochondria. Biochem. J. 1980;186(1):111–117. doi: 10.1042/bj1860111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baradaran R., Berrisford J.M., Minhas G.S., Sazanov L.A. Crystal structure of the entire respiratory complex I. Nature. 2013;494(7438):443–448. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zickermann V., Wirth C., Nasiri H., et al. Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex I. Science. 2015;347(6217):44–49. doi: 10.1126/science.1259859. [DOI] [PubMed] [Google Scholar]
- 38.Fiedorczuk K., Letts J.A., Degliesposti G., Kaszuba K., Skehel M., Sazanov L.A. Atomic structure of the entire mammalian mitochondrial complex I. Nature. 2016;538(7625):406–410. doi: 10.1038/nature19794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blaza J.N., Vinothkumar K.R., Hirst J. Structure of the deactive state of mammalian respiratory complex I. Structure. 2018;26(2):312–319.e3. doi: 10.1016/j.str.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu J., Vinothkumar K.R., Hirst J. Structure of mammalian respiratory complex I. Nature. 2016;536(7616):354–358. doi: 10.1038/nature19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chouchani E.T., Methner C., Nadtochiy S.M., et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013;19(6):753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yano T., Dunham W.R., Ohnishi T. Characterization of the delta muH+-sensitive ubisemiquinone species (SQ(Nf)) and the interaction with cluster N2: new insight into the energy-coupled electron transfer in complex I. Biochemistry. 2005;44(5):1744–1754. doi: 10.1021/bi048132i. [DOI] [PubMed] [Google Scholar]
- 43.Efremov R.G., Sazanov L.A. The coupling mechanism of respiratory complex I - a structural and evolutionary perspective. Biochim. Biophys. Acta. 2012;1817(10):1785–1795. doi: 10.1016/j.bbabio.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 44.Stephan D.W., Erker G. Frustrated Lewis pairs: metal-free hydrogen activation and more. Angew. Chem. Int. Ed. Engl. 2010;49(1):46–76. doi: 10.1002/anie.200903708. [DOI] [PubMed] [Google Scholar]
- 45.Berrisford J.M., Sazanov L.A. Structural basis for the mechanism of respiratory complex I. J. Biol. Chem. 2009;284(43):29773–29783. doi: 10.1074/jbc.M109.032144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans R.M., Brooke E.J., Wehlin S.A., et al. Mechanism of hydrogen activation by [NiFe] hydrogenases. Nat. Chem. Biol. 2016;12(1):46–50. doi: 10.1038/nchembio.1976. [DOI] [PubMed] [Google Scholar]
- 47.Welch G.C., San Juan R.R., Masuda J.D., Stephan D.W. Reversible, metal-free hydrogen activation. Science. 2006;314(5802):1124–1126. doi: 10.1126/science.1134230. [DOI] [PubMed] [Google Scholar]
- 48.Medda R., Padiglia A., Pedersen J.Z., Floris G. Evidence for alpha-proton abstraction and carbanion formation involving a functional histidine residue in lentil seedling amine oxidase. Biochem. Biophys. Res. Commun. 1993;196(3):1349–1355. doi: 10.1006/bbrc.1993.2401. [DOI] [PubMed] [Google Scholar]
- 49.Hernández-Ortega A., Lucas F., Ferreira P., Medina M., Guallar V., Martínez A.T. Role of active site histidines in the two half-reactions of the aryl-alcohol oxidase catalytic cycle. Biochemistry. 2012;51(33):6595–6608. doi: 10.1021/bi300505z. [DOI] [PubMed] [Google Scholar]
- 50.Carr S.B., Evans R.M., Brooke E.J., et al. Hydrogen activation by [NiFe]-hydrogenases. Biochem. Soc. Trans. 2016;44(3):863–868. doi: 10.1042/BST20160031. [DOI] [PubMed] [Google Scholar]
- 51.Sazanov L.A., Hinchliffe P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science. 2006;311(5766):1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 52.Zwicker K., Galkin A., Dröse S., Grgic L., Kerscher S., Brandt U. The Redox-Bohr group associated with iron-sulfur cluster N2 of complex I. J. Biol. Chem. 2006;281(32):23013–23017. doi: 10.1074/jbc.M603442200. [DOI] [PubMed] [Google Scholar]
- 53.Tocilescu M.A., Zickermann V., Zwicker K., Brandt U. Quinone binding and reduction by respiratory complex I. Biochim. Biophys. Acta. 2010;1797(12):1883–1890. doi: 10.1016/j.bbabio.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 54.Kashani-Poor N., Zwicker K., Kerscher S., Brandt U. A central functional role for the 49-kDa subunit within the catalytic core of mitochondrial complex I. J. Biol. Chem. 2001;276(26):24082–24087. doi: 10.1074/jbc.M102296200. [DOI] [PubMed] [Google Scholar]
- 55.Fiedorczuk K., Sazanov L.A. Fiedorczuk., K, Sazanov LA. Mammalian mitochondrial complex I structure and disease-causing mutations. Trends Cell Biol. 2018;28(10):835–867. doi: 10.1016/j.tcb.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 56.Stuehr D.J., Kwon N.S., Nathan C.F., Griffith O.W., Feldman P.L., Wiseman J. N omega-hydroxy-L-arginine is an intermediate in the biosynthesis of nitric oxide from L-arginine. J. Biol. Chem. 1991;266(10):6259–6263. [PubMed] [Google Scholar]
- 57.Mitchell P. The protonmotive Q cycle: a general formulation. FEBS Lett. 1975;59(2):137–139. doi: 10.1016/0014-5793(75)80359-0. [DOI] [PubMed] [Google Scholar]
- 58.Crofts A.R., Holland J.T., Victoria D., et al. The Q-cycle reviewed: How well does a monomeric mechanism of the bc(1) complex account for the function of a dimeric complex? Biochim. Biophys. Acta. 2008;1777(7-8):1001–1019. doi: 10.1016/j.bbabio.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wikström M.K., Berden J.A. Oxidoreduction of cytochrome b in the presence of antimycin. Biochim. Biophys. Acta. 1972;283(3):403–420. doi: 10.1016/0005-2728(72)90258-7. [DOI] [PubMed] [Google Scholar]
- 60.Lapointe J., Stepanyan Z., Bigras E., Hekimi S. Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived Mclk1+/- mice. J. Biol. Chem. 2009;284(30):20364–20374. doi: 10.1074/jbc.M109.006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y., Hekimi S. Understanding Ubiquinone. Trends Cell Biol. 2016;26(5):367–378. doi: 10.1016/j.tcb.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 62.Wang Y., Oxer D., Hekimi S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat. Commun. 2015;6:6393. doi: 10.1038/ncomms7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Acín-Pérez R., Fernández-Silva P., Peleato M.L., Pérez-Martos A., Enriquez J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell. 2008;32(4):529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 64.Genova M.L., Lenaz G. Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta. 2014;1837(4):427–443. doi: 10.1016/j.bbabio.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 65.Althoff T., Mills D.J., Popot J.L., Kühlbrandt W. Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 2011;30(22):4652–4664. doi: 10.1038/emboj.2011.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Blaza J.N., Serreli R., Jones A.J., Mohammed K., Hirst J. Kinetic evidence against partitioning of the ubiquinone pool and the catalytic relevance of respiratory-chain supercomplexes. Proc. Natl. Acad. Sci. USA. 2014;111(44):15735–15740. doi: 10.1073/pnas.1413855111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dröse S., Zwicker K., Brandt U. Full recovery of the NADH:ubiquinone activity of complex I (NADH:ubiquinone oxidoreductase) from Yarrowia lipolytica by the addition of phospholipids. Biochim. Biophys. Acta. 2002;1556(1):65–72. doi: 10.1016/S0005-2728(02)00307-9. [DOI] [PubMed] [Google Scholar]
- 68.Ono H., Nishijima Y., Adachi N., et al. A basic study on molecular hydrogen (H2) inhalation in acute cerebral ischemia patients for safety check with physiological parameters and measurement of blood H2 level. Med. Gas Res. 2012;2(1):21. doi: 10.1186/2045-9912-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nakashima-Kamimura N., Mori T., Ohsawa I., Asoh S., Ohta S. Molecular hydrogen alleviates nephrotoxicity induced by an anti-cancer drug cisplatin without compromising anti-tumor activity in mice. Cancer Chemother. Pharmacol. 2009;64(4):753–761. doi: 10.1007/s00280-008-0924-2. [DOI] [PubMed] [Google Scholar]
- 70.Albracht S.P. Nickel hydrogenases: in search of the active site. Biochim. Biophys. Acta. 1994;1188(3):167–204. doi: 10.1016/0005-2728(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 71.Pershad H.R., Duff J.L., Heering H.A., Duin E.C., Albracht S.P., Armstrong F.A. Catalytic electron transport in Chromatium vinosum [NiFe]-hydrogenase: application of voltammetry in detecting redox-active centers and establishing that hydrogen oxidation is very fast even at potentials close to the reversible H+/H2 value. Biochemistry. 1999;38(28):8992–8999. doi: 10.1021/bi990108v. [DOI] [PubMed] [Google Scholar]
- 72.Ostojic S.M. Does H2 alter mitochondrial bioenergetics via GHS-R1a activation? Theranostics. 2017;7(5):1330–1332. doi: 10.7150/thno.18745. [DOI] [PMC free article] [PubMed] [Google Scholar]