

Summary
Exploration of predictive descriptors for the performance of electrocatalytic oxygen evolution reaction (OER) is significant for material development in many energy conversion processes. In this work, we used high-throughput density functional theory (DFT) calculations to systematically investigate the OER performance of thirty kinds of isolated transition metal atoms-doped ultrathin MoS2 nanosheets (M-UMONs). The results showed that the OER activity could be a function of the decorated transition metal-sulfur (M-S) bond orders with a volcanic-shaped correlation, and a strong correlation could be found when the difference of the M-S bond orders and corresponding metal-oxygen (M-O) bond orders were taken into consideration, implying that the difference in M-S and M-O bond orders could be a predictive descriptor of OER activity for M-UMON system. This successful result also implies this calculation-based method for the exploring of descriptors would also provide a new promising avenue for the discovery of high-performance OER catalysts.
Subject Areas: Computational Chemistry, Nanomaterials, Energy Materials
Graphical Abstract
Highlights
-
•
A predictive descriptor of OER activity for M-UMONs was proposed
-
•
Calculation and experiments were combined to explore the descriptor
-
•
A series of monatomic catalyst was prepared via a universal method
-
•
High-throughput calculation was applied to shorten the development cycle
Computational Chemistry; Nanomaterials; Energy Materials
Introduction
The design of high-performance catalysts for energy conversion and storage is a high priority in light of the popular pursuit of sustainable energy (Bergmann et al., 2018, Chia and Pumera, 2018, Gray, 2009, Kim et al., 2018, Roger et al., 2017). As a critical process, oxygen evolution reaction (OER) is usually the rate-limiting step for such energy conversions since the kinetics of OER are often unsatisfactory due to the four-electron transfer process, which usually requires a large overpotential to drive the reaction (Lewis and Nocera, 2006, Kanan and Nocera, 2008, Han et al., 2018, Silva et al., 2018, Wang et al., 2018a, Wang et al., 2018b, Liu et al., 2018a, Liu et al., 2018b). As a result, the large overpotential ineluctably results in low efficiency. Over the past few years, reports show that different noble-metal-based catalysts have been found to significantly reduce the overpotential of OER. However, these reported noble-metal based catalysts have been limited in their practical applicability owing to their high cost, which hinders the development of energy conversion devices (Xie et al., 2018, Chen et al., 2018, Yu et al., 2018, Guo et al., 2018).
An ideal catalyst should possess both high activity and low cost. For this purpose, a great deal of research has been performed to seek highly active materials based on earth-abundant elements. Recently, supported monatomic catalysts have drawn increasing attention because of their high utilization of active sites and the resulting high performance of the OER (Li et al., 2018, Fei et al., 2018, Yan et al., 2018, Chen et al., 2017) and have been considered as promising alternative electrocatalysts for the OER. However, how to rapidly obtain these desired monatomic OER catalysts without tedious “trial and error” remains a problem. An easily obtainable descriptor is urgently required for the rapid screening of desirable high-performance materials (Huang et al., 2017, Jacobs et al., 2018, Wang et al., 2018a, Wang et al., 2018b, Lin et al., 2017, Fang et al., 2017, Gu et al., 2018). By means of a descriptor, we can rapidly evaluate OER activity since obtaining the descriptor value is usually much faster than calculating the Gibbs free energy via frequency calculations. This method would significantly shorten the calculation time and thus reduce the calculation cost. Before experimentally preparing the catalysts, we can first screen out the desired materials quickly based on the descriptor, avoiding tedious trial and error and improve research efficiency. Hence, we seek to better understand the correlation between electrochemical performance and chemical structure by means of high-throughput density functional theory (DFT) calculations owing to their successful applications in material screening and then obtain a credible descriptor of OER activity for surface monatomic materials to predict and design highly active monatomic catalysts, that is, the metal atoms-doped ultrathin MoS2 nanosheets (M-UMONs) detailed in this work. Fortunately, previously reported research provides us with a good reference. For examples, surface oxygen binding energy (Rossmeisl et al., 2007, Man et al., 2011), the enthalpy of a lower to higher oxide transition (Trasatti, 1980), and the 3d electron number (Bockris and Otagawa, 1984) are reported as successful OER activity descriptors. Although these proposed descriptors are not straightforward in their practical application (Suntivich et al., 2011a, Suntivich et al., 2011b), their exploration method and processes offer us much inspiration.
In this work, the credibility of our calculations of theoretical overpotentials was first validated when we explored the descriptor based on the DFT calculations. For the validation purpose, more than ten kinds of M-UMONs were prepared and systematically characterized, and the electrochemical experimental results confirmed our calculations. Based on this, different kinds of M-UMONs were theoretically investigated by means of using high-throughput DFT calculations. Our calculation results demonstrated a volcanic-shaped correlation between the activity and metal-sulfur (M-S) bond orders. As the differences between the M-S bond orders and corresponding metal-oxygen (M-O) bond orders increased, the OER performance would decline, meaning that peak activity is predicted to occur when the bond order difference is minimal. More importantly, this successful exploration of the descriptor of OER performance for M-UMONs systems implies this calculation-based method for the exploring of descriptors would provide new promising avenues of research for the development of high-performance OER catalysts.
Results and Discussion
Exploring descriptors usually requires a significant amount of data points to ensure the accuracy and reliability of the results. Using more traditional methods, data points are obtained from experimental results, requiring a huge quantity of synthesis and measurement procedures, which are time and resource consuming. A more efficient way of exploring descriptors is urgently desired. The emergence of high-throughput calculations offers an opportunity for solving this problem, which can rapidly generate a large number of credible data such as overpotentials that can be measured experimentally, as well as gathering electronic structure parameters that are difficult to obtain experimentally.
Before using high-throughput calculations, a reasonable initial model structure was constructed, so a series of pre-calculations could be performed on eleven kinds of M-UMONs, to identify the binding configurations (Figure S1 and Table S1), frontier orbitals calculations (Figure S2), electrostatic potential (ESP) distribution calculations (Figures S3 and S4), and adsorption energy calculations (Figure S5). These pre-calculations showed that the Mo-top binding configuration is energetically preferred and the activity mainly comes from the surface metal atoms, which agree with previous reports (Liu et al., 2017). For verification purposes, the eleven kinds of M-UMONs were prepared and systematically characterized using powder X-ray diffraction (Figure S6), transmission electron microscopy (Figure S7), atomic force microscopy (Figure S8), inductively coupled plasma-mass spectrometry (ICP-MS) (Table S2), and high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) (Figure 1). These measurements highlight the successful preparation of the different M-UMONs, so the experimental data for these samples are credible. Additionally, the Mo-top binding configurations were experimentally observed in the HAADF-STEM images (Figure 1), in which the Mo atoms are distributed in a hexagonal pattern, and the supported transition metal atoms are located at the Mo-top sites, experimentally confirming the calculation results. In addition, no obvious aggregation was observed, verifying the atomic dispersion of the transition metal atoms. Collectively, the aforementioned results confirmed that the Mo-top binding configuration is the most stable and the activity is mainly from the surface metal atoms. Based on these results, all the subsequent calculations were based on the surface metal sites with Mo-top binding configuration.
Figure 1.

HAADF-STEM Images of the (002) Planes for Different M-UMONs and MoS2 Ultrathin Nanosheets
The red color represents transition metal atoms supported on Mo-top sites, yellow for Mo atoms, and green for S atoms and the background. Scale bar, 1.0 nm. Since the contrast of atom columns is approximately proportional to the square of the atomic number in HAADF-STEM images, only the heavy transition metal atoms, namely, the Mo atoms and supported transition metal atoms, are visible as bright spots.
In this work, the OER performance was appraised according to the overpotential since it is the key parameter for evaluating activity (Hai et al., 2018), and high-throughput DFT calculations were used to obtain the theoretical overpotentials for the subsequent exploration of the descriptors for OER performance. It should be noted that our calculated theoretical overpotentials results were first validated by the electrochemical measurements of the aforementioned eleven kinds of M-UMONs before using high-throughput calculations (Figures S9 and S10); thus, the use of the calculated theoretical overpotentials to explore the descriptors is a valid method. Since the substrate MoS2 is a kind of 2D ultrathin nanosheet, the activity may also come from the edge sites (Friebel et al., 2015), so pure MoS2 nanosheets were also tested, and their negligible OER activity indicated that the OER activity of different M-UMONs are mainly the result of supported single metal atoms. The DFT calculation results of theoretical overpotentials represented by η are shown in Figure 2. (See theoretical calculations in Supplemental Information). As shown, the overpotentials for the M-UMONs decorated with transition metal elements in VIII group, such as Ni, Fe, and Co, are lower than that of the others and therefore could offer better performance. This result agrees well with the fact that proposed catalysts constructed based on VIII group transition metal elements have better performance toward OER compared with other transition metal elements-based materials (Gao et al., 2014, Liu et al., 2018a, Liu et al., 2018b, Roy et al., 2018). Meanwhile, in each period, the activity first increases until reaching the maximum at the VIII group and then drops again in relation to the rising of the atomic number of supported transition metal atoms. This apparent pattern implies that an inflection point exists in the position of the VIII group, which inspired us to investigate the correlation within the data to find a straightforward descriptor.
Figure 2.

The High-throughput DFT Calculated Theoretical Overpotentials of Different Transition Metal-Based M-UMONs
Lanthanide series were not calculated in this work (red color). Unit: V.
To investigate the activity-structure relationship for the M-UMONs system and then obtain a straightforward descriptor, a series of correlation analyses was conducted. To fully account for all kinds of possible descriptors, the local density of states (LDOS), electronic localization function (ELF), energy level of d-bands edges at the surface metal sites, deformation electron density, Fermi level, bond strength, and atomic charge were all taken into consideration and then empirically analyzed by rational inference, as detailed later. According to previous reports the specific structure of the M-UMONs and the OER processes (i.e., the active sites are the isolated metal atoms and the OH∗ bonded with these surface metal sites), the bond orders that are associated with the surface metal sites, the atomic charge at the surface metal sites, and certain d-band electronic structure parameters show greater promise of becoming the desired descriptors compared with other indexes (Hossain et al., 2019), that is to say, LDOS and ELF could neither accurately reflect the interaction between substrates and surface metal sites nor show the deformation electron density since it could only show the difference of electron density before and after bonding. Based on this, our correlation with theoretical overpotentials analysis mainly focuses on these screened possible descriptors, including bond population calculations, charge population calculations, and a series of electronic configuration parameters such as d-band edge and d-band electron number. The d-band center, d-band edge position, etc. were subsequently ruled out as descriptors as they are tedious in application (Suntivich et al., 2011a, Suntivich et al., 2011b). The atomic charge was also ruled out owing to its weak correlation with the theoretical overpotential as according to our calculation results, namely, more positive charges at the active sites do not necessarily result in better performance, even though OER is an oxidation reaction. The bond orders that are associated with the active sites, which could reflect the interaction strength between the active sites and MoS2 supporter or reaction substrate, were expected to give a proper description of the OER performance (Hossain et al., 2019). This was subsequently confirmed by our bond population analysis, as shown in Figure 3. The distribution of M-S bond orders is similar to that of the theoretical overpotentials (Figure 2), that is, in each period, the M-S bond orders always possess a certain value (∼0.85) at the VIII group, at which the corresponding M-UMONs offer the smallest theoretical overpotential. It can also be seen that the theoretical overpotentials would go higher with the deviation of M-S bond orders from the value at the VIII group, whenever the bond orders increase or decline. This result agrees well with the well-known Sabatier principles, in which a proper bond strength is required to achieve higher activity. It also should be noted that the bond order in this work was the Mayer bond order due to its transferability for different basis sets (Table S3). More specially, the Mayer bond order is defined below:
Here, BAB is the bond orders between atom A and atom B and Pα, Pβ are the density matrices for spin α and β. The bond order value could be directly obtained from the population analysis.
Figure 3.

Averaged M-S Bond Orders of Different Kinds of M-UMONs
The averaged M-S bond orders values are displayed below the associated element symbol, and the attached boxes were mapped with different colors for different ranges.
Collectively, the above-mentioned results indicated that the more the M-S bond orders deviate from this value, the higher the theoretical overpotentials would be. This result indicates the volcanic-shaped correlation between the OER activity and the M-S bond orders, and the peak activity should be located at a certain value.
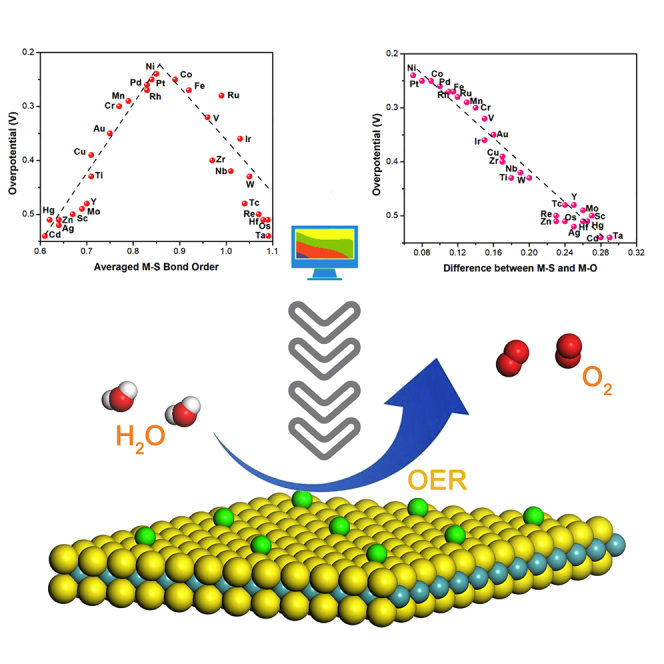
To investigate this relationship, the correlation between M-S bond orders and theoretical overpotentials was further analyzed. As shown in Figure 4A, the theoretical overpotential of different M-UMONs is a volcanic-shaped function of the M-S bond orders, where the OER activities are compared through the theoretical overpotential to trigger the reaction. The volcanic-shaped activity trend might be explained as follows. The surface-supported transition metal atoms participate in the σ-bonding with both adsorbates (M-O bond) and sulfur atoms (M-S bond) during the OER processes, so the M-S bond strength could greatly influence the binding strength of the intermediates and thus the activity (Suntivich et al., 2011a, Suntivich et al., 2011b, Liu et al., 2017). According to the data shown on the left branch of the volcano, the weaker M-S bond strength limited the OER performance owing to the relatively strong M-O bonding, which is unfavorable for the dissociation and desorption of the intermediates. The weaker M-S bond strength would become the bottleneck that restricted the electron transfer between the surface and the MoS2 substrate. As for the right branch of the volcano, the M-S bond is too strong, which is unfavorable for the adsorption of the intermediates, and the weaker M-O bond strength would become the dominant factor restricting the electron transfer between the O∗ and surface metal atoms. These results showed that the peak activity would occur when there is minimal difference between the M-S bond orders and M-O bond orders. Hence, further analysis was conducted from the perspective of the interaction of M-S and M-O bond orders with the theoretical overpotentials, and it was finally found that their differences were linearly correlated with the theoretical overpotentials. As shown in Figure 4B, an obvious linear decay of activity occurred as the difference between the M-S and M-O bond orders increased (see Figure S11 for M-O bond orders), confirming that the bond order difference can greatly impact the adsorption and influence the reaction's activity. Meanwhile, taking the bond order difference as the descriptor would be more credible. That is, for a specified kind of M-UMONs, the M-S and M-O bond orders were all derived from the same optimized structure and the same calculated parameters, and their difference, namely, the difference between M-S and M-O bond orders should reasonably make sense. Furthermore, in terms of the M-UMONs in the left branch, the G4 value is the maximum value of the OER process, indicating that the potential determining step (PDS) is the deprotonation of OOH∗ (Figure S12). Besides, the G2 and G5 values of the left branch are clearly higher than those of right branch, indicating that the weaker M-S bond is unfavorable for the dissociation and desorption of the intermediates. When it comes to the right branch, the G3 value is the maximum value, demonstrating that the PDS is the adsorption of OH- to form the O-O bond in OOH adsorbates. Also, the G1 values of the right branch are significantly higher than those of the left branch, reflecting that the M-S bond that is too strong is unfavorable for the adsorption of the intermediates.
Figure 4.

The Correlation between Bond Orders and Calculated Overpotential and Experimental Overpotential
(A) The relation between the theoretical overpotentials and averaged M-S bond orders.
(B) The correlation between the theoretical overpotentials and the difference between M-S and M-O bond orders.
(C) Experimental observed relations between the OER activity, defined by the onset overpotential at the current density of 5.0 mA cm−2, and the averaged M-S bond orders.
(D) Experimental correlation between the OER activity and the difference between the M-S and M-O bond orders. The black dashed lines are for reference only.
Our descriptor was intuitively validated by the electrochemical measurements of the aforementioned eleven kinds of M-UMONs. The experimental OER activity tendency experimentally confirmed the volcanic-shaped correlation between the M-S bond order and the OER activity (Figure 4C). Additionally, these results also indicate that, as the M-S bond orders get closer to the M-O bond orders, this could enhance the OER activity (Figure 4D). Moreover, four kinds of M-UMONs were tested at the constant overpotential of 250 mV to evaluate their robustness. These results showed that the stability of M-UMONs during the OER process (Figure S13) has a positive correlation with the M-S bond orders, that is, a higher M-S bond order leads to better stability, which could be attributed to the more stable M-S bond with higher bond orders, preventing the loss of supported metal atoms. Even though only four samples were experimentally measured, their M-S bond orders were distributed at different ranges, which shows our results to be representative and credible. Besides, several kinds of M-UMONs, such as Ni-UMONs, Pt-UMONs, and Co-UMONs, could offer comparable OER performance or even outperform other commonly used OER catalysts, including Pt/C and RuO2 (Figure S14). These results also confirmed that the construction of M-UMONs is a practical pathway to develop high-performance OER catalysts.
In conclusion, this work suggests that the relationship between M-S bond orders and corresponding M-O bond orders could significantly influence the OER activity for the M-UMONs system, and systematic investigations confirmed that their difference is a credible descriptor of OER activity for M-UMONs. Decreasing the bond order difference between M-S and M-O for enhancing the activity of M-UMONs materials is a promising strategy to create high-performance electrochemical catalysts for developing high-efficiency energy conversion devices. Furthermore, the successful practice in exploring the descriptor of OER activity for M-UMONs materials demonstrated that the calculation-based method is a practical pathway to obtain a credible descriptor of OER performance for fast materials screening and designing.
Limitation of the Study
The descriptor is sufficiently validated only for the M-UMONs system, and further research is needed for other systems. So far, we have started the study of the transferability of the bond order descriptor for other host materials, and it would be reported in our next research.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (No. 2016YFB0701100), the National Natural Science Foundation of China (51702013), and the Fundamental Research Funds for the Central Universities (FRF-BD-18-013A). Not OER, United States.
Author Contributions
G.H. designed the study. G.H. and X.H. carried out the sample synthesis and electrochemical measurements. G.H. and H.G. performed the electron microscopy characterization. G.H., G.Z., Y.L., and W.D. finished the DFT calculations and analysis. G.H., X.H., G.W., and G.Z. wrote the manuscript. G.H. conducted the AFM and ICP-MS measurements. W.D., G.Z., X.H., and G.W. reviewed the data and revised portions of the manuscript, triggering helpful discussions.
Declaration of Interests
The authors declare no competing interests.
Published: October 25, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.10.001.
Contributor Information
Guixia Zhao, Email: guixiazhao@163.com.
Ge Wang, Email: gewang@mater.ustb.edu.cn.
Data and Code Availability
The crystal structure of molybdenum disulfide reported in this paper could be accessed from the Crystallography Open Database (COD, no. 1010993).
Supplemental Information
References
- Bergmann A., Jones T.E., Moreno E.M., Teschner D., Chernev P., Gliech M., Reier T., Dau H., Strasser P. Unified structural motifs of the catalytically active state of Co(oxyhydr)oxides during the electrochemical oxygen evolution reaction. Nat. Catal. 2018;1:711–719. [Google Scholar]
- Bockris J.O.'M., Otagawa T. The electrocatalysis of oxygen evolution on perovskites. J. Electrochem. Soc. 1984;131:290–302. [Google Scholar]
- Chen Z., Mitchell S., Vorobyeva E., Leary R.K., Hauert R., Furnival T., Ramasse Q.M., Thomas J.M., Midgley P.A., Dontsova D. Stabilization of single metal atoms on graphitic carbon nitride. Adv. Funct. Mater. 2017;27:1605785. [Google Scholar]
- Chen D., Zhang H., Li Y., Pang Y., Yin Z., Sun H., Zhang L.-C., Wang S., Saunders M., Barker E., Jia G. Spontaneous formation of noble-and heavy-metal-free alloyed semiconductor quantum rods for efficient photocatalysis. Adv. Mater. 2018;30:1803351. doi: 10.1002/adma.201803351. [DOI] [PubMed] [Google Scholar]
- Chia X., Pumera M. Characteristics and performance of two-dimensional materials for electrocatalysis. Nat. Catal. 2018;1:909–921. [Google Scholar]
- Fang M., Dong G., Wei R., Ho J.C. Hierarchical nanostructures: design for sustainable water splitting. Adv. Energy Mater. 2017;7:1700559. [Google Scholar]
- Fei H., Dong J., Feng Y., Allen C.S., Wan C., Volosskiy B., Li M., Zhao Z., Wang Y., Sun H. General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities. Nat. Catal. 2018;1:63–72. [Google Scholar]
- Friebel D., Louie M.W., Bajdich M., Sanwald K.E., Cai Y., Wise A.M., Cheng M.-J., Sokaras D., Weng T.-C., Alonso-Mori R. Identification of highly active Fe sites in (Ni,Fe)OOH for electrocatalytic water splitting. J. Am. Chem. Soc. 2015;137:1305–1313. doi: 10.1021/ja511559d. [DOI] [PubMed] [Google Scholar]
- Gao M., Sheng W., Zhuang Z., Fang Q., Gu S., Jiang J., Yan Y. Efficient water oxidation using nanostructured α-Nickel-hydroxide as an electrocatalyst. J. Am. Chem. Soc. 2014;136:7077–7084. doi: 10.1021/ja502128j. [DOI] [PubMed] [Google Scholar]
- Gray H.B. Powering the planet with solar fuel. Nat. Chem. 2009;1:7. doi: 10.1038/nchem.141. [DOI] [PubMed] [Google Scholar]
- Gu Z., Zhang L., Wen B., An X., Lan H., Liu L.-M., Chen T., Zhang J., Cao X., Tang J. Efficient design principle for interfacial charge separation in hydrogen-intercalated nonstoichiometric oxides. Nano Energy. 2018;53:887–897. [Google Scholar]
- Guo Y., Tang J., Wang Z., Kang Y.-M., Bando Y., Yamauchi Y. Elaborately assembled core-shell structured metal sulfides as a bifunctional catalyst for highly efficient electrochemical overall water splitting. Nano Energy. 2018;47:494–502. [Google Scholar]
- Hai G., Jia X., Zhang K., Liu X., Wu Z., Wang G. High-performance oxygen evolution catalyst using two-dimensional ultrathin metal-organic frameworks nanosheets. Nano Energy. 2018;44:345–352. [Google Scholar]
- Han X., Wu X., Deng Y., Liu J., Lu J., Zhong C., Hu W. Ultrafine Pt nanoparticle-decorated pyrite-type CoS2 nanosheet arrays coated on carbon cloth as a bifunctional electrode for overall water splitting. Adv. Energy Mater. 2018;8:1800935. [Google Scholar]
- Hossain M.D., Liu Z., Zhuang M., Yan X., Xu G.-L., Gadre C.A., Tyagi A., Abidi I.H., Sun C.-J., Wong H. Rational design of graphene-supported single atom catalysts for hydrogen evolution reaction. Adv. Energy Mater. 2019;9:1803689. [Google Scholar]
- Huang Z.-F., Wang J., Peng Y., Jung C.-Y., Fisher A., Wang X. Design of efficient bifunctional oxygen reduction/evolution electrocatalyst: recent advances and perspectives. Adv. Energy Mater. 2017;7:1700544. [Google Scholar]
- Jacobs R., Mayeshiba T., Booske J., Morgan D. Material discovery and design principles for stable, high activity perovskite cathodes for solid oxide fuel cells. Adv. Energy Mater. 2018;8:1702708. [Google Scholar]
- Kanan M.W., Nocera D.G. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+ Science. 2008;321:1072–1075. doi: 10.1126/science.1162018. [DOI] [PubMed] [Google Scholar]
- Kim N., Sa Y.J., Yoo T.S., Choi S.R., Afzal R.A., Choi T., Seo Y.-S., Lee K.-S., Hwang J.Y., Choi W.S. Oxygen-deficient triple perovskites as highly active and durable bifunctional electrocatalysts for oxygen electrode reactions. Sci. Adv. 2018;4:eaap9360. doi: 10.1126/sciadv.aap9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis N.S., Nocera D.G. Powering the planet: chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. U S A. 2006;103:15729–15735. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Zhang Y.-Y., Guo H., Huang L., Lu H., Lin X., Wang Y.-L., Du S., Gao H.-J. Epitaxial growth and physical properties of 2D materials beyond graphene: from monatomic materials to binary compounds. Chem. Soc. Rev. 2018;47:6073–6100. doi: 10.1039/c8cs00286j. [DOI] [PubMed] [Google Scholar]
- Lin C.-Y., Zhang L., Zhao Z., Xia Z. Design principles for covalent organic frameworks as efficient electrocatalysts in clean energy conversion and green oxidizer production. Adv. Mater. 2017;29:1606635. doi: 10.1002/adma.201606635. [DOI] [PubMed] [Google Scholar]
- Liu G., Robertson A.W., Li M.M.-J., Kuo W.C.H., Darby M.T., Muhieddine M.H., Lin Y.-C., Suenaga K., Stamatakis M., Warner J.H., Tsang S.C.E. MoS2 monolayer catalyst doped with isolated Co atoms for the hydrodeoxygenation reaction. Nat. Chem. 2017;9:810–816. doi: 10.1038/nchem.2740. [DOI] [PubMed] [Google Scholar]
- Liu L., Su H., Tang F., Zhao X., Liu Q. Confined organometallic Au1Nx single-site as an efficient bifunctional oxygen electrocatalyst. Nano Energy. 2018;46:110–116. [Google Scholar]
- Liu K., Zhang C., Sun Y., Zhang G., Shen X., Zou F., Zhang H., Wu Z., Wegener E.C., Taubert C.J. High-performance transition metal phosphide alloy catalyst for oxygen evolution reaction. ACS Nano. 2018;12:158–167. doi: 10.1021/acsnano.7b04646. [DOI] [PubMed] [Google Scholar]
- Man I.C., Su H.-Y., Calle-Vallejo F., Hansen H.A., Martínez J.I., Inoglu N.G., Kitchin J., Jaramillo T.F., Nørskov J.K., Rossmeisl J. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem. 2011;3:1159–1165. [Google Scholar]
- Roger I., Shipman M.A., Symes M.D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017;1:0003. [Google Scholar]
- Rossmeisl J., Qu Z.-W., Zhu H., Kroes G.-J., Nørskova J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007;607:83–89. [Google Scholar]
- Roy C., Sebok B., Scott S.B., Fiordaliso E.M., Sørensen J.E., Bodin A., Trimarco D.B., Damsgaard C.D., Vesborg P.C.K., Hansen O. Impact of nanoparticle size and lattice oxygen on water oxidation on NiFeOxHy. Nat. Catal. 2018;1:820–829. [Google Scholar]
- Silva G.C., Fernandes M.R., Ticianelli E.A. Activity and stability of Pt/IrO2 bifunctional materials as catalysts for the oxygen evolution/reduction reactions. ACS Catal. 2018;8:2081–2092. [Google Scholar]
- Suntivich J., Gasteiger H.A., Yabuuchi N., Nakanishi H., Goodenough J.B., Yang S.-H. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal–air batteries. Nat. Chem. 2011;3:546–550. doi: 10.1038/nchem.1069. [DOI] [PubMed] [Google Scholar]
- Suntivich J., May K.J., Gasteiger H.A., Goodenough J.B., Yang S.-H. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science. 2011;334:1383–1385. doi: 10.1126/science.1212858. [DOI] [PubMed] [Google Scholar]
- Trasatti S. Electrocatalysis by oxides-attempt at a unifying approach. J. Electroanal. Chem. 1980;111:125–131. [Google Scholar]
- Wang D., Sheng T., Chen J., Wang H.-F., Hu P. Identifying the key obstacle in photocatalytic oxygen evolution on rutile TiO2. Nat. Catal. 2018;1:291–299. [Google Scholar]
- Wang H.-F., Tang C., Zhang Q. A review of precious-metal-free bifunctional oxygen electrocatalysts: rational design and applications in Zn−air batteries. Adv. Funct. Mater. 2018;28:1803329. [Google Scholar]
- Xie M., Xiong X., Yang L., Shi X., Asiri A.M., Sun X. An Fe(TCNQ)2 nanowire array on Fe foil: an efficient non-noble-metal catalyst for the oxygen evolution reaction in alkaline media. Chem. Commun. (Camb.) 2018;54:2300–2303. doi: 10.1039/c7cc09105b. [DOI] [PubMed] [Google Scholar]
- Yan H., Zhao X., Guo N., Lyu Z., Du Y., Xi S., Guo R., Chen C., Chen Z., Liu W. Atomic engineering of high-density isolated Co atoms on graphene with proximal-atom controlled reaction selectivity. Nat. Commun. 2018;9:3197. doi: 10.1038/s41467-018-05754-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F., Zhou H., Huang Y., Sun J., Qin F., Bao J., Goddard W.A., III, Chen S., Ren Z. High-performance bifunctional porous non-noble metal phosphide catalyst for overall water splitting. Nat. Commun. 2018;9:9551. doi: 10.1038/s41467-018-04746-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystal structure of molybdenum disulfide reported in this paper could be accessed from the Crystallography Open Database (COD, no. 1010993).