

Abstract
Background
Previous research has shown that there is an association between galectin‐3 (gal‐3) protein and cardiovascular pathology. The aim of this study was to investigate the effects of rs2274273 and rs17128183 on genetic susceptibility to advanced carotid atherosclerosis (CA) and its complications. The rs2274273 has been singled out as the lead SNP of the haplotype block containing LGALS‐3 (gal‐3 gene) associated with gal‐3 circulating levels, while rs17128183 constitutes a potentially functional SNP of the same hap‐block. We further sought to determine whether these genetic variants have an impact on the expression of LGALS‐3 mRNA in human carotid atherosclerotic plaque tissue.
Methods
The study encompassed 300 control subjects and 485 patients with advanced CA who had undergone carotid endarterectomy. Rs2274273, rs17128183, and LGALS‐3 relative mRNA expression was detected by means of real‐time PCR (TaqMan® technology).
Results
There were no statistically significant associations of the investigated genetic variants with susceptibility to advanced CA, nor did we find any associations in terms of ultrasonographically defined plaque phenotypes. The relative expression of LGALS‐3 mRNA proved to be significantly higher in carriers of the rare alleles (P = 0.039) for both genetic variants.
Conclusion
Our exploratory results suggest that while rs2274273 and rs17128183 bear no association with the risk of advanced CA or CA‐related complications, these genetic variants are likely to affect LGALS‐3 expression levels. In order to reach a definitive conclusion on the role played by rs2274273 and rs17128183 in advanced CA, our results should be further validated.
Keywords: atherosclerosis, carotid plaque, gene expression, LGALS‐3, rs17128183, rs2274273
Introduction
Atherosclerosis is one of the leading causes of death and morbidity worldwide; it is predicted to become “the number one killer” by 2020 1. All stages of disease, from its inception, through its progression to its final complications, are known to be mediated by inflammation 2. Galectin‐3 (gal‐3) has been widely recognized as a biomarker of heart failure (HF) and vascular fibrosis 3, 4, and has recently been included in “2013 ACCF/AHA Guideline for the Management of Heart Failure” 5. It is a multifunctional, pleiotropic protein shown to promote vascular inflammation by chemoattraction of monocytes and activation of macrophages, thus leading to atherosclerotic plaque progression 6, 7. In addition, gal‐3 has been identified as an important factor in the remodeling of the vascular wall 3. It is well known that collagen type I and III represent major components of the vascular wall (intima) in all stages of atherosclerotic lesion development 8. As demonstrated in previous studies, gal‐3 overexpression in primary vascular smooth muscle cells (VSMCs) results in increased synthesis of collagen type I, while the same does not apply to collagen type III 3. Moreover, collagen type I is associated with migration of VSMCs, differentiation of monocytes and uptake of lipids by macrophages 9, 10. It is thus reasonable to assume that gal‐3 could represent an important factor leading not only to cardiac remodeling but also to vascular remodeling as well.
Upregulation of gal‐3 has been described both in rodent models of atherosclerotic disease 11, 12 and in human atherosclerotic lesions 13. The inhibition of gal‐3 with modified citrus pectin has been shown to reduce the plaque volume in ApoE ‐/‐ mice 14. Thus far, increased levels of plasma gal‐3 have been correlated with conventional cardiovascular risk factors 15, although the strength of these correlations was weak. When compared to control subjects or to asymptomatic patients without carotid plaques, circulating gal‐3 levels were found to be higher in asymptomatic patients with carotid plaques 16. The most recent study in the field has identified gal‐3 as an independent predictive marker for cerebrovascular events in female subjects who had undergone carotid endarterectomy (CEA) 17. Only a handful of studies have examined the role of gal‐3 in carotid atherosclerosis (CA), focusing almost exclusively on protein levels. Two studies investigating LGALS‐3 mRNA expression in different plaque phenotypes have reported controversial data 7, 18. No studies thus far have attempted to explore the possible effects of rs2274273 and rs17128183, two candidate genetic variants, on LGALS‐3 mRNA expression.
Human gal‐3, weighting about 30 kDa 19, is encoded by LGALS‐3 gene, located on chromosome 14 at locus q21–q22, in a unique 300 kb long haplotype block of Caucasians. Genetic association studies of LGALS‐3 are scarce. Recently, in the only genome‐wide association study (GWAS) of gal‐3 levels, rs2274273 has been identified as the lead SNP of a haplotype block containing LGALS‐3 gene. The study reported an association of the rare allele with a decrease in gal‐3 circulating levels 20. However, its authors emphasized that their results must be taken with caution, given the possibility of a false‐positive association. In the course of our research, we have examined the entire hap‐block in RegulomeDB database in order to detect other, potentially functional, SNPs. Rs17128183, located 16 kb upstream of LGALS‐3, was singled out as the most likely functional SNP in this hap‐block 21. It is in absolute linkage disequilibrium (LD) with rs4652 (r 2 = 1, D′ = 1), the most investigated non‐synonymous LGALS‐3 SNP, and in almost absolute LD with rs2274273 (r 2 = 0.91, D′ = 1). Rs4652 has previously been investigated with regard to overall survival rates in patients with glioma 22 and in terms of its association with rheumatoid arthritis 23. To date, none of these genetic variants have been analyzed in association with any atherosclerotic phenotype.
In the light of the above, the aim of this study was to analyze the association of genetic variants rs2274273 and rs17128183 with susceptibility to advanced CA and its complications. In addition, our goal was to determine whether these variants have an impact on LGALS‐3 mRNA expression levels in human carotid atherosclerotic plaque tissue.
Materials and Methods
Study Population
The study group consisted of 785 participants. All subjects were unrelated Caucasians of European descent originating from Serbia; the ethnicity of the study population was homogeneous (Serbian).
Four hundred and eighty‐five subjects were recruited among patients admitted for CEA to the Clinic for Vascular and Endovascular Surgery within the Clinical Center of Serbia in Belgrade Serbia, between 2008 and 2009. The patients displayed evidence of carotid plaque presence in internal carotid artery (ICA) and/or in common carotid artery (CCA). The exclusion criteria for all patients were as follows: (a) history of previous CEA (possible restenosis); (b) carotid kinking; (c) carotid aneurysm; (d) tumors; (e) chronic inflammatory and autoimmune diseases; (f) renal failure and (g) occurrence of cerebrovascular insult (CVI) within 6 months prior to sample collection.
Three hundred individuals who were undergoing an annual medical checkup at the Occupational Medical Center in Belgrade, Serbia, were recruited as control subjects. Clinical, ultrasound, and electrocardiographic (ECG) examinations performed on control subjects did not show any evidence of CA, cerebrovascular or cardiovascular disease, chronic inflammatory diseases, renal failure, or diabetes mellitus.
A complete medical history was compiled for each individual enrolled in the study. The collected data included smoking and drinking habits, presence of diabetes, coronary artery disease (CAD), peripheral arterial occlusive disease, and the use of medication (antihypertensive drugs, oral hypoglycemic drugs, insulin). Subjects already diagnosed with diabetes mellitus (fasting glucose level of ≥7.0 mmol/l) and taking either insulin or oral hypoglycemic drugs were characterized as having diabetes mellitus. Those with previous myocardial infarction or stable angina pectoris, evaluated by means of selective coronarography used either to confirm or to reveal CAD, were characterized as having coronary heart disease (CHD). Peripheral artery disease (PAD) was diagnosed as an ankle brachial index (ABI) lower than 0.90. Hypertension was defined based on systolic blood pressure ≥140 mmHg, diastolic blood pressure ≥90 mmHg, and/or in terms of ongoing treatment with an antihypertensive medication.
All biochemical analyses were performed in the hospital laboratory following standard laboratory procedures.
The study was approved by the ethics committees of the medical centers involved and each subject gave written informed consent to participate in the study.
Ultrasound Assessment of Carotid Arteries
Ultrasound assessment of bilateral carotid arteries was performed using the high‐resolution B‐mode ultrasound Acuson Antares™ system (Siemens, Munich, Germany). The degree of carotid stenosis was calculated by North American Symptomatic Carotid Endarterectomy Trial method (NASCET) 24. Both the highest peak systolic velocities and the end‐diastolic velocities were recorded, as well as ICA/CCA carotid artery ratio. All patients included in the study had stenosis >70%. In cases where plaques were obscured due to acoustic shadowing, patients were excluded from the study. Atherosclerotic plaque was defined as a focal widening relative to adjacent segments, evidenced by a protrusion into the lumen and/or by localized roughness with an increased echogenicity. With regard to ultrasound carotid measurements, intraclass correlation coefficient for inter‐rater and intra‐rater reliability was 0.916 and 0.968, respectively. CA was defined as the presence of atherosclerotic plaques in ICA and/or CCA. Duplex ultrasound analysis was performed to classify different plaque types as hypoechogenic (dominantly echolucent) and hyperechogenic (dominantly echogenic) in accordance with the Gray‐Weale criteria 25. Additionally, B mode was used to analyze the surface irregularity of plaques (irregular 0.4–2 mm). Plaque ulceration was defined as an irregularity larger than 2 mm. All patients underwent surgery within 2 weeks following the ultrasound measurements. Per‐operative validation of the extent of stenosis was performed. The concordance between ultrasound measurements and per‐operative findings in defining the stenosis >70% was found to be 100%.
Genetic Analysis
Genomic DNA was extracted from whole blood samples collected with EDTA using a standardized BloodPrep® DNA Chemistry isolation kit (Applied Biosystems, Forester City, CA) on ABI PRISM ™ 6100 Nucleic Acid PrepStation (Applied Biosystems) or by proteinase K/phenol extraction method 26. Genetic variants rs2274273 and rs17128183 were detected by means of real‐time PCR (ABI 7500, Applied Biosystems, Foster City, CA) using TaqMan® SNP Genotyping Assays (C_16181227_10 and C_33685190_10, respectively) purchased from and tested by Applied Biosystems (Forester City, CA). Each PCR reaction contained 120 ng of DNA.
Quantitative Real‐Time Reverse Transcription‐PCR (Real‐Time RT‐PCR)
Total RNA was extracted from segments of frozen plaque tissue specimens with the largest plaque burden using TRIZOL reagent (Life Technologies) in compliance with the manufacturer's instructions. The quantity of RNA was assessed using NanoDrop® ND‐1000 spectrophotometer (Thermo Scientific, Wilmington, Delaware). The structural integrity of RNA was evaluated by formaldehyde gel electrophoresis. Out of 56 human carotid plaque specimens collected by endarterectomy, 39 specimens yielded a total RNA of satisfactory quality and were converted to cDNA. One microgram of RNA was treated with DNAse I (Fermentas, Lithuania) and reverse transcription was performed using a First strand cDNA synthesis kit with oligo‐dT18 primer (Fermentas, Lithuania) in compliance with the manufacturer's instructions. The real‐time PCR was performed in duplicate on ABI Real‐time 7500 system (Applied Biosystems, Foster City, CA). The detection of LGALS‐3 gene expression was executed using the predeveloped TaqMan® gene expression assay Hs00173587_m1 (Applied Biosystems, Foster City, CA), with the forward primer located in exon 3, the reverse in exon 4, and the probe spanning an exon–exon boundary. The detection of the internal reference, peptidylprolyl isomerase A (cyclophilin A), was performed using the predeveloped TaqMan® gene expression assay Hs99999904_m1 (Applied Biosystems, Foster City, CA). The significance of any differences in mRNA expression levels based on the genotype of the investigated genetic variants, the gender of the subjects, and the atherosclerotic plaque types (hypoechogenic and hyperechogenic), was tested by means of REST 2009 software (Qiagen, Hilden, Germany), a tool particularly suitable for comparing the relative gene expression levels between two sample groups as it provides prompt and reliable results in the context of gene expression analysis.
Statistical Analysis
Allelic frequencies and genotype distributions were estimated by the gene counting method. All observed differences in allele and genotype frequencies between the analyzed groups, as well as any deviations from Hardy–Weinberg equilibrium, were estimated by chi‐square (χ2) test. The odds ratio (OR), with its 95% confidence interval (CI), was used as a measure of strength of association between the studied genetic variants and CA. Means of normally distributed continuous variables were compared by unpaired t‐test or analysis of variance (ANOVA), while medians of skewed continuous variables were compared using nonparametric Mann–Whitney U test or Kruskal–Wallis ANOVA. Values of continuous variables were expressed as mean ± standard deviation (SD). Statistical analysis was performed using the software package Statistica Version 8 (StatSoft Inc, Tulsa, OK). The relative LGALS‐3 mRNA expression levels were analyzed by means of REST 2009, a relative expression software tool that applies a pairwise randomization and bootstrapping technique (http://rest.gene-quantification.info) 27. We used Bonferroni correction for multiple testing, for analyzing the two genetic variants within the same study group, and for testing based on gender. Consequently, we considered P values <0.0125 as statistically significant. Sample size calculation was performed using PS software (v3.0.43) (Vanderbilt University School of Medicine, Department of Biostatistics, Nashville, TN) 28.
Results
Characteristics of the Study Population
The main characteristics of control subjects and patients with advanced CA are shown in Table 1. When compared with control subjects, patients with advanced CA were characterized by significantly higher body mass index (BMI), total cholesterol (TC), low‐density lipoprotein cholesterol (LDLC), and triglyceride (TG) levels, as well as by significantly lower high‐density lipoprotein cholesterol (HDLC) levels. They were also significantly older, and within their group, there was a significantly higher proportion of smokers and hypertensive individuals. About 40% of the participants were female in both groups.
Table 1.
Main Characteristics of Controls and Patients with Advanced CA
| Variable | Controls | Patients | P value |
|---|---|---|---|
| n = 300 | n = 485 | ||
| BMI, kg/m2 | 25.2 ± 3.7 | 26.6 ± 3.3 | <0.001a |
| Age, years | 54.2 ± 14.3 | 66.0 ± 7.9 | <0.001a |
| TC, mmol/l | 5.59 ± 1.30 | 5.75 ± 1.26 | 0.05a |
| TG, mmol/l | 1.59 ± 1.12 | 1.89 ± 1.26 | <0.001a |
| HDLC, mmol/l | 1.49 ± 0.88 | 1.21 ± 0.35 | <0.001a |
| LDLC, mmol/l | 3.28 ± 1.22 | 3.70 ± 1.07 | <0.001a |
| Gender F/M, % | 44.1 / 54.9 | 39.6 / 60.4 | nsb |
| Hypertension, % | 27.0 | 86.5 | <0.001b |
| Smokers, % | 54.7 | 71.6 | <0.001b |
| DMT II, % | 7.1 | 21.6 | <0.05b |
| CVI, % | 0 | 19.9 | N/A |
| Symptomatic patients, % | 0 | 58.4 | N/A |
| TIA, % | 0 | 21.8 | N/A |
| Treatment with ACE inhibitors, % | 0 | 88.8 | N/A |
Values are mean ± SD for: Body Mass Index (BMI), age, Total Cholesterol (TC), Triglycerides (TG), High‐Density Lipoproteins Cholesterol (HDLC) and Low‐Density Lipoproteins Cholesterol (LDLC); DMT II, diabetes mellitus type 2; CVI, cerebrovascular insult; TIA, transient ischemic attack; ns, non significant; N/A, not applicable;.
Mann–Whitney U test was used to compare the values between controls and CA patients, for continuous variables with a skewed distribution.
Chi‐square test was used for comparison of the categorical variables
P values <0.05 were considered statistically significant.
Association of rs2274273 and rs17128183 with Advanced CA
Genotype distributions of the investigated genetic variants were in Hardy–Weinberg equilibrium (P > 0.05). Both rs2274273 and rs17128183 were identified in Caucasians from Serbia and were in strong, but not absolute, LD (r 2 = 0.88, D′ = 0.99).
As presented in Table 2, the distributions of genotype and allele frequencies in control subjects and patients with advanced CA were similar for rs2274273 and rs17128183: no statistically significant differences were seen. We additionally analyzed the genotype and allele frequencies based on gender. We found no statistically significant differences between control subjects and patients with advanced CA when stratified by gender for either of the genetic variants (Table 3). In order to reliably reject the null hypothesis, the study would need to encompass 18,118 patients and 11,233 control subjects, shown by retrospectively calculated sample size (for: controls to patients ratio of 0.62; probability of exposure among controls of 0.0933; OR of 1.12 and study power of 80%, at a significance level of 0.05).
Table 2.
Genotype and Allele Frequencies of rs2274273 (C/T) and rs17128183 (A/G) Genetic Variants in Controls and Patients with Advanced CA
| Controls n (%) | Patients n (%) | P value | OR (95% CI) | |
|---|---|---|---|---|
| rs2274273 C/T | ||||
| Genotypes | n = 300 | n = 485 | ||
| CC | 123 (41.00) | 203 (41.86) | 0.26 | 1 |
| CT | 149 (49.67) | 220 (45.36) | 1.06 (0.85–1.32) | |
| TT | 28 (9.33) | 62 (12.78) | 1.12 (0.73–1.74) | |
| Allele frequencies | ||||
| C | 0.66 | 0.65 | ||
| T | 0.34 | 0.35 | 1.06 (0.78–1.43) | |
| rs17128183 A/G | ||||
| Genotypes | n = 278 | n = 448 | ||
| AA | 107 (38.49) | 176 (39.29) | 0.53 | 1 |
| AG | 139 (50.00) | 209 (46.65) | 1.04 (0.83–1.30) | |
| GG | 32 (11.51) | 63 (14.06) | 1.08 (0.69–1.69) | |
| Allele frequencies | ||||
| A | 0.63 | 0.63 | ||
| G | 0.37 | 0.37 | 1.04 (0.76–1.42) | |
OR, crude odds ratio; CI, confidence interval; P, Pearson Chi‐square test, P value was corrected for multiple testing of two genetic variants, and P < 0.0125 was considered statistically significant.
Table 3.
Genotype and Allele Frequencies of rs2274273 (C/T) and rs17128183 (A/G) Genetic Variants in Controls and Patients with Advanced CA, Divided by Gender
| Controls, n (%) | Patients, n (%) | P value | OR (95% CI) | |
|---|---|---|---|---|
| Males | ||||
| rs2274273 C/T | n = 165 | n = 294 | ||
| CC | 64 (38.79) | 120 (40.82) | 1 | |
| CT | 85 (51.52) | 133 (45.24) | 0.28 | 1.05 (0.79–1.40) |
| TT | 16 (9.70) | 41 (13.95) | 1.10 (0.62–1.96) | |
| Alleles | ||||
| C | 0.65 | 0.63 | ||
| T | 0.35 | 0.37 | 1.05 (0.71–1.56) | |
| rs17128183 A/G | n = 149 | n = 271 | ||
| AA | 53 (35.57) | 106 (39.11) | 1 | |
| AG | 80 (53.69) | 123 (45.39) | 0.20 | 1.03 (0.76–1.38) |
| GG | 16 (10.74) | 42 (15.5) | 1.05 (0.58–1.91) | |
| Alleles | ||||
| A | 0.62 | 0.62 | ||
| G | 0.38 | 0.38 | 1.03 (0.68–1.55) | |
| Females | ||||
| rs2274273 C/T | n = 135 | n = 191 | ||
| CC | 59 (43.7) | 83 (43.46) | 1 | |
| CT | 64 (47.41) | 87 (45.55) | 0.82 | 1.06 (0.75–1.48) |
| TT | 12 (8.89) | 21 (10.99) | 1.12 (0.57–2.20) | |
| Alleles | ||||
| C | 0.67 | 0.66 | ||
| T | 0.33 | 0.34 | 1.05 (0.66–1.68) | |
| rs17128183 A/G | n = 129 | n = 177 | ||
| AA | 54 (41.86) | 70 (39.55) | 1 | |
| AG | 59 (45.74) | 86 (48.59) | 0.88 | 1.04 (0.74–1.46) |
| GG | 16 (12.40) | 21 (11.86) | 1.08 (0.55–2.14) | |
| Alleles | ||||
| A | 0.65 | 0.64 | ||
| G | 0.35 | 0.36 | 1.04 (0.65–1.67) | |
OR, crude odds ratio; CI, confidence interval; P, Pearson Chi‐square test, P value was corrected for multiple testing of two genetic variants, and P < 0.0125 was considered statistically significant.
No association of the investigated genetic variants with ultrasonographically defined atherosclerotic plaque phenotypes (hyperechogenic vs. hypoechogenic) (rs2274273: P = 0.79; rs17128183: P = 0.76) or with clinical endpoint (CVI) (rs2274273: P = 0.27; rs17128183: P = 0.29) was observed.
Relative mRNA Expression of LGALS‐3 in Carotid Plaques
We analyzed the relative LGALS‐3 mRNA expression in carotid atherosclerotic plaque tissue specimens (n = 39) based on the genotypes of the investigated genetic variants. All 39 specimens had an identical distribution of genotypes regardless of the variant. The relative expression of LGALS‐3 mRNA regarding rs2274273 (CC vs. CT+TT) and/or rs17128183 (AA vs. AG+GG) was found to be significantly higher in carriers of the rare alleles (mean factor = 1.405, S. E. range = 0.770–2.509, P = 0.039) (Fig. 1). We also compared the relative expression of LGALS‐3 mRNA in the carotid atherosclerotic plaque tissue based on two criteria: (a) different plaque phenotypes as determined by ultrasonographic examination (hyperechogenic vs. hypoechogenic), and (b) different gender of the subjects (male vs. female). The relative expression levels of LGALS‐3 mRNA were not significantly different in hyperechogenic (n = 25) vs. hypoechogenic (n = 14) carotid plaques (mean factor = 0.904, S. E. range = 0.444–1.801, P = 0.541), nor did we observe any significant difference between the plaques of men (n = 25) vs. women (n = 14) (mean factor = 1.185, S. E. range = 0.669–2.219, P = 0.273).
Figure 1.
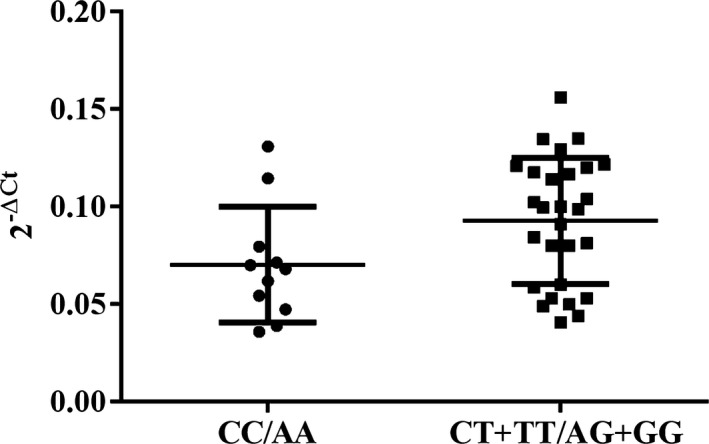
Relative LGALS‐3 mRNA expression in the carotid plaque tissues ex vivo, according to rs2274273 (CC vs. CT+TT) and/or rs17128183 (AA vs. AG+GG), presented as 2‐ΔCt value for each tissue specimen. Quantitative real‐time PCR was performed on 39 endarterectomized human carotid plaque tissue specimens to quantify the LGALS‐3 and housekeeping Cyclophilin A relative gene expression levels. For the quantification of each PCR product, the threshold cycle (Ct) was used. The delta Ct value was calculated from the difference in the Ct of the gene of interest and that of the housekeeping gene. Data are presented as 2‐ΔCt, with mean value for both groups (CC/AA and CT+TT/AG+GG) ± SD. Relative expression levels of LGALS‐3 mRNA calculated using the REST software were significantly higher in carriers of the CT+TT or AG+GG (n = 28) comparing to carriers of the CC or AA (n = 11) (fold induction = 1.405, SE range = 0.770–2.509, P = 0.039).
Discussion
Previous research has shown that there is an association between gal‐3 protein and pathology of cardiovascular diseases. Thus far, no studies have explored a possible association between genetic variants located in LGALS‐3 locus, or in its vicinity, and atherosclerosis. In order to investigate the impact of two candidate genetic variants potentially affecting gal‐3 levels—rs2274273 and rs17128183—on the risk of advanced carotid atherosclerosis, we performed a case–control study in Serbian population. Additionally, we have analyzed whether these two variants could affect LGALS‐3 mRNA expression in carotid plaque tissue.
We did not observe any statistically significant association of the two investigated variants with advanced CA or with different plaque phenotypes as defined by ultrasonographic examination. Considering that the literature data suggest higher gal‐3 plasma concentration levels in women than in men 15, 29, we additionally analyzed the allele and genotype frequencies based on gender. We did not establish a significant difference in the distribution of either genotype or allele frequencies. In this study, the two variants were in strong LD, although not in an absolute one. For both variants, the frequencies of genotypes in control subjects from Serbia corresponded to the frequencies reported in 1000 Genomes Project 30. Previously, these two genetic variants have been examined within the GWASs only. Rs2274273 has been investigated in association with hypertension and diabetes type 1 31, 32, while rs17128183 has been analyzed in association with diabetes type 1 and 2 32, 33, and systolic blood pressure 34. The results obtained did not allow to identify these two SNPs as prognostic markers of diabetes or blood pressure levels. In a recent GWAS study 20, rs2274273 was singled out as the lead variant associated with circulating levels of gal‐3; however, no validation or replication studies were conducted.
Circulating gal‐3 levels were increased in asymptomatic patients with carotid plaques as opposed to control subjects or patients without carotid plaques 16. Interestingly, even when significantly higher than normal values, all levels of circulating gal‐3 fell within the range of low risk for heart failure, proposed by commercially available blood kit tests. Nevertheless, in a 5‐year follow‐up study of PAD patients, the same authors associated gal‐3 circulating levels above the median (4.75 ng/ml) with all‐cause and cardiovascular mortality 16. Increased circulating levels of gal‐3 have been further associated with all‐cause mortality in the general population 15, heart failure 4, 35, myocardial infarction 36, and cerebrovascular events in female subjects after the CEA 17. In the course of our research, we sought to determine whether rs2274273 and rs17128183 genetic variants were related to previous CVI: having analyzed the data in our patient group, both overall and based on gender, we found no significant association. Our study did not have a prospective design, and the absence of cerebrovascular events within 6 months prior to CEA was defined as one of the inclusion criteria.
In the study correlating rs2274273 with circulating levels of gal‐3 mentioned above, the authors warned that their results should be interpreted with caution, seeing that the non‐synonymous SNP, rs4652, which demonstrates a strong linkage disequilibrium with rs2274273, is located within the epitope of the tracer antibody used for the galectin‐3 assay 20. In the same study, they also found that both genetic variants bore an association with LGALS‐3 gene expression levels. Consequently, while the association with circulating gal‐3 could be a false positive, rs2274273 can reasonably be assumed to have a functional effect. In the course of our research, we examined the entire haplotype block containing LGALS‐3 gene in RegulomeDB database in order to find other, potentially functional, SNPs. According to RegulomeDB, rs17128183 is likely to affect the binding of regulatory proteins and could therefore be related to the expression of the target gene. Moreover, this genetic variant demonstrates a high LD with both rs2274273 and rs4652. In the light of these findings, we proceeded to analyze the relative LGALS‐3 mRNA expression levels in carotid plaque tissue specimens with regard to rs2274273 and rs17128183 genotypes.
Using microarray analysis, gal‐3 was identified as an abundant transcript in atherosclerotic lesions obtained post‐CEA 37. We have also found LGALS‐3 mRNA to be abundantly expressed in human carotid plaque tissue specimens. In our study, carotid plaques of patients who carried the genotypes containing the rare alleles of both investigated variants were characterized by significantly higher levels of LGALS‐3 mRNA compared to the wild‐type homozygotes. De Boer et al. 20 have shown that a decrease in circulating gal‐3 levels was associated with the rare allele of rs2274273. A possible explanation for this discrepancy could be a false‐positive association established between a decrease in gal‐3 levels and the rare allele. On the other hand, it is known that mRNA and protein levels do not have to correlate well 38. In addition, the rare allele of rs4652 (which is in almost absolute LD with rs2274273) has been described as significantly associated with decreased cognitive function at old age 39, as well as with rheumatoid arthritis 23.
Previous research has shown that LGALS‐3 mRNA and gal‐3 protein levels were upregulated in unstable, rather than stable, regions of CEA specimens (n = 12), as classified based on macroscopic morphological features 7. In contrast, a recent study has questioned a positive interplay between gal‐3 and carotid plaque instability, suggesting that gal‐3 was not expressed only by inflammatory cells infiltrating the unstable plaque regions but also by VSMCs in more stable, macrocalcified plaques 18. The authors found that LGALS‐3 mRNA expression levels were slightly, but significantly, lower in less stable, microcalcificated carotid plaques (n = 18 per group) 18. In the course of our research, we examined approximately the same number of plaque samples ultrasonographically defined as hyperechogenic or hypoechogenic without finding a statistically significant difference in their LGALS‐3 mRNA expression levels. It is possible to assume that after plaque development gal‐3 protein functions as a protective agent. Previous research has shown that plaque tissue in patients on long‐term statin treatment was characterized by increased gal‐3 protein levels, which in turn could be related to plaque stabilization 40. Only three out of 39 patients whose carotid plaque tissue was used for analysis in this study were receiving statin therapy and their inclusion did not bias the result. Nevertheless, further studies involving a larger number of tissue samples and applying a more detailed histological classification of plaque types are required in order to fully elucidate the effects of gal‐3 on carotid plaque stability.
One of the major limitations of this study is a relatively small sample size. For this reason, our results should be viewed as exploratory rather than conclusive. It is well known that the power necessary to reject the null hypothesis can be provided only by large multicenter studies; we therefore consider the replication of our results indispensable. Even if a positive association was to be established using a larger cohort of subjects, we suggest, based on our current findings that the effects of rs2274273 and rs17128183 genetic variants on advanced carotid atherosclerosis would prove to be rather moderate. In this context, and given that increased circulating gal‐3 levels have been associated with heart failure and cardiovascular mortality, it would be of great interest to use Mendelian randomization approach in order to further elucidate whether these two variants have a true causal effects on carotid atherosclerosis through their impact on gal‐3 levels.
Another limitation of our study was the fact that we investigated only two SNPs in the vicinity of LGALS‐3 locus. This may be insufficient to evaluate the overall effect of the haplotype block containing LGALS‐3. In later studies, the tag SNPs that cover the genotypic variance of this haplotype block should be investigated in association with advanced CA and its complications. Also, the result obtained regarding the LGALS‐3 mRNA expression based on the genotypes should be validated using a larger sample size. Further studies investigating the interplay between genetic variants, gene expression, and circulating gal‐3 in cardiovascular diseases are needed.
Our exploratory results suggest that while rs2274273 and rs17128183 bear no association with the risk of developing advanced CA or CA‐related complications, these genetic variants are likely to affect LGALS‐3 expression levels. A lack of studies dedicated to either or both genetic variants precludes us from drawing definitive conclusions. Replication and validation of our results are required in order to elucidate the role played by rs2274273 and rs17128183 in advanced carotid atherosclerosis.
Acknowledgments
This study was funded by the Serbian Ministry of Education, Science and Technological Development, Grant No. III41028.
References
- 1. Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet 2006;367:1747–1757. [DOI] [PubMed] [Google Scholar]
- 2. Hansson GK, Robertson AK, Soderberg‐Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol 2006;1:297–329. [DOI] [PubMed] [Google Scholar]
- 3. Calvier L, Miana M, Reboul P, et al. Galectin‐3 mediates aldosterone‐induced vascular fibrosis. Arterioscler Thromb Vasc Biol 2013;33:67–75. [DOI] [PubMed] [Google Scholar]
- 4. van Kimmenade RR, Januzzi JL Jr, Ellinor PT, et al. Utility of aminoterminal pro‐brain natriuretic peptide, galectin‐3, and apelin for the evaluation of patients with acute heart failure. J Am Coll Cardiol 2006;48:1217–1224. [DOI] [PubMed] [Google Scholar]
- 5. Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA Guideline for the Management of Heart Failure. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013;62:e147–e239. [DOI] [PubMed] [Google Scholar]
- 6. Sano H, Hsu DK, Yu L, et al. Human galectin‐3 is a novel chemoattractant for monocytes and macrophages. J Immunol 2000;165:2156–2164. [DOI] [PubMed] [Google Scholar]
- 7. Papaspyridonos M, McNeill E, de Bono JP, et al. Galectin‐3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arterioscler Thromb Vasc Biol 2008;28:433–440. [DOI] [PubMed] [Google Scholar]
- 8. Katsuda S, Okada Y, Minamoto T, Oda Y, Matsui Y, Nakanishi I. Collagens in human atherosclerosis: Immunohistochemical analysis using collagen typespecific antibodies. Arterioscler Thromb 1992;12:494–502. [DOI] [PubMed] [Google Scholar]
- 9. Stringa E, Knäuper V, Murphy G, Gavrilovic J. Collagen degradation and plateletderived growth factor stimulate the migration of vascular smooth muscle cells. J Cell Sci 2000;113:2055–2064. [DOI] [PubMed] [Google Scholar]
- 10. Wesley RB II, Meng X, Godin D, Galis ZS. Extracellular matrix modulates macrophage functions characteristic to atheroma: Collagen type I enhances acquisition of resident macrophage traits by human peripheral blood monocytes in vitro. Arterioscler Thromb Vasc Biol 1998;18:432–440. [DOI] [PubMed] [Google Scholar]
- 11. Nachtigal M, Legrand A, Sasiela W, Watson SD, Fowler SD. Gene expression in macrophage foam cells of carrageenan‐induced granulomas in hypercholesterolemic rabbits. FASEB J 1994;8:A393. [Google Scholar]
- 12. Arar C, Gaudin JC, Capron L, Legrand A. Galectin‐3 gene (LGALS3) expression in experimental atherosclerosis and cultured smooth muscle cells. FEBS Lett 1998;430:307–311. [DOI] [PubMed] [Google Scholar]
- 13. Nachtigal M, Al‐Assaad Z, Mayer EP, Kim K, Monsigny M. Galectin‐3 expression in human atherosclerotic lesions. Am J Pathol 1998;152:1199–1208. [PMC free article] [PubMed] [Google Scholar]
- 14. MacKinnon AC, Liu X, Hadoke PW, Miller MR, Newby DE, Sethi T. Inhibition of galectin‐3 reduces atherosclerosis in apolipoprotein E‐deficient mice. Glycobiology 2013;23:654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Boer RA, van Veldhuisen DJ, Gansevoort RT, et al. The fibrosis marker galectin‐3 and outcome in the general population. J Intern Med 2012;272:55–64. [DOI] [PubMed] [Google Scholar]
- 16. Madrigal‐Matute J, Lindholt JS, Fernandez‐Garcia CE, et al. Galectin‐3, a Biomarker Linking Oxidative Stress and Inflammation With the Clinical Outcomes of Patients With Atherothrombosis. J Am Heart Assoc 2014;3:e000785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Edsfeldt A, Bengtsson E, Asciutto G, et al. High Plasma Levels of Galectin‐3 Are Associated with Increased Risk for Stroke after Carotid Endarterectomy. Cerebrovasc Dis 2016;41:199–203. [DOI] [PubMed] [Google Scholar]
- 18. Menini S, Iacobini C, Ricci C, et al. The galectin‐3/RAGE dyad modulates vascular osteogenesis in atherosclerosis. Cardiovasc Res 2013;100:472–480. [DOI] [PubMed] [Google Scholar]
- 19. Menon RP, Hughes RC. Determinants in the N‐terminal domains of galectin‐3 for secretion by a novel pathway circumventing the endoplasmic reticulum–Golgi complex. Eur J Biochem 1999;264:569–576. [DOI] [PubMed] [Google Scholar]
- 20. de Boer RA, Verweij N, van Veldhuisen DJ, et al. A genome‐wide association study of circulating galectin‐3. PLoS ONE 2012;7:e47385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012;22:1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen HJ, Zheng ZC, Yuan BQ, Liu Z, Jing J, Wang SS. The effect of galectin‐3 genetic variants on the susceptibility and prognosis of gliomas in a Chinese population. Neurosci Lett 2012;518:1–4. [DOI] [PubMed] [Google Scholar]
- 23. Hu CY, Chang SK, Wu CS, Tsai WI, Hsu PN. Galectin‐3 gene (LGALS3) +292C allele is a genetic predisposition factor for rheumatoid arthritis in Taiwan. Clin Rheumatol 2011;30:1227–1233. [DOI] [PubMed] [Google Scholar]
- 24. North American Symptomatic Carotid Endarterectomy Trial Collaborators . Beneficial effect of carotid endarterectomy in symptomatic patients with high‐grade carotid stenosis. N Engl J Med 1991;325:445–453. [DOI] [PubMed] [Google Scholar]
- 25. Gray‐Weale AC, Graham JC, Burnett JR, Byrne K, Lusby RJ. Carotid artery atheroma: Comparison of preoperative B‐mode ultrasound appearance with carotid endarterectomy specimen pathology. J Cardiovasc Surg (Torino) 1998;29:676–681. [PubMed] [Google Scholar]
- 26. Kunkel LM, Smith KD, Boyer SH, et al. Analysis of human Y chromosome‐specific reiterated DNA in chromosome variants. Proc Natl Acad Sci USA 1977;74:1245–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group‐wise comparison and statistical analysis of relative expression results in real‐time PCR. Nucleic Acids Res 2002;30:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dupont WD, Plummer WD Jr. Power and sample size calculations: A review and computer program. Control Clin Trials 1990;11:116–128. [DOI] [PubMed] [Google Scholar]
- 29. Ho JE, Liu C, Lyass A, et al. Galectin‐3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol 2012;60:1249–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. The 1000 Genomes Project Consortium . An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Salvi E, Kutalik Z, Glorioso N, et al. Genome‐wide association study using a high‐density single nucleotide polymorphism array and case‐control design identifies a novel essential hypertension susceptibility locus in the promoter region of endothelial NO synthase. Hypertension 2012;59:248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barrett JC, Clayton DG, Concannon P, et al. Type 1 Diabetes Genetics Consortium. Genome‐wide association study and meta‐analysis finds over 40 loci affect risk of type 1 diabetes. Nat Genet 2009;41:703–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Diabetes Genetics Initiative of Broad Institute of Harvard and MIT , Lund University , Novartis Institutes of BioMedical Research , et al. Genome‐wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007;316:1331–1336. [DOI] [PubMed] [Google Scholar]
- 34. Strachan DP, Rudnicka AR, Power C, Shepherd P, Fuller E. Lifecourse influences on health among British adults: Effects of region of residence in childhood and adulthood. Int J Epidemiol 2007;36:522–531. [DOI] [PubMed] [Google Scholar]
- 35. de Boer RA, Voors AA, Muntendam P, van Gilst WH, van Veldhuisen DJ. Galectin‐3: A novel mediator of heart failure development and progression. Eur J Heart Fail 2009;11:811–817. [DOI] [PubMed] [Google Scholar]
- 36. Sanchez‐Mas J, Lax A, Asensio‐Lopez MC, et al. Galectin‐3 expression in cardiac remodeling after myocardial infarction. Int J Cardiol 2014;172:e98–e101. [DOI] [PubMed] [Google Scholar]
- 37. Papaspyridonos M, Smith A, Burnand KG, et al. Novel candidate genes in unstable areas of human atherosclerotic plaques. Arterioscler Thromb Vasc Biol 2006;26:1837–1844. [DOI] [PubMed] [Google Scholar]
- 38. Maier T, Güell M, Serrano L. Correlation of mRNA and Protein in Complex Biological Samples. FEBS Lett 2009;583:3966–3973. [DOI] [PubMed] [Google Scholar]
- 39. Trompet S, Jukema W, Mooijaart SP, et al. Genetic variation in galectin‐3 gene associates with cognitive function at old age. Neurobiol Aging 2012;33:2232.e1–2232.e9. [DOI] [PubMed] [Google Scholar]
- 40. Kadoglou NPE, Sfyroeras GS, Spathis A, et al. Galectin‐3, carotid plaque vulnerability, and potential effects of statin therapy. Eur J Vasc Endovasc Surg 2015;49:4–9. [DOI] [PubMed] [Google Scholar]