

Abstract
Background
Duchenne and Becker muscular dystrophy (DMD/BMD) are X‐linked recessive disorders caused by mutation in dystrophin gene. We reported 3‐year clinic experience from a single hospital in Shanghai using multiplex ligation dependent probe amplification (MLPA) assay to detect DMD mutations.
Methods
Four hundred and fifty‐one males and 184 females, who were clinically diagnosed as DMD/BMD patients or carriers at our hospital's outpatient clinic, were collected and performed with MLPA to detect DMD gene mutations.
Results
Seventeen novel mutation points not reported in the Leiden Muscular Dystrophy pages were identified in this study. We found that the most frequent deletion spots ranged from exon45 to exon52, and exon2, exon19 were the two most frequently detected duplication spots.
Conclusion
The results of our study confirmed MLPA as an efficient clinical method for detecting DMD gene mutations in DMD/BMD patients. Single exon mutation detected by MLPA should be verified by other methods, and we should emphasize that only precise clinical molecular diagnosis can lead to the feasibility of prenatal diagnosis.
Keywords: DMD, BMD, MLPA, mutation, clinical diagnosis
INTRODUCTION
Duchenne and Becker muscular dystrophy (DMD/BMD) are X‐linked recessive disorders caused by mutation in the DMD gene. DMD [MIM 310200] is the most common muscle disorder in children, with a reported incidence of 1 in 3,500 liveborn males 1. BMD [MIM 300376] is caused by mutations in the same gene, but with later onset and mild progression, with a reported incidence of 1 in 18,500 liveborn males 2. Both dystrophies are characterized by progressive symmetric muscular weakness.
The DMD is the largest gene among the identified human genes located at Xp21, spanning 2.4 MB of a genomic sequence, and corresponding to about 0.1% of the total human genome 3, 4. The gene contains 79 exons and encoded a 14.6 kb mRNAs, which is expressed in the skeleton, muscles, and brain 5, 6. The majority of identified mutations are deletions, accounting for approximately 60∼65% of DMD and 85% of BMD mutations, and duplications have been observed in 5∼15% mutations 3, 4, 7. The remaining causes are small mutations such as indel, point mutation, or splicing mutation 4.
The diagnosis of DMD or BMD is based on the clinical severity of the patient. DMD refers to the more severe phenotype with delayed walking in early childhood, pseudohypertrophy of muscles, a sky‐high serum creatine kinase level, and progressive deterioration in muscle power, and most patients become confined to a wheelchair by the age of 12. BMD is a milder phenotype, most BMD patients remain ambulatory beyond the age of 12 8. The mutations in the DMD gene that disrupt the open reading frame (out of frame) will cause the premature abortion of the synthesis of the dystrophin protein, thus leading to the severe DMD phenotype. Some mutations are “in‐frame,” which conserved the reading frame, a truncated but mostly functional dystrophin is produced, and caused less‐severe BMD phenotype. According to the “reading frame” hypothesis, any mutation that does not affect the reading frame should produce a BMD phenotype 9. In many occasions, when the patient came to see a doctor, he was too young to be defined as either DMD or BMD—at that time the reading frame rule is ultra‐important—it will have guiding significance on the prediction of life expectancies for the patient.
Many methods are available for detecting deletions and duplications in the DMD gene. Southern blot using cDNA probe 10, 11, multiplex PCR (mPCR), quantitative mPCR 12, 13, and high‐resolution melting curve analysis 14 was reportedly used in detecting DMD mutations. The most commonly used method is mPCR that allows for 90∼95% of deletions to be detected in male patients. Since this method is only semiquantitative, neither duplications nor female carriers can be reliably identified 3, 15. Multiplex ligation dependent probe amplification (MLPA) is a technique that had first been reportedly used in DMD analysis in 2004 by Schwartz 16. Parallel comparison between mPCR and MLPA had been made and reported from many clinic labs, MLPA was reported more superior 13, 15, 17, 18. Since then MLPA has been increasingly used for deletion/duplication screening of the DMD gene in male patients and female carriers worldwide 15, 16, 19.
In this study, we reported genetic analysis of DMD gene by using MLPA method. We emphasized on several new mutations found in our patients using MLPA method, and also reported the importance of MLPA, which should be combined with other technologies to establish a reliable clinical diagnosis of DMD gene mutations.
MATERIALS AND METHODS
Sample Collection and DNA Extraction
This study included 451 male and 184 female patients seeking help at outpatient clinic of genetic counseling and prenatal diagnosis center—Department of Shanghai Xinhua Hospital—between 2009 and 2013. These 451 male patients were diagnosed with DMD/BMD based on typical clinical presentation, elevated creatine kinase levels (most over 10,000 U/l), or have a family history. Four hundred and fourteen of them were younger than 15 years at the time of diagnosis, and among these 184 female patients, 15 of them were clinically diagnosed as DMD/BMD, the others were asymptomatic females who had given birth to at least one DMD/BMD patient before or had close relatives diagnosed with DMD/BMD.
All subjects were given written informed consent. Genomic DNA was extracted from leukocytes of 2 ml peripheral blood sample using QIAamp DNA blood Mini Kit (QIAGEN, Hilden, Germany). The quality of the DNA was monitored by NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, US), the working density of the DNA was adjusted to 50∼70 ng/μl. Workflow of clinical diagnosis of DMD gene mutations in our lab is shown in Figure 1.
Figure 1.
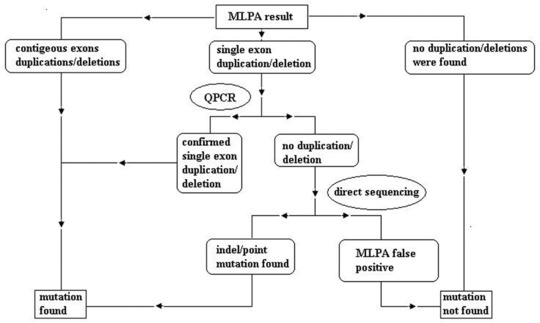
Workflow for clinical DMD mutation test.
Multiplex Ligation Dependent Probe Amplification
The MLPA reactions were performed to screen all the exons of DMD gene using two SALSA MLPA probe mix sets: P034 and P035 (MRC‐Holland, Amsterdam, Netherlands) 20. According to the manufacturer's instruction, 5 μl of the working DNA was used for each sample. At least two normal control samples were included in each reaction, for every six samples there was at least one normal control sample. The carboxyfluorescein (FAM)‐labeled MLPA PCR products were separated by capillary electrophoresis on ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, California, US). The sizes of the exon‐specific amplified fragments were identified according to their migration relative to the GeneScan Rox‐500 size standard (Applied Biosystems) using GeneMapper version 4.0 software. Relative amounts of PCR products were determined using Coffalyser software provided online by the manufacturer (www.mlpa.com).
Confirmation of an Ambiguous MLPA Result by PCR, Q‐PCR, and Direct Sequencing
The MLPA results from both male and female patients were initially assessed visually for the detection of deletion and duplication. Absence or duplication of the DMD‐specific peaks corresponding to two or more contagious exons were regarded as reliable, and no further investigation was performed. The absence/duplication of only one DMD peak was further examined by quantitative PCR (Q‐PCR) using SYBR® Premix Ex Taq™ (TaKaRa, China) on Applied Biosystems® 7500 Real‐Time PCR Systems. If the MLPA result and Q‐PCR result are not corroborated, a direct sequencing on ABI 3730xl DNA Analyzer will follow up. Both PCR and Q‐PCR primers used for each specific DMD exon were according to the published sequences in the Leiden database (http://www.dmd.nl). Sequences were compared against the NCBI reference sequence NG_012232.1.
RESULT
MLPA Mutation Detected in Male Patients
Among the 451 male patients, 245 (54.3%) were found to have exon(s) deletions, 48 (10.6%) were found to have exon(s) duplications, and through single exon PCR and sequencing, we found seven (1.5%) indels. The percentage of deletions and duplications was similar to the commonly quoted percentage in the literature, about 60∼65% and 5∼15%, respectively 3, 4, 7. The total detection rate is [(245 + 48 + 7)/425] 66.5%, which is close to the freshly released literature concerning Chinese population, 70.56% 21.
Within the 245 deletion pattern male patients, single‐exon deletions were the most frequent, representing up to (48/245) 19.6%. The most frequent deletion spots ranged from exon45 to exon52, which have been detected over 70 times among these 245 patients; the most frequent deletion exon was exon49, which have been detected 116 times (47%) (Fig. 2).
Figure 2.
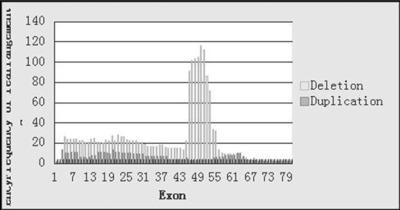
Deletion and duplication frequency of each exon in the DMD gene.
In the 48 duplication pattern male patients, single‐exon duplication was also the most frequent, which accounted for (13/48) 27%. Exon2 and exon19 were the two most frequently detected duplication spots, both detected 13 times among these 48 duplication pattern male patients (27%) (Fig. 2). Seven indels were detected through follow‐up sequencing in which single‐exon mutations were detected by MLPA.
MLPA Mutation Detected in Female Patients
Among the 184 female patients, 15 were symptomatic female patients, and the rest were potential asymptomatic female carriers of DMD. For the 15 female symptomatic patients, three were detected with heterozygous deletion.
Among the 169 potential female carriers, 152 had history of giving birth to known DMD mutation patient or having close relations with known DMD mutation, 17 of them had history of giving birth to an untested DMD patient.
Referring to the 152 females who had given birth to identified DMD mutation patients, 75 (49.3%) showed the same mutation as the identified patients and 77 (50.7%) showed no mutations through MLPA test. Among the 17 females who had given birth to untested DMD patients, six were found to have deletions in DMD, 11 showed no mutation through MLPA test.
Novel Mutations Found Through MLPA Test
Seventeen novel mutation points (not reported in the Leiden Muscular Dystrophy pages, http://www.dmd.nl) were identified in this study, the mutation patterns are listed in Table 1. The MLPA results were reproducible, the repeated MLPA analysis yielded identical findings in all subjects.
Table 1.
Novel Mutations and Patients Information List
| Case no. | Age (years/months) | Sex | Mut pat | Involved exon(s) | CK (U/l) | Clinical information | Clinical diagnosis | Reading frame |
|---|---|---|---|---|---|---|---|---|
| 1 | 14 years | M | Del | 14∼51 | 11,098 | GMP; difficulty in walking, easy to fall down; at age of 13 years, cannot stand up after crouch; Gower+ | BMD | IF |
| 2 | 3 years 4 months | M | Del | 14∼27 | Up | Mild GMP walk, climbing stairs, running, jumping all ok at the time of test | BMD | IF |
| 3 | 9 years | M | Del | 31∼45 | Up | Lower limbs are faint | DMD | OF |
| 4 | 4 years | M | Del | 8∼30 | 14,046 | GMP; difficulty in climbing stairs | DMD | OF |
| 5 | 3 years | M | Del | 14∼36 | Up | NK | BMD | IF |
| 6 | 2 years | M | Del | 47∼64 | 6,543 | NK | BMD/DMD too young | OF |
| 7 | 10 years | M | Del | 5∼9 | >10,000 | GMP; winged shoulders | BMD | IF |
| 8 | 6 years | M | Del | 5∼15 | NK | NK | BMD | IF |
| 9 | 3 years 6 months | M | Del | 42 + 45∼50 | Up | GMP; difficulty in climbing stairs; not have much strength from very early age; Gower+ | DMD | OF |
| 10 | 7 months | M | Del | 3∼39 | 9,569 | GMP; MD | BMD/DMD too young | IF |
| 11 | 3 years | M | Dup | 2∼25 | NK | NK | DMD | OF |
| 12 | 5 years | M | Dup | 14∼37 | >10,000 | Difficulty in walking, easy to fall down | DMD | IF |
| 13 | 7 years | M | Dup | 14∼37 | NK | GMP; hypotonic; difficulty in walking, easy to fall down | DMD | IF |
| 14 | 5 years | M | Dup | Dp427m + Dp427c + 45∼55 | 23,430 | GMP; MD; difficulty in climbing stairs; cannot crouch | DMD | NK |
| 15 | 10 years and 32 years | M&F* | Dup | 53∼55 + 57∼59 | NK | Start walking at 26 months, speak at 3+ years; muscle weakness; cannot crouch | DMD/carrier | OF |
| 16 | 4 years 8 months | M | Indel | 50: c.7302_7309del | 18,431 | GMP | DMD | OF |
| 17 | 8 years | M | Indel | 44: c.6385_6388del | NK | Have difficulty in walking | DMD | OF |
| 18 | 2 years 7 months and 27 years | M&F* | Indel | 34: c.4750_4751del | NK | Family history of DMD | DMD/carrier | OF |
Mut pat, mutation pattern; F*, mother of the male patient; M, male; GMP, gastrocnemius muscle pseudohypertrophy; MD, myogenic damage; IF, in‐frame; OF, out of frame; NK, not known; age, age at test; Gower+, a clinical sign for use of arms and hands from a sitting position to a standing position by grasping and pulling on body parts from the knees to hips until they are in an erect position.
Phenotype and Reading Frame Check
All the deletion(s) and duplication(s) we found underwent reading frame check on http://www.dmd.nl. Most of the mutations fitted the reading frame rules (not including those patients who were too young to be classified as DMD or BMD). But in several cases, the reading frame result did not accord to clinical diagnosis. We listed those phenotype and reading frame rule discrepancy patients in Table 2.
Table 2.
Phenotype and Reading Frame Rule Discrepancy Patients List
| Case no. | Age (years/months) | Sex | Mut pat | Involved exon(s) | CK (U/l) | Clinical information | Clinical phenotype | Reading frame |
|---|---|---|---|---|---|---|---|---|
| 1 | 3 years | M | Del | 10∼13 | NK | GMP; difficulty in climbing stairs; difficulty in standing up after crouch | DMD | IF |
| 2 | 18 years | M | Del | 45∼54 | 15,100 | GMP; cannot walk | Mild DMD | OF |
| 3 | 19 years | M | Del | 46∼52 | 1,297 | Difficulty in walking; at 23 years, on the wheelchair; GMP; MD | Mild DMD | OF |
| 4 | 3 years 7 months | M | Dup | 3∼4 | 9,763 | Difficulty in climbing stairs; GMP; MD | DMD | IF |
| 5 | 4 years 5 months | M | Dup | 19∼44 | 958 | Difficulty in walking; on wheelchair at age 12; GMP | Mild DMD | IF |
| 6 | 7 years | M | Dup | 45∼57 | NK | Difficulty in climbing stairs; cannot run; GMP; MD | DMD | IF |
| 7* | 5 years | M | Dup | 14∼37 | >10,000 | Difficulty in walking, easy to fall down | DMD | IF |
| 8* | 7 years | M | Dup | 14∼37 | NK | GMP; hypotonic; difficulty in walking, easy to fall down | DMD | IF |
DISCUSSION
In this study, we agreed with several other studies that MLPA technology has great advantage in detecting both deletion and duplication mutations of DMD gene. Our results indicated that the detection rate of MLPA is 66.45%, same as former reported. Through MLPA test, we found many novel mutations in DMD gene that caused DMD/BMD. If we used mPCR method to detect the patients, all the duplications and indel mutations could be missed. So, we strongly supported that MLPA should be considered as an initial clinical test for suspected DMD/BMD patients 15, 19, 21.
There were still 151 male patients (33.5%) among whom MLPA did not detect a mutation. For these patients, whole gene sequencing can be further used for DMD gene defects detection. Next‐generation sequencing technology (NGST) seems to be a very promising technology when their cost could come down to a suitable level for clinical test 22. In our lab, we are working on sequencing these patients using NGST, the result will be published later.
Among those women who had given birth to a mutation‐identified patient, 49.3% of them carried the same mutation and 50.7% did not carry that mutation. For those in whom no mutation had been found, there are two probable explanations: either the proband's mutation is de novo or there is a germline mosaic that exists. In our study, we eliminated the percentage of possible germline mosaic, which is estimated about 4.3% among those nonmutation mothers 23, 24; we got 46.6% de novo mutation increase in our patients. This rate is much higher than reported human gene de novo mutation rates, which is around one‐third 25, 26. So, we assumed that DMD might have some other mutation mechanism that makes the de novo mutation rate much higher. This needs to be further explored and more follow‐up data collection is required.
In our study we found that the most frequent deletion exon was exon49, it is not the same as Leiden‐reported exon47, but exon47 is also a very frequent deletion exon in our study. The most frequent duplication exons were exon2 and exon19. These three kinds of mutations add up to 47% of deletions and 54% of duplications found. As 46.6% of mutations we found in this study were de novo, we thought that these most frequent exons could be a good target point for noninvasive prenatal screening test for DMD del/dup mutation screening.
The reading frame rule is quite helpful in clinical evaluation of the patients at an early age. Our result shows that most of the time, the reading frame rule is correct; physicians can give explanation of the disease process to the patient based on it. But sometimes, there will be discrepancy between the clinical phenotype and expectations based on the reading frame rule. We now classify the discrepancy cases in Table 2 into three groups, and give out conceivable reasons on how this might happened. (1) Four different types of “in‐frame” duplications with clinical DMD phenotype. Duplication is harder to define than deletion; without sequencing, we are not sure where exactly do these extra exons insert. We assumed that the duplications might interrupt the translation of the dystrophin. (2) One “in‐frame” deletion led to DMD phenotype. This could be due to small mutation not detectable by MLPA, which occurred simultaneously with the deletion. If two jumping deletions can occur in one patient (Table 1, case 9), then this can also happen. (3) Two deletions that we found had “out‐of‐frame” change and got milder DMD phenotype. This could be caused by a nature‐generated frame‐restoring exon(s) skipping. The patient “exon 45∼54 deletions” could be restored by skipping exon44. The other “exon 46∼52 deletions” patient, 45 + 51 double skip exon could restore the partial function of dystrophin. This nature self‐restore mechanism detected in real patients gave the foundation support of the most promising gene therapy of DMD 27. In conclusion, final classification of DMD or BMD is determined by clinical phenotype, reading frame rule is a useful tool for clinical consultation; discrepancy between these two indicated a potential change in the expression of the mutated DMD.
In China, all the physicians know that DMD/BMD is a genetic neuromuscular disease, but not all of them are aware that only precise molecular diagnosis could permit future prenatal diagnosis. Sometimes when patients seeking help at our prenatal diagnosis center followed their local physicians’ direction, they have already carried a gestation over 16 weeks. And that will give an extra anxiety to the pregnant women because they have to wait extra time to get precise molecular diagnosis first. And in some cases no prenatal diagnosis is allowed to be processed, and then the pregnant women could be depressed. So here, we strongly suggest that when there are likely DMD/BMD patients, physicians better suggest the patients to take a precise molecular test first.
Although in China, people are more and more aware of the importance of prenatal screening, till now only Down's screening is officially recommended by the government and knowledged by the population. There are so many genetic diseases that could be avoided by prenatal screening when clinical technology is well established. Now for DMD, technology is ready—maybe in future in China—DMD/BMD could be prioritized as a prenatal screening project as in other countries 18, 28.
REFERENCES
- 1. Emery AE. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul Disord 1991;1(1):12–29. [DOI] [PubMed] [Google Scholar]
- 2. Bushby KM, Thambyayah M, Gardner‐Medwin D. Prevalence and incidence of Becker muscular dystrophy. Lancet 1991;337(8748):1022–1024. [DOI] [PubMed] [Google Scholar]
- 3. Prior TW, Bridgeman SJ. Experience and strategy for the molecular testing of Duchenne muscular dystrophy. J Mol Diagn 2005;7(3):317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2(12):731–740. [DOI] [PubMed] [Google Scholar]
- 5. Hoffman EP, Brown RH, Jr. , Kunkel LM. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987;51(6):919–928. [DOI] [PubMed] [Google Scholar]
- 6. Nobile C, Marchi J. A refined restriction map of YAC clones spanning the entire human dystrophin gene. Mamm Genome 1994;5(9):566–571. [DOI] [PubMed] [Google Scholar]
- 7. Hu XY, Ray PN, Murphy EG, Thompson MW, Worton RG. Duplicational mutation at the Duchenne muscular dystrophy locus: Its frequency, distribution, origin, and phenotypegenotype correlation. Am J Hum Genet 1990;46(4):682–695. [PMC free article] [PubMed] [Google Scholar]
- 8. Lo IF, Lai KK, Tong TM, Lam ST. A different spectrum of DMD gene mutations in local Chinese patients with Duchenne/Becker muscular dystrophy. Chin Med J (Engl) 2006;119(13):1079–1087. [PubMed] [Google Scholar]
- 9. Monaco AP, Bertelson CJ, Liechti‐Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988;2(1):90–95. [DOI] [PubMed] [Google Scholar]
- 10. Darras BT, Blattner P, Harper JF, Spiro AJ, Alter S, Francke U. Intragenic deletions in 21 Duchenne muscular dystrophy (DMD)/Becker muscular dystrophy (BMD) families studied with the dystrophin cDNA: Location of breakpoints on HindIII and BglII exon‐containing fragment maps, meiotic and mitotic origin of the mutations. Am J Hum Genet 1988;43(5):620–629. [PMC free article] [PubMed] [Google Scholar]
- 11. Hiraishi Y, Kato S, Ishihara T, Takano T. Quantitative Southern blot analysis in the dystrophin gene of Japanese patients with Duchenne or Becker muscular dystrophy: A high frequency of duplications. J Med Genet 1992;29(12):897–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ioannou P, Christopoulos G, Panayides K, Kleanthous M, Middleton L. Detection of Duchenne and Becker muscular dystrophy carriers by quantitative multiplex polymerase chain reaction analysis. Neurology 1992;42(9):1783–1790. [DOI] [PubMed] [Google Scholar]
- 13. Madania A, Zarzour H, Jarjour RA, Ghoury I. Combination of conventional multiplex PCR and quantitative real‐time PCR detects large rearrangements in the dystrophin gene in 59% of Syrian DMD/BMD patients. Clin Biochem 2010;43(10–11):836–842. [DOI] [PubMed] [Google Scholar]
- 14. Almomani R, van der Stoep N, Bakker E, den Dunnen JT, Breuning MH, Ginjaar IB. Rapid and cost effective detection of small mutations in the DMD gene by high resolution melting curve analysis. Neuromuscul Disord 2009;19(6):383–390. [DOI] [PubMed] [Google Scholar]
- 15. Sansovic I, Barisic I, Dumic K. Improved detection of deletions and duplications in the DMD gene using the multiplex ligation‐dependent probe amplification (MLPA) method. Biochem Genet 2013;51(3–4):189–201. [DOI] [PubMed] [Google Scholar]
- 16. Schwartz M, Duno M. Improved molecular diagnosis of dystrophin gene mutations using the multiplex ligation‐dependent probe amplification method. Genet Test 2004;8(4):361–367. [DOI] [PubMed] [Google Scholar]
- 17. Itto AB, Hamzi K, Bellayou H, Itri M, Slassi I, Nadifi S. Evolution of molecular diagnosis of Duchenne muscular dystrophy. J Mol Neurosci 2013;50(2):314–316. [DOI] [PubMed] [Google Scholar]
- 18. Verma PK, Dalal A, Mittal B, Phadke SR. Utility of MLPA in mutation analysis and carrier detection for Duchenne muscular dystrophy. Indian J Hum Genet 2012;18(1):91–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Janssen B, Hartmann C, Scholz V, Jauch A, Zschocke J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: Potential and pitfalls. Neurogenetics 2005;6(1):29–35. [DOI] [PubMed] [Google Scholar]
- 20. Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation‐dependent probe amplification. Nucleic Acids Res 2002;30(12):e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang J, Li SY, Li YQ, et al. MLPA‐based genotype‐phenotype analysis in 1053 Chinese patients with DMD/BMD. BMC Med Genet 2013;14:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wei X, Dai Y, Yu P, et al. Targeted next‐generation sequencing as a comprehensive test for patients with and female carriers of DMD/BMD: A multi‐population diagnostic study. Eur J Hum Genet 2014;22(1):110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Helderman‐van den Enden AT, de Jong R, den Dunnen JT, et al. Recurrence risk due to germ line mosaicism: Duchenne and Becker muscular dystrophy. Clin Genet 2009;75(5):465–472. [DOI] [PubMed] [Google Scholar]
- 24. Helderman‐van den Enden AT, van den Bergen JC, Breuning MH, et al. Duchenne/Becker muscular dystrophy in the family: Have potential carriers been tested at a molecular level? Clin Genet 2011;79(3):236–242. [DOI] [PubMed] [Google Scholar]
- 25. Haldane JB. The rate of spontaneous mutation of a human gene. 1935. J Genet 2004;83(3):235–244. [DOI] [PubMed] [Google Scholar]
- 26. Nachman MW. Haldane and the first estimates of the human mutation rate. J Genet 2004;83(3):231–233. [DOI] [PubMed] [Google Scholar]
- 27. Aartsma‐Rus AvOGJ. Antisense‐mediated exon skipping: A versatile tool with therapeutic and research applications. RNA 2007;13(10):1609–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Helderman‐van den Enden AT, Madan K, Breuning MH, et al. An urgent need for a change in policy revealed by a study on prenatal testing for Duchenne muscular dystrophy. Eur J Hum Genet 2013;21(1):21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]