

Abstract
Background
In case of clinical analysis, especially in blood lead (Pb‐B) and cadmium (Cd‐B) determination, the accuracy and precision of the method are crucial. The objective of this article is to present a simple and useful method for Pb‐B and Cd‐B determination using ICP‐MS (inductively coupled plasma‐mass spectrometry) as well as GF‐AAS (graphite furnace‐atomic absorption spectrometry).
Methods
The principle of the method is based on the deproteinization of blood samples by addition of 5% nitric acid that eliminates the presence of the protein in the samples, thereby excluding the influence of the organic matrix on the result determinations. A comparison of the two techniques ICP‐MS and GF‐AAS was established for Pb and Cd determinations in the same 40 blood samples collected from lead workers.
Results
The results showed that validation parameters for ICP‐MS and GF‐AAS were similar, however better for ICP‐MS for Pb‐B determinations. The detection limit (3×SD) for Pb‐B determinations for ICP‐MS and GF‐AAS was, respectively, 0.16 and 1.0 μg/l, and for Cd‐B it was, respectively, 0.08 and 0.02 μg/l. Correlation coefficients (rs) for comparable Pb‐B and Cd‐B determinations, using these two techniques, showed very good statistically significant correlations and were r = 0.9988, P < 0.0001 for Pb‐B and r = 0.9949, P < 0.0001 for Cd‐B.
Conclusions
The obtained results indicate that the method of deproteinization of blood samples is still the best way to eliminate spectral interferences and influence of the organic matter. The elaborated method is especially dedicated to clinical laboratories and determined low concentrations of lead and cadmium in biological samples.
Keywords: biological monitoring analysis, determination, lead, cadmium, blood, environmental, occupational exposure
INTRODUCTION
Biological monitoring plays a complementary role in occupational hygiene and has been an important tool of health surveillance in the European countries. Measurements for biological monitoring have been recognized to be very important for the protection and promotion of health for environmentally exposed people and workers exposed to hazardous chemicals 1, 2, 3, 4. In lead exposure, biological monitoring is based on measurements of blood lead (Pb‐B) levels and the concentration of one of the markers of early health effects of exposure: delta‐aminolevulinic acid in urine or zinc protoporphyrin in blood 5, 6. In cadmium (Cd) exposure, concentration of Cd in blood (Cd‐B) and Cd in urine (Cd‐U) are measured.
In Poland, the Regulation of the Minister of Health and Welfare made it obligatory to perform the Pb‐B determinations for workers’ occupational exposure to lead 7, 8. Biological Exposure Index (BEI) has been developed as the main guideline in workers’ protection against adverse health effects 6, 7, 8. The value of 500 μg/l Pb‐B has been adopted for workers’ occupational exposure to lead 6, 7, 8. In accordance with EU Directive 97/24/EC, determining Pb‐B is compulsory throughout the European Union for employees exposed to lead 9. For workers’ occupational exposure to Cd, the BEI of 5 μg/l Cd‐B has been adopted 10.
In case of clinical analysis, especially in Pb‐B and Cd‐B determination, the accuracy and precision of the method are crucial. Decision on the conditions of exposure or diagnosis of health status of the people occupationally and environmentally exposed may be influenced by the used method.
Until recently, the graphite furnace‐atomic absorption spectrometry (GF‐AAS) technique as a mono‐elemental method of metal determination has been extensively used for Pb and Cd analysis in many laboratories. High sensitivity and selectivity for specific elements allowed using this technique wherever precision determination was important, especially in clinical analysis.
Presently, the most powerful technique applied for measurements of trace and ultratrace elements in biological matrices has been inductively coupled plasma‐mass spectrometry (ICP‐MS). This technique enables fast and parallel multi‐element determination with many advantages such as less spectral interference, wide dynamic range, and lower detection limit (DL) than GF‐AAS and the ability to analyze isotopic ratio.
Many authors have described methods for metal determination in blood using ICP‐MS technique. The blood matrix requires extensive sample pretreatment prior to analysis. Usually, organic matrix (e.g., proteins) presented in blood sample should be previously removed by the digestion of samples with mineral acids 11, 12, 13.
A much‐easier approach was simple dilution of the sample with an appropriate diluent. Many authors, as a pretreatment method, used sample dilution with, for example, purified water, acid (nitric or hydrochloric), Triton X‐100, and butanol 13, 14; or ammonia solution with EDTA and with Triton X‐100 15, 16; or ammonia solution with Triton X‐100 17; or nitric acid combined with Triton X‐100 18. Fifty‐fold dilution with a TMAH/HCl mixture and addition of Rh as an internal standard 14 was also possible. Some of authors also used different internal standards, for example, Ir 16, 18.
The analysis after dilution was possible, however in the case of blood samples the simple dilution was insufficient to remove organic substances and the matrix influence on results of determinations. This was a serious problem with serial and routine determinations of blood samples.
The potential risk of contamination during blood‐sample collection and treatment requires contamination‐free and simple analytical procedures that have to be optimized and applied in the context of human biomonitoring, where the testing of a large number of samples was usually involved.
One of those underestimated methods for determinations of Pb‐B and Cd‐B is Stoeppler and Brandt method 19, 20 based on deproteinization of blood by nitric acid. This method can be successfully used for determinations with ICP‐MS and GF‐AAS techniques. The validation of this method is described and the analytical performance is demonstrated by internal and external quality assurance. This simple, accurate, and precise method, reducing manipulation and thus exogenous contamination, allows fast determination of Pb and Cd in blood samples, which is particularly important for laboratories performing clinical analysis.
The method validation presented in this article, with regard to the external and internal quality control based on the results of the Shewhart chart and interpretation of the results according to the Wheeler criteria, may be a model to develop other analytical methods. The aim of this work is to present a simple and useful method for Pb‐B and Cd‐B determination using ICP‐MS as well as GF‐AAS.
MATERIALS AND METHODS
Reagents and Chemicals
The reagents and chemicals used included 5% and 20% HNO3 (Nitric Acid‐Merck, Darmstadt, Germany), deionized water (Millipore, Milli‐Q‐Elix3, ≤18 MΩ), element stock solutions—Cd 1000 μg/l AAS/ICP and Lead 1000 μg/l AAS/ICP, and Trace Metal Analysis Standard, “Baker Instra‐Analyzed.”
Certified reference material (CRM) was BCR 634 Lyophilised Human Blood (European Community Measurements and Testing, Institute for Reference Materials and Measurements (IRMM), Belgium), RM SeronormTM, Trace Elements Whole Blood (Sero AS, Norway, Human‐based matrix), and Recipe—ClinChek (Munich, Germany): Whole Blood Control—level I and level II.
Instrumentation
ICP‐MS Perkin Elmer ELAN DRC‐e (Sciex, Wellesley, MA) was used to optimize and validate method of Pb and Cd determination in blood samples. ICP‐MS system was equipped with Meinhard concentric glass nebulizer and Cyclonic spray chamber for sample introduction and nickel‐based interface cones (skimmer and sampler). Argon was employed as the plasma gas of 99.9995% purity (Linde Group, Poland). Details of operation instrumental conditions and measurement parameters are presented in Table 1. Measurements were made in standard mode without Dynamic Reaction Cell (DRC).
Table 1.
Operating Conditions for Pb‐B and Cd‐B Determinations Using ICP‐MS Perkin Elmer ELAN DRC‐e
| Parameter | Setting |
|---|---|
| RF power | 1,075 Kw |
| Nebulizer gas flow | Typically 0.8–1.0 min−1 |
| Plasma gas flow | 15 l min−1 |
| Auxiliary gas flow | 1.2 l min−1 |
| Cones | Ni |
| Detector mode | Dual |
| Measurement units | Counts per second |
| Autolens | On |
| Mass monitored | 204Pb+206Pb+207Pb+208Pb;114Cd |
| Blank subtraction | Before standard |
| Curve type | Linear throw zero |
| Sample units | μg/l−1 |
| Sweeps/reading | 20 |
| Readings/replicate | 1 |
| replicates | 3 |
| Dwell time | 50 |
| Integrator time | 1,000 |
| Correction equations Cd | 114Cd‐(0.027250 × 118Sn) |
GF‐AAS Perkin Elmer Analyst 600 (Shelton, CT) with a transversely heated graphite atomizer (THGA with L'vov platform) eliminated most interferences and longitudinal Zeeman‐effect correction for improved accuracy and detection. The instrument settings and furnace THGA heating program for Pb‐B and Cd‐B determination are summarized in Tables 2 and 3.
Table 2.
Operating Conditions for Pb‐B and Cd‐B Determinations Using on GF‐AAS, Perkin Elmer AAnalyst 600
| Parameters | Pb | Cd |
|---|---|---|
| Wavelength | 283.3 nm | 228.8 nm |
| Lamp current | 10 mA | 4 mA |
| Slit width | 0.7 nm | |
| Signal type | Zeeman AA | |
| Signal measurements | Peak area | |
| Standard, sample volume | 20 μl | |
| Standard, sample replication | 2 | |
| Read time | 3.0 sec | |
| Injection temperature | 100°C | |
| Argon flow rate | 0.25 l min‐1 | |
| Tube | THGA with L'vov platform | |
Table 3.
Furnace (THGA) Parameters for Pb‐B and Cd‐B Determinations
| Temperature (°C) | Ramp (°C/s) | ||||||
|---|---|---|---|---|---|---|---|
| Step | Pb‐B | Cd‐B | Pb‐B | Cd‐B | Time (s) | Gas flow rate (ml/min) | Command |
| 1. | 110 | 110 | 3.0 | 1.0 | 20.0 | 250 | |
| 2. | 170 | 170 | 10.0 | 10.0 | 30.0 | 250 | |
| 3. | 500 | 250 | 5.0 | 30.0 | 30.0 | 250 | |
| 4. | 1,350 | 1,450 | 0 | 0 | 3.0 | 0 | TC, RD |
| 5. | 2,400 | 2,400 | 1.0 | 1.0 | 2.0 | 250 | |
TC, temperature control; RD, read.
Other equipment used during the sample preparation included Venosafe plastic tubes (Li‐Heparine, Terumo, Europe), Polypropylene tube (volume 4 ml, Equimed, Poland), Mixer (Maxi Mix II; Barnstead/Thermolyne; Dubuque, IA 52001), and Centrifuge (Centrifuge GS‐15 R, Beckman Inc., Palo Alto, CA).
Sample Collection and Preparation
The comparison of two techniques for Pb‐B and Cd‐B determination was based on 40 blood samples collected from workers occupationally exposed to lead, employees in battery‐manufacturing plant.
A total of 4 ml blood sample was taken by venipuncture using Venosafe vacuum tubes certified for metal determination. Immediately after sampling, the blood had to be thoroughly mixed to avoid clotting during storage. The samples before analysis were stored in refrigerator at <4°C, in a case of a long‐time storage they should be placed in freezer at a temperature <20°C.
To compare the two techniques, our own modification of Stoeppler and Brandt method 21 was used. The principle of the method is based on deproteinization of blood samples by nitric acid. A quantity of 1.6 ml 5% HNO3 in ultrapure quality was poured into a polypropylene tube (volume 4 ml) and then a 0.4 ml venous blood sample was added to avoid premature precipitation. Immediately after blood sampling, the tube was vigorously shaken using mixer. The samples should be left for at least 1 h at room temperature and next centrifuged for 15 min /speed 11500 at 4°C. After centrifugation, the supernatant was transferred to autosampler tubes and Pb and Cd were analyzed by GF‐AAS or ICP‐MS. All vessels used during the analysis were pre‐washed with 20% nitric acid and deionized water. Pipet tips were swilled once with 5% HNO3 and then two times with deionized water.
For metals analyzed by ICP‐MS, higher dilution factor was used. Blood specimens (0.4 ml blood + 1.6 ml 5% HNO3) were additionally diluted (1+4) with deionized water.
The concentrations of samples were calculated in relation to calibration curve prepared by adding appropriate volumes of metals aqueous standard solutions to bovine/cow's blood.
Calibration
The GF‐AAS and ICP‐MS methods were calibrated with matrix‐matched calibration standards prepared in cow's blood by addition of Pb and Cd aqueous standard solution. The demanded aqueous standard solutions were prepared by appropriate dilution single‐element stock solutions: Cd and Pb.
The calibration working range was adjusted to the concentrations expected in the environmental and occupational samples. The two elements’ calibration curve included five points of different concentrations of lead and Cd, and blood blank and reagent blank were prepared. Before making calibration, cow's blood was tested for metal content to ensure that endogenous levels of target analytes were low. A few series of calibration curves on bovine blood were prepared before and kept in freezer (<200) till analysis.
Calibrations and RMs samples were treated in the same way as investigated samples. The same values of samples and nitric acid were used for pretreatment of blood samples.
Internal Quality Control
Many RMs are commercially available and many of them can be used in analysis of blood samples. In this work for internal quality control CRMs BCR 634 Lyophilised Human Blood, 1.4 ± 0.4 μg Cd/l and 46 ± 5 μg Pb/l, were used. Additionally, analytical procedure was verified by applying RMs Seronorm, Trace Elements Whole Blood—6.0 ± 0.2 μg Cd/l and 396 ± 65 μg Pb/l—as well as Recipe—ClinChek: Whole Blood Control: 1.3 μg Cd/l (range 0.97 ÷ 1.7 μg/l) and 78.0 μg Pb/l (level I, range 64 ÷ 84 μg/l) and level II 4.2 μg Cd/l (range 3.3 ÷ 5.1 μg/l) and 290.0 μg Pb/l (range 246 ÷ 334 μg/l).
Prior to analysis the commercial control materials were prepared according to the recommendation of the manufacturer and treated in the same way as examined sample. Control materials were determined after reconstitution of the freeze‐dried material. For routine analysis, control samples were always analyzed after calibration and after every 20th sample.
External Quality Control
As regards the Pb‐B and Cd‐B determinations, the method has been tested in two external quality assessment schemes; first organized by the U.K National External Quality Assessment Servis UK NEQAS (Wolfson EQA Laboratory) and the second by the German External Quality Assessment Scheme (G‐EQUAS), according to the Guidelines of the German Federal Medical Council. The aim of both external quality control schemes was to assess the laboratories’ competence to determine selected metals in two (or three) different concentrations for subjects’ environmental and occupational exposure.
Accreditation
Laboratory has been accredited according to PN/EN ISO/IEC 17025 to perform Cd‐B and Pb‐B determinations using GF‐AAS and ICP‐MS in the fields of occupational medicine and environmental health.
RESULTS AND DISCUSSION
Validation Parameters
Validation method is a routine process that demonstrates that analytical procedures are suitable for intended use. The analytical method is described by the validation parameters 22, 23.
Validation parameters were assessed for ICP‐MS as well as GF‐AAS techniques. According to Polish/European standard PN‐EN ISO/IEC 17025 24, validations parameters such as linearity, working range, DL, limit of quantitation (QL), bias, recovery, intermediate precision, and uncertainty (as an expended uncertainty) were calculated.
DL and QL were calculated as three times and six times of standard deviation for reagent blank 25 The DL of applied method for Pb and Cd was as follows: for ICP‐MS 0.16 and 0.08 μg/l and for GF‐AAS 1.0 and 0.02 μg/l. The results indicated that DL for Pb‐B determination using ICP‐MS was about six times lower (0.16 vs. 1.00) and inversely for Cd‐B determination four times higher (0.08 vs. 0.02). QL for ICP‐MS was 0.32 μg/l (Pb‐B) and 0.16 μg/l (Cd‐B), for GF‐AAS 2.0 μg/l and 0.04 μg/l, respectively. However, very low DL was suitable for the determination of even very low concentrations of Pb and Cd in blood of the people environmental exposure. Computed DL for ICP‐MS for Pb and Cd was in line with values reported by other authors and amounted 0.09 ÷ 0.2 μg/l for Cd and 0.5 ÷ 1.0 μg/l for Pb 18, 26, 27.
In whole the calibration range signal should be proportional to concentration of analyte (r = 0.999). Calibration range for ICP‐MS for Pb‐B was 50 to 500 μg/l where slope (a) ranged within 209.0 ÷ 291.7, (r) 0.9998 ÷ 0.9999, and intercept (b) = 0. For Cd‐B range fits between 1 ÷ 8 μg/l and (a) 166.9 ÷196.3; (r) 0.9995 ÷ 0.9999. For GF‐AAS for Pb‐B slope (a) ranged within 0.00111 ÷ 0.00160, (r) 0.9993 ÷ 0.9996; and for Cd‐B (a) 0.0194 ÷ 0.0235, (r) 0.9993 ÷ 0.9999, (b) = 0.
The working range of this method was designed by lowest and highest concentration of metal in the sample for which it has been established that the procedure has an appropriate level of precision, accuracy, and linearity. The working range of analytical methods was appointed by QL and highest concentration in calibration range. For ICP‐MS technique working range for Pb‐B and Cd‐B was, respectively, 0.32 to 500 μg/l, 0.16 to 8.0 μg/l and for GF‐AAS 2.0 to 500 μg/l, 0.04 to 8.0 μg/l.
Intermediate precision within working range for Pb‐B determination using ICP‐MS and GF‐AAS was on the same level and amounted 5.2% and 5.0%, respectively; however, for Cd‐B it was two times lower than for GF‐AAS (3.1%) and ICP‐MS (7.5%).
Currently, the most important factor is uncertainty, which in contrast to validation parameters, characterized the result and not analytical method. Usually, expanded uncertainty expressed as mathematical formula U = u c × k (u c, combined uncertainty; k, cover factor 2) is calculated 28.
The combined uncertainty may consist of bias, intermediate precision, systematic error, and other (evaluating each component) such uncertainty of the calibration curve 28. Uncertainty for Pb‐B in two techniques was similar and for ICP‐MS and GF‐AAS amounted to 6.5% versus 5%, and for Cd‐B it much better for GF‐AAS (16.5% vs. 9%).
Comparing validation parameters for these techniques, it could be stated that ICP‐MS has been comparable to GF‐AAS; however, ICP‐MS was better for Pb determination and GF‐AAS for Cd measurements. Nevertheless, this trend does not affect the quality and accuracy of the results.
Internal Quality Control
Internal quality control is one of the components of quality assurance. RMs (with no certified values) and CRMs (values are certified by a technically valid procedure, accompanied by documentation that is issued by a certifying body) are usually used in internal quality control. Regularly used control materials are an important part of assessment of analytical methods. Results of daily used internal quality control in laboratory should be registered on control card (Shewhart's Chart) and compared with target value and standard deviation (target value ± 2SD = 95.5% – warning limits/region, ± 3SD = 97.5% – control/action limits/region, which if exceeded require immediate action) 28, 29. User has been informed about these parameters (for CRM), based on certification report supplied by the manufacturer. In the case of RM, user should calculate himself “true value” and SD based at least on 20 determinations of control material on 20 different working days 25.
Shewhart's control graph card displays visually the results of control samples as a function of time or analytical run number. Each result should be marked on graph card and should be considered according to four Wheeler's Criteria 25, 30. The result is out of control if 1 one value is outside of the control limits (±3SD) 2, two of three consecutive values outside the warning limits (±2SD) but for one side (above or below) of central line, 3 four of five consecutive values between ±1SD and ±2SD, but for one side (above or below) of central line and 4 eight consecutive values on one side of the central line (above or below) ±1SD 29. Conducting control card allows us to keep track of random and systematic errors. The determinations of CRM or RM have been repeated a few times every day at least twice in each analysis series. Figure 1 presents Shewhart's control graph card for determination of Pb‐B using ICP‐MS.
Figure 1.
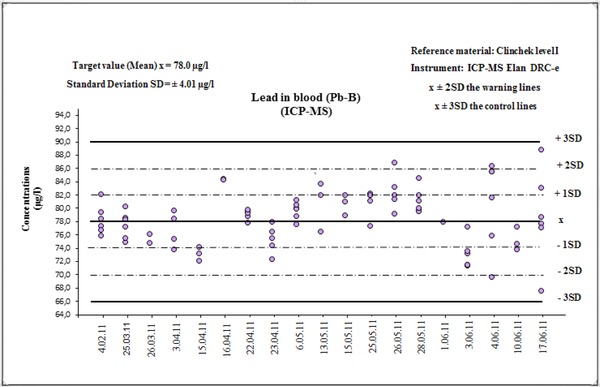
Shewhart Chart for Pb‐B determinations in RM—ClinChek level I.
Control materials may be used also to assess validation parameters, for example, verify precision and bias of analytical methods. Based on daily used or certified or noncertified reference materials, parts of validation parameters were calculated. The obtained results are summarized in Table 4, including validation parameters calculated for ICP‐MS and Table 5 for GF‐AAS. Both methods were validated using references materials ClinChek, Seronorm, and CRM BCR. The counted results for ICP‐MS (Table 4) indicated that for Pb‐B determination in RM, bias was within 1.2% and 4.5%, recovery 99% and 105%. For Cd, these values were 2.7% and 7.4%, recovery 93% and 103%. For GF‐AAS, bias and recovery assessed for Seronorm for Pb‐B determination were, respectively, 0.76% and 99% and the same for Cd‐B: 0.78% and 99% (Table 5).
Table 4.
Validation Parameters Calculated on the Basis of Reference Materials for the Method of Pb‐B and Cd‐B Determinations for ICP‐MS Technique
| Standard reference material | Reference concentration (μg/l) × (range/ uncertainty) | Determined concentration (μg/l) | Bias % | Recovery % | Intermediate precision% |
|---|---|---|---|---|---|
| Pb‐B (ClinChek level I) n = 83 | 78 (64 ÷84) | 79 ± 4.1 | 1.2 | 101 | 5.2 |
| Pb‐B (ClinChek level II) n = 10 | 290 (246 ÷ 334) | 286 ± 5.4 | 1.4 | 99 | 1.9 |
| Pb‐B (Seronorm II) n = 72 | 396 ± 62 | 414 ±16.7 | 4.5 | 105 | 4.0 |
| Pb‐B (BCR 634) n = 45 | 46 ± 5 | 47.4 ±2.34 | 3.0 | 103 | 4.9 |
| Cd‐B (ClinChek level I) n = 13 | 1.3 (0.97 ÷ 1.7) | 1.3 ± 0.08 | 2.7 | 100 | 6.2 |
| Cd‐B (ClinChek level II) n = 15 | 4.2 (3.3 ÷ 5.1) | 3.9 ± 0.2 | 7.4 | 93 | 4.1 |
| Cd‐B (Seronorm II) n = 15 | 6.0 ± 0.2 | 6.2 ± 0.4 | 3.3 | 103 | 6.2 |
| Cd‐B (BCR 634) n = 45 | 1.4 ± 0.4 | 1.2 ± 0.12 | 14.3 | 86 | 9.4 |
Table 5.
Validation Parameters Calculated on the Basis of Reference Materials for the Method of Pb‐B and Cd‐B Determinations for GF‐AAS Technique
| Standard reference material | Reference concentration (μg/l) × (range/ uncertainty) | Determined concentration (μg/l) | Bias% | Recovery % | Intermediate precision% |
|---|---|---|---|---|---|
| Pb‐B (Seronorm II) n = 19 | 396 (372 ÷ 414) | 393 ± 21.8 | 0.76 | 99 | 5.5 |
| Pb‐B (BCR 634) n = 8 | 46.0 ± 5.0 | 44.3 ± 1.57 | 3.69 | 96 | 3.5 |
| Cd‐B (Seronorm II) n = 16 | 6.0 (5.6 ÷ 6.4) | 6.35 ± 0.196 | 0.78 | 99 | 3.1 |
| Cd‐B (BCR 634) n = 21 | 1.4 ± 0.4 | 1.43 ± 0.12 | 4.28 | 102 | 8.3 |
The most useful reference material for internal quality control was BCR with certified true values. Using ICP‐MS technique and considering bias and recovery, comparable validation parameters were obtained for Pb‐B (3.0% and 103%) and a little bit worse for Cd‐B (14.3% and 86%). When we considered results obtained using GF‐AAS (bias and recovery) for Pb‐B and Cd‐B, they were similar and amounted, respectively, 3.7% and 4.3% and 96% and 102%.
For both techniques, values calculated for BCR were good enough to make a positive acceptance. However, better parameters were obtained for GF‐AAS; it seems that GF‐AAS (Tables 4 and 5) produced more stable results, especially for Cd‐B determinations.
The comparison of reference concentrations (target values) with measured values, for all materials used for the control, showed a good agreement for Pb‐B and Cd‐B determinations. Also for control materials with low metal concentration (Cd in ClinChek–level I and BCR) results were very satisfactory. For both metals (Pb and Cd), good recoveries were achieved.
The validation parameters such as bias, recovery, and intermediate precision assessed for GF‐AAS and ICP‐MS based on standard reference materials demonstrated good agreement between two methods. Based on validation parameters calculated for both techniques, we could conclude that ICP‐MS is as a useful technique for Pb and Cd determination in blood samples (in the same way) as GF‐AAS.
In the present study, the obtained results were similar to the results reported by Palmer et al. 18. The principle of Palmer's method, quite different than ours, was based on 50‐time blood‐sample dilution using diluent solution consisting of 0.5% nitric acid, Rh, Ir as internal standard, and Triton X‐100. However, our findings are in accordance with Palmer's results not only for the DL but also to intermediate precision. The values amounted for ICP‐MS for Pb‐B and Cd‐B, in our study, were 1.9 ÷ 5.2 and 4.1 ÷ 9.4 μg/l, and in Palmer's study were 1.4 ÷ 2.8 and 3.1 ÷10.1 μg/l.
External Quality Control
Participation in at least one EQAS is required in the process of laboratory accreditation. EQAS gives laboratories opportunity to confirm the quality of results receiving by laboratory 24.
In this article, the accuracy of the method for Pb‐B and Cd‐B determination has been verified through the participation of laboratory in two EQASs: UK NEQAS and G‐EQUAS.
In UK NEQAS, organizers send, in every round, to participants three samples with different concentrations four times per year and in G‐EQUAS two samples covered environmental concentrations and occupational medical range, 31, 32 once a year. Frequency depends on the character of QC. The German QC, mostly, has been opened toward accreditation of laboratories and this scheme supported the laboratories certification. UK QC has been dedicated to regularly control the quality of results and to verify and improve laboratory performance 33.
The results for participation of laboratory in these two external quality control schemes using ICP‐MS or GF‐AAS revealed good correlation between reference/target values and the determined control samples concentrations.
In UK NEQAS, for three rounds, bias for Pb‐B results ranged from 2.4% to 12.4% and for Cd‐B from 2.9% to 7.6%, whereas in the G‐EQUAS bias for Pb‐B was between 1.1% and 4.5% for occupational levels and 2.7% to 9.1% for environmental concentrations and for Cd‐B from 0% to 6.3% and 1.6% to 5% (results not shown).
The results of external quality control proved undoubtedly correctness of the applied method as well as positively verified them.
Analysis of Blood Samples
The comparison of the two techniques, that is, ICP‐MS and GF‐AAS have been carried out on the basis of Pb‐B and Cd‐B determinations in the same 40 blood samples workers occupational exposure to lead. The samples were determined within the framework of the program of health care workers exposed occupationally 34, 35. The trace metals were measured in the whole blood. Determinations were conducted under the conditions developed for ICP‐MS and GF‐AAS and described above. Using ICP‐MS, Pb and Cd were determined parallel from the same sample, on GF‐AAS sequentially one after the other.
Relationship between Pb‐B and Cd‐B determination in the same 40 blood samples using ICP‐MS and GF‐AAS is presented in Figures 2 and 3. The comparison indicated good agreement between two used techniques. The linear regressions of analysis were described as y = ax + b, where values obtained by ICP‐MS were taken as a (y) dependent variable and GF‐AAS values as an independent variable (x). The statistically significant correlation (P < 0.0001) was found for Pb‐B as well as Cd‐B determination for two considered techniques. Relationship was described by the regression equation for Pb‐B: y = 1.0148x + 3.5786 (r = 0.9998, P < 0.0001; Fig. 2) and for Cd‐B y = 1.0117x + 0.0761 (r = 0.9949, P < 0.0001) (Fig. 3). The correlation coefficients were high and statistically significant in all cases. Although compatibility for two techniques for both Pb‐B and Cd‐B determinations were very good, results for Cd‐B showed better compliance than Pb‐B. The results of the correlation analysis indicated that (a) slope values were close to 1.000 (Pb = 1.0148, Cd = 1.0117), the value of (b) intercepts varied (Pb = 3.5786, Cd = 0.0761) indicating that as well as ICP‐MS and GF‐AAS might give values a little bit higher comparing to GF‐AAS.
Figure 2.
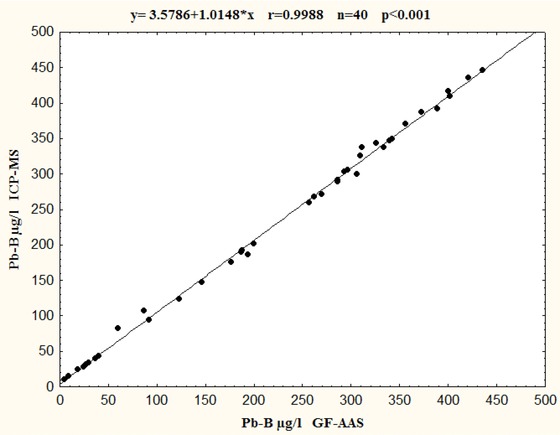
Comparison of GF‐AAS and ICP‐MS techniques for Pb‐B determinations in the same 40 blood samples.
Figure 3.
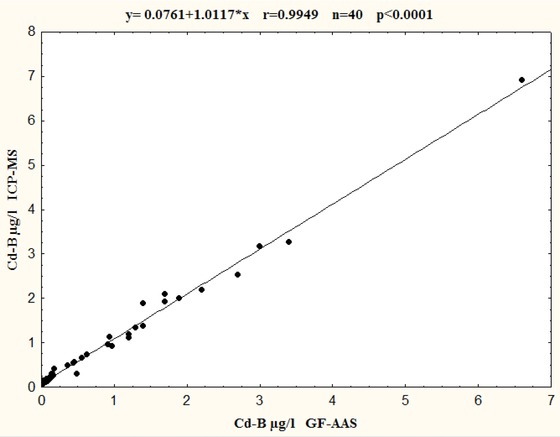
Comparison of GF‐AAS and ICP‐MS techniques for Cd‐B determinations in the same 40 blood samples.
However, excellent results obtained in the internal quality control as well as external quality control gave us the doubtlessness that the results of Pb‐B and Cd‐B determination were accurate and precise and developed method may be applied to determine low concentrations of metals within biological monitoring analysis.
CONCLUSION
This paper presents simple and fast method of lead and Cd determination in blood samples using two techniques ICP‐MS and GF‐AAS. The principle of this method consists of deproteinization of blood samples by nitric acid and is based on a forgotten and not often used Stoeppler and Brandt publication. It relies on the use of nitric acid and its excellent deproteinization properties, minimizing spectral interferences and influence of the organic matter on determination metals in blood samples. This is especially important when using GF‐AAS, where the influence of the matrix can change the results of the determination.
The obtained results showed that two techniques may be successfully used to determination Pb and Cd in blood. However, ICP‐MS is faster than GF‐AAS and allows parallel determination of the two elements in one blood sample. ICP‐MS enables us to determine lead at the lower range of concentrations due to lower DL. Other validation parameters are comparable, however a bit worse for Cd‐B.
The elaborated method, validated and verified by participation in quality control, is especially dedicated to clinical laboratories and all others performing Pb and Cd determination in the analysis of biological monitoring for health protection of people environmental and occupational exposed to lead.
This method should be popularized in all laboratories performing routine analysis of Pb‐B and Cd‐B using ICP‐MS but especially in those equipped with GF‐AAS.
The above‐presented method may also be successfully used for manganese and nickel determination or for determination og any other metal, provided that the metal is not bound to plasma proteins.
CONFLICT OF INTEREST
The authors have declared no conflict of interest.
ACKNOWLEDGMENTS
The authors thank Prof. Wojciech Wasowicz, Head of Department of Toxicology and Carcinogenesis, for critical reading of the manuscript and for helpful discussion. We also thank Malgorzata Gontarek, Elzbieta Hiler, and Wieslaw Kuszka for technical assistance.
Grant sponsor: National Science Centre; Grant number: 2011/01/B/NZ 7/01325.
REFERENCES
- 1. Angerer J, Gündel J. Biomonitoring and occupational medicine. Possibilities and limitations. Ann Ist Super Sanità 1996;32:199–206. [PubMed] [Google Scholar]
- 2. IPCS. Biomarkers and Risk Assessment: Concepts and Principles . Environmental Health Criteria 155. Geneva: World Health Organization; 1993. Available at: http://www.inchem.org/documents/ehc/ehc/ehc155.htm. [Google Scholar]
- 3. U.S. Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry. Toxicological Profile for Lead. Atlanta, Georgia, 2007. Available at: http://www.atsdr.cdc.gov/toxprofiles/tp13.pdf. [Google Scholar]
- 4. WHO, Regional Office for Europe. Health Risks of Heavy Metals from Long‐Range Transboundary Air Pollution in Cadmium, Lead. Copenhagen, Denmark: 2007. Available at: http://www.euro.who.int/__data/assets/pdf_file/0007/78649/E91044.pdf.
- 5. Jakubowski M, Trzcinka‐Ochocka M, Razniewska G. Biological Monitoring of Occupational and Environmental Exposure to Metals – Methods of Determination, Interpretation of Results. Lodz: Nofer Institute of Occupational Medicine; 2000. [Google Scholar]
- 6. Jakubowski M, Marek K, Piotrowski JK, Iżycki J. Recommendations for the Diagnosis and Medical Prevention of Lead Poisoning. Lodz: Nofer Institute of Occupational Medicine; 1997. [Google Scholar]
- 7. Regulation of the Minister of Health and Social Welfare of 30 May 1996 on the medical examinations of employees, the scope of preventive health care workers and medical purposes provided for in the Labour Code Laws, 1996, No. 69, item 322. Available at: http://www.saws.pl/asp/pliki/Przepisy/1996-69-332.pdf.
- 8. Regulation of the Minister of Health of 30 December 2004 on health and safety at work regulations, apply to workplace chemicals. In Acts 2005, No. 11, item. 86. Available at: http://www.ciop.pl/10621.html.
- 9. Council Directive 98/24/EC of 7 April 1998 on the protection of the health and safety of workers from the risks related to chemical agents at work (fourteenth individual Directive within the meaning of Article 16 (1) of Directive 89/391/EEC). European Agency for Safety and Health at Work. Available at: https://osha.europa.eu/en/legislation/directives/exposure-to-chemical-agents-and-chemical-safety/osh-directives/75.
- 10. Central Institute for Labour Protection. Interdepartmental Commission for Maximum Admissible Concentrations and Intensities for Agents Harmful to Health in the Working Environment . Warsaw: CIOP; /PIB; 2003. [Google Scholar]
- 11. Krachler M, Irgolic KJ. The potential of inductively coupled plasma mass spectrometry (ICP‐MS) for the simultaneous determination of trace elements in whole blood, plasma and serum. J Trace Elem Med Biol 1999;13:157–169. [DOI] [PubMed] [Google Scholar]
- 12. Thermo Scientific. Determination of Trace Elements in Clinical Samples by High Resolution ICP‐MS. Application Note: 30003. 2007.
- 13. Fukui Y, Ohashi F, Sakuragi S, Moriguchi J, Ikeda M. Comparative evaluation of GFAAS and ICP‐MS for analyses of cadmium in blood. Ind. Health 2011;49:338–343. [DOI] [PubMed] [Google Scholar]
- 14. Goullé JP, Mahieu L, Castermant J, et al. Metal and metalloid multi‐elementary ICP‐MS validation in whole blood, plasma, urine and hair: Reference values. Forensic Sci Int 2005;153:39–44. doi: 10.1016/j.forsciint.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 15. Wahlen R, Evans L, Turner J, Hearn R. The use of collision/reaction cell ICP‐MS for the determination of elements in blood and serum samples. Spectroscopy 2005;20:84–89. [Google Scholar]
- 16. Krachler M, Heisel CH, Kretzer JPH . Validation of ultratrace analysis of Co, Cr, Mo and Ni in whole blood, serum and urine using ICP‐SMS. J Anal At Spectrom 2009;24:605–610. doi: 10.1039/b821913c. [DOI] [Google Scholar]
- 17. Heitland P, Köster HD. Biomonitoring of 37 trace elements in blood samples from inhabitants of northern Germany by ICP‐MS. J Trace Elem Med Biol 2006;20:253–262. doi: 10.1016/j.forsciint.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 18. Palmer CD, Lewis ME Jr., Geraghty CM, Barbosa F Jr., Parsons PJ. Determination of lead, cadmium and mercury in blood for assessment of environmental exposure: A comparison between inductively coupled plasma‐mass spectrometry and atomic absorption spectrometry. Spectrochim Acta Part B At Spectrosc 2006;61:980–990. doi: 10.1016/j.sab.2006.09.001. [DOI] [Google Scholar]
- 19. Stoeppler M, Brandt K, Rains TC. Contributions to automated trace analysis. Part II. Rapid method for the automated determination of lead in whole blood by electrothermal atomic‐absorption spectrophotometry. Analyst 1978;103:713–722. doi: 10.1039/an9780300714. [DOI] [PubMed] [Google Scholar]
- 20. Stoeppler M, Brandt K. Contributions to automated trace analysis. Part V. Determination of cadmium in whole blood and urine by electrothermal atomic‐absorption spectrophotometry. Fresenius J Anal Chem 1980;300:372–380. doi: 10.1007/bf01154738. [DOI] [PubMed] [Google Scholar]
- 21. Raźniewska G, Trzcinka‐Ochocka M. Methods for determining lead and cadmium in blood; cadmium, copper, nickel and chromium in urine using flameless atomic absorption spectrometry. Med Pr 1995;66:347–358. [PubMed] [Google Scholar]
- 22. ICH Q2B (The International Conference of Harmonization), Validation of Analytical Procedures: Methodology, adopted in 1996, Geneva Q2B, in 2005 incorporated in Q2(R1). Available at: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf.
- 23. ICH Q2A (The International Conference of Harmonization), Text on validation of analytical procedures: Definitions and terminology, March 1995. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073381.pdf.
- 24.EN ISO/IEC 17025:2005 General requirements for the competence of testing and calibration laboratories. European Standard Norm, Brussels, 2005.
- 25. Dobecki M. Quality Assurance in Chemical Analysis. Lodz, Poland: Nofer Institute of Occupational Medicine; 1998. [Google Scholar]
- 26. Barany E, Bergdahl IA, Bratteby LE, et al. Trace element levels in whole blood and serum from Swedish adolescents. Sci. Total Environ 2002;286:129–141. doi: 10.1016/s0048-9697(01)00970-6. [DOI] [PubMed] [Google Scholar]
- 27. Barany E, Bergdahl IA, Schütz A, Skerfving S, Oskarsson A. Inductively coupled plasma mass spectrometry for direct multi‐element analysis of diluted human blood and serum. J Anal At Spectrom 1997;12:1005–1009. doi: 10.1039/a700904f. [DOI] [Google Scholar]
- 28. Eurachem/CITAC Guide CG 4, Quantifying Uncertainty in Analytical Measurement, Second Edition, QUAM‐2000. Available at: http://www.measurementuncertainty.org/mu/QUAM2000‐1.pdf.
- 29. Funk W, Dammann V, Donnevert G. Quality assurance in analytical chemistry. Edited by Philippe Quevauviller VCH Verlagssgesellschaft mbH, D‐69451 Weinheim, 1995, Germany. Available at: http://onlinelibrary.wiley.com/doi/10.1002/9783527615216.fmatter/pdf.
- 30. Wheeler JD, Chambers DS. Understanding Statistical Process Control, second edition. Knoxville, TN: SPC Press; 1992. [Google Scholar]
- 31. Schaller KH, Angerer J, Drexler H. Quality assurance of biological monitoring in occupational and environmental medicine. J Chromatogr B 2002;778:403–417. doi: 10.1016/s1570-0232(02)00171-x. [DOI] [PubMed] [Google Scholar]
- 32. Schaller KH, Angerer J, Weltle D, Drexler H. External quality assurance program for biological monitoring in occupational and environmental medicine. Rev Environ Health 2001;16:223–232. [DOI] [PubMed] [Google Scholar]
- 33. UK NEQAS Report and Directory, 4th Edition, 2000, UK NEQAS office PO Box 401, Sheffield S5 7YZ.
- 34. Trzcinka‐Ochocka M, Jakubowski M, Razniewska G. Assessment of occupational exposure to lead in Poland. Med Pr 2005;56:395–404. [PubMed] [Google Scholar]
- 35. Trzcinka‐Ochocka M, Jakubowski M, Nowak U. Effectiveness of preventive actions for lead exposed workers: An assessment based on biological monitoring. Med Pr 2006;57:537–542. [PubMed] [Google Scholar]