

Abstract
Background
Previous studies have suggested that adipokines play a role in inflammatory bowel disease by inducing proinflammatory cytokines, but it is uncertain whether visfatin is causally involved in ulcerative colitis (UC). We evaluated visfatin levels in patients who presented with UC flares before and after treatment.
Methods
In this cohort study, we assessed 31 patients with UC in the activation period and remission in the same patients after treatment, and a healthy control group, consisting of 29 persons, at a single academic medical centre between 2010 and 2013. Disease severity was evaluated clinically using Trulove and Witt's criteria.
Results
Serum visfatin levels did not vary according to the extent of disease and were significantly higher in patients in the activation period (7.77 ± 2.41 ng/ml) than in remission (6.18 ± 2.04 ng/ml) and the healthy controls (6.54 ± 2.20 ng/ml; P < 0.01 and < 0.05, respectively). In a comparison of patients in the inactive period with the control group, there was no statistically significant difference (P > 0.05). To assess activation of the disease, a visfatin cut‐off point for active UC was determined as 6.40, with sensitivity, specificity, positive predictive value (PPV) and negative predictive values (NPV) of 72%, 52%, 66.7% (43.0–85.4) and 50.0% (29.1–70.9), respectively.
Conclusions
The visfatin level was higher in the active group than in post‐treatment remission and the healthy control group. Sensitivity and specificity were similar to other inflammatory markers for assessing clinical activity, which did not improve clinical outcomes in patients with acute respiratory distress syndrome (ARDS). These findings did not provide a rationale for assessment of UC activation.
Keywords: visfatin, inflammatory bowel disease, ulcerative colitis, adipokine
INTRODUCTION
Adipose tissue is an anatomical term for loose connective tissue, composed mostly of adipocytes. Adipocytes are the main cellular components of white adipose tissue (WAT). They not only control energy homeostasis by storing and mobilising triacylglycerols but also secrete numerous bioactive substances that regulate metabolism, called adipokines or adipocytokines. Since the first identification of an adipokine, which was named leptin, in 1994, many types of adipokine have been shown to be derived from WAT. They can act locally within adipose tissue, but can also play a larger role in the regulation of physiological and pathological processes, such as eating behaviour, insulin sensitivity, haemostasis, blood pressure, metabolism, immunity and inflammation 1, 2, 3, 4, 5.
Ulcerative colitis (UC) is a chronic, relapsing gastrointestinal disease accompanied by extraintestinal manifestations involving various organs and systems (e.g., joints, skin, liver, eye, mouth and coagulation). The aetiology and pathogenesis are unknown, but are likely to be multifactorial. Disturbed gut microbiota composition, genetic predisposition and modulating immune system activation all play roles in the pathogenesis of UC 6, 7, 8.
Recent research has highlighted the important role of cytokines. Alterations in cytokine regulation may result in local ischaemia, which can mediate epithelial cell cytotoxicity, apoptosis and epithelial barrier dysfunction. Proinflammatory cytokines that are produced by macrophages, lymphocytes and colonic epithelial cells, such as interleukins (IL‐1, IL‐2, IL‐5, IL‐6, IL‐12, IL‐21, IL‐23) and tumour necrosis factor alpha (TNF‐α) are involved in the pathogenesis of UC, providing potential therapeutic targets 9, 10. Visfatin is a recently identified adipokine, secreted primarily by visceral WAT. It is also produced by various cells, and levels are elevated in the systemic circulation of patients with various diseases such as Behçet's disease, rheumatoid arthritis, chronic viral hepatitis B, nonalcoholic fatty liver disease and cardiovascular disease 11, 12, 13, 14, 15.
Several proinflammatory and immune‐regulatory cytokines are up‐regulated in patients with UC, and a similar cytokine profile is induced by visfatin, suggesting that visfatin may play a role in the emergence of UC. Therefore, serum visfatin levels also may correlate with the magnitude of inflammation in UC. We investigated the relationship between visfatin and UC, and whether visfatin levels could be used in clinical practice to monitor and/or predict disease activity, whether a therapeutic intervention is associated with a reduction in serum visfatin levels.
MATERIALS AND METHODS
Medical records of patients with active UC admitted to Erciyes University, Faculty of Medicine, Department of Gastroenterology and Hepatology, Kayseri, Turkey, from January 2010 to July 2013 were examined prospectively. The diagnosis of UC was made using standard clinical, endoscopic and histological criteria 16. The disease activity was determined at admission and post‐treatment with Trulove–Witt Activity Index (TWAI) 17. In total, 31 active UC patients with ages 18–65 who were admitted to the outpatient and inpatient clinic were included. Clinical disease activity was defined using TWAI, consisting of eight clinical items scored from 0 to 21 points; stools per 24 h (0–4), abdominal pain or cramping (0–3), abdominal tenderness (0–3), general well‐being (0–5), visible blood in stool (0–3), nocturnal diarrhoea (0–1), faecal incontinence (0–1) and the need for anti‐diarrhoeal drugs (0–1). The patients with a score of 4 or higher on admission were defined as having active disease. Patients with active UC were treated with azathioprine, 5‐aminosalicylic acid based medications, enemas, TNF‐α inhibitors and prednisone. Patients with a score of 4 or below after treatment were defined as being in remission.
A total of 66 patients were enrolled. Of these, 31 patients were included in the study. Patients were excluded if they had undergone anti‐inflammatory therapies, including steroid, azathioprine or combinations of these treatments, or if they were pregnant, or had undergone intestinal surgery, or suffered from diabetes mellitus (DM), OGTT disorders, coronary artery disease (CAD), malignancy, hypertension, autoimmune connective tissue disorders, chronic kidney disease or had evidence of active or chronic infection, probably because of increased visfatin levels.
The study was approved by the Clinical Research Ethics Committee of Erciyes University. Informed consent was obtained from all participants.
All the participants’ height and weight values were measured and their body mass indexes (BMIs) were calculated. Serum samples were collected from the patients during active disease and remission. White blood cell (WBC), erythrocyte sedimentation rate (ESR) and C‐reactive protein (CRP) parameters were measured. WBC analysis was performed using ADVIA 2120Ai (Siemens, Berlin, Germany) automatic whole blood counter. ESR was determined manually using Westergren method. CRP levels were measured using an immunoturbidometric assay (Architect C16000, Abbott Laboratories).
Visfatin was analysed using a commercially available ELISA kit (Phoenix Peptides, Belmont, CA). Assay sensitivity was 2 ng/ml and the inter‐assay and intra‐assay coefficients of variation were < 10% and < 5%, respectively.
Statistical Analysis
Shapiro–Wilk's and Levene's tests were used for assessing the normality and variance homogeneity of data. Independent sample t‐tests, paired t‐tests, the Mann–Whitney U‐test and Wilcoxon's non‐parametric tests were used for the comparison of continuous variables, and χ2 analyses were used for categorical variables. Values are expressed as frequencies and percentages, means ± standard deviation or medians and 25th to 75th percentiles.
Receiver‐operating characteristic (ROC) curves were prepared for the WBC, CRP, sedimentation and N/L variables and the areas under the ROC curve with 95% CIs were calculated and compared. Optimal cut‐off values were determined. Sensitivity, specificity, positive predictive rate, negative predictive rate and accuracy rate diagnosing measures were calculated (with 95% CIs), and kappa tests were performed for each variable. Analyses were conducted using the SPSS software (ver. 15.0; SPSS Inc., Chicago, IL). P values < 0.05 were considered to indicate statistical significance.
RESULTS
In total, 31 patients with UC and 29 control subjects were enrolled. There were 9 females and 22 males in the UC group, and 15 females and 14 males in the control group (P = 0.028). The median disease duration in UC patients was 2 years. The demographic characteristics and laboratory values of patients and control subjects are summarised in Table 1. There was no statistically significant difference in the ages or BMI of the UC and control groups. The mean visfatin levels of the control, inactive and active UC patients were 6.54 ± 2.20, 6.18 ± 2.04 and 7.77 ± 2.41, respectively (P < 0.05). The serum visfatin levels of active patients were significantly higher than inactive UC patients and controls. Table 2 shows the serum visfatin levels and other laboratory values of the UC patients during the active period and remission. There was no significant difference between the groups with active disease versus in remission, or between those in remission and controls, in WBC counts or ESR after appropriate therapy (P = 0.65 and 0.61, respectively).
Table 1.
Demographic Characteristics and Laboratory Values of Controls and Ulcerative Colitis Patients
| Variable | Control (n = 29) | UC (n = 31) | P value |
|---|---|---|---|
| Gender (M/F) | 14 (48.3.)/15 (51.7) | 22 (71)/9 (29) | 0.028 |
| Age (years) | 40.79 ± 11.28 | 43.61 ± 12.12 | 0.56 |
| Disease duration (years) | 2.00 (1.00–6.00) | ||
| BMI (kg/m2) | 26.1 (22.40–28.50) | 26.7 (23.0–6.0) | 0.78 |
Values are expressed as n (%), means ± SD or medians (25th to 75th percentiles).
Table 2.
Laboratory Values of Ulcerative Colitis Patients According to Disease Activity
| Variable | Active (n = 31) | Remission (n = 31) | P value |
|---|---|---|---|
| WBC (per mm3) | 8,105 ± 2,869 | 7,697 ± 1,968 | 0.42 |
| Sedimentation (mm/h) | 17 (9.00–28.50) | 11 (4.75–21.25) | 0.61 |
| CRP (mg/l) | 5.04 (3.34–10.40) | 3.45 (3.30–7.96) | 0.65 |
| Visfatin (ng/ml) | 7.71 ± 2.43 | 6.18 ± 2.04 | 0.01 |
| Severity index (TWAI) | 11 (9.00–12.00) | 2 (1.00–3.00) | < 0.001 |
Values are expressed as n (%), means ± SD or medians (25th to 75th percentiles). WBC, white‐cell count; CRP, C‐reactive protein; TWAI, Trulove–Witt Activity Index.
ROC curve analysis suggested that the optimum visfatin cut‐off level for active UC was 6.40, with a sensitivity, specificity, PPV and NPV of 72%, 52%, 66.7% (43.0–85.4) and 50.0% (29.1–70.9), respectively (Fig. 1). The same analyses for other inflammatory markers are summarised in Table 3. There was no significant pair‐wise difference in area under the curve (AUC) values (P > 0.05).
Figure 1.
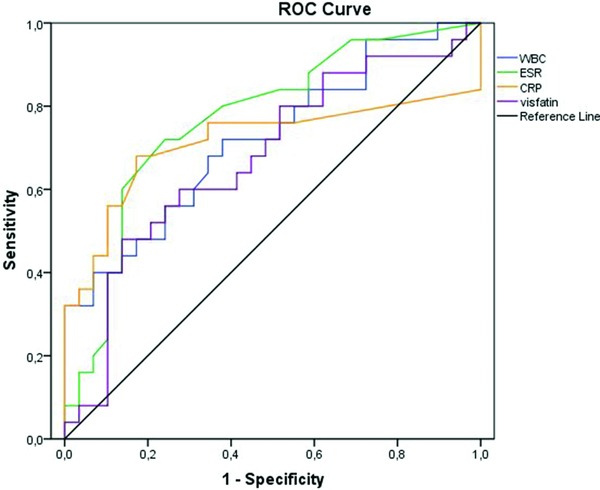
Comparison of ROC curves for WBC, CRP, sedimentation and visfatin. AUC values were 0.72 (0.58–0.85), 0.71 (0.56–0.87), 0.77 (0.64–0.90) and 0.67 (0.53–0.82), respectively, and there was no significant pair‐wise difference (P > 0.05).
Table 3.
Diagnosing Measures Test Results of WBCs, CRP, Sedimentation and Visfatin Variables for the Determined Cut‐Off Values in the Detection of Ulcerative Colitis Activity
| Diagnosing measures | ||||
|---|---|---|---|---|
| SEN | SPE | PPR | NPR | |
| Variables and cut‐off values | (%) | (%) | (%) | (%) |
| WBC (>6,915/mm3) | ||||
| 72 | 62 | 64 | 67 | |
| CRP (>3.4 mg/l) | 76 | 66 | 75 | 73 |
| Sedimentation (>11.5 mm/h) | 72 | 76 | 73 | 69 |
| Visfatin (>6.40) | 72 | 52 | 59 | 58 |
SEN, sensitivity; SPE, specificity; PPR, positive predictive rate; NPR, negative predictive rate.
Ten patients were classified as having extensive and 21 patients as left‐sided disease, according to the endoscopic examination on study entry. On the initial examination, only four patients were classed as having proctitis; these patients were included in the left‐sided group to compare inflammatory parameters between two groups with active disease. There was no significant difference between inflammation parameters or disease extent between these groups (P > 0.05).
DISCUSSION
In this small cohort trial, we examined severe flares in UC patients who had not received previous steroid or azothiopurine therapy. We found that patients who had active UC disease had higher visfatin values than their counterparts in remission after anti‐inflammatory therapy. But our findings did not provide a rationale for assessment of UC activation.
UC is an inflammatory disease of the gastrointestinal tract; its cause remains unknown, but the disease seems to be multifactorial and polygenic. Alterations in the ingredient of the gut microbiota, mucosal intolerance against microbial load, dysregulation of the mucosal immune response and autoimmunity are associated with susceptibility to UC. Research is currently focused on its immunopathogenesis. These studies have resulted in progress in understanding the process of the disease and the identification of several immunological markers that may play important roles in treatment modalities. Some of the molecules may be valuable as indicators of disease activity and severity.
Many studies of inflammatory bowel disease (IBD) have revealed the regulation of proinflammatory cytokine pathways and their roles in systemic and organ‐specific inflammation. In recent years, particularly, studies have included adipokines, evaluating their role in inflammation and their contribution to the pathogenesis of IBD. Obesity and systemic micro‐inflammation are strongly associated with disturbed circulating adipokine levels 18, 19. Elevated plasma leptin concentrations have been observed during acute phases of inflammation in experimental colitis, despite decreased body weight, anorexia and enhanced release of TNF‐α. Leptin has also been correlated with the degree of inflammation 20. Furthermore, the importance of abnormalities in fat in the mesentery in IBD, including abnormal hypertrophy of adipose tissue, was reported by Gambero et al. in a study of rats with experimental colitis. These changes were site‐specific and were not observed in other mesentery fat depots. Modified mesenteric fat content surrounding the gut, the so‐called ‘wrapping’ or ‘creeping’ fat, produced different adipocytokine profiles (leptin and adiponectin), suggesting a role in the pathogenesis of IBD 21. In this respect, one of the factors studied was visfatin. We searched the literature using PubMed for a combination of the terms ‘IBD’, ‘visfatin’ and ‘UC’. Only three relevant reports providing evidence of a relationship between visfatin and UC were identified. In one of these, Moschen et al. showed that visfatin induced the production of inflammatory cytokines and may be considered a new proinflammatory adipocytokine. Plasma visfatin was found to be elevated in patients with IBD and its mRNA expression was increased significantly in the colonic tissue of IBD colitis patients versus healthy controls 22. Another study investigated five circulating adipokines (leptin, resistin, visfatin, RBP‐4 and adiponectin) and glucose homeostasis in patients with inactive and active IBD. Visfatin alone was increased in active disease versus remission 23. The study by Waluga et al., which is similar to ours, investigated serum adipokine levels in IBD patients before treatment and after achieving clinical remission. Baseline serum concentrations of visfatin were significantly higher in subjects with UC than in healthy controls. None of the previously reported studies, between active and inactive groups, failed to detect a significant difference in visfatin levels 22, 23, 24. In this cohort study, involving patients with active UC, we compared levels before treatment of the flare with anti‐inflammatory and/or immunosuppressive therapy, and the post‐treatment remission phase and a healthy control group. Unlike other studies, treatment decreased visfatin levels in the remission group 22, 24. Although not diagnostic, elevation of CRP and ESR in IBD are well‐known markers for determining and monitoring disease activity. However, surprisingly, their levels did not show a difference among the groups (P = 0.65 and 0.61, respectively).
Previous studies have suggested that visfatin affects peripheral blood mononuclear cells, induces proinflammatory cytokine production, inhibits neutrophil apoptosis and is associated with low bone mass and mRNA expression of visfatin in inflamed intestinal mucosa 25, 26. Other reported effects include nuclear transcription factors, such as nuclear factor kappa B (NF‐κB), which plays a key role in the regulation of immune responses. Studies have shown that visfatin up‐regulated NF‐κB p65 (RelA) DNA‐binding activity in human leukocytes by p38 and MEK‐1 pathways 27, 28. The pathogenesis of UC must take into consideration the findings of those studies. Taken together, our results and these previous reports suggest that visfatin plays a role in the activation but not the pathogenesis of UC.
Our study had several limitations. First, we assessed visfatin levels only in the blood of patients with a diagnosis of active UC and achieved remission with a specific anti‐inflammatory therapy. Visfatin may be implicated in the disease process. The role of visfatin mRNA levels in inflamed and uninflamed tissue remains unclear. Second, Moschen et al. showed that visfatin had a proinflammatory effect in CD14+ monocytes and CD19+ B cells, by inducing chemokine production 23, 29, and also increasing IL‐1β, TNF‐α and, especially, IL‐6 levels. We did not determine the signalling pathway of visfatin or examine proinflammatory cytokine levels.
In conclusion, we report here a connection between UC flares and visfatin levels. Visfatin levels may reflect disease activity. The role of visfatin in UC flares should be investigated in further research.
CONFLICT OF INTEREST
All the authors declare that they have no conflict of interest.
REFERENCES
- 1. Zhao ZJ, Zhu QX, Chen KX, et al. Energy budget, behavior and leptin in striped hamsters subjected to food restriction and refeeding. PLoS One 2013;8(1):e54244. doi: 10.1371/journal.pone.0054244. Epub 2013 Jan 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesityrelated insulin resistance. J Clin Invest 2003;112:1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Romacho T, Sánchez‐Ferrer CF, Peiró C. Visfatin/NAMPT: An adipokine with cardiovascular impact. Mediators Inflamm 2013;2013:946427. doi: 10.1155/2013/946427. Epub 2013 Jun 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fantuzzi, G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol 2005;115:911–919. [DOI] [PubMed] [Google Scholar]
- 5. Weisberg SP, McCann D, Desai M, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sartor RB. Microbial influences in inflammatory bowel disease. Gastroenterology 2008;134:577–94. [DOI] [PubMed] [Google Scholar]
- 7. Heel DA. The genetic of chronic inflammatory diseases. Hum Mol Genet 2009;18:R101–R106. [DOI] [PubMed] [Google Scholar]
- 8. Baumgart CD, Carding SR. Inflammatory bowel disease: Cause and immunobiology. Lancet 2007;369:1627–1640. [DOI] [PubMed] [Google Scholar]
- 9. Mayer L, Shlien R. Evidence for function of Ia molecules on gut epithelial cells in man. J Exp Med 1987;166:1471–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kolls JK, Lindén A. Interleukin‐17 family members and inflammation. Immunity 2004;21(4):467–476. [DOI] [PubMed] [Google Scholar]
- 11. Sezen H, Okumus S, Pehlivan Y, et al. Visfatin levels in Behcet's disease. Inflammation 2012;35(2):405–408. doi: 10.1007/s10753-011-9328-2 [DOI] [PubMed] [Google Scholar]
- 12. Chen X, Lu J, Bao J, et al. Adiponectin: A biomarker for rheumatoid arthritis? Cytokine Growth Factor Rev 2013;24(1):83–89. doi: 10.1016/j.cytogfr.2012.07.004. Epub 2012 Aug 19. [DOI] [PubMed] [Google Scholar]
- 13. Romacho T, Cercas E, Carraro R, Sánchez‐Ferrer CF, Peiró C. Visfatin/NAMPT: An adipokine with cardiovascular impact. Mediators Inflamm 2013;2013:946427. doi: 10.1155/2013/946427. Epub 2013 Jun 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yüksel E, Akbal E, Koçak E, et al. The relationship between visfatin, liver inflammation, and acute phase reactants in chronic viral hepatitis B. Wien Klin Wochenschr 2015. (April 9) [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 15. Akbal E, Koçak E, Taş A, et al. Visfatin levels in nonalcoholic fatty liver disease. J Clin Lab Anal 2012;26(2):115–119. doi: 10.1002/jcla.21491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dignass A, Eliakim R, Magro F, et al. Second European evidence‐based consensus on the diagnosis and management of ulcerative colitis part 1: Definitions and diagnosis. J Crohns Colitis 2012;6(10):965–990. doi: 10.1016/j.crohns.2012.09.003. Epub 2012 Oct 3. [DOI] [PubMed] [Google Scholar]
- 17. Lichtiger S, Present DH, Kornbluth A, et al. Cyclosporine in severe ulcerative colitis refractory to steroid therapy. N Engl J Med 1994;330(26):1841–1845. [DOI] [PubMed] [Google Scholar]
- 18. Olszanecka‐Glinianowicz M, Chudek J, Kocełak P, et al. Body fat changes and activity of tumor necrosis factor α system—A 5‐year follow up study. Metabolism 2011;60:531–536. [DOI] [PubMed] [Google Scholar]
- 19. Olszanecka‐Glinianowicz M, Zahorska‐Markiewicz B, Janowska J, et al. Serum concentrations of nitric oxide, tumor necrosis factor (TNF)‐alpha and TNF‐soluble receptors in women with overweight and obesity. Metabolism 2004;53:1268–1273. [DOI] [PubMed] [Google Scholar]
- 20. Barbier M, Cherbut C, Aubé AC, et al. Elevated plasma leptin concentrations in early stages of experimental intestinal inflammation in rats. Gut 1998;43:783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gambero A, Maróstica M, Abdalla Saad MJ, et al. Mesenteric adipose tissue alterations resulting from experimental reactivated colitis. Inflamm Bowel Dis 2007;13:1357–1364. [DOI] [PubMed] [Google Scholar]
- 22. Valentini L, Wirth EK, Schweizer U, et al. Circulating adipokines and the protective effects of hyperinsulinemia in inflammatory bowel disease. Nutrition 2009;25(2):172–181. doi: 10.1016/j.nut.2008.07.020. Epub 2008 Oct 11. [DOI] [PubMed] [Google Scholar]
- 23. Moschen AR, Kaser A, Enrich B, et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol 2007;178(3):1748–1758. [DOI] [PubMed] [Google Scholar]
- 24. Waluga M, Hartleb M, Boryczka G. Serum adipokines in inflammatory bowel disease. World J Gastroenterol 2014;20(22):6912–6917. doi: 10.3748/wjg.v20.i22.6912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jia SH, Li Y, Parodo J, et al. Pre‐B cell colony‐enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J Clin Invest 2004;113:1318–1327 [PMID: 15124023, doi: 10.1172/JCI19930]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moschen AR, Geiger S, Gerner R, et al. Pre‐B cell colony enhancing factor/NAMPT/visfatin and its role in inflammation‐related bone disease. Mutat Res 2010;690:95–101. [DOI] [PubMed] [Google Scholar]
- 27. Ghosh S, Karin M. Missing pieces in the NF‐_B puzzle. Cell 2002;109(Suppl.):S81–S96. [DOI] [PubMed] [Google Scholar]
- 28. Kumar S, Boehm J, Lee JC. p38 MAP kinases: Key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2003;2:717–726. [DOI] [PubMed] [Google Scholar]
- 29. Esche C, Stellato C, Beck LA. Chemokines: Key players in innatand adaptive immunity. J Invest Dermatol 2005;125(4):615–628. [DOI] [PubMed] [Google Scholar]