

Abstract
Constructive machine learning aims to create examples from its learned domain which are likely to exhibit similar properties. Here, a recurrent neural network was trained with the chemical structures of known cell‐migration modulators. This machine learning model was used to generate new molecules that mimic the training compounds. Two top‐scoring designs were synthesized, and tested for functional activity in a phenotypic spheroid cell migration assay. These computationally generated small molecules significantly increased the migration of medulloblastoma cells. The results further corroborate the applicability of constructive machine learning to the de novo design of druglike molecules with desired properties.
Keywords: chemoinformatics, chemotaxis, drug discovery, neural networks, phenotypic screening
1. Introduction
The collection of methods enabling computational de novo design has recently been expanded by techniques utilizing machine learning, rather than explicit chemical transformations, for the construction of potentially novel molecules.1, 2 Certain classes of artificial neural networks, especially recurrent neural networks (RNN), are particularly suitable for this purpose.3, 4, 5, 6 These machine learning models are trained with collections of chemical structures, often represented as SMILES strings.5 After successful training, an RNN can sample new SMILES strings from the learned training data distribution, such that the generated molecules possess features of the training compounds.
We previously applied an RNN with long short‐term memory cells (LSTM7), which was trained with bioactive small molecules from ChEMBL,8, 9 and by fine‐tuning with small sets of known bioactives (transfer learning), reoriented to generate target‐specific compounds. Prospective applications demonstrated that nuclear receptor modulators and natural‐product‐inspired bioactive molecules which were synthetically accessible and confirmed as having in vitro activity against the desired targets, could be obtained using this approach.10, 11 Here, we apply this constructive machine learning approach to the computational design of cell migration modulators.
Small molecules affecting cellular migration and chemotaxis are of interest both as tool compounds for studying the metastatic behavior of malignant cells, and as potential drugs, e. g. for stem cell mobilization.12 The often‐complex biochemical networks controlling these processes are known to heavily involve the chemokine receptors.13 These regulate a range of signaling pathways with G‐protein or arrestin dependent branches, in a manner dependent on receptor polymerization state.14, 15 The G‐protein coupled receptors (GPCRs) and their endogenous chemokine ligands are vital for immunoregulation and have also been linked to cancer pathology and prognosis.16, 17 The chemokine CXCL12, and its receptors CXCR4 and ACKR3, plays a central role in the metastasis of malignant cells.18, 19 CXCR4 antagonists have been developed as cell migration modulators, including the approved drug Plerixafor (AMD3100) for stem cell mobilization.12
Here, we use a constructive RNN model5 to generate small molecule cell migration inducers. A pre‐trained RNN was fine‐tuned with a set of known chemokine receptor ligands. The candidate molecules suggested by the constructive machine learning approach were prioritized based on their pharmacophore similarity to the fine‐tuning set, and two of the de novo designs were synthesized. These compounds induced pronounced medulloblastoma cell migration in a phenotypic spheroid invasion assay (SIA).20 RNAi mediated knockdown of CXCR4 abrogated the migration‐inducing effects, indicating the relevance of this chemokine receptor for the observed phenotypic activity. These results corroborate the applicability of constructive machine intelligence to the task of generating new molecules with desired biological activity.
2. Results and Discussion
For computational de novo design of cell migration inducers we employed a pre‐trained RNN model that had successfully been used for generating bioactive new chemical entities in previous studies.10, 11 This model was fine‐tuned by transfer‐learning using a collection of compounds exhibiting CXCR4 antagonistic activity in a variety of in vitro test systems with a potency threshold of <1 μM (“template” collection). As the RNN was pre‐trained on SMILES strings with a maximum length of 74 characters, this cutoff was also applied to the template collection (Figure S1). To promote structural diversity in the computer‐generated molecular structures, the templates were selected to avoid bias towards certain scaffolds. For the most frequently occurring atom scaffolds (Murcko scaffolds)21 (where the scaffolds were contained in at least eight of the known CXCR4 antagonists), the three most active molecules were selected for RNN fine‐tuning, and from the scaffolds with two to eight representatives, the two most active compounds were included. The fine‐tuning set contained 25 structurally diverse molecules with annotated CXCR4 antagonistic bioactivity22, 23, 24, 25, 26, 27, 28, 29, 30 (Table S1). Of these 25 molecules, two had previously been assessed for CXCL12‐induced cell migration inhibition, one of which had an activity (IC50) of 58 nM.22
After transfer learning, a total of 1000 SMILES strings were sampled from the fine‐tuned RNN model, of which 911 were chemically valid and 792 were unique. To prioritize compounds for synthesis and in vitro pharmacological evaluation, we analyzed the pharmacophore similarity of the designs and the templates using the Chemically Advanced Template Search (CATS) descriptors.31 CATS descriptors represent chemical structures by means of topologically cross‐correlated pharmacophore features.31 The de novo molecular designs were ranked according to their decreasing average distance to the 25 template molecules used for model fine‐tuning. Compounds 1 and 2 were selected for synthesis based on their ranking position and building block availability.
Compound 1 was prepared from the commercially available building block 3 in a linear three‐step strategy with 68 % overall yield (Figure 1). A two‐step Staudinger reaction involving the formation of azide 4 from alcohol 3 followed by reduction with triphenylphosphine/water afforded intermediate tetrahydroisoquinoline‐8‐amine (5). Subsequent reaction of 3 with 2‐phenylacetyl chloride (6) yielded compound 1. Since the generative model was not trained to capture stereochemical information, this aspect was also not considered in the synthesis of 1. Compound 2 was synthesized from isocyanate 7 and amine 8 under microwave irradiation in a single step (Figure 1).
Figure 1.
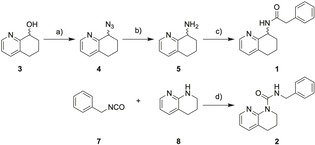
Synthesis of computer‐generated designs 1 and 2. Reagents & conditions: a) MsCl, NaN3, 4‐DMAP, CH2Cl2, DMF, −4 °C→rt, 4 h, 82 %; b) PPh3, H2O, THF, rt, 16 h, 90 %; c) 2‐phenylacetyl chloride (6), 4‐DMAP, THF, 0 °C→rt, 60 min., 92 %. d) neat, μw 80 °C, 1 h, 75 %.
To study the effects of compounds 1 and 2 on cell migration, we employed a phenotypic spheroid invasion assay20 (Figure 2) with a medulloblastoma cell line (DAOY) expressing CXCR4 (Figure S2). In this system, the CXCR4 antagonist AMD3100 induced significant cell migration which was abrogated by siRNA‐mediated silencing of the GPCR. Non‐targeting siRNA had no effect. These results suggest a CXCR4‐dependent pro‐migratory function of AMD3100. Apparently, this effect of AMD3100 is phenocopied when compounds 1 or 2 are applied. Both compounds dose‐dependently induced cell migration with greater efficacy than AMD3100 (Figure 2b).
Figure 2.
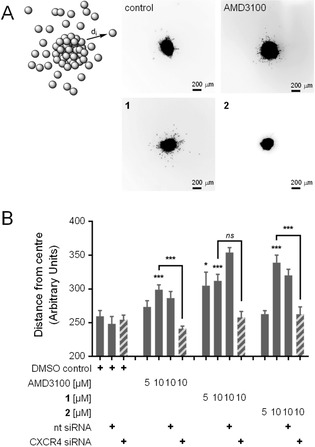
Phenotypic spheroid invasion assay (SIA). (a) Examples of microscopy images showing the effects of compounds 1 and 2, the untreated control, and AMD3100. (b) Activity of computational designs 1 and 2 in the SIA. CXCR4 antagonist AMD3100 for comparison. Cell migration was induced by compounds 1, 2, and the known CXCR4 antagonist AMD3100. This effect was abrogated by siRNA mediated CXCR4 silencing. Non‐targeting (nt) siRNA had no effect. Results are given as mean±S.E.M.; N=3 independent repetitions with n=3 technical replicates each; *p<0.05, **p<0.01, ***p<0.001 (one‐way ANOVA with Kruskal‐Wallis test).
Silencing of CXCR4 by RNAi abolished the activity of AMD3100 as well as designs 1 and 2 (Figure S3), thereby confirming the involvement of this chemokine receptor in the observed effects. Closer inspection of cell migration activators 1 and 2 in a functional assay for CXCR4‐mediated effects on cAMP levels revealed no activity at 50 μM, thus not showing AMD3100’s inhibitory effect on cAMP production. Of note, the RNN fine‐tuning set contained compounds that induce calcium mobilization through CXCR4, and exert anti‐HIV activity. Our in vitro tests, in contrast, were focused on compound effects on intracellular cAMP levels, and on cell mobility. The discrepancy between the observed readouts suggests that, at least partially, other, CXCR4‐dependent pathways are responsible for the observed effects of cell migration activators 1 and 2.
In an attempt to assess the potential co‐involvement of other target proteins in the observed effects of 1 and 2 on cell migration, we predicted off‐targets for both compounds with the SPiDER32 and TIGER33 software programs. These computational tools employ pre‐trained self‐organizing maps to identify pharmacophore similarities in query compounds with known reference ligands of pharmacological targets.33 The predicted targets were assessed for their potential involvement in cell migration or chemotaxis according to the respective Gene Ontology34, 35 definitions. Positively predicted targets with known relevance in cell migration, and which are expressed in the DAOY cells used in the phenotypic assay, were selected for the further in vitro characterization of compounds 1 and 2 (Figure 3, Table S2). While no relevant activities (≥25 % effect at 50 μM) were observed for compound 1, compound 2 slightly modulated the GPCRs somatostatin sst1, dopamine receptor D2S and chemokine receptor CCR10. The most prominent activity of 2 (partial agonism) was observed for the D2S receptor. This preliminary observation is in agreement with the known importance of dopamine D2 receptors in cancer cell migration and invasion36. This working hypothesis is further supported by target predictions with TIGER software for the RNN fine‐tuning set of compounds. Three (12 %) of these CXCR4 modulators were also predicted to interact with dopamine receptors, specifically with D2 and D3 subtypes.
Figure 3.
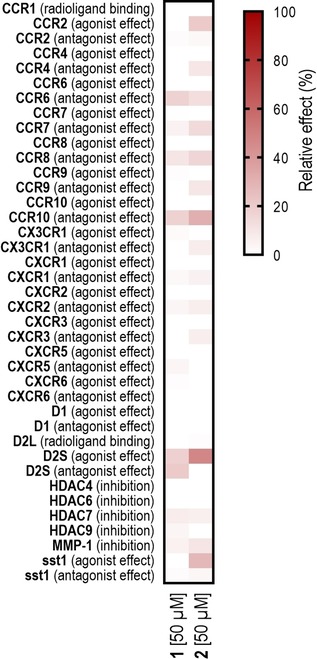
Functional profiling of compounds 1 and 2 on proteins involved in the regulation of cell migration. Data are presented as the mean of two technical replicates. Activities exceeding 25 % were considered relevant. Compound 2 activated dopamine D2S receptor (47 %), antagonized CCR10 (31 %) and activated sst1 receptor (28 %). No relevant activities were observed for compound 1. Individual activity values are presented in Table S2.
3. Conclusions
The neural network employed for compound design was able to capture activity‐relevant features from a collection of template compounds and autonomously construct new molecules with the desired bioactivity. The computer‐generated compounds 1 and 2 were synthetically accessible with standard chemistry in rapid procedures and with high yields. Both tested compounds achieved the desired phenotypic effect and provide starting points for the systematic development of synthetic cell‐migration modulators. Thus, in addition to generating molecules with isofunctional activity on a well‐defined protein target, as shown previously, the machine learning approach was capable of generating de novo designs with isofunctional phenotypic effects. This result further corroborates the potential of constructive machine learning to provide innovative chemical hypotheses for drug discovery.
Experimental Section
Technical implementation. The RNN model was implemented in Python (3.6.1), using Tensorflow (1.4.1) and Keras (2.0.8) libraries. The generative runs were executed on a medium‐grade Linux Workstation (Ubuntu 16.04) equipped with Intel(R) Core(TM) i7‐6850 K CPU (3.60GHz, 32Gb DDR4 RAM), and a NVIDIA 1080 Ti 12GB (ASUS ROG) consumer‐grade GPU. All further calculations were performed on Macintosh Workstation (OS X Yosemite, 10.10.5, 2×2.26 GHz Quad‐Core Intel Xeon with 48GB memory). MOE 2016.08 (The Chemical Computing Group, Montreal, Canada) was used for the standardization of molecules. Murcko scaffolds21 were described using Python (3.5.3) with RDKit (2016.03.4), seaborn (0.7.1), matplotlib (2.0.0) and pandas (0.19.3) libraries.
Recurrent neural network. The general model structure and model training was performed as described in our earlier work.5 Fine‐tuning of the model was performed for five epochs using the template collection of CXCR4 ligands. Stereochemical information was not taken into account during RNN training and fine‐tuning.
Training data. The ChEMBL dataset used for the RNN training is described in ref. 5. For model fine‐tuning, CXCR4 ligands were selected from several sources.8, 9, 22, 23, 24, 25, 26, 27, 28, 29, 30, 37, 38, 39, 40, 41 Compounds were considered if they were reported as active in a functional assay with a potency <1 μM. In case several activities for one compound were reported, the strongest activity was kept. The molecular structures were canonicalized using MOE2016.
CATS descriptor. Chemically advanced template search (CATS) is a 2D descriptor encoding pharmacophoric features dependent on topological distances.31 All computationally generated molecules and scaffolds were standardized to be neutral. CATS descriptors31 were calculated using in‐house software (SpeedCATS, implemented as Knime (2.12.02) node) with default parameterization (distance=10, scaling=’types’).
Scaffold analysis. Molecules were neutralized using MOE 2016.08. Murcko decomposition was performed, keeping the atomic information but generalizing the compound to a framework21. The RDKit library function MurckoScaffold.GetScaffoldForMol() in python3 was used.
SPiDER target prediction. The molecules were standardized to nominal pH 7 (MOE 2016.08). Molecules supplied in SMILES format were converted to molecules using the ’Mol From Smiles’ node, if molecules were stored in SDF format, they were loaded directly. SpeedCats software was used for the CATS descriptor32 calculation and joined in a vector. SpeedCats settings were applied as for the CATS calculations. The respective MOE node was used to calculate the structural descriptors. The MOE‐descriptors were stored in a vector as well and both descriptor vectors selected in the SPIDER node to calculate the predictions. Predictions with a p‐value <0.05 were considered as positive.
TIGER target prediction. Target predictions were performed as described previously.42 Compound structures were neutralized for computational analysis. Targets predicted with TIGER scores>1 were considered meaningful and kept for further analysis.
Chemical synthesis and analytics. All chemicals and solvents were reagent grade and used without further purification, unless specified otherwise. All reactions were conducted in oven‐dried glassware under argon‐atmosphere and in absolute solvents. NMR spectra were recorded on a Bruker AV 400 spectrometer (Bruker Corporation, Billerica, MA, USA). Chemical shifts (δ) are reported in ppm relative to TMS as reference; approximate coupling constants (J) are given in Hertz (Hz). Mass spectra were obtained on an Advion expression CMS (Advion, Ithaka, NY, USA) equipped with an Advion plate express TLC extractor (Advion) using electrospray ionization (ESI). High‐resolution mass spectra were recorded on a Bruker maXis ESI−Qq‐TOF‐MS instrument (Bruker). Compound purity was analyzed by HPLC on a VWR LaChrom ULTRA HPLC (VWR, Radnor, PA, USA) equipped with a MN EC150/3 NUCLEODUR C18 HTec 5 μ column (Machery‐Nagel, Düren, Germany) using a gradient (gradient 1: H2O/MeCN 95 : 5+0.1 % formic acid isocratic for 5 min to H2O/MeCN 5 : 95+0.1 % formic acid after additional 25 min and H2O/MeCN 5 : 95+0.1 % formic acid isocratic for additional 5 min; gradient 2: H2O/MeCN 70 : 30+0.1 % formic acid isocratic for 3 min to H2O/MeCN 5 : 95+0.1 % formic acid after additional 9 min H2O/MeCN 5 : 95+0.1 % formic acid isocratic for additional 2 min) at a flow rate of 0.5 ml min−1, temperature of 27 °C and UV‐detection at 245 nm and 280 nm. All final compounds for biological evaluation had a purity >95 % (area‐under‐the‐curve for UV245 and UV280 peaks).
8‐Azido‐5,6,7,8‐tetrahydroisoquinoline (4)
5,6,7,8‐Tetrahydroquinoline‐8‐ol (3, 746 mg, 5.00 mmol, 1.00 eq) was dissolved in a mixture of methylene chloride (abs., 45 ml) and DMF (abs., 5 ml), and 4‐DMAP (1.22 g, 10.0 mmol, 2.00 eq.) and sodium azide (975 mg, 15.0 mmol, 3.00 eq) were added. The mixture was cooled to −4 °C and methanesulfonyl chloride (780 μl, 1.15 g, 10.0 mmol, 2.00 eq) was added slowly. The mixture was allowed to warm to room temperature and stirred for 4 h. Sodium hydroxide solution (1 M, 100 ml) and hexane (100 ml) were then added, phases were separated and the aqueous layer was extracted with hexane (2×100 ml). The combined organic layers were washed with brine (200 ml), dried over magnesium sulfate, and the solvents were evaporated in vacuum. The crude product was purified by column chromatography using methylene chloride/methanol (97 : 3) as mobile phase to obtain the title compound as yellow oil (715 mg, 82 %). 1H NMR (400 MHz, Chloroform‐d) δ=1.69–2.06 (m, 2H), 2.07–2.23 (m, 2H), 2.27–2.38 (m, 1H), 2.66–2.87 (m, 2H), 7.08 (dd, J=7.8, 4.7 Hz, 1H), 7.33–7.40 (m, 1H), 8.42 (ddd, J=4.7, 1.8, 0.9 Hz, 1H) ppm. 13C NMR (101 MHz, Chloroform‐d) δ=17.45, 28.02, 32.47, 58.98, 123.21, 132.15, 137.55, 147.85, 154.52 ppm. MS (ESI+): m/z 175.4 ([M+H]+).
5,6,7,8‐Tetrahydroquinoline‐8‐amine (5)
8‐Azido‐5,6,7,8‐tetrahydroisoquinoline (4, 696 mg, 4.00 mmol, 1.00 eq) and triphenylphosphine (315 mg, 5.00 mmol, 1.20 eq) were dissolved in THF (40 ml) and water (4 ml) was added. The mixture was stirred at room temperature for 16 h. The solvents were then evaporated in vacuum and the crude product was purified by column chromatography using methylene chloride/methanol (95 : 5) as mobile phase to obtain the title compound as yellow oil (533 mg, 90 %). 1H NMR (400 MHz, Chloroform‐d) δ=1.59–1.78 (m, 2H), 1.85–1.94 (m, 1H), 2.09–2.20 (m, 1H), 2.63–2.81 (m, 2H), 3.95 (dd, J=7.7, 5.4, 1H), 7.00 (ddd, J=7.7, 4.8, 0.7, 1H), 7.30 (ddt, J=7.8, 1.9, 1.0, 1H), 8.34 (ddt, J=4.7, 1.7, 0.8, 1H) ppm. 13C NMR (101 MHz, Chloroform‐d) δ=29.04, 31.96, 39.07, 51.44, 121.76, 129.17, 131.64, 136.80, 149.22 ppm. MS (ESI+): m/z 149.1 ([M+H]+).
2‐Phenyl‐N‐(5,6,7,8‐tetrahydroquinoline‐8‐yl)acetamide (1)
5,6,7,8‐Tetrahydroquinoline‐8‐amine (4, 38 mg, 0.25 mmol, 1.00 eq) was dissolved in THF (abs., 5 ml), 4‐DMAP (61 mg, 0.50 mmol, 2.00 eq) was added, the mixture was cooled to 0 °C and 2‐phenylacetylchloride (5, 43 μl, 50 mg, 0.33 mmol, 1.30 eq.) was added dropwise under stirring. The mixture was allowed to warm and stirred for 60 min at room temperature. Sodium hydroxide solution (1 M, 5 ml) and ethyl acetate (5 ml) were added, phases were separated and the aqueous layer was washed with brine (10 ml). The organic layer was dried over magnesium sulfate and the solvents were evaporated in vacuum. The crude product was purified by column chromatography using methylene chloride/methanol (97 : 3) as mobile phase and subsequently washed with boiling hexane to obtain the title compound as colorless solid (61 mg, 92 %). 1H NMR (400 MHz, DMSO‐d 6) δ=1.69–1.99 (m, 4H), 2.67–2.85 (m, 2H), 3.43 (d, J=14.0 Hz, 1H), 3.47 (d, J=14.0 Hz, 1H), 4.90 (q, J=6.0 Hz, 1H), 7.18–7.26 (m, 2H), 7.27–7.33 (m, 4H), 7.54 (dd, J=7.7, 1.7 Hz, 1H), 8.36–8.46 (m, 2H) ppm. 13C NMR (101 MHz, DMSO‐d 6) δ=19.31, 28.43, 30.14, 42.79, 49.38, 122.77, 126.65, 128.58, 129.44, 133.26, 137.19, 137.30, 147.50, 155.99, 169.61 ppm. MS(ESI+) m/z 267.0 ([M+H]+). HRMS(ESI+) m/z calculated 267.1492 for C17H19N2O found 267.1486 ([M+H]+). HPLC (gradient 1): RT 12.79 min.
N‐Benzyl‐3,4‐dihydro‐1,8‐naphthyridine‐1(2H)‐carboxamide (2)
1,2,3,4‐Tetrahydro‐1,8‐naphthyridine (8, 50 mg, 0.37 mmol; 1.0 eq.) and (isocyanatomethyl)benzene (7, 0.5 ml, 463 mg, 3.5 mmol; 10 eq.) were mixed in a 5 ml microwave vial. The mixture was irradiated for 5 minutes at 50 °C, followed by 1 hour at 80 °C. 1 M sodium hydroxide solution and ethyl acetate were then added, phases were separated, and the aqueous layer was extracted twice with ethyl acetate. The combined organic layers were dried with MgSO4 and concentrated in vacuum. The crude product was purified by column chromatography using hexane/ethyl acetate (6 : 4) as mobile phase to obtain the title compound as colorless oil (74 mg, 75 %). 1H NMR (400 MHz, DMSO‐d 6) δ=1.87–1.78 (m, 2H), 2.79 (t, J=6.3 Hz, 2H), 3.92–3.83 (m, 2H), 4.46 (d, J=5.8 Hz, 2H), 6.96 (dd, J=7.4, 4.9 Hz, 1H), 7.28–7.18 (m, 1H), 7.37–7.28 (m, 4H), 7.60–7.53 (m, 1H), 8.13–8.05 (m, 1H), 10.90 (t, J=5.9 Hz, 1H) ppm.13C NMR (101 MHz, DMSO‐d 6) δ=20.94, 27.51, 43.26, 43.45, 116.94, 121.89, 126.65, 126.98, 128.33, 138.32, 140.10, 143.41, 152.17, 155.77 ppm. HRMS(ESI+) m/z calculated 268.1444 for C17H19N2O found 268.1441 ([M+H]+). HPLC (gradient 2): RT 11.55 min.
Spheroid invasion assay. The spheroid invasion assay (SIA) using the medulloblastoma tumour cell line DAOY was performed as described previously20 to observe effects of 1 and 2 on cell migration. DAOY cells stably expressing lifeact (LA) Enhanced green fluorescent protein (EGFP) produced by lentiviral transduction with pLenti‐LA‐EGFP were used for SIA. In brief, 1000 DAOY LA‐EGPF cells per 100 μl per well were seeded in a 96 well cell‐repellent 96 well microplate (650790, Greiner Bio‐one). The cells were incubated overnight at 37 °C to form spheroids. 70 μl of the medium was removed from each well, and remaining medium with spheroid was overlaid with 2.5 % (final concentration) of ice cold bovine collagen 1 (5005‐B, Advanced BioMatrix, San Diego, CA, USA). The collagen is allowed to polymerize for one hour at 37 °C. Following the polymerization of collagen, fresh serum free medium was added to the cells and then treated with 10 μM (final concentration) of the compounds. The embedded cells were allowed to invade the 3D collagen matrix for 24 hours, after which they were fixed with 4 % PFA and stained with Hoechst. Images were acquired on an Axio Observer 2 mot plus fluorescence microscope (Zeiss, Munich, Germany) using a 5x objective. The extent of cell invasion was determined as the average of the distance invaded by the cells from the center of the spheroid as using automated cell dissemination counter (aCDc). D′Agostino and Pearson normality test followed by unpaired t‐test were performed using GraphPad Prism version 7.00 for Apple (GraphPad Software, La Jolla California USA).
Functional cAMP assay. Compounds were tested in a single‐concentration assay at 50 μM. CXCR4 (86‐0007P), were assayed in functional assays testing their agonistic and antagonistic activity respectively measuring cAMP levels. Assays were provided at a fee‐for‐service basis by DiscoverX (Fremont, CA, USA).
Target panel screening. Compounds were tested in a single‐concentration assay at 50 μM. CCR2 (2497, 2501), CCR4 (4229, 4230), CCR6 (4231, 4232), CCR7 (4233, 4234), CCR8 (4235, 4236), CCR9 (4237, 4238), CCR10 (4227, 4228), CX3CR1 (4239, 4240), CXCR1 (4241, 4242), CXCR2 (4243, 4244), CXCR3 (4245, 4246), CXCR5 (4247, 4248), CXCR6 (4249, 4250), D1 (1685, 1686), D2 S (2566, 2569), sst1 (2253, 2254) were assayed in functional assays testing their agonistic and antagonistic activity respectively. CCR1 (361), and D2 L (1405) were assayed in binding assays using scintillation counting. MMP‐1 (510), HDAC4 (2493), HDAC6 (2495), HDAC7 (2610), and HDAC9 (2611) were assayed in enzyme‐based assays using fluorimetry. Assays were provided at a fee‐for‐service basis by Cerep (Celle l′Evescault, France).
Conflict of interest
The authors declare the following competing interests: G.S. declares a potential financial conflict of interest in his role as life science industry consultants and cofounder of inSili.com GmbH, Zurich. D.M. was financially supported by an ETH Zurich Postdoctoral Fellowship (grant no. 16–2 FEL‐07).
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank Lukas Friedrich, Berend Huisman, Claudia Neuhaus and Sarah Haller for technical support. The authors thank Ryan Byrne for proof reading of the manuscript. This research was financially supported by the Swiss National Science Foundation (grant no. CR3212_159737 to G.S.), the Novartis Forschungsstiftung (FreeNovation: AI in Drug Discovery), and the ETH RETHINK initiative. inSili.com GmbH kindly provided access to the TIGER software.
D. Bruns, D. Merk, K. Santhana Kumar, M. Baumgartner, G. Schneider, ChemistryOpen 2019, 8, 1303.
Contributor Information
PD Dr. Martin Baumgartner, Email: martin.baumgartner@kispi.uzh.ch.
Prof. Dr. Gisbert Schneider, Email: gisbert.schneider@pharma.ethz.ch.
References
- 1. Schneider G., Nature Mach. Intell. 2019, 1, 128–130. [Google Scholar]
- 2. Hessler G., Baringhaus K. H., Molecules 2018, 23, 2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Schneider G., Clark D., Angew. Chem. 2019, 131, 10906–10917; [Google Scholar]
- 3b. Schneider G., Clark D., Angew. Chem. Int. Ed. 2019, 58, 10792–10803. [DOI] [PubMed] [Google Scholar]
- 4. Olivecrona M., Blaschke T., Engkvist O., Chen H., J. Cheminform. 2017, 9, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gupta A., Mueller A. T., Huisman B. J. H., Fuchs J. A., Schneider P., Schneider G., Mol. Inf. 2018, 37, 1700111. [Google Scholar]
- 6. Segler M. H. S., Kogej T., Tyrchan C., Waller M., ACS Cent. Sci. 2018, 4, 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hochreiter S., Schmidhuber J., Neural Comput. 1997, 9, 1735–1780. [DOI] [PubMed] [Google Scholar]
- 8. Gaulton A., Bellis L. J., Bento A. P., Chambers J., Davies M., Hersey A., Light Y., McGlinchey S., Michalovich D., Al-Lazikani B., Overington J. P., Nucleic Acids Res. 2012, 40, D1100–D1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bento A. P., Gaulton A., Hersey A., Bellis L. J., Chambers J., Davies M., Krüger F. A., Light Y., Mak L., McGlinchey S., Nowotka M., Papadatos G., Santos R., Overington J. P., Nucleic Acids Res. 2014, 42, D1083–D1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Merk D., Grisoni F., Friedrich L., Schneider G., Commun. Chem. 2018, 1, 68. [Google Scholar]
- 11. Merk D., Friedrich L., Grisoni F., Schneider G., Mol. Inf. 2018, 37, 1700153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Clercq E., Biochem. Pharmacol. 2009, 77, 1655–1664. [DOI] [PubMed] [Google Scholar]
- 13. Rubin J. B., Semin. Cancer Biol. 2009, 19, 116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sachpatzidis A., Benton B. K., Manfredi J. P., Wang H., Hamilton A., Dohlman H. G., Lolis E., J. Biol. Chem. 2003, 278, 896–907. [DOI] [PubMed] [Google Scholar]
- 15. Levoye A., Balabanian K., Baleux F., Bachelerie F., Lagane B., Blood 2009, 113, 6085–6093. [DOI] [PubMed] [Google Scholar]
- 16. Murphy P. M., Annu. Rev. Immunol. 1994, 12, 593–633. [DOI] [PubMed] [Google Scholar]
- 17. Ma Q., Jones D., Borghesani P. R., Segal R. A., Nagasawa T., Kishimoto T., Bronson R. T., Springer T. A., Proc. Natl. Acad. Sci. USA 1998, 95, 9448–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mantovani A., Immunol. Today 1999, 20, 254–257. [DOI] [PubMed] [Google Scholar]
- 19. Burger J. A., Kipps T. J., Blood 2006, 107, 1761–1767. [DOI] [PubMed] [Google Scholar]
- 20. Kumar K. S., Pillong M., Kunze J., Burghardt I., Weller M., Grotzer M. A., Schneider G., Baumgartner M., Sci. Rep. 2015, 5, 15338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bemis G. W., Murcko M. A., J. Med. Chem. 1996, 39, 2887–2893. [DOI] [PubMed] [Google Scholar]
- 22. Thoma G., Beerli C., Bigaud M., Bruns C., Cooke N. G., Streiff M. B., Zerwes H.-G., J. Med. Chem. 2008, 51, 7915–7920. [DOI] [PubMed] [Google Scholar]
- 23. Skerlj R. T., Bridger G. J., Kaller A. I., McEachern E. J., Crawford J. B., Zhou Y., Atsma B., Langille J., Nan S., Veale D., Wilson T., Harwig C., Hatse S., Princen K., De Clercq E., Schols D., J. Med. Chem. 2010, 53, 3376–3388. [DOI] [PubMed] [Google Scholar]
- 24. Miller J. F., Gudmundsson K. S., D′Aurora Richardson L., Jenkinson S., Spaltenstein A., Thomson M., Wheelan P., Bioorg. Med. Chem. Lett. 2010, 20, 3026–3030. [DOI] [PubMed] [Google Scholar]
- 25. Skerlj R., Bridger G., McEachern E., Harwig C., Smith C., Wilson T., Veale D., Yee H., Crawford J., Skupinska K., Wauthy R., Yang W., Zhu Y., Bogucki D., Di Fluri M., Langille J., Huskens D., De Clercq E., Schols D., Bioorg. Med. Chem. Lett. 2011, 21, 262–266. [DOI] [PubMed] [Google Scholar]
- 26. Skerlj R., Bridger G., McEachern E., Harwig C., Smith C., Kaller A., Veale D., Yee H., Skupinska K., Wauthy R., Wang L., Baird I., Zhu Y., Burrage K., Yang W., Sartori M., Huskens D., De Clercq E., Schols D., Bioorg. Med. Chem. Lett. 2011, 21, 1414–1418. [DOI] [PubMed] [Google Scholar]
- 27. Truax V. M., Zhao H., Katzman B. M., Prosser A. R., Alcaraz A. A., Saindane M. T., Howard R. B., Culver D., Arrendale R. F., Gruddanti P. R., Evers T. J., Natchus M. G., Snyder J. P., Liotta D. C., Wilson L. J., ACS Med. Chem. Lett. 2013, 4, 1025–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao J., Huang Y., Liu D., Chen Y., Bioorg. Med. Chem. Lett. 2015, 25, 4950–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang H., Kang D., Huang B., Liu N., Zhao F., Zhan P., Liu X., Eur. J. Med. Chem. 2016, 114, 65–78. [DOI] [PubMed] [Google Scholar]
- 30. Das D., Maeda K., Hayashi Y., Gavande N., Desai D. V., Chang S. B., Ghosh A. K., Mitsuya H., Antimicrob. Agents Chemother. 2015, 59, 1895–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reutlinger M., Koch C. P., Reker D., Todoroff N., Schneider P., Rodrigues T., Schneider G., Mol. Inf. 2013, 32, 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reker D., Rodrigues T., Schneider P., Schneider G., Proc. Natl. Acad. Sci. USA 2014, 111, 4067–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schneider G., Schneider P., Expert Opin. Drug Discovery 2017, 12, 271–277. [DOI] [PubMed] [Google Scholar]
- 34.The Gene Ontology Consortium, Nucleic Acids Res 2017, 45, D331–D338. [DOI] [PMC free article] [PubMed]
- 35. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G. M., Ashburner M., Nat. Genet. 2000, 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang H., Wu K., Ma J., Du Y., Cao C., Nie Y., Int. Immunopharmacol. 2016, 39, 113–120. [DOI] [PubMed] [Google Scholar]
- 37. Mysinger M. M., Weiss D. R., Ziarek J. J., Gravel S., Doak A. K., Karpiak J., Heveker N., Shoichet B. K., Volkman B. F., Proc. Natl. Acad. Sci. USA 2012, 109, 5517–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oishi S., Kuroyanagi T., Kubo T., Montpas N., Yoshikawa Y., Misu R., Kobayashi Y., Ohno H., Heveker N., Furuya T., Fujii N., J. Med. Chem. 2015, 58, 5218–5225. [DOI] [PubMed] [Google Scholar]
- 39. Zachariassen Z. G., Thiele S., Berg E. A., Rasmussen P., Fossen T., Rosenkilde M. M., Våbenø J., Haug B. E., Bioorg. Med. Chem. 2014, 22, 4759–4769. [DOI] [PubMed] [Google Scholar]
- 40. Zachariassen Z. G., Karlshøj S., Haug B. E., Rosenkilde M. M., Våbenø J., J. Med. Chem. 2015, 58, 8141–8153. [DOI] [PubMed] [Google Scholar]
- 41. Ha H., Debnath B., Odde S., Bensman T., Ho H., Beringer P. M., Neamati N., J. Chem. Inf. Model. 2015, 55, 1720–1738. [DOI] [PubMed] [Google Scholar]
- 42.
- 42a. Schneider P., Schneider G., Angew. Chem. 2017, 129, 11678–11682; [Google Scholar]; Angew. Chem. Int. Ed. 2017, 56, 11520–11524; [DOI] [PubMed] [Google Scholar]
- 42b. Schneider P., Schneider G., Angew. Chem. Int. Ed. 2017, 56, 11520–11524; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11678–11682. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary