

Abstract
Inhibition of phosphodiesterase 4 (PDE4) produces neuroprotective effects against cerebral ischemia. However, the involved mechanism remains unclear. Augmentation of endoplasmic reticulum (ER) stress promotes neuronal apoptosis, and excessive oxidative stress is an inducer of ER stress. The present study aimed to determine whether suppression of ER stress is involved in the protective effects of PDE4 inhibition against cerebral ischemia. We found that exposing HT-22 cells to oxygen-glucose deprivation (OGD) significantly activated ER stress, as evidenced by increased expression of the 78-kDa glucose-regulated protein (GRP78), phosphorylated eukaryotic translation-initiation factor 2α (eIF2α), and C/EBP-homologous protein (CHOP). Overexpression of PDE4B increased ER stress, while knocking down PDE4B or treatment with the PDE4 inhibitor, FCPR03, prevented OGD-induced ER stress in HT-22 cells. Furthermore, FCPR03 promoted the translocation of nuclear factor erythroid 2-related factor 2 (Nrf-2) from the cytoplasm to the nucleus. Importantly, the Nrf-2 inhibitor, ML385, blocked the inhibitory role of FCPR03 on OGD-induced ER stress. ML385 also abolished the protective role of FCPR03 in HT-22 cells subjected to OGD. Knocking down heme oxygenase-1 (HO-1), which is a target of Nrf-2, also blocked the protective role of FCPR03, enhanced the level of reactive oxygen species (ROS), and increased ER stress and cell death. We then found that FCPR03 or the antioxidant, N-Acetyl-l-cysteine, reduced oxidative stress in cells exposed to OGD. This effect was accompanied by increased cell viability and decreased ER stress. In primary cultured neurons, we found that FCPR03 reduced OGD-induced production of ROS and phosphorylation of eIF2α. The neuroprotective effect of FCPR03 against OGD in neurons was blocked by ML385. These results demonstrate that inhibition of PDE4 activates Nrf-2/HO-1, attenuates the production of ROS, and thereby attenuates ER stress in neurons exposed to OGD. Additionally, we conclude that FCPR03 may represent a promising therapeutic agent for the treatment of ER stress-related disorders.
Keywords: PDE4, Cerebral ischemia, Nrf-2, Oxidative stress, ER stress
Graphical abstract
Highlights
-
•
Overexpression of PDE4 increased ER stress under both basal and OGD conditions.
-
•
Inhibition of PDE4 reduced ER stress and neuronal apoptosis in neurons exposed to OGD.
-
•
PDE4 inhibition activated Nrf-2, and increased the level of antioxidant enzyme HO-1.
-
•
Inhibition of Nrf-2 attenuated the role of FCPR03 on ER stress and cell viability.
-
•
HO-1 knockdown abolished the effects of FCPR03 on ER stress and ROS production.
1. Introduction
Cerebral ischemic stroke is a prevalent neurological disease that is usually caused by cerebrovascular occlusion, which leads to an interrupted or reduced supply of blood to the brain. The incidence of ischemic stroke has increased globally each year and is the second-leading cause of death and the third-leading cause of disability [1]. Currently, recombinant tissue plasminogen activator (r-tPA) is the only drug approved by the US FDA for stroke treatment. However, r-tPA needs to be injected within 4.5 h of stroke onset. Additionally, administration of r-tPA tends to aggravate preconditions of aberrant bleeding [2]. All of these factors have limited the clinical application of r-tPA. Hence, it is necessary to further investigate the mechanisms responsible for neuronal injury caused by ischemia/reperfusion and to explore effective interventions based on specific targets.
Phosphodiesterase 4 (PDE4) is an enzyme that specifically hydrolyzes cyclic adenosine monophosphate (cAMP) and, thus, regulates the concentration of intracellular cAMP [3]. PDE4 is highly expressed in neurons, inflammatory cells, and epithelial cells, and is involved in diverse pulmonary, dermatological, and central nervous system diseases [3]. Inhibition of PDE4 in these cells leads to the accumulation of cAMP, which concomitantly activating protein kinase A (PKA) and the exchange protein directly activated by cAMP (Epac). PKA is the upstream protein kinase of the cAMP-response element binding protein (CREB)/brain-derived neurotrophic factor (BDNF) signaling pathway, and Epac is involved in the activation of protein kinase B (Akt) [4,5]. PDE4 has been implicated in a number of physiological and pathological processes through regulating various signaling pathways that allow cells to respond to various stimuli [6,7]. Our previous studies have indicated that inhibition of PDE4 enhances intracellular concentrations of cAMP, protects against neuronal apoptosis and limits neuroinflammation in the central nervous system [[8], [9], [10], [11]]. In recent years, the role of PDE4 in the development of ischemic stroke has aroused extensive attention [12,13]. The first-generation PDE4 inhibitor rolipram has been used as a tool to study the role of PDE4 in ischemic brain injury. The results have shown that inhibition of PDE4 reduces the damage of the blood-brain barrier, the release of inflammatory factors, and neuronal apoptosis [14]. Rolipram has also been shown to significantly increase the survival of newborn hippocampal neurons after ischemia [15]. In one of our earlier studies, we confirmed that inhibition of PDE4 ameliorated learning and memory deficits in rats subjected to global cerebral ischemia [16]. Our recent study revealed that inhibition of PDE4 by the novel PDE4 inhibitor FCPR03 alleviated brain damage and improved motor functions in rats following cerebral ischemia/reperfusion injury [17]. These data suggest that inhibition of PDE4 is a potential therapeutic strategy to reduce brain injury following cerebral ischemia, although the underlying mechanisms remain largely unknown.
Cerebral ischemia/reperfusion triggers a complicated series of pathological events that leads to neuronal damage. Despite decades of intense research, our understanding of the causal pathways of post-ischemic neuronal death remains incomplete. Recently, the involvement of endoplasmic reticulum (ER) stress has aroused extensive attention [18,19]. The ER is an important organelle where secreted and transmembrane proteins are synthesized and correctly folded into three-dimensional conformations. The above dynamic process requires highly precise regulation to maintain protein homeostasis [20]. Any stimuli that break the above equilibrium will trigger the unfolded protein response (UPR) and the occurrence of ER stress [21]. Under physiological conditions, ER-transmembrane signaling molecules, such as protein kinase-like endoplasmic reticulum kinase (PERK), is inactive through binding to the 78-kDa glucose-regulated protein (GRP78), which serves as an ER stress sensor [22]. Under the condition of ER stress, GRP78 is released from PERK. PERK is then autophosphorylated and activated. Activated PERK phosphorylates the eukaryotic translation-initiation factor 2α (eIF2α) to attenuate further protein synthesis [23]. This process is beneficial for the survival of cells [24]. However, when the duration of stimuli is too long or the stress is too severe, over-activated ER stress promotes the expression of pro-apoptotic proteins, including C/EBP-homologous protein, (CHOP) [24]. Ultimately, under these conditions, cells undergo apoptosis. Collectively, ER stress is presumed to be a pro-survival event under mild to moderate stress conditions, while prolonged periods of ER stress trigger apoptosis.
A number of pathologic conditions lead to acute ER stress and activate the UPR, including nutrient deprivation, calcium overloading and overproduction of free radicals [25]; all of these conditions are viewed as contributors to neuronal damage induced by cerebral ischemia. Current studies support that ER stress is an essential step in the progression of cerebral ischemia-induced neuronal injury, and modulation of ER stress exerts protective effects against ischemic stroke and offers the prospect of novel stroke therapies [26]. Our previous study showed that inhibition of PDE4 reduced neuronal cell death and improved motor functions in an animal model of middle cerebral artery occlusion (MCAO) [17]. Importantly, we found that the PDE4 inhibitor FCPR03 suppressed the excessive production of ROS in neurons subjected to oxygen-glucose deprivation (OGD) [17]. ROS is an inducer of ER stress. Ischemia/reperfusion leads to substantial production of ROS, and excessive ROS causes oxidative damage to lipids and proteins. Aggregation of oxidized proteins aggravates the misfolding or unfolding of proteins, which contributes to the occurrence of ER stress [27]. Interestingly, a recent study indicated that deletion of PDE4 reduced ER stress in neurons and promoted the recovery of hindlimb locomotion following spinal cord injury in mice [28]. Inspired by these findings, we sought to determine whether inhibition of PDE4 exerts protective effects through inhibiting ER stress in neurons. In the present study, we hypothesized that specific inhibition of PDE4 would protect neurons against OGD-induced cell death. We also hypothesized that the protective effects of PDE4 inhibition would be mediated through suppressing the production of ROS and the subsequent activation of ER stress. In the present study, we exposed HT-22 neuronal cells and primary cultured neurons to OGD and examined the expression of canonical ER stress markers, ROS production, apoptosis, and potential signaling pathways. We also verified the role of PDE4 inhibition in rats subjected to MCAO. Collectively, we demonstrate that PDE4 played an important role in the development of ER stress and concomitant neuronal death following OGD. Importantly, we also found that the novel PDE4 inhibitor FCPR03 was a potent neuroprotective agent against cerebral-ischemia-induced brain insults.
2. Materials and methods
2.1. Materials
Antibodies against caspase-3 (#9662), phosphorylated PERK (p-PERK, Thr980, #3179), PERK (#3192), phosphorylated eIF2α (p-eIF2α, Ser51, #3597), eIF2α (#2103), GRP78 (#3183), CHOP (#2895) and HO-1 (#43966) were obtained from Cell signing technology corporation (Massachusetts, USA). Anti-β-tubulin (#ab6046), anti-β-actin (#ab8226) and anti-Nrf-2 (#ab137550) were purchased from Abcam (Cambridge, USA). Goat anti-Rabbit IgG DyLight 488 (#A23220) and Goat anti-Rabbit IgG Dylight 549 (#A23320) were obtained from Abbkine (Abbkine Scientific Co., Ltd, Wuhan, China). Pierce™ Rapid Gold BCA Protein Assay Kit (#A53225), high glucose DMEM (#11965092), glucose-free DMEM (#11966025), fetal bovine serum (FBS, #10099141), Neurobasal-A (#10888022), B27 (#17504044), glutamine (#25030081), Lipofectamine™ 2000 Transfection Reagent (#11668019), Opti-MEM® Reduced-Serum Medium (#31985088), Live Cell Imaging Solution (#A14291DJ), and CellROX Deep Red Reagent (#C10422) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). FCPR03 was obtained from Southern medical university (Guangzhou, Guangdong, China) [29,30]. ML385 (#T4360) and N-Acetyl-l-cysteine (NAC, #T0875) were purchased from TargetMol (Wellesley, MA, USA). Protoporphyrin IX zinc (II) (ZnPP, #MB4231) was purchased from Meilun Biotechnology (Dalian, Shenyang, China). Small interfering RNAs (si-RNAs) specific for HO-1 and PDE4B were obtained from GenePharma (Shanghai, China). PDE4B plasmid was constructed by Shuangquan Biotechnology (Guangzhou, Guangdong, China). Lipid peroxidation malondialdehyde (MDA) Assay Kit (#S0131) and Nuclear and Cytoplasmic Protein Extraction Kit (#P0027) were purchased from Beyotime Biotechnology (Shanghai, China). Protease inhibitor cocktail (#FD1001), phosphatase inhibitor cocktail (#FD1002) and RIPA buffer (#FD009) were obtained from Fude Biological Technology (Hangzhou, Zhejiang, China). Immoblilon PVDF membranes (#ISEQ00010) and Immobilon Western Chemiluminescent HRP Substrate (#WBKLS0100) were purchased from Merck Co., (Darmstadt, Germany). The Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Molecular Technologies (#CK04; Tokyo, Japan).
2.2. Cell culture
HT-22 hippocampal neuronal cells were purchased from Merck (Darmstadt, Germany) and were cultured in DMEM containing 10% FBS [17]. The cells were cultivated at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air.
For the culturing of primary cortical neurons, 12 newborn one-day-old Sprague-Dawley (SD) rats were used. Cultivation of primary cortical neurons were obtained from the cerebral cortices of one-day-old SD rats. After decapitation, the cerebral cortices were dissected and digested with 0.125% trypsin for 5 min at 37 °C. Then, the digestion was terminated by adding a complete medium and cells were dispersed with a Pasteur pipette and seeded in plates or glass-bottom Petri dishes coated with poly-l-lysine. Then, 4 h later, the medium was replaced with Neurobasal-A contained 2% B27 and 1% glutamine.
2.3. Oxygen and glucose deprivation cell model
Oxygen and glucose deprivation were performed in HT-22 neuron cell lines and neurons as previously reported [17]. Briefly, cells were washed once with PBS and the medium was then replaced with glucose-free DMEM. Then, cells were transferred to an anaerobic chamber (Billups-Rothenberg, Del Mar, CA, USA) containing a mixture gas composed of 95% N2 and 5% CO2, while the control group was maintained in an aerobic environment (95% air and 5% CO2). After OGD, cells were returned to aerobic environment and the medium was replaced with DMEM containing glucose.
2.4. Cell viability assay
Cell viability was determined by using the Cell Counting Kit-8 (CCK-8). For HT-22 cells, cells were seeded in 48-well plates at the density of 8,000 cells per well. The next day, after pretreatment with FCPR03 for 1 h, HT-22 cells were subjected to 6 h of OGD followed by 24 h of re-oxygenation. Then, 20 μl of CCK-8 was added per well and incubated at 37 °C. Finally, the absorbance at 450 nm was detected by a microplate reader (Biotech, Winooski, VT, USA).
For primary cortical neurons, cells were seeded in 48-well plates at a density of 40,000 cell per well. The processing was conducted at 7–10 day after seeding. Briefly, neurons, pretreated with FCPR03, were washed with PBS and the medium was changed to DMEM without glucose. After 1 h of OGD, cells were transferred to neurobasal-A medium for 24 h of re-oxygenation. Then, 20 μl of CCK-8 was added per well and incubated at 37 °C. Finally, the absorbance at 450 nm was detected by a microplate reader.
2.5. Extraction of cytoplasmic and nuclear protein
After the treatment described above, cytosolic and nuclear proteins were detached by using the Nuclear and Cytoplasmic Protein Extraction Kit. After protein quantification, the variation of Nrf-2 expression was detected via Western blotting.
2.6. Transfection of small interfering RNA (siRNA)
Knockdown of PDE4B and HO-1 was conducted by using specific siRNA synthesized by GenePharma (Shanghai, China). The sequences of si-RNAs for HO-1 were as follows: sense: 5′-CAGAUCAGCACUAGCUCAUTT-3′, antisense: 5′-AUGAGCUAGUGCUGAUCUGTT-3’. The sequences of siRNA for PDE4B were as follows: sense: 5′-CCUGCAAGAAGAAUCAUAUTT-3′, antisense: 5′-AUAUGAUUCUUCUUGCAGGTT-3’. Briefly, cells were grown to 80–90% confluency at the time of transfection. The siRNAs and lipofectamine 2000 transfection reagent were mixed in opti-MEM for 20 min. Then, HT-22 cells were incubated in these mixtures. At 6 h following the transfection, the medium was changed to DMEM containing 1% FBS. Then, 18–24 h later, cells underwent OGD.
2.7. PDE4B plasmid transfection
Overexpression of PDE4B was conducted by using a PDE4B plasmid. One day before transfection, HT-22 cells were seeded in 12-well-plates at a density of 80–90% confluency. The following day, plasmid-transfection complexes were prepared. Specifically, 1 μg of the PDE4B plasmid and the vector were transfected into HT-22 cells with 3 μl of Lipofectamine 2000, according to the manufacturer's protocol. Subsequently, 8–10 h following the transfection, the medium was changed to DMEM containing 1% FBS. Finally, 18–24 h later, cells underwent OGD.
2.8. Detection of reactive oxygen species (ROS)
HT-22 cells were seeded overnight in glass-bottom petri dishes at a density of 70% confluency. HT-22 cells, pretreated with FCPR03 or a vehicle, were then subjected to OGD. After 6 h of OGD, the HT-22 cells were incubated with CellROX Deep Red Reagent for 30 min in DMEM. Then, the medium was replaced with Live Cell Imaging Solution. Finally, intracellular ROS was measured with a confocal microscope (Nikon ECLIPSE Ti, Tokyo, Japan). The average fluorescent intensity was analyzed by Image J (NIH, Bethesda, Maryland, USA).
Primary cultured cortical neurons were processed at the seventh day after seeding. Primary cultured neurons, pretreated with FCPR03 or a vehicle, were subjected to OGD. After 1 h of OGD, primary cultured cortical neurons were incubated with CellROX Deep Red Reagent for 30 min in neurobasal-A. Then, cells were fixed with 4% paraformaldehyde for 15 min. Finally, intracellular ROS was measured with a confocal microscope. The average fluorescent intensity was analyzed by Image J.
2.9. Detection of malondialdehyde (MDA) level
The MDA level was measured with the Lipid Peroxidation MDA Assay Kit (#S0131, Beyotime). In brief, after the treatment described above, HT-22 cells were washed once with PBS and were then lysed. The lysates were centrifugated at 12000 rpm for 10 min. Then, the supernatant was collected for the detection of MDA. Finally, the absorbance at 532 nm was detected by a microplate reader (Biotech, Winooski, VT, USA) and the MDA level was normalized by the protein concentration in each sample.
2.10. Immunohistochemistry
For the visualization of p-eIF2α and Nrf-2, immunofluorescent experiments were performed. HT-22 cells were seeded in glass-bottom petri dishes. After treatment, cells were fixed with 4% paraformaldehyde and were then permeabilized with 0.5% Triton X-100. Then, non-specific binding sites were blocked with 5% BSA in PBS for 1 h and cells were then incubated with either a p-eIF2α antibody (1:200) or a Nrf-2 antibody (1:200) at 4 °C overnight. After washing three times with PBST (5 min each time), cells were incubated with the secondary conjugated antibodies, DyLight 488 or Dylight 549, at 4 °C for 4 h. Then, the cells were washed three times with PBST. Finally, the cells were counterstained with DAPI and imaged with a confocal microscope (Nikon ECLIPSE Ti, Tokyo, Japan). For the quantification of Nrf-2 in the nucleus, the green fluorescence intensity (Nrf-2) in the nucleus was analyzed using Image J software.
2.11. Western blotting
After cells were subjected to OGD or control conditions, cells were frozen at −80 °C overnight and were then lysed in RIPA buffer (#FD009, FUDE) containing 1% protease inhibitor (#FD1001, FUDE) and 1% phosphatase inhibitor (#FD1002, FUDE) on ice. Then, the lysates were ultrasound pyrolysed and then centrifuged. The supernatant was collected and the concentration of protein was measured by using a BCA protein assay kit (#A53225, Thermo Fisher Scientific). After normalization based on the protein level in each sample, the lysates were boiled at 100 °C in a dry bath incubator. Protein extracts were separated by SDS-polyacrylamide gel electrophoresis (8, 10, and 12%) at 80 V and were transferred to 0.22-μm PVDF membranes (#ISEQ00010, Merck) in an ice-water bath at 100 V for 100 min. Then, the PVDF membranes were blocked with 5% skim milk for 1 h at room temperature and were then incubated with primary antibodies overnight at 4 °C. After incubation with primary antibodies, the membranes were washed four times with TBST (5 min each time) and incubated with secondary antibodies conjugated with HRP for 4 h at 4 °C followed by four washes with TBST (5 min each time). Finally, protein bands were detected using the Western Chemiluminescent HRP Substrate and were either scanned with a multifunctional imaging analysis system (ProteinSimple, California, USA) or exposed to X-ray films. Densitometric analysis was conducted by using Image J.
2.12. Statistical analysis
All of the data are expressed as the mean ± standard deviation (SD) and were analyzed by analysis of variance followed by the Tukey's post-hoc test. Data were analyzed using SPSS 20.0 (SPSS Inc., Armonk, NY, USA). The figures were plotted by GraphPad Prism 8.0.1 (GraphPad Software, La Jolla, CA, USA). Significance was defined as P < 0.05. Each experiment was conducted at least in triplicate.
3. Results
3.1. OGD treatment induces significant ER stress in HT-22 neuronal cells
First, an in vitro model of cerebral ischemia in HT-22 neuronal cells was established by OGD. Cells were exposed to OGD for various durations (1, 3, and 6 h). We then investigated the stimulatory roles of OGD on ER stress. GRP78, p-PERK, p-eIF2α and CHOP were used as indicators of ER stress. Western blotting assay was employed to measure the levels of the above proteins. Treatment with OGD time-dependently increased the expression of GRP78 (Fig. 1A and B), p-PERK (Fig. 1C and D), p-eIF2α (Fig. 1E and F), and CHOP (Fig. 1G and H) in HT-22 cells. We also assayed the cell viability of HT-22 cells. Cells were subjected to OGD for 1–6 h, followed by reperfusion for 24 h, and a CCK-8 assay was applied to determine cell viability. We found that treatment of HT-22 cells with OGD for 1, 3 or 6 h followed by 24 h of reperfusion induced significant decreases in cell survival (Fig. 1I). These data suggest that significant ER stress occurred in the early stage of OGD, and that this phenomenon was accompanied by decreased cell viability.
Fig. 1.

OGD-induced ER-stress and loss of cell viability in HT-22 cells. (A, C, E, G) HT-22 cells were subjected to 1, 3, or 6 h of OGD. The levels of GRP78, p-PERK, PERK, p-eIF2α, eIF2α, and CHOP were detected by Western blotting. (B, D, F, H) The relative levels of GRP78/β-tubulin (n = 3), p-PERK/PERK (n = 3), p-eIF2α/eIF2α (n = 3), and CHOP/β-tubulin (n = 3) were measured by semiquantitative analysis of the Western blots. (I) HT-22 cells were subjected to 1, 3, or 6 h of OGD followed by 24 h of re-oxygenation, and cell viability was detected by a CCK-8 assay (12 duplications from three independent experiments, n = 3). Results are expressed as the mean ± SD, **P < 0.01 versus the indicated group.
3.2. Overexpression of PDE4B enhances ER stress in HT-22 cells
We next examined the effects of PDE4 on ER stress. PDE4B is highly expressed in neurons. Our previous study showed that knocking down PDE4B exerted protective effects in neurons [9]. In the present study, we first transfected a PDE4B plasmid and a corresponding empty vector into HT-22 cells using lipofectamine 2000. PDE4B overexpression was examined by Western blotting (Fig. 2A and B). Under the condition of normoxia, PDE4B overexpression significantly increased the levels of phosphorylated eIF2α and GRP78 in HT-22 cells (Fig. 2C–E), suggesting that ER stress was activated. We then further investigated the effect of PDE4B overexpression on ER stress under OGD. Similarly, overexpression of PDE4B under OGD also increased the expression of ER-stress-related proteins (Fig. 2F–H). These findings suggest that PDE4 promoted the occurrence of ER stress in neuronal cells under both normoxia and OGD conditions.
Fig. 2.

Overexpression of PDE4B exacerbates OGD-triggered ER stress in HT-22 cells. (A, B) HT-22 cells were transfected with an empty plasmid (vector) or PDE4B plasmid. At 24 h after transfections, the overexpression of PDE4B was verified by Western blotting. The relative level of PDE4B/β-tubulin was measured by semiquantitative analysis of the Western blots (n = 3). (C, D, E) HT-22 cells were transfected with an empty vector or a PDE4B plasmid. At 24 h after transfections, the protein levels of GRP78, p-eIF2α, and eIF2α were determined by Western blotting. The relative levels of GRP78/β-tubulin and p-eIF2α/eIF2α were measured by semiquantitative analysis of the Western blots (n = 3). (F, G, H) HT-22 cells transfected with an empty vector or a PDE4B plasmid were subjected to 3 h of OGD, and protein levels of GRP78, p-eIF2α, and eIF2α were determined by Western blotting. The relative levels of GRP78/β-tubulin and p-eIF2α/eIF2α were measured by semiquantitative analysis of the Western blots (n = 4). Results are expressed as the mean ± SD, **P < 0.01 versus the indicated group.
3.3. PDE4 knockdown inhibits OGD-triggered ER stress in HT-22 cells
The above results clarified that overexpression of PDE4B promoted ER stress. Next, we further knocked down the expression of PDE4B through transfecting cells with PDE4B-specific siRNA and investigated the proteins related to ER stress and apoptosis in HT-22 cells. The knock-down efficiency was confirmed by Western blotting in our previous study [9]. We found that the expression of p-eIF2α, GRP78, and CHOP decreased in the cells after knocking down PDE4B (Fig. 3A–F). We then investigated cell viability in OGD-treated HT-22 cells transfected with control siRNA or PDE4B siRNA. We found that transfection with PDE4B siRNA alone had no effect on cellular survival. OGD treatment decreased cell viability to 54.64 ± 5.31% (P < 0.01), while PDE4 siRNA increased cell viability to 70.11 ± 12.2% (P < 0.01, Fig. 3G). Correspondingly, OGD increased the expression of cleaved caspase 3 (Fig. 3H and I), while knockdown of PDE4B inhibited the level of intracellular cleaved caspase 3 (Fig. 3H and I). These results indicate that knocking-down the expression of PDE4B suppressed ER stress and decreased cellular apoptosis in neuronal cells subjected to OGD.
Fig. 3.

Silencing of PDE4B ameliorates OGD-triggered ER stress in HT-22 cells. (A, C, E) HT-22 cells were transfected with negative control si-RNA (NC) or si-RNA for PDE4B (si-PDE4B). At 18–24 h after transfection, the cells were subjected to 6 h of OGD. The protein levels of GRP78, p-eIF2α, eIF2α, and CHOP were determined by Western blotting. (B, D, F) The relative levels of GRP78/β-tubulin (n = 4), p-eIF2α/eIF2α (n = 5) and CHOP/β-tubulin (n = 4) were measured by semiquantitative analysis of the Western blots. (G) HT-22 cells transfected with NC or si-PDE4B were subjected to OGD for 6 h and re-oxygenation for 24 h, and then the cell viability was detected by a CCK-8 assay. (12 duplications from three independent experiment, n = 3). (H, I) HT-22 cells were processed as described above, and the protein levels of cleaved-caspase 3 were determined by Western blotting. The relative level of cleaved-caspase 3/pro-caspase 3 was measured by semiquantitative analysis of the blots (n = 4). Results are expressed as the mean ± SD, *P < 0.05, **P < 0.01 versus the indicated group.
3.4. Inhibition of PDE4 activity by FCPR03 reduces OGD-triggered ER stress
Our previous study showed that the PDE4 inhibitor, FCPR03, was effective in blocking neuronal damage induced by cerebral ischemia both in vitro and in vivo [17]. However, whether FCPR03 exerts its protective role through attenuating ER stress has remained unknown. We previously demonstrated that knocking down the expression of PDE4B ameliorated OGD-induced ER stress and provided subsequent neuroprotection. In the present study, we aimed to investigate the effect of the PDE4 inhibitor, FCPR03 (with high selectivity for PDE4B), on ER stress and to explore the underlying mechanisms. As shown in Fig. 4, OGD significantly stimulated the expression of ER-stress-associated proteins (Fig. 4), while administration of the PDE4 inhibitor FCPR03 (20 μM) blocked the OGD-induced increase in GRP78 (Fig. 4A and B), p-eIF2α (Fig. 4D and E), p-PERK (Fig. 4F and G), and CHOP (Fig. 4H and I) in HT-22 cells. Immunocytochemistry was used to further investigate the expression of p-eIF2α in HT-22 cells, and the results showed that OGD robustly increased the fluorescence of p-eIF2α, which was partially reversed by FCPR03 (Fig. 4C). These results demonstrate that inhibition of PDE4 by FCPR03 attenuated OGD-induced ER stress in HT-22 cells.
Fig. 4.

FCPR03 ameliorates OGD-triggered ER stress. (A, D, F, H) HT-22 cells pretreated with FCPR03 (20 μM) for 1 h were subjected to 6 h of OGD. The protein levels of p-eIF2α, eIF2α, GRP78, p-PERK, PERK, and CHOP were determined by Western blotting. (C) HT-22 cells pretreated with FCPR03 (20 μM) for 1 h were subjected to 6 h of OGD. After fixing, cells were stained with p-eIF2α (red) and DAPI (blue). The representative images were detected by confocal microscopy. (B, E, G, I) The relative levels of p-eIF2α/eIF2α (n = 5), GRP78/β-tubulin (n = 5), p-PERK/PERK (n = 3) and CHOP/β-actin (n = 5) were determined by semiquantitative analysis of the Western blots. Results are expressed as the mean ± SD, *P < 0.05, **P < 0.01 versus the indicated group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.5. Inhibition of PDE4 promotes the translocation of Nrf-2 from the cytoplasm into the nucleus under OGD
OGD induces substantial production of ROS. Our previous study indicated that inhibition of PDE4 decreased the production of ROS in neuronal cells under OGD [17]. Nrf-2 is a transcription factor that increases the transcription of antioxidant proteins through binding to antioxidant-response element (ARE) in the nucleus. In view of the important role of Nrf-2 in ER stress, we next investigated whether inhibition of PDE4 promotes the nucleus translocation of Nrf-2. Immunocytochemical staining was used to show the sub-cellular location of Nrf-2 in HT-22 cells. As shown in Fig. 5A and B, we found that OGD treatment increased the nuclear localization of Nrf-2, and treatment with FCPR03 further increased the nuclear localization of Nrf-2. Nuclear and cytoplasmic proteins were then separated and the Nrf-2 levels were determined by Western blotting. We found that OGD treatment significantly decreased the level of Nrf-2 in the cytoplasm (P < 0.01, Fig. 5C and E). Interestingly, FCPR03 increased the expression of Nrf-2 in the cytoplasm. We continued to examine the level of Nrf-2 in the nucleus. In contrast, OGD increased the entry of Nrf-2 into the nucleus (Fig. 5D and F). Consistent with the results of cellular immunofluorescent staining, FCPR03 further enhanced the expression of Nrf-2 in the nucleus (Fig. 5D and F), suggesting that PDE4 inhibition enhanced Nrf-2 translocation into the nucleus.
Fig. 5.

FCPR03 promotes translocation of Nrf-2 into the nucleus and increases the level of Nrf-2 in the cytoplasm after OGD. (A) HT-22 cells pretreated with FCPR03 (20 μM) for 1 h were subjected to 6 h of OGD. After fixing, cells were stained with Nrf-2. The intracellular localization of Nrf-2 was visualized by confocal microscopy. (B) The relative fluorescence intensity of Nrf-2 in the nucleus was quantified using image J (18 fields from three independent experiments, n = 3). (C, D, E, F) HT-22 cells pretreated with FCPR03 for 1 h were subjected to 6 h of OGD. The change of cytosolic and nuclear Nrf-2 protein levels were detected by Western blotting. The relative levels of Nrf-2/β-tubulin and Nrf-2/Histone H3 were measured by semiquantitative analysis of the Western blots (n = 5). Results are expressed as the mean ± SD, *P < 0.05, **P < 0.01 versus the indicated group.
3.6. Nrf-2 inhibition attenuates the inhibitory effects of FCPR03 on ER stress and apoptosis
We showed that the PDE4 inhibitor, FCPR03, increased the nuclear localization of Nrf-2. Next, we further investigated whether inhibition of Nrf-2 could block the effects of FCPR03 on ER stress and apoptosis under OGD. As shown in Fig. 6A–F, OGD significantly induced the expression of ER-stress-related proteins, while FCPR03 reversed the effects of OGD and reduced ER stress. As expected, the Nrf-2 inhibitor ML385 (5 μM), blocked the action of FCPR03, and the levels of ER stress-related proteins increased significantly after administration of ML385 (Fig. 6A–F). These results suggest that Nrf-2 plays a key role in the regulation of PDE4 on ER stress. Similarly, we investigated whether ML385 blocked the role of PDE4 inhibition on apoptosis. Fig. 6G–I shows that OGD treatment significantly reduced cellular viability significantly (54.34 ± 8.05%, P < 0.01, Fig. 6G and H). The PDE4 inhibitor, FCPR03, increased cell viability to 71.07 ± 8.16% (P < 0.01), and reduced the level of cleaved caspase 3 (P < 0.01). Notably, the cytoprotective effect of FCPR03 was blocked by ML385 (Fig. 6G–I). ML385 reduced cell viability to 50.84 ± 7.34% (P < 0.01). These data suggest that Nrf-2 was involved in the neuroprotective effect of FCPR03.
Fig. 6.

Protective effects of FCPR03 in ameliorating ER stress and reducing apoptosis are abolished by ML385. (A, C, E) HT-22 cells pre-treated with the Nrf-2 inhibitor ML385 (5 μM), were treated with FCPR03 (20 μM) for 1 h followed by 6 h of OGD. The protein levels of GRP78, p-eIF2α, eIF2α, and CHOP were determined by Western blotting. (B, D, F) The relative protein levels of GRP78/β-tubulin (n = 4), p-eIF2α/eIF2α (n = 3) and CHOP/β-tubulin (n = 4) were measured by semiquantitative analysis of the Western blots. (G) HT-22 cells pre-treated with the Nrf-2 inhibitor, ML385, were treated with FCPR03 for 1 h followed by OGD for 6 h and re-oxygenation for 24 h, and then the cell viability was detected by a CCK-8 assay (12 duplications from three independent experiment, n = 3). (H, I) HT-22 cells were processed as described above, and the variation of cleaved-caspase 3 was determined by Western blotting. The relative level of cleaved-caspase 3/pro-caspase 3 was measured by semiquantitative analysis of the Western blots (n = 4). Results are expressed as the mean ± SD, *P < 0.05, **P < 0.01 versus the indicated group.
3.7. Knockdown of HO-1 attenuates the inhibitory effects of FCPR03 on ER stress and increases the production of ROS
HO-1 is a downstream target gene of Nrf-2. Nrf-2 enhances the transcription of HO-1 through binding to ARE in the nucleus [31]. In the present study, we also confirmed that knocking down the expression of Nrf-2 reduced the protein level of HO-1 in HT-22 cells under OGD (Supplementary Fig. S1). HO-1 is known to be a crucial anti-oxidant protein. We showed that Nrf-2 was involved in the inhibitory effect of FCPR03 on OGD-triggered ER stress and cellular injury. We then further examined if HO-1 was involved in this process. Cells were transfected with specific HO-1 si-RNA. Then, at 24 h after transfection, cells were treated with FCPR03 and were exposed to OGD. We found that FCPR03 significantly increased the expression of HO-1 in HT-22 cells (Fig. 7A), which was consistent with the finding that FCPR03 increased the level of Nrf-2 in the nucleus (Fig. 5). FCPR03 reduced the levels of ER stress-related proteins, such as GRP78 and p-eIF2α. In contrast, knockdown of HO-1 blocked the role of FCPR03 and significantly increased the expression of these ER stress-related proteins (Fig. 7C–F). Since Nrf-2/HO-1 signaling is a canonical pathway for ameliorating oxidative stress, we next examined the production of ROS in HT-22 neuronal cells transfected with HO-1 si-RNA. Our results indicated that knockdown of HO-1 blocked the anti-oxidant effects of FCPR03 and increased the production of ROS in cells subjected to OGD (Fig. 7G and H). Similarly, FCPR03 increased cell viability (Fig. 7I) and reduced the level of the apoptotic protein, cleaved caspase 3, while knockdown of HO-1 blocked the anti-apoptotic effect of FCPR03 (Fig. 7J and K). In addition, to further confirm the involvement of HO-1, we also investigated the role of the HO-1 inhibitor, ZnPP, on the expression of ER-related proteins. Similar to the results showing in Fig. 7, inhibition of HO-1 attenuated the inhibitory role of FCPR03 on the expression of both GRP78 and p-eIF-2α (Supplementary Fig. S2). ZnPP also blocked the protective role of FCPR03 and decreased cell viability in HT-22 neuronal cells subjected to OGD (Supplementary Fig. S3). These results indicate that HO-1 was involved in the protective effect of FCPR03 ameliorating ER stress.
Fig. 7.

Protective effects of FCPR03 in ameliorating ER stress and reducing apoptosis are abolished by silencing of HO-1. (A, C, E) HT-22 cells were transfected with a negative control (NC) or siRNA of HO-1 (si–HO–1). At 24 h after transfections, cells were pretreated with FCPR03 for 1 h and subjected to 6 h of OGD. The protein levels of HO-1, GRP78, p-eIF2α, and eIF2α were determined by Western blotting. (B, D, F) The relative levels of HO-1/β-tubulin (n = 3), GRP78/β-tubulin (n = 3) and p-eIF2α/eIF2α (n = 3) were measured by semiquantitative analysis of the Western blots. (G, H) HT-22 cells transfected with NC or si–HO–1 were pretreated with FCPR03 for 1 h, subjected to 6 h of OGD, and were incubated with CellROX Deep Red Reagent (5 μM) for 30 min. Finally, the intracellular ROS was detected by confocal microscopy. Intracellular ROS was quantified by Image J (18 fields from three independent experiments, n = 3). (I) HT-22 cells transfected with NC or si–HO–1 were pretreated with FCPR03 for 1 h and subjected to OGD for 6 h and re-oxygenation for 24 h. Then, cell viability was measured by a CCK-8 assay (12 duplications from three independent experiments, n = 3). (J, K) HT-22 cells were processed as described above, and the protein level of cleaved-caspase3 was determined by Western blotting. The relative level of cleaved-caspase 3/pro-caspase 3 was measured by semiquantitative analysis of the Western blots (n = 3). Results are expressed as the mean ± SD, *P < 0.05, **P < 0.01 versus the indicated group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.8. FCPR03 reduces OGD-induced oxidative stress and ER stress similar to that of a free radical scavenger
Nrf-2/HO-1 is an intracellular endogenous antioxidant system. Oxidative stress can trigger ER stress [27]. We showed that FCPR03 activated Nrf-2/HO-1 and reduced ROS production. We next investigated whether removal of ROS by the free radical scavenger, NAC, would exert a similar effect to that of FCPR03. HT-22 cells were subjected to OGD in the presence of NAC (2.5 mM) or FCPR03 (20 μM), and the level of ROS was determined by the CellROX™ Deep Red reagent. We found that NAC exerted similar effects to these of FCPR03 in reducing the production of ROS (Fig. 8A and B). In addition, OGD treatment caused a significant increase in lipid peroxide MDA (Fig. 8C, P < 0.01), FCPR03 and NAC antagonized the oxidative stress triggered by OGD and restored the level of MDA (Fig. 8C). Next, we examined whether NAC could reduce OGD-induced ER stress. As shown in Fig. 8D–G, OGD induced the expression of GRP78 and p-eIF2α, while NAC blocked the role of OGD and significantly reduced the level of GRP78 (P < 0.05) and p-eIF2α (P < 0.01). Consistently, NAC and FCPR03 played similar effects in increasing cell viability in HT-22 cells subjected to OGD. OGD treament decreased cell viability to 41.3 ± 8.80%, while NAC and FCPR03 increased cell viability to 57.88 ± 14.63% (P < 0.01) and 58.47 ± 5.93% (P < 0.01), respectively. (Fig. 8H). Taken together, these results indicate that FCPR03 exhibited a similar effect to that of a free radical scavenger and, importantly, that removal of free radicals is beneficial to reduce OGD-triggered ER stress and subsequent neuronal death.
Fig. 8.

FCPR03 reduces intracellular ROS and represses ER stress in OGD-treated HT-22 cells. (A, B) HT-22 cells pretreated with FCPR03 (20 μM) or N-Acetyl-l-cysteine (NAC, 2.5 mM) for 1 h were subjected to 6 h of OGD and were incubated with CellROX Deep Red Reagent (5 μM) for 30 min. Finally, intracellular ROS was detected by confocal microscopy. Intracellular ROS was quantified by Image J (18 fields from three independent experiments, n = 3). (C) HT-22 cells pretreated with FCPR03 or NAC for 1 h were subjected to 6 h of OGD. Lipid peroxidation in HT-22 cells was measured by the level of malondialdehyde (MDA). (D, E, F, G) HT-22 cells pretreated with NAC for 1 h were subjected to 6 h of OGD. The protein levels of GRP78, p-eIF2α, and eIF2α were determined by Western blotting. The relative levels of GRP78/β-tubulin and p-eIF2α/eIF2α were determined by semiquantitative analysis of the Western blots (n = 3). (H) HT-22 cells pretreated with FCPR03 or NAC for 1 h were subjected to OGD for 6 h and re-oxygenation for 24 h, and then cell viability was detected by a CCK-8 assay (12 duplications from three independent experiments, n = 3). Results are expressed as the mean ± SD, *P < 0.05, **P < 0.01 versus the indicated group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.9. Inhibition of PDE4 reduces ROS production and ER stress in primary cultured neurons
We showed that knockdown or inhibition of PDE4 reduced ROS and ER stress in HT-22 neuronal cells exposed to OGD. Next, we validated these results in primary cultured cortical neurons. Cultured neurons were identified by immunocytochemical staining of neuronal nuclei (NeuN), and the ratio of NeuN-positive cells was more than 85% (Supplementary Fig. S4). First, p-eIF2α was used as a representative marker to indicate ER stress. Western blotting showed that FCPR03 significantly decreased the level of p-eIF2α in primary cultured neurons (Fig. 9 A and B, P < 0.01). Next, similar to the results obtained in HT-22 neuronal cells, OGD treatment caused substantial production of ROS in primary neurons, while FCPR03 was effective in reducing the intracellular ROS (Fig. 9C and D). We then tested the effects of FCPR03 on cell viability and the involvement of Nrf-2 in primary cultured neurons. We found that cell viability was significantly decreased in neurons subjected to OGD treatment (56.87 ± 9.79%), while the PDE4 inhibitor, FCPR03, blocked the role of OGD and enhanced cell viability (71.07 ± 5.66%, Fig. 9E, P < 0.01). Consistently, the Nrf-2 inhibitor, ML385, abolished the role of FCPR03 on cell viability (51.62 ± 12.41, Fig. 9E, P < 0.01). These results suggest that inhibition of PDE4 in primary cultured neurons reduced ROS production, ER stress, and apoptosis. Additionally, these findings suggest that Nrf-2 was potentially involved in these processes. Next, we examined the role of PDE4 inhibition on ER stress in vivo. A focal cerebral ischemia/reperfusion model in rats was induced by MCAO for 2 h, followed by reperfusion for 24 h. The experimental procedure and sample preparation were the same as those used in our previously published work [17]. We found that the protein levels of GRP78, p-eIF2α, and CHOP were significantly increased in the penumbra of the ipsilateral hemisphere (Supplementary Fig. S5), while intraperitoneal administration of FCPR03 (5 mg/kg) at 2 h after MCAO restored the levels of GRP78 (P < 0.01), p-eIF2α (P < 0.01), and CHOP (P < 0.01).
Fig. 9.

FCPR03 ameliorates ER stress and reduces intracellular ROS caused by OGD in primary cortical neurons. (A, B) Primary cortical neurons pre-treated with FCPR03 (10 μM) for 1 h were subjected to OGD for 1 h and re-oxygenation for 4 h. The protein level of p-eIF2α was determined by Western blotting. The relative levels of p-eIF2α/eIF2α was determined by semiquantitative analysis of the Western blots (n = 5). (C, D) Primary cortical neurons pretreated with FCPR03 for 1 h were subjected to 1 h of OGD and were incubated with CellROX Deep Red Reagent (5 μM) for 30 min. Finally, intracellular ROS was detected by confocal microscopy. Intracellular ROS was quantified by Image J (18 fields from three independent experiments, n = 3). (E) Primary cortical neurons pre-treated with the Nrf-2 inhibitor, ML385 (5 μM), were treated with FCPR03 (10 μM) for 1 h followed by 1 h of OGD and 24 h of re-oxygenation, and then the cell viability was detected by a CCK-8 assay (12 duplications from three independent experiments, n = 3). Results are expressed as the mean ± SD, **P < 0.01 versus the indicated group. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
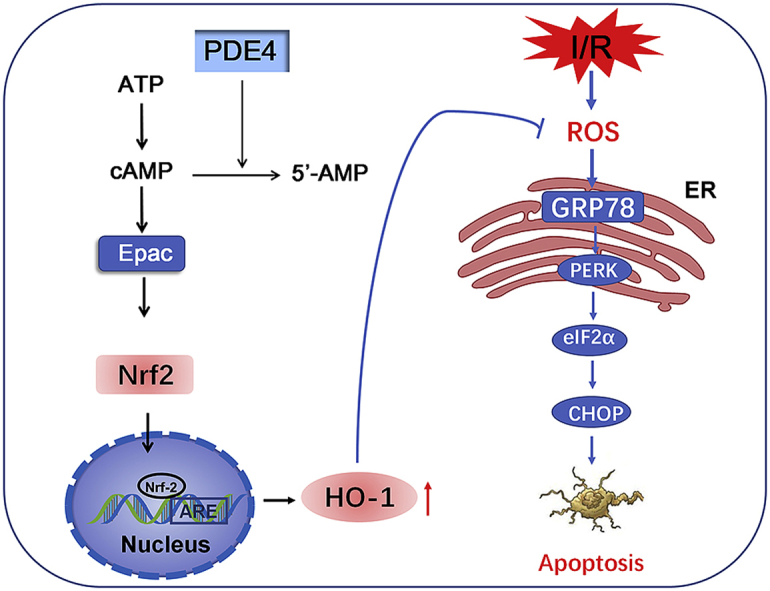
In the current study, we showed for the first time that inhibition of PDE4 reduced OGD-triggered ER stress and apoptosis in neuronal cells and, importantly, that this protective effect was mediated by activating Nrf-2/HO-1 signaling and subsequently suppressing oxidative stress. These conclusions are based on the following observations: (1) exposure of neuronal cells to OGD caused significant ER stress at the early stage of OGD; (2) overexpression of PDE4 increased ER stress under both basal and OGD conditions; (3) knockdown of PDE4 or treatment with the PDE4 inhibitor, FCPR03, reduced ER stress and neuronal apoptosis in HT-22 neuronal cells exposed to OGD; (4) FCPR03 increased the nuclear localization of Nrf-2, and inhibition of Nrf-2 attenuated the role of FCPR03 on ER stress and cell viability; (5) FCPR03 increased the expression of HO-1, whereas HO-1 knockdown abolished the inhibitory effects of FCPR03 on ER stress, ROS production, and neuronal apoptosis; (6) FCPR03 was effective in reducing ROS, and scavenging ROS was beneficial in reducing ER stress; and (7) these findings were verified in primary cultured neurons and in an MCAO animal model. A summary of the protective effects of PDE4 in ameliorating OGD-triggered ER stress and potential related signaling pathways are shown in Fig. 10.
Fig. 10.

Inhibition of PDE4 protects neurons against ER stress via activation of the Nrf-2/HO-1 pathway. Under the condition of cerebral ischemia-reperfusion (I/R), excessive production of ROS triggers ER stress, and activates PERK/eIF2α/CHOP pathway. This leads to cellular apoptosis following cerebral ischemia. Inhibition of PDE4 increases the intracellular concentration of cAMP, which subsequently activates Nrf-2. In the nucleus, Nrf-2 binds to ARE and promotes the expression of HO-1, thereby enhancing the scavenge of ROS, reducing secondary ER stress and neuronal apoptosis.
Cerebral ischemia-reperfusion injury is a trigger to induce and initiate ER stress in neurons. Excessive ER stress induces neuronal apoptosis, and in turn aggravates neuronal damage caused by cerebral ischemia [32]. Under this circumstance, GRP78 acts as a sensor and activates PERK and IRE1α, after which downstream signaling molecules such as eIF2α, ATF4, and CHOP are activated [33]. In the present study, we established an OGD cellular model and found that GRP78 was up-regulated at the early stage of hypoxia (3 h) and continued to increase in a time-dependent manner (Fig. 1A and B). GRP78 binds to PERK at rest and is inactive. When ER stress occurs, GRP78 is activated by dissociation from PERK, and PERK also activates and phosphorylates the downstream target, eIF2α. eIF2α is a translational regulator that is activated by phosphorylation. We found that OGD treatment led to the activation of both PERK and eIF2α (Fig. 1C–F). Activated eIF2α promotes the translation of ATF4, which further promotes the expression of CHOP [34]. As expected, we found that the expression of CHOP was significantly increased in the absence of oxygen for 3–6 h (Fig. 1G and H). When stimuli persist, ER stress exceeds the neuronal compensatory capacity, and high expression of CHOP activates the Caspase-3 apoptotic cascade, which promotes apoptosis. Our present study also confirmed that hypoxia and reoxygenation for 24 h significantly decreased the cell viability (Fig. 1I). In the present study, we did not examine the expression of ATF4, but it is predicted that OGD treatment also increases the level of ATF4, as ATF4 is activated by the GRP78/PERK/eIF2α pathway.
PDE4 is highly expressed in neurons [35]. A genome-wide search found that the PDE4 gene is highly associated with ischemic stroke [36]. To investigate the protective effects and underlying mechanisms of PDE4 inhibition against ischemic stroke, we established in vitro and in vivo models to mimic the pathology of stroke. We found that overexpression of PDE4 significantly increased ER stress under basal and OGD conditions, as evidenced by increased levels of GRP78 and p-eIF2α/eIF2α (Fig. 2). Consistently, knockdown of PDE4 or inhibition of PDE4 decreased ER stress and down-regulated the expression of ER stress-related proteins, such as GRP78, p-eIF2α and CHOP. Importantly, knockdown PDE4 increased cell viability and decreased the level of cleaved caspase 3 under OGD. These data are consistent with our previous findings that inhibition of PDE4 by FCPR03 enhances cellular survival in neurons subjected to OGD [17]. Additionally, we also verified that cerebral ischemia-reperfusion significantly increased the levels of GRP78, p-eIF2α/eIF2α, and CHOP in the penumbra of the ipsilateral hemisphere of rats subjected to MCAO, whereas administration of PDE4 inhibitors reduced ER stress (Supplementary Fig. S5). These data are further supported by the results that inhibition of PDE4 reduced cerebral infarct size and ameliorated neurological deficits in rats following MCAO [17]. These results suggest that neuroprotective effects of PDE4 inhibition in models of cerebral ischemia is mediated through inhibiting ER stress. Collectively, for the first time, we linked the protective effects of PDE4 inhibition in neurons with ER stress suppression. Inspired by these findings, we were interested to further study by which molecules PDE4 inhibition suppressed ER stress.
Nrf-2 is widely expressed in the brain and is one of the most important antioxidant defense mechanisms within cells [37]. Under physiological conditions, Nrf-2 binds to Keap1 in the cytoplasm. Under the condition of cerebral ischemia and reperfusion, excessive oxidative stress promotes the separation of Keap1 with Nrf-2, and activates Nrf-2 [38]. Activated Nrf-2 translocates to the nucleus and binds to ARE, which initiates transcription of multiple downstream anti-oxidant genes, such as HO-1 [39]. Therefore, Nrf-2 is a key transcription factor maintaining redox homeostasis inside cells. Previous studies have shown that Nrf-2 possesses cytoprotective effects in various models of ischemia-reperfusion-induced brain injury and renal injury [40,41]. More importantly, administration of an Nrf-2 activator is effective at reducing oxidative stress damage [41]. When cerebral ischemia occurs, the production of ROS is significantly increased, and antioxidant defense is usually insufficient to antagonize the substantially increased ROS. Excessive oxidative stress directly triggers the occurrence of ER stress, which subsequently increases the expression of HO-1 in cells. The increase in HO-1 expression is a compensative auto-regulatory mechanism or a negative-feedback mechanism that is supposed to scavenge ROS and attenuate cell death caused by ER stress [42]. In the present study, we found that the amount of Nrf-2 in the nucleus of HT-22 neuronal cells was increased following OGD, while FCPR03 further increased the level of Nrf-2 in the nucleus. Interestingly, the Nrf-2 inhibitor, ML385, attenuated the neuroprotective effect of FCPR03, decreased cell viability and increased the expression of cleaved caspase 3. Consistently, ER-stress-related proteins such as GRP78, CHOP and p-eIF-2α/eIF-2α were increased by ML385 as well. These data suggest that the neuroprotective effects of PDE4 inhibition against OGD depends on the presence of Nrf-2. We further knocked down the expression of HO-1 in cells by specific siRNA. The results demonstrated that the effects of the PDE4 inhibitor was weakened, and that ER stress-related proteins were up-regulated. These results suggest that the Nrf-2/HO-1 system is involved in the neuroprotective effect of PDE4 inhibition against OGD, and that its effect is highly correlated with the suppression of ER stress.
HO-1 shows efficiency in inhibiting ER stress. Mechanistically, HO-1 is distributed in the ER. HO-1 is able to regulate the redox homeostasis of the ER [43], and thus provides a redox microenvironment suitable for the folding of newly synthesized proteins or misfolded molecules. Alternatively, HO-1 may also reduce ER stress through enhancing autophagy [44]. Hence, it is worthwhile for future studies to further investigate how HO-1 affects OGD-triggered ER stress. Additionally, another phenomenon worth elucidating is how PDE4 regulates Nrf-2. Recent studies have shown that Nrf-2 activation is not solely dependent on the Keap1 pathway. Non-Keap1-dependent pathways, such as PI3K/Akt and GSK3β, are also involved in the regulation of Nrf-2 [45,46]. Studies have shown that the expression of GSK3β is increased in ischemic brain injury [47]. Consistent with this finding, we also found that an inhibitor of GSK3β exerted neuroprotective effects in an ischemic hypoxic animal model [48]. Activated GSK3β may inhibit the activation of Nrf-2 by both direct and indirect ways. GSK3β may directly phosphorylate Nrf-2, which would result in degradation and decreased localization of Nrf-2 in the nucleus [49]. Additionally, activated GSK3β may promote the translocation of Nrf-2 from the nucleus to the cytoplasm and its subsequent degradation through the phosphorylation of Fyn [50]. Interestingly, our recent study found that inhibition of PDE4 by FCPR03 inactivates GSK3β through increasing cAMP and the subsequent activation of the Epac/Akt pathway in neurons exposed to OGD and in rats subjected to MCAO [17]. Hence, it is highly possible that inhibition of PDE4 promotes the nuclear translocation of Nrf-2 through promoting the phosphorylation of Akt and GSK3β.
In the present study, we demonstrated that PDE4 inhibitor FCPR03 protected neurons against oxygen-glucose deprivation-induced endoplasmic reticulum stress. We would like to point out that compared with the first-generation PDE4 inhibitor rolipram, FCPR03 is a novel PDE4 inhibitor with better PDE4 inhibitory activity. FCPR03 has an IC50 (Inhibitory Concentration 50%) value of 60 nM, while rolipram has an IC50 value of 550 nM [29]. Importantly, rolipram is characterized for its potential to trigger nausea and vomiting, while FCPR03 had no effect on inducing emesis in rodents at its effective doses [29]. Hence, FCPR03 might be a promising compound for the intervention of neurological disorders, such as ischemic stroke.
5. Conclusions
In conclusion, the present study demonstrates that inhibition of PDE4 protects against ER stress–induced cellular damage by activating the Nrf-2/HO-1 pathway, thereby restoring ER homeostasis and preventing neuronal death. In addition, our present findings suggest that PDE4 may serve as a therapeutic target for the intervention of cerebral ischemia. Moreover, our results suggest that FCPR03 may represent a promising candidate for the treatment of ER stress-mediated neurological diseases.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Acknowledgments
This work was supported by National Natural Science Foundation of China (No. 81773698), the Program for Changjiang Scholars and Innovative Research Team in University (IRT_16R37), Science and Technology Program of Guangdong (Nos. 2018B030334001 and 2015B020211007), Science and Technology Program of Guangzhou (No. 201604020112), and Undergraduate Training Programs for Innovation and Entrepreneurship, Guangdong, China (S201912121168).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101342.
Contributor Information
Jiangping Xu, Email: jpx@smu.edu.cn.
Haitao Wang, Email: wht821@smu.edu.cn.
Author contribution
BX, YQ, DL, NC, JW, LJ (Lan Jiang) and LJ (Limei Jie) performed the experiments and analyzed the data. BX, HW and JX drafted the manuscript. HW and JX designed research project, supervised experiments and critically reviewed the manuscript. ZZ provided FCPR03 and revised the manuscript. All authors have given their final approval for the manuscript.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Colpo G.D., Venna V.R., McCullough L.D., Teixeira A.L. Systematic review on the involvement of the kynurenine pathway in stroke: pre-clinical and clinical evidence. Front. Neurol. 2019;10:778. doi: 10.3389/fneur.2019.00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yaghi S., Boehme A.K., Dibu J., Leon Guerrero C.R., Ali S., Martin-Schild S., Sands K.A., Noorian A.R., Blum C.A., Chaudhary S., Schwamm L.H., Liebeskind D.S., Marshall R.S., Willey J.Z. Treatment and outcome of thrombolysis-related hemorrhage: a multicenter retrospective study. JAMA Neurol. 2015;72:1451–1457. doi: 10.1001/jamaneurol.2015.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H., Zuo J., Tang W. Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front. Pharmacol. 2018;9:1048. doi: 10.3389/fphar.2018.01048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly M.P. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell. Signal. 2018;42:281–291. doi: 10.1016/j.cellsig.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwak H.J., Park K.M., Choi H.E., Chung K.S., Lim H.J., Park H.Y. PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways. Cell. Signal. 2008;20:803–814. doi: 10.1016/j.cellsig.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Gianello V., Salvi V., Parola C., Moretto N., Facchinetti F., Civelli M., Villetti G., Bosisio D., Sozzani S. The PDE4 inhibitor CHF6001 modulates pro-inflammatory cytokines, chemokines and Th1- and Th17-polarizing cytokines in human dendritic cells. Biochem. Pharmacol. 2019;163:371–380. doi: 10.1016/j.bcp.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Santiago A., Soares L.M., Schepers M., Milani H., Vanmierlo T., Prickaerts J., Weffort de Oliveira R.M. Roflumilast promotes memory recovery and attenuates white matter injury in aged rats subjected to chronic cerebral hypoperfusion. Neuropharmacology. 2018;138:360–370. doi: 10.1016/j.neuropharm.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 8.Guo H., Cheng Y., Wang C., Wu J., Zou Z., Niu B., Yu H., Wang H., Xu J. FFPM, a PDE4 inhibitor, reverses learning and memory deficits in APP/PS1 transgenic mice via cAMP/PKA/CREB signaling and anti-inflammatory effects. Neuropharmacology. 2017;116:260–269. doi: 10.1016/j.neuropharm.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Zhong J., Xie J., Xiao J., Li D., Xu B., Wang X., Wen H., Zhou Z., Cheng Y., Xu J., Wang H. Inhibition of PDE4 by FCPR16 induces AMPK-dependent autophagy and confers neuroprotection in SH-SY5Y cells and neurons exposed to MPP(+)-induced oxidative insult. Free Radic. Biol. Med. 2019;135:87–101. doi: 10.1016/j.freeradbiomed.2019.02.027. [DOI] [PubMed] [Google Scholar]
- 10.Yu H., Zhong J., Niu B., Zhong Q., Xiao J., Xie J., Lin M., Zhou Z., Xu J., Wang H. Inhibition of phosphodiesterase 4 by FCPR03 alleviates chronic unpredictable mild stress-induced depressive-like behaviors and prevents dendritic spine loss in mice hippocampi. Int. J. Neuropsychopharmacol. 2019;22:143–156. doi: 10.1093/ijnp/pyy092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong J., Yu H., Huang C., Zhong Q., Chen Y., Xie J., Zhou Z., Xu J., Wang H. Inhibition of phosphodiesterase 4 by FCPR16 protects SH-SY5Y cells against MPP(+)-induced decline of mitochondrial membrane potential and oxidative stress. Redox Biol. 2018;16:47–58. doi: 10.1016/j.redox.2018.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H., Gaur U., Xiao J., Xu B., Xu J., Zheng W. Targeting phosphodiesterase 4 as a potential therapeutic strategy for enhancing neuroplasticity following ischemic stroke. Int. J. Biol. Sci. 2018;14:1745–1754. doi: 10.7150/ijbs.26230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yasmeen S., Akram B.H., Hainsworth A.H., Kruuse C. Cyclic nucleotide phosphodiesterases (PDEs) and endothelial function in ischaemic stroke. Rev. Cell Signal. 2019;61:108–119. doi: 10.1016/j.cellsig.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Kraft P., Schwarz T., Gob E., Heydenreich N., Brede M., Meuth S.G., Kleinschnitz C. The phosphodiesterase-4 inhibitor rolipram protects from ischemic stroke in mice by reducing blood-brain-barrier damage, inflammation and thrombosis. Exp. Neurol. 2013;247:80–90. doi: 10.1016/j.expneurol.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 15.Sasaki T., Kitagawa K., Omura-Matsuoka E., Todo K., Terasaki Y., Sugiura S., Hatazawa J., Yagita Y., Hori M. The phosphodiesterase inhibitor rolipram promotes survival of newborn hippocampal neurons after ischemia. Stroke. 2007;38:1597–1605. doi: 10.1161/STROKEAHA.106.476754. [DOI] [PubMed] [Google Scholar]
- 16.Li L.X., Cheng Y.F., Lin H.B., Wang C., Xu J.P., Zhang H.T. Prevention of cerebral ischemia-induced memory deficits by inhibition of phosphodiesterase-4 in rats. Metab. Brain Dis. 2011;26:37–47. doi: 10.1007/s11011-011-9235-0. [DOI] [PubMed] [Google Scholar]
- 17.Xu B., Wang T., Xiao J., Dong W., Wen H.Z., Wang X., Qin Y., Cai N., Zhou Z., Xu J., Wang H. FCPR03, a novel phosphodiesterase 4 inhibitor, alleviates cerebral ischemia/reperfusion injury through activation of the AKT/GSK3beta/beta-catenin signaling pathway. Biochem. Pharmacol. 2019;163:234–249. doi: 10.1016/j.bcp.2019.02.023. [DOI] [PubMed] [Google Scholar]
- 18.Jakaria M., Azam S., Haque M.E., Jo S.H., Uddin M.S., Kim I.S., Choi D.K. Taurine and its analogs in neurological disorders: focus on therapeutic potential and molecular mechanisms. Redox Biol. 2019;24:101223. doi: 10.1016/j.redox.2019.101223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anuncibay-Soto B., Perez-Rodriguez D., Santos-Galdiano M., Font-Belmonte E., Ugidos I.F., Gonzalez-Rodriguez P., Regueiro-Purrinos M., Fernandez-Lopez A. Salubrinal and robenacoxib treatment after global cerebral ischemia. Exploring the interactions between ER stress and inflammation. Biochem. Pharmacol. 2018;151:26–37. doi: 10.1016/j.bcp.2018.02.029. [DOI] [PubMed] [Google Scholar]
- 20.Needham P.G., Guerriero C.J., Brodsky J.L. Chaperoning endoplasmic reticulum-associated degradation (ERAD) and protein conformational diseases. Cold Spring Harb. Perspect. Biol. 2019;11 doi: 10.1101/cshperspect.a033928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z., Zhang L., Zhou L., Lei Y., Zhang Y., Huang C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2018 doi: 10.1016/j.redox.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. 2005;35:373–381. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 23.Cnop M., Toivonen S., Igoillo-Esteve M., Salpea P. Endoplasmic reticulum stress and eIF2alpha phosphorylation: the Achilles heel of pancreatic beta cells. Mol. Metab. 2017;6:1024–1039. doi: 10.1016/j.molmet.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang M., Wey S., Zhang Y., Ye R., Lee A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxidants Redox Signal. 2009;11:2307–2316. doi: 10.1089/ars.2009.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malhotra J.D., Kaufman R.J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxidants Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 26.Xin Q., Ji B., Cheng B., Wang C., Liu H., Chen X., Chen J., Bai B. Endoplasmic reticulum stress in cerebral ischemia. Neurochem. Int. 2014;68:18–27. doi: 10.1016/j.neuint.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 27.Zeeshan H.M., Lee G.H., Kim H.R., Chae H.J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016;17:327. doi: 10.3390/ijms17030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myers S.A., Gobejishvili L., Saraswat Ohri S., Garrett Wilson C., Andres K.R., Riegler A.S., Donde H., Joshi-Barve S., Barve S., Whittemore S.R. Following spinal cord injury, PDE4B drives an acute, local inflammatory response and a chronic, systemic response exacerbated by gut dysbiosis and endotoxemia. Neurobiol. Dis. 2019;124:353–363. doi: 10.1016/j.nbd.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Z.Z., Cheng Y.F., Zou Z.Q., Ge B.C., Yu H., Huang C., Wang H.T., Yang X.M., Xu J.P. Discovery of N-alkyl catecholamides as selective phosphodiesterase-4 inhibitors with anti-neuroinflammation potential exhibiting antidepressant-like effects at non-emetic doses. ACS Chem. Neurosci. 2017;8:135–146. doi: 10.1021/acschemneuro.6b00271. [DOI] [PubMed] [Google Scholar]
- 30.Zou Z.Q., Chen J.J., Feng H.F., Cheng Y.F., Wang H.T., Zhou Z.Z., Guo H.B., Zheng W., Xu J.P. Novel phosphodiesterase 4 inhibitor FCPR03 alleviates lipopolysaccharide-induced neuroinflammation by regulation of the cAMP/PKA/CREB signaling pathway and NF-kappaB inhibition. J. Pharmacol. Exp. Ther. 2017;362:67–77. doi: 10.1124/jpet.116.239608. [DOI] [PubMed] [Google Scholar]
- 31.Wang J., Hu X., Jiang H. The Nrf-2/ARE-HO-1 axis: an important therapeutic approach for attenuating myocardial ischemia and reperfusion injury-induced cardiac remodeling. Int. J. Cardiol. 2015;184:263–264. doi: 10.1016/j.ijcard.2015.02.073. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H.Y., Wang Z.G., Lu X.H., Kong X.X., Wu F.Z., Lin L., Tan X., Ye L.B., Xiao J. Endoplasmic reticulum stress: relevance and therapeutics in central nervous system diseases. Mol. Neurobiol. 2015;51:1343–1352. doi: 10.1007/s12035-014-8813-7. [DOI] [PubMed] [Google Scholar]
- 33.Li Y., Zhang Y., Fu H., Huang H., Lu Q., Qin H., Wu Y., Huang H., Mao G., Wei Z., Liao P. Hes1 knockdown exacerbates ischemic stroke following tMCAO by increasing ER stress-dependent apoptosis via the PERK/eIF2alpha/ATF4/CHOP signaling pathway. Neurosci. Bull. 2019 doi: 10.1007/s12264-019-00411-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han J., Back S.H., Hur J., Lin Y.H., Gildersleeve R., Shan J., Yuan C.L., Krokowski D., Wang S., Hatzoglou M., Kilberg M.S., Sartor M.A., Kaufman R.J. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishi A., Kuroiwa M., Miller D.B., O'Callaghan J.P., Bateup H.S., Shuto T., Sotogaku N., Fukuda T., Heintz N., Greengard P., Snyder G.L. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J. Neurosci. 2008;28:10460–10471. doi: 10.1523/JNEUROSCI.2518-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munshi A., Roy S., Thangaraj K., Kaul S., Babu M.S., Jyothy A. Association of SNP41, SNP56 and a novel SNP in PDE4D gene with stroke and its subtypes. Gene. 2012;506:31–35. doi: 10.1016/j.gene.2012.06.079. [DOI] [PubMed] [Google Scholar]
- 37.Bellaver B., Souza D.G., Souza D.O., Quincozes-Santos A. Hippocampal astrocyte cultures from adult and aged rats reproduce changes in glial functionality observed in the aging brain. Mol. Neurobiol. 2017;54:2969–2985. doi: 10.1007/s12035-016-9880-8. [DOI] [PubMed] [Google Scholar]
- 38.Wu G., Zhu L., Yuan X., Chen H., Xiong R., Zhang S., Cheng H., Shen Y., An H., Li T., Li H., Zhang W. Britanin ameliorates cerebral ischemia-reperfusion injury by inducing the Nrf2 protective pathway. Antioxidants Redox Signal. 2017;27:754–768. doi: 10.1089/ars.2016.6885. [DOI] [PubMed] [Google Scholar]
- 39.Loboda A., Damulewicz M., Pyza E., Jozkowicz A., Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016;73:3221–3247. doi: 10.1007/s00018-016-2223-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang M.Y., Yu Q.L., Huang Y.S., Yang G. Neuroprotective effects of andrographolide derivative CX-10 in transient focal ischemia in rat: involvement of Nrf2/AE and TLR/NF-kappaB signaling. Pharmacol. Res. 2019;144:227–234. doi: 10.1016/j.phrs.2019.04.023. [DOI] [PubMed] [Google Scholar]
- 41.Nezu M., Souma T., Yu L., Suzuki T., Saigusa D., Ito S., Suzuki N., Yamamoto M. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int. 2017;91:387–401. doi: 10.1016/j.kint.2016.08.023. [DOI] [PubMed] [Google Scholar]
- 42.Khakurel A., Park P.H. Globular adiponectin protects hepatocytes from tunicamycin-induced cell death via modulation of the inflammasome and heme oxygenase-1 induction. Pharmacol. Res. 2018;128:231–243. doi: 10.1016/j.phrs.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 43.Gall T., Balla G., Balla J. Heme, heme oxygenase, and endoplasmic reticulum stress-A new insight into the pathophysiology of vascular diseases. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20153675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cernigliaro C., D'Anneo A., Carlisi D., Giuliano M., Marino Gammazza A., Barone R., Longhitano L., Cappello F., Emanuele S., Distefano A., Campanella C., Calvaruso G., Lauricella M. Ethanol-mediated stress promotes autophagic survival and aggressiveness of colon cancer cells via activation of Nrf2/HO-1 pathway. Cancers (Basel) 2019;11 doi: 10.3390/cancers11040505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duan J., Cui J., Yang Z., Guo C., Cao J., Xi M., Weng Y., Yin Y., Wang Y., Wei G., Qiao B., Wen A. Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK-3beta/Nrf2 signaling. J. Neuroinflammation. 2019;16:24. doi: 10.1186/s12974-019-1406-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai H., Wang P., Mao H., Mao X., Tan S., Chen Z. Dynorphin activation of kappa opioid receptor protects against epilepsy and seizure-induced brain injury via PI3K/Akt/Nrf2/HO-1 pathway. Cell Cycle. 2019;18:226–237. doi: 10.1080/15384101.2018.1562286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen B.H., Ahn J.H., Park J.H., Shin B.N., Lee Y.L., Kang I.J., Hong S., Kim Y.H., Jeon Y.H., Kim I.H., Cho J.H., Lee T.K., Lee J.C., Won M.H., Cho J.H., Moon J.B. Transient cerebral ischemia alters GSK-3beta and p-GSK-3beta immunoreactivity in pyramidal neurons and induces p-GSK-3beta expression in astrocytes in the gerbil hippocampal CA1 area. Neurochem. Res. 2017;42:2305–2313. doi: 10.1007/s11064-017-2245-5. [DOI] [PubMed] [Google Scholar]
- 48.Wang H., Huang S., Yan K., Fang X., Abussaud A., Martinez A., Sun H.S., Feng Z.P. Tideglusib, a chemical inhibitor of GSK3beta, attenuates hypoxic-ischemic brain injury in neonatal mice. Biochim. Biophys. Acta. 2016;1860:2076–2085. doi: 10.1016/j.bbagen.2016.06.027. [DOI] [PubMed] [Google Scholar]
- 49.Mathur A., Pandey V.K., Kakkar P. Activation of GSK3beta/beta-TrCP axis via PHLPP1 exacerbates Nrf2 degradation leading to impairment in cell survival pathway during diabetic nephropathy. Free Radic. Biol. Med. 2018;120:414–424. doi: 10.1016/j.freeradbiomed.2018.04.550. [DOI] [PubMed] [Google Scholar]
- 50.Xin Y., Bai Y., Jiang X., Zhou S., Wang Y., Wintergerst K.A., Cui T., Ji H., Tan Y., Cai L. Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 via stimulating the Akt/GSK-3ss/Fyn pathway. Redox Biol. 2018;15:405–417. doi: 10.1016/j.redox.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.