

Abstract
Background
Rapid and comprehensive pathogen identification is crucial in zoonotic influenza diagnosis.
Methods
By optimizing the design of primers and probes and reverse‐transcriptase polymerase chain reaction (RT‐PCR) conditions, we achieved simultaneous detection of multiple influenza and zoonotic influenza viruses, including H1N1, H5N1, and H9N2 strains, in a one‐step, quantitative real‐time RT‐PCR array (rRT‐PCR array) of RNA from multiple influenza strains utilizing a single set of conditions for RT‐PCR amplification. The target sequences from all targeted zoonotic influenza viruses were cloned into recombinant RNA virus particles, which were used to evaluate sensitivity, specificity, and reproducibility of the zoonotic influenza viruses RT‐PCR array.
Results
The detection limit of the array was shown to be between 100 and 101 copies per reaction, and the standard curve demonstrated a linear range from 10 to 106 copies. Thus, the analytical sensitivity of this zoonotic influenza viruses RT‐PCR array is 10–100 times higher than conventional RT‐PCR. Specificity of the one‐step zoonotic influenza viruses RT‐PCR array was verified by comparison of results obtained with retroviral‐like particles (RVPs), which contained RNA from isolates of seasonal influenza viruses, zoonotic influenza viruses, and other pathogens known to cause acute respiratory disease.
Conclusion
The high sensitivity, rapidity, reproducibility, and specificity of this zoonotic influenza viruses rRT‐PCR array has been verified as being sufficient to detect the presence of multiple zoonotic influenza viruses in a single assay. The zoonotic influenza viruses RT‐PCR array might provide rapid identification of emergent zoonotic influenza viruses strains during influenza outbreaks and disease surveillance initiatives.
Keywords: zoonotic influenza virus, H1N1, H5N1, H9N2, one step real‐time RT‐PCR array
INTRODUCTION
Influenza A viruses belong to Orthomyxoviridae, a family of enveloped, negative‐sense, segmented, single‐stranded RNA viruses. Genetic reassortment of avian, swine, and human influenza viruses could cause novel viruses containing animal virus genes transmitting to humans 1, 2, 3. The two major antigens used to distinguish between strains of influenza A are surface antigens, hemagglutinin (HA or H), a protein that causes red blood cells to agglutinate, and neuroaminidase (NA or N), an enzyme that cleaves the glycosidic bond of neuraminic acid. There are multiple subtypes of both H and N proteins expressed on influenza envelopes, each with distinct amino acid sequence characteristics. However, each influenza virus typically expresses a single H subtype and N subtype, so influenza strains are often classified according to their expressed H & N subtypes 4, 5, 6, 7. This combination of the H subtype and N subtype not only helps to identify different strains of influenza A, but it provides clues to the origin of the virus, which may be zoonotic. For example, influenza A viral subtypes H1N1 H1N2, H2N1, H3N1, H3N2, and H2N3 are commonly associated with influenza infections in pigs. Other influenza A viral subtypes, including H9N2 and H5N1, are almost exclusively found in avian populations 1, 3, 8, 9, 10.
While H1N2 and H3N2 influenza A subtypes are commonly seen in seasonal influenza disease surveys, during the flu season of 2009 “swine flu,” a number of human cases was caused by influenza with an H1N1 subtype 6, 11, 12. More recent influenza virus epidemic in Asia and Europe, caused by H5N1 and H9N2 subtypes, respectively, indicates that zoonotic influenza viruses are capable of infecting humans directly 4, 13, 14. The prevalence and pandemic potential of zoonotic influenza has raised concern among public health authorities.
Influenza A viruses of the H1N1 subtype are known to infect birds and pigs. However, several incidences of influenza in the 20th century have been attributed to the H1N1 subtype, including an outbreak in the United States in 1976 and a zoonotic infection in Wisconsin (USA) in 1988 during a swine influenza outbreak 1. Also, the global “Spanish flu” pandemic of 1918 appears to have been due to the H1N1 subtype 4, 16. This pandemic was particularly virulent and had a high mortality rate, leading to 50–100 million people dead worldwide. In the 21st century, a new outbreak of H1N1 in the human population emerged 2. This zoonotic, emergent H1N1 viral subtype demonstrated an exceptionally rapid geographical spread with sustained human–human transmission. This led the World Health Organization (WHO) to declare this event the first influenza virus pandemic of the 21st century on 11 June 2009. Although the threat to human population from H1N1 is significant, previous exposures of the human population to H1N1 viral subtypes provide some inherent immunity 4, 10, 15, 17. This is evidenced by the fact that, at the present time, H1N1, H1N2, and H3N2 are the only known influenza A virus subtypes commonly circulating among humans 3.
It is feared that zoonotic influenza outbreak associated with a novel subtype could be much more virulent or have a much higher mortality rate in the human population. Fortunately, most zoonotic influenza infections in humans are not readily transmissible to other humans. However, should a particular zoonotic strain gain human‐to‐human transmissibility, an influenza pandemic of zoonotic origin could be much more virulent, due to a lack of immunity to the novel influenza virus in the general population 17, 18, 19, 20.
The threat posed by highly pathogenic avian influenza A H5N1 viruses to humans is also significant. A total of 499 human cases in 15 countries with a high case fatality rate (approximately 60%) had been reported and confirmed by laboratory testing 11, 14, 21, 22. Although the H9N2 has a lower fatality rate than H5N1, the WHO earlier warned that H9N2 could trigger a global influenza outbreak in humans and, therefore, needs to be closely monitored. H9N2 subtype of avian influenza in China is currently still the main type of bird flu pandemic 13, 23, 24. H7N7 virus is another highly pathogenic avian influenza virus (AIV) that has been reported to be responsible for isolated zoonotic infections in China, but it had not been isolated or confirmed by laboratory tests 25, 26. Most recently, in early 2013, reports of zoonotic infections by an H7N9 strain of AIV has been reported with a fairly high mortality rate 6 deaths in 14 cases reported on the eastern coast of China, as of early April 2013)
The diagnosis of zoonotic influenza is made according to epidemiological history, clinical manifestations, and laboratory test results. Ideally, it should be differential diagnosis of influenza versus the common cold, severe acute respiratory syndrome (SARS), infectious mononucleosis syndrome, cytomegalovirus infection, bacterial pneumonia (associated with Streptococcus pneumonia in roughly 50% of cases, Haemophilus influenzae in 20%, Chlamydophila pneumoniae in 13%, and Mycoplasma pneumoniae in 3% of cases, as well as other bacteria in rare instances), viral pneumonia, fungal pneumonia, and other diseases 6, 25, 27, 28. However, the most convincing evidence of influenza infection is positive detection of influenza viral proteins or genes by immunologic or polymerase chain reaction (PCR) detection methods, respectively.
A variety of techniques had been used for the detection and identification of influenza viruses. Many standard laboratory methods depend upon viral isolation and growing isolated virus particles in tissue or chicken embryos (in fertilized eggs) 4. Once the virus is isolated and grown in sufficient quantities, further tests can be performed. Hemagglutination inhibition (HI), an immunologic test procedure, is commonly used for subtyping and identifying antigenic variation of influenza A viruses 29, 30, 31. HI is a very sensitive assay, but it depends upon the presence of infectious particles in the original sample and upon successful extraction and preservation of viable viral specimens, as well as a 1‐ to 2‐week incubation period is needed for growing influenza virus particles. Additionally, laboratory safety regulations that require viral isolation of highly pathogenic AIV be performed in P2 or P3 laboratory (biosafety level 2 or 3, respectively) 5. Therefore, the HI method has limited usefulness in most emergency cases. More rapid tests, based on antigen detection by enzyme‐linked immunosorbent assay or direct immunofluorescence, are carried out for the detection of the virus as well. Although the sensitivity and specificity of these techniques make them suitable for many diagnostic applications, they have limited applications in detection of influenza A virus infection of zoonotic origin, as novel virus antigens may remain undetected, resulting in false‐negative diagnoses 20, 29, 32.
PCR is one of the most sensitive and specific techniques that have been developed for typing and subtyping of influenza viruses. Identification of influenza viral type, subtype, and virulence of different strains by DNA sequencing and structure analysis of PCR‐generated amplicons can be accomplished using template DNA or RNA obtained from clinical patient samples or virus isolated and grown in embryonic culture 33, 34. However, PCR‐based detection of pathogenic microorganisms has its own difficulties and limitations. First, it is well known that the sensitivity and specificity of a PCR assay is highly dependent on the sequence of the target gene(s), primer sequences, DNA extraction procedures, and the specific PCR techniques and PCR product detection methods employed. Also, standard methods of PCR do not represent a quantitative analysis of the nucleic acid template. To solve this latter problem, quantitative, fluorescence‐based real‐time PCR assays (often referred to as qPCR or RT‐PCR) have been developed in different formats 27, 28, 35. In quantitative PCR techniques, quantification is determined based upon the number of amplification cycles necessary to generate a fluorescent signal intensity high enough to be significantly above the background, that is, a signal threshold. The number of cycles required to reach the signal threshold is often referred to as the Ct or Cq (“t” referring to threshold and “q” to quantitation). The relationship between the Ct value and the template concentration is inversed. Thus, a higher Ct value corresponds to a larger number of amplification cycles, indicating a lower initial concentration of template, and vice versa. The change in Ct (ΔCt) can be used to describe differences in template concentration when comparing samples Ct values to that of a known standard. These methods exhibit good sensitivity, broad dynamic range, and are capable of detecting all influenza virus subtype 9, 30, 33, 36. However, a PCR‐based assay that can be used to analyze multiple DNA/RNA amplicons, representing multiple influenza virus subtypes, in a large volume of samples had yet to be developed 34.
PCR “gene arrays,” (PCR methods based on the fluorescence‐based real‐time PCR (RT‐PCR) assay and intended to amplify multiple amplicons in parallel), are newer, miniaturized, and automatic techniques that provide a rapid, simultaneous analysis of multiple amplicons in large number of samples. Its outstanding feature is its integration of multiple PCR reactions in parallel, miniaturization (low reaction volumes), high throughput, and automated detection of fluorescent signal intensities after every amplification cycle and generation of amplification plots and calibration curves (for quantification) 37, 38. By detecting the fluorescent signal intensity of multiple amplicons simultaneously, an RT‐PCR array could determine the presence and abundance of low‐abundance DNA and/or RNA from various microorganisms simultaneously. This would allow researchers to distinguish between multiple potential pathogen species, strains and/or, subtypes that might be present in the samples in a single rapid, sensitive, and high‐throughput assay 28, 33, 37, 39. Therefore, this technology has great potential for epidemiological screening for microbial pathogens.
In this study, influenza type A virus specific primer/probe sets were designed for the rapid detection of influenza virus A, while specific primer pairs and fluorogenic probes for the identification of zoonotic viruses, such as the zoonotic influenza virus H1N1, H5N1, and H9N2 subtypes, were also designed to create a novel, one‐step, real‐time PCR array system, which could be used for qualitative and quantitative analysis of zoonotic influenza viral subtypes in a simple experiment and that could provide a highly specific, highly sensitive method for zoonotic influenza virus detection.
MATERIALS AND METHODS
Viruses and Samples
The reference‐laboratory strains used in this study were obtained from the Chinese Centers for Disease Control and Prevention (CDC), in the FuJian province of China, including zoonotic influenza virus subtypes (H1N1, H5N1, and H9N2), endemic seasonal influenza virus subtypes (parainflueza/influenza A/H1N1, influenza A/H3N2, influenza B/Victoria, and influenza B/Yamagata), Legionella pneumophila, Brucella species, and Streptococcus suis. RNA/DNA was extracted from each sample using silicon‐based spin columns, according to the manufacturer's instruction. Briefly, 140 μl of clinical samples was loaded on each spin column for RNA/DNA isolation, and the RNA/DNA bound to the spin column was eluted with 40 μl of elution buffer.
Primers and Probes
All virus primers and/or probes were designed by using the published sequences of the endemic seasonal influenza viruses and zoonotic influenza virus subtypes (the latter being defined in this study as H1N1, H5N1, and H9N2). To assure specificity of the primers, an alignment of similar genes from related organisms was performed using National Center for Biotechnology Information's Basic Local Alignment Search Tool (BLAST®) queries to the GenBank database. Two primer and probe sets for each virus (Table 1) were designed using the Primer 3.0 or Primer 5.0 software. In our study, the best possible PCR primers and probes had been designed through a series of strict principles. To allow for using a single annealing temperature for every well in each PCR array, only primer pairs with similar GC contents, melting temperature (Tm), and other chemical and physical properties are used, and all of the amplified fragments were limited to 95–150 bp. Detection probes were labeled with FAM at 5′ and BHQ at 3′ (Table 1).
Table 1.
Sequences of Primers and Probes used in rRT‐PCR Array
| Target/gene | Oligonucleotide | Nucleotide sequence (5′→3′) | Amplicon size (bp) |
|---|---|---|---|
| HA | HA‐f | CTTGTCTTTAGCCATTCCATGAG | 140 |
| HA‐r | CTTCTAACCGAGGTCGAAACGT | ||
| HA‐p | (FAM)‐CCTCTAGATCGGTGTTCTTCCCTGCA‐(BHQ) | ||
| H1N1/NA | H1N1‐f | TCAGAATGCAGATACATGTTTTTGT | 126 |
| H1N1‐r | CATATGTCTGATTTACCCAAGTGTT | ||
| H1N1‐p | (FAM)‐TACAGCAAGAAGTTCAAGCCGGAAATAGC‐(BHQ) | ||
| H5/HA | H5‐f | ACGGCCTCAAACTGAGTGTTC | 89 |
| H5‐r | GCAGACAAAGAATCCACTCAAA | ||
| H5‐p | (FAM)‐CAATAGATGGAGTCACCAATAAGGTCAAC‐(BHQ) | ||
| H9/HA | H9‐f | CAGATTTGTTGCACACAGCATC | 110 |
| H9‐r | GCTACCAGTCAACAAACTCCACAG | ||
| H9‐p | (FAM)‐CCCATTGTGCTCTGTGTGGAGCAATTC‐(BHQ) |
Forward (f) and reverse (r) primers and probes (p) are indicated.
Detection probes were labeled with FAM at 5′ and BHQ at 3′.
Description of the Zoonotic Influenza Virus Real‐Time PCR Array
Since nucleic acid detection is the only available method for the determination of suspected zoonotic influenza virus cases at present, the array was designed to validate and enhance ability of detecting zoonotic influenza virus subtypes H1N1, H5N1, amd H9N2, while avoiding misdiagnosis of acute respiratory infections (ARI) caused by other Influenza A virus subtypes and other pathogens. Target‐specific primer pairs and probes were added to appropriate PCR wells, as intended to detect their respective targets. Also added to each well was a set of universal primers and probe specific for a region of HA gene that is strictly conserved for most influenza A sequences available, as well as sets of primers and probes for H1N1, H5, and H9 serum subtypes. The four‐tube assay also took into consideration the specificity and sensitivity of the tests. The first tube contained primers for the influenza A HA gene to screen for the presence of any zoonotic influenza virus subtype, while the other three tubes engaged sets of primers and probes for the H1N1, H5N1, and H9N2 subtypes to screen for these zoonotic influenza virus subtypes. We optimized the assay to use a single, uniform set of real‐time PCR conditions to achieve the high levels of sensitivity, specificity, and reproducibility expected of real‐time PCR for each target in the zoonotic influenza virus RT‐PCR array.
Real‐Time PCR (RT‐PCR) Conditions
Total RNA was extracted using an E.Z.N.A.® Viral RNA Kit (Omega, Guangzhou, China) in accordance with the manufacturer's protocol. Purified RNA was frozen at −80°C in aliquots. One‐step real‐time PCR (RT‐PCR) analysis was performed using Applied Biosystems 7500 Real‐time PCR System. RT‐PCR reactions were performed in capillary tubes in 25 μl volumes with 10 pM primers and 5 pM probe each reaction. The reaction mixture, containing reaction buffer, MgCl2 buffer, dNTPs, DTT, RNase‐free H2O, and M‐MLV and Hotstart Taqman® DNA polymerase, was incubated with 5 μl RNA sample per capillary tube. In vitro transcribed RNA virus particles containing the target sequence were introduced as the positive control sample and H2O was utilized as negative control sample. Before DNA amplification cycles were begun, the RNA templates were reverse transcribed to produce the corresponding dsDNA templates during a two‐step reverse transcription procedure: 40°C for 15 min and 50°C for 30 min. DNA templates for the gene targets were then subjected to an initialization step (1 cycle at 94°C for 10 min), followed by 40 cycles of amplification: denaturation at 94°C for 30 s and annealing and elongation at 58°C for 45 s.
In Vitro Nucleic Acid Synthesis and Standard RNA Virus Standard Production
To validate the zoonotic influenza virus RT‐PCR array, RNA virus standards were constructed by inserting a synthetic nucleic acid fragment containing the target sequences into pLNCX, a retroviral expression vector, according to the manufacturer's instructions (Clontech). An invalid coding fragment was also cloned to pLNCX as the negative control. Gene sequence was confirmed by gene sequencing (Applied Biosystems, USA). The plasmid pLNCX‐flu and pLNCX‐NA was propagated in Escherichia coli DH5α cells, and serial ten‐fold dilutions were made to obtain samples containing 108–100 plasmids per microliter. These dilutions were performed both to generate the standard curve for the real‐time PCR and to evaluate the titer of the recombination virus.
To create a cell line that could generate the virus‐like particles containing the target sequence stably, the pLNCX‐flu and pLNCX‐NA were co‐transfected in GP2–293 cells with pVSVG vector, and the cells were cultured with G418 selective medium for 7 days. The genes encoding the viral gag and pol proteins are stably integrated into GP2–293., but because the VSV‐G envelope protein causes toxicity by fusing cellular membranes, it must be expressed transiently from pVSV‐G during viral packaging. Virus production was confirmed by PCR and RT‐PCR 48 hours later, and the virus titer was evaluated once a week for three months by comparison of a standard curve generated by real‐time PCR using the diluted plasmid as the template.
Validation of the Sensitivity, Reproducibility, and Specificity of the Assay
It is important to address the zoonotic influenza virus RT‐PCR array's specificity and sensitivity with samples obtained from natural specimens. The laboratory influenza virus isolates of zoonotic influenza virus subtypes H1N1, H5N1, H9N2 were detectable using the zoonotic influenza virus RT‐PCR array. The specificity of the zoonotic influenza virus RT‐PCR array was confirmed by direct sequencing of the PCR amplicons from the positive controls (obtained from reference laboratory influenza A virus isolates of zoonotic influenza virus subtypes H1N1, H5N1, and H9N2).
To test the specificity of the array, the primers and probes for zoonotic influenza virus were tested using real‐time PCR under optimal conditions with templates containing nucleic acid sequences from other pathogens that typically cause ARI, such as isolates of endemic seasonal influenza viruses in China (A/N1N1, A/H3N2, B/Victoria, and B/Yamagata), L. pneumophila, Brucella species, and S. suis PCR amplicons obtained with these isolates were also verified by DNA sequencing. H2O was utilized as a negative control.
To establish the limit of quantization (LOQ) of the assay, recombinant virus‐like particles (RVLPs) containing 106, 105, 104, 103, 102, and 101 copies RNA per sample were used as standard samples and run in triplicate. Samples containing 10 copies (101) and one copy per sample (100) were also tested to estimate the limit of detection (LOD) of the assay. We also compared the sensitivity of the combined zoonotic influenza virus RT‐PCR “array” technique with those obtained using individual real‐time PCR (IRTP) or standard PCR techniques.
RNA virus standards with 107, 105, and 103 copies per sample were used to evaluate the coefficients of variation (CVs) of the zoonotic influenza virus RT‐PCR array and IRTP techniques. Intra‐ and interassay CVs for Ct values were both included.
Data Analysis
The data and standard curves were obtained with in vitro transcribed RNA virus particles containing the target sequence. Raw data were exported from the ABI 7500 software v.2.0.5 into Microsoft Excel 2007 (Microsoft, Redmond, Washington, USA) for graphical and statistical analysis. Results were expressed as means (average) and standard deviation (SD). The intra‐ and interassay variations were expressed as CV, based on the mean Ct values. Standard curve analysis was performed to determine the LOD. The LOD was determined as 95% probability of obtaining a positive result. Correlation coefficients (R) were calculated for linearity data.
RESULTS
Standard RNA Virus Like Particle Preparation
To validate the extraction and/or PCR procedures, in vitro transcribed RNA virus particles containing the target sequence were introduced into the array and H2O was utilized as a blank control (Fig. 1). DNase was added to RNA virus particles prior to RT‐PCR to avoid DNA contamination. The standards for each assay were prepared in duplicate to assess self‐reproducibility. The reaction condition of one step real‐time RT‐PCR was optimal and uniformed to detection of three kinds of high pathogenic AIV through an experiment. Ct values for the four zoonotic influenza virus positive controls ranged from 12 to 20, while Ct values for the negative control approached 40 cycles (i.e., practically undetectable).
Figure 1.
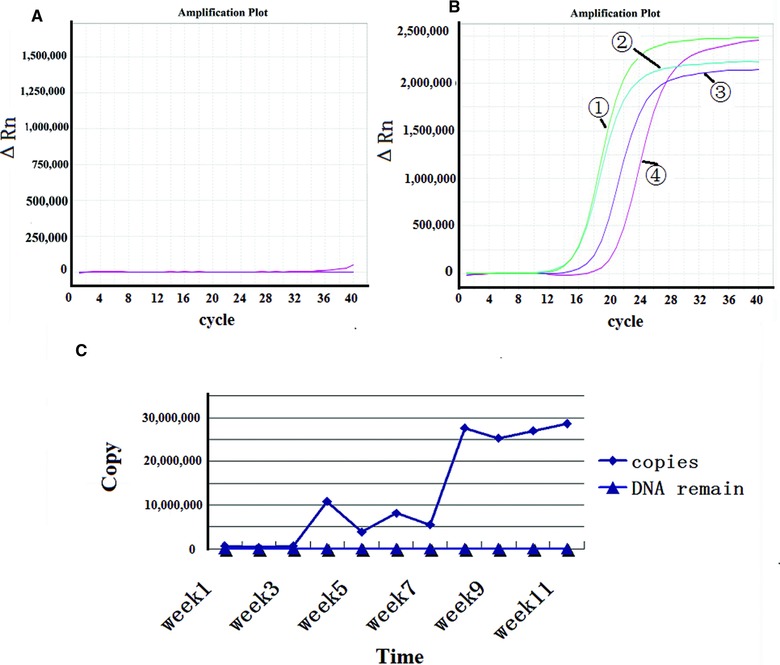
Identification of recombination RNA‐virus‐like particles. (A) PCR results for RNA virus like particles. (B) RT‐PCR results for RNA virus like particles. ①‐HA gene for all influenza A virus, ②‐H1N1subtype, ③‐H5N1 subtype, and ④‐H9N2 subtype. (C) The virus titer was validated by real‐time PCR with standard curve, and the virus titer were almost 107 copies per milliliter.
Sensitivity
In order to optimize the sensitivities of the zoonotic influenza virus, RT‐PCR array, recombinant RNA virus particles (rRVPs) containing the target sequences were prepared and tested by using the zoonotic influenza virus RT‐PCR array. The LOD was calculated using serial 10‐fold dilutions of the rRVP standard. Figure 2 presents the experiments that indicate a linear correlation between the Log (base 10) of the gene copy number and the Ct, with a regression line showing a slope of −3.43 (R 2 = 0.998), −3.468(R 2 = 1), −3.747 (R 2 = 0.966), −3.274 (R 2 = 0.965) for HA, H1N1, H5N1, and H9N2, respectively. The LOD of the rRVP standards corresponded to 101 or 100 RNA copies per reaction (Table 2). These results demonstrated that the zoonotic influenza virus RT‐PCR array demonstrated high sensitivity in detecting all four zoonotic influenza virus subtypes tested. We also compared the sensitivity of our zoonotic influenza virus RT‐PCR array with results using the same primers, probes, and templates using IRTP and standard PCR techniques. The sensitivity of standard PCR techniques was about 102 or 103 copies per reaction. Thus, the zoonotic influenza virus RT‐PCR array had 10‐fold advantage in sensitivity of zoonotic influenza virus detection compared to standard PCR. No significance has been observed between our zoonotic influenza virus RT‐PCR array compared with individual real‐time RT‐PCR results for the zoonotic influenza virus H1N1, H5N1, H9N2 subtypes.
Figure 2.
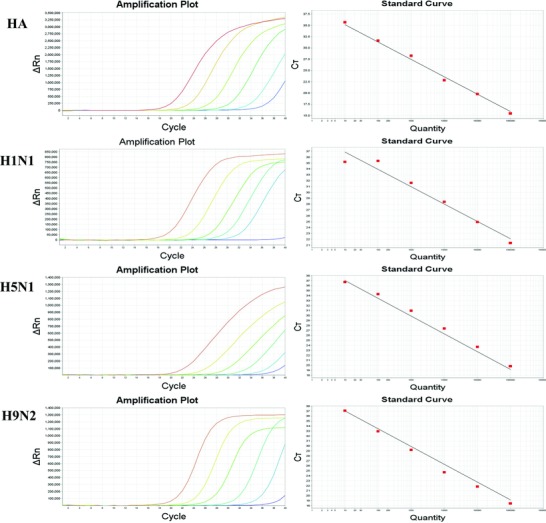
Standard curve of each virus of the RT‐PCR array. Tenfold dilution series of RNA virus like particles (from 105–101 copies/μl) was plotted against the threshold cycle. The coefficient of determination (R2) and equation of regression curve (y) calculated.
Table 2.
Validation of rRT‐PCR Array Sensitivity
| Ct value | ||||
|---|---|---|---|---|
| Assay | Number of RNA (copies/reaction tube) | RT‐PCR array | Individual RT‐PCR | Standard PCR |
| HA | 100,000 | 21.79 | 21.19 | + |
| 10,000 | 25.54 | 26.07 | + | |
| 1,000 | 30.78 | 29.77 | + | |
| 100 | 34.16 | 33.84 | − | |
| 10 | 37.67 | 37.23 | − | |
| 1 | − | − | − | |
| H1N1 | 100,000 | 19.50 | 19.77 | + |
| 10,000 | 22.50 | 22.61 | + | |
| 1,000 | 25.13 | 26.48 | + | |
| 100 | 28.34 | 28.30 | − | |
| 10 | 35.37 | 34.97 | − | |
| 1 | − | − | − | |
| H5N1 | 100,000 | 21.77 | 21.46 | + |
| 10,000 | 25.33 | 25.43 | + | |
| 1,000 | 29.13 | 29.38 | + | |
| 100 | 33.54 | 32.73 | + | |
| 10 | 37.08 | 36.36 | − | |
| 1 | − | − | − | |
| H9N2 | 100,000 | 19.97 | 20.14 | + |
| 10,000 | 23.28 | 23.49 | + | |
| 1,000 | 27.33 | 27.41 | + | |
| 100 | 31.24 | 31.38 | − | |
| 10 | 36.04 | 35.18 | − | |
| 1 | − | − | − | |
Positive (+) and negative detections (−) are indicated
Cross‐Reactivity and Specificity Testing
The specificity of our zoonotic influenza virus RT‐PCR array was examined with the reference laboratory influenza virus isolates of zoonotic influenza virus (H1N1, H5N1, and H9N2) and other pathogens that are also known to cause ARI, including influenza virus isolates of endemic seasonal influenza viruses in China (A/N1N1, H3N2, B/Victoria, and B/Yamagata), L. pneumophila, Brucella species, and S. suis from the Sino Centers for Disease Control and Prevention (CDC), in the FuJian province of China. No cross‐reactivity had been observed in the array as shown in Table 3. Three reference laboratory clinical samples for H1N1, H5N1, and H9N2 virus, respectively, were used to validate the RT‐PCR array system, All samples also showed positive results when tested using the zoonotic influenza virus RT‐PCR array platform. The specificity of the zoonotic influenza virus RT‐PCR array was also confirmed by direct sequencing of the PCR amplicons from each assay well. BLAST analysis of the PCR products confirmed that the amplicons of all assays were specific for their intended targets (data not shown). These results indicated that the ability to detect mixed zoonotic influenza virus infections is valuable.
Table 3.
Validation of rRT‐PCR and AIV RT‐PCR Array Specificities to Different Pathogens
| AIV RT‐PCR array results | |||||
|---|---|---|---|---|---|
| Pathogen isolate | Serum subtype | HA | H1N1 | H5N1 | H9N2 |
| Influenza | |||||
| A/Swine /Shanghai/07/1999 | H1N1 | + | + | − | − |
| A/Chiken/Fujian/1/2001 | H5N1 | + | − | + | − |
| A/Duck/Fujian/107/2007 | H9N2 | + | − | − | + |
| A/ Swine/Fujian/ 01/ 2009 | H1N1 | + | − | − | − |
| A/Chiken/Yunnan/1145/2005 | H3N2 | + | − | − | − |
| B(Victoria)/Shanghai/361/2002 | NA | − | − | − | − |
| B(Yamagata)/Zhejiang/02/2002 | NA | − | − | − | − |
| L. pneumophila/Fujian/05/2005 | NA | − | − | − | − |
| Brucella melitensis/ Fujian /06/2009 | NA | − | − | − | − |
| Streptococcus suis/Fujian/07/2010 | NA | − | − | − | − |
+, positive detection; −, negative; NA, Not Applicable.
Reproducibility
The intra‐ and inter‐assay coefficients of variation (CV) were calculated using three dilutions of rRVP, corresponding to 107, 105, and 103 copies per sample, in order to simulate positive samples with a wide range of concentrations (Table 4 presents the copy number, Ct values, intraassay CV, and interassay CV based on the optimized primers and probes used in our zoonotic influenza virus RT‐PCR array. The SD and CV values for detection of zoonotic influenza virus subtypes H1N1, H5N1, and H9N2 were in triplicate by real‐time PCR, revealing that the our zoonotic influenza virus RT‐PCR array was reliable and accurate.
Table 4.
Intra‐ and Interassay Reproducibility of the rRT‐PCR and AIV RT‐PCR Arrays
| Intraassay | Interassay | ||||||
|---|---|---|---|---|---|---|---|
| Assay | Number of RNA copies | Mean Ct | SD | CV (%) | Mean Ct | SD | CV (%) |
| HA | 106 | 18.95 | 0.325 | 1.71 | 19.31 | 0.629 | 3.26 |
| 104 | 27.78 | 0.241 | 0.87 | 27.62 | 0.617 | 2.23 | |
| 102 | 34.88 | 0.096 | 0.28 | 34.86 | 0.181 | 0.51 | |
| H1N1 | 106 | 15.86 | 0.341 | 2.15 | 15.98 | 0.235 | 1.47 |
| 104 | 22.50 | 0.401 | 1.78 | 23.06 | 0.742 | 3.21 | |
| 102 | 28.30 | 0.453 | 1.6 | 29.13 | 0.979 | 3.36 | |
| H5N1 | 106 | 18.08 | 0.118 | 0.65 | 18.81 | 0.631 | 3.35 |
| 104 | 25.33 | 0.278 | 1.1 | 25.62 | 0.625 | 2.44 | |
| 102 | 32.20 | 0.780 | 2.42 | 33.22 | 0.792 | 2.38 | |
| H9N2 | 106 | 21.61 | 0.306 | 1.42 | 21.226 | 0.908 | 4.28 |
| 104 | 28.77 | 0.158 | 0.55 | 28.559 | 0.580 | 2.03 | |
| 102 | 35.77 | 0.173 | 0.48 | 35.556 | 0.300 | 0.84 | |
DISCUSSION
The epidemics of zoonotic influenza in Asia and more recently, in some parts of Europe, have caused considerable public concern and raised the need for a means of rapid determination of viruses subtypes in cases of zoonotic disease outbreaks (1, 8).
PCR arrays are the most reliable—and most sensitive—tools available for analyzing the presence and abundance of various specific microorganisms in a focused, multi‐targeted panel 33, 34. High‐quality primer/probe design and master mix formulations enable the RT‐PCR array format to amplify multiple gene‐specific products simultaneously under uniform cycling conditions. In this report, we describe the development of a novel zoonotic influenza virus detection method, based on real‐time PCR array using Taqman® polymerase. that enabled multiple zoonotic influenza virus subtypes targets to be distinguished simultaneously in a single assay (36, 40).
Successful design of a PCR array requires selecting the primers and probes capable of being used with the same, uniform PCR amplification conditions. First, we designed several sets of universal primers and probes specific for the influenza virus HA gene. Second, we designed distinct sets of primers and probes for conserved regions of three zoonotic influenza virus subtypes (H1N1, H5N1, and H9N2). Finally, we optimized the real‐time quantitative PCR array system by choosing primers and probes sets that were compatible for use under uniform PCR conditions.
To achieve reliable results of the PCR detection, a strict quality control standard for quantitative determination of the virus is extremely important. Currently, the fluorescent quantitative RT‐PCR detection of RNA virus is always evaluated by using the plasmid, RNA fragment as standard. While the fatal disadvantage of these kinds of standards was either unstable or there was failure in monitoring the virus lysis and transcript effect of the RNA 40, 41, 42, 43.
Therefore, the second purpose of this study is to create an appropriate standard to validate the transcription and amplication of zoonotic influenza virus RNA sequences in the RT‐PCR reaction. In this report, the target sequences from all viruses packaged in recombinant virus‐like particles (rRVPs) could also be contributed by PCR amplification of the corresponding DNA fragment. However, the original template DNA concentration in the supernatant is very low cell culture and was easily removed in our study by the addition of DNase prior to PCR amplification. Our assay was easily adapted to a quantitative format, by using rRVPs to create a standard curve to which results from unknown samples could be compared.
We also used these rRVPs as standard to evaluate the sensitivity of our RT‐PCR array platform compared with IRTP or standard PCR techniques. Using the rRVP standards, the detection limits for zoonotic influenza A virus subtypes H1N1, H5N1, and H9N2 in this RT‐PCR arrays were validated and ranged from 100–101 copies/ml. These limits were similar to that previously published by others using IRTP assays. We used an IRTP kit (Daan Gene Co., Guangzhou, China) and standard PCR methods to detect the same target of same standard sample. The results indicate that no difference of the sensitivity had been observed between IRTP and our array, whereas it is much better than the standard PCR.
These results indicate our real‐time RT‐PCR array detected three different subtypes of a highly pathogenic zoonotic influenza virus in a simple assay at the same level in sensitivity and specificity as the IRTP.
The suitability of the zoonotic influenza viruses RT‐PCR array test described in this study as a diagnostic and epidemiological tool for zoonotic influenza viruses detection was confirmed by testing isolated strains and clinical samples. The RNA virus standard with 107, 105, and 103 copies was used to evaluate the reliability of the zoonotic influenza viruses RT‐PCR array. Results indicate that our test has a good reproducibility, as shown by a low inter‐ and intraassay CV.
In summary, our zoonotic influenza viruses RT‐PCR array could offer an efficient, flexible, and reliable platform for identification of several zoonotic influenza viruses subtypes (H1N1, H5N1, and H9N2) in a single assay. The addition of an RNA virus‐like particle control would further improve the assay as a diagnostic tool, as each sample could be checked for the quality of the nucleic acid extraction, transcription, and PCR performance.
CONFLICT OF INTEREST
No conflict of interest exits in the submission of this article, and the article was approved by all authors for publication.
ACKNOWLEDGMENTS
This study was sponsored by the following foundation programs: National Science and Technology Major Project of China (2009ZX10601).
Grant sponsor: National Science and Technology Major Project of China; grant number: 2009ZX10601
REFERENCES
- 1. Alexander DJ. Avian influenza viruses and human health. Dev Biol (Basel) 2006;124:77–84. [PubMed] [Google Scholar]
- 2. Capua I, Alexander DJ. Human health implications of avian influenza viruses and paramyxoviruses. Eur J Clin Microbiol` Infect Dis 2004;23(1):1–6. [DOI] [PubMed] [Google Scholar]
- 3. Cong Y, Wang G, Guan Z, et al. Reassortant between human‐like H3N2 and avian H5 subtype influenza A viruses in pigs: A potential public health risk. PLoS ONE 2010;5(9):e12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hrnjakovic‐Cvjetkovic I, Cvjetkovic D, Jerant‐Patic V, et al. [Avian influenza viruses–new causative agents of human infections]. Med Pregl 2006;59(1–2):29–32. [DOI] [PubMed] [Google Scholar]
- 5. Kaye D, Pringle CR. Avian influenza viruses and their implication for human health. Clin Infect Dis 2005;40(1):108–112. [DOI] [PubMed] [Google Scholar]
- 6. McCaughey C. Influenza: A virus of our times. Ulster Med J 2010;79(2):46–51. [PMC free article] [PubMed] [Google Scholar]
- 7. Schulze M, Nitsche A, Schweiger B, Biere B. Diagnostic approach for the differentiation of the pandemic influenza A(H1N1)v virus from recent human influenza viruses by real‐time PCR. PLoS ONE 2010;5(4):e9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Capua I, Alexander DJ. Ecology, epidemiology and human health implications of avian influenza viruses: Why do we need to share genetic data? Zoonoses Public Health 2008;55(1):2–15. [DOI] [PubMed] [Google Scholar]
- 9. He J, Bose ME, Beck ET, et al. Rapid multiplex reverse transcription‐PCR typing of influenza A and B virus, and subtyping of influenza A virus into H1, 2, 3, 5, 7, 9, N1 (human), N1 (animal), N2, and N7, including typing of novel swine origin influenza A (H1N1) virus, during the 2009 outbreak in Milwaukee, Wisconsin. J Clin Microbiol 2009;47(9):2772–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ison MG, Lee N. Influenza 2010–2011: Lessons from the 2009 pandemic. Cleve Clin J Med 2010;77(11):812–820. [DOI] [PubMed] [Google Scholar]
- 11. Van Kerkhove MD, Mumford E, Mounts AW, et al. Correction: Highly pathogenic avian influenza (H5N1): Pathways of exposure at the animal‐human interface, a systematic review. PLoS ONE;2012;6(1):e14582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shu B, Wu K‐H, Emery S, et al. Design and performance of the CDC real‐time reverse transcriptase PCR swine flu panel for detection of 2009 A (H1N1) pandemic influenza virus. J Clin Microbiol 2011;49(7):2614–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu ZQ, Ji J, Zuo KJ, et al. Cloning and phylogenetic analysis of hemagglutinin gene of H9N2 subtype avian influenza virus from different isolates in China during 2002 to 2009. Poult Sci 2010;89(6):1136–1143. [DOI] [PubMed] [Google Scholar]
- 14. Van Kerkhove MD, Mumford E, Mounts AW, et al. Highly pathogenic avian influenza (H5N1): Pathways of exposure at the animal‐human interface, a systematic review. PLoS ONE 2011;6(1):e14582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bateman AC, Kieke BA, Irving SA, Meece JK, Shay DK, Belongia EA. Effectiveness of monovalent 2009 pandemic influenza A virus subtype H1N1 and 2010–2011 trivalent inactivated influenza vaccines in Wisconsin during the 2010–2011 influenza season. J Infect Dis 2013;207(8):1262–1269. [DOI] [PubMed] [Google Scholar]
- 16. Moore CL, Smagala JA, Smith CB, et al. Evaluation of MChip with historic subtype H1N1 influenza A viruses, including the 1918 “Spanish Flu” strain. J Clin Microbiol 2007;45(11):3807–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haque N, Bari MS, Bilkis L, et al. Swine flu: A new emerging disease. Mymensingh Med J 2010;19(1):144–149. [PubMed] [Google Scholar]
- 18. Figuie M. Global health risks and cosmopolitisation: From emergence to interference. Sociol Health Illn 2013;35(2):227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abadia G, Hars J. [Prevention of avian flu]. Influenza aviaire: un point d'actualite. Rev Prat 2007;57(11 Suppl):33–36. [PubMed] [Google Scholar]
- 20. Heeney JL. Zoonotic viral diseases and the frontier of early diagnosis, control and prevention. J Intern Med 2006;260(5):399–408. [DOI] [PubMed] [Google Scholar]
- 21. Lu C‐y, Lu J‐h, Chen W‐q, et al. Potential infections of H5N1 and H9N2 avian influenza do exist in Guangdong populations of China. Chin Med J (Engl) 2008;121(20):2050–2053. [PubMed] [Google Scholar]
- 22. Liu Y, Li Q, He Y‐X, et al. [The firstly confirmed pregnant woman case of avian influenza A (H5N1) by etiological research in China]. Bing Du Xue Bao 2007;23(6):429–433. [PubMed] [Google Scholar]
- 23. Ben Shabat M, Meir R, Haddas R, et al. Development of a real‐time TaqMan RT‐PCR assay for the detection of H9N2 avian influenza viruses. J Virol Methods 2010;168(1–2):72–77. [DOI] [PubMed] [Google Scholar]
- 24. Kang W, Pang W, Hao J, Zhao D. Isolation of avian influenza virus (H9N2) from emu in China. Ir Vet J 2006;59(3):148–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wong SSY, Yuen K‐Y. Avian influenza virus infections in humans. Chest 2006;129(1):156–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bertran K, Perez‐Ramirez E, Busquets N, et al. Pathogenesis and transmissibility of highly (H7N1) and low (H7N9) pathogenic avian influenza virus infection in red‐legged partridge (Alectoris rufa). Vet Res 2011;42(1):24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakurai A, Shibasaki F. Updated values for molecular diagnosis for highly pathogenic avian influenza virus. Viruses 2012;4(8):1235–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pasick J. Advances in the molecular based techniques for the diagnosis and characterization of avian influenza virus infections. Transbound Emerg Dis 2008;55(8):329–338. [DOI] [PubMed] [Google Scholar]
- 29. Coonrod JD, Karathanasis P, Betts RF, Donofrio JC. Enzyme‐linked immunosorbent assay of core antigens for clinical diagnosis of influenza. J Med Virol 1988;25(4):399–409. [DOI] [PubMed] [Google Scholar]
- 30. Atmar RL, Baxter BD, Dominguez EA, Taber LH. Comparison of reverse transcription‐PCR with tissue culture and other rapid diagnostic assays for detection of type A influenza virus. J Clin Microbiol 1996;34(10):2604–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rowe T, Abernathy RA, Hu‐Primmer J, et al. Detection of antibody to avian influenza A (H5N1) virus in human serum by using a combination of serologic assays. J Clin Microbiol 1999; 37(4):937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meseko CA, Oladokun AT, Ekong PS, et al. Rapid antigen detection in the diagnosis of highly pathogenic avian influenza (H5N1) virus in Nigeria. Diagn Microbiol Infect Dis 2010;68(2):163–165. [DOI] [PubMed] [Google Scholar]
- 33. Li J, Chen S, Evans DH. Typing and subtyping influenza virus using DNA microarrays and multiplex reverse transcriptase PCR. J Clin Microbiol 2001;39(2):696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schweiger B, Zadow I, Heckler R, Timm H, Pauli G. Application of a fluorogenic PCR assay for typing and subtyping of influenza viruses in respiratory samples. J Clin Microbiol 2000;38(4):1552–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Romero JR, Newland JG. Diagnosis of viral encephalitides: Zoonotic‐associated viruses. Pediatr Infect Dis J 2006;25(8):741–742. [DOI] [PubMed] [Google Scholar]
- 36. Wong ML, Medrano JF. Real‐time PCR for mRNA quantitation. Biotechniques 2005;39(1):75–85. [DOI] [PubMed] [Google Scholar]
- 37. Belgrader P, Benett W, Hadley D, et al. Rapid pathogen detection using a microchip PCR array instrument. Clin Chem 1998;44(10):2191–2194. [PubMed] [Google Scholar]
- 38. Chou P, Milne TJ. Real‐time PCR focused‐gene array profiling of gingival and periodontal ligament fibroblasts. Methods Mol Biol 2010;666:373–383. [DOI] [PubMed] [Google Scholar]
- 39. Pierik A, Zwanenburg C, Moerland E, Broer D, Stapert H, van den Brule AJC. Rapid genotyping of human papillomavirus by post‐PCR array‐based hybridization techniques. J Clin Microbiol 2011;49(4):1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. WalkerPeach CR, Winkler M, DuBois DB, Pasloske BL. Ribonuclease‐resistant RNA controls (Armored RNA) for reverse transcription‐PCR, branched DNA, and genotyping assays for hepatitis C virus. Clin Chem 1999;45(12):2079–2085. [PubMed] [Google Scholar]
- 41. Yoo SH, Hong SH, Jung SR, et al. Application of bovine viral diarrhoea virus as an internal control in nucleic acid amplification tests for hepatitis C virus RNA in plasma‐derived products. J Microbiol 2006;44(1):72–76. [PubMed] [Google Scholar]
- 42. Jorgensen PA, Neuwald PD. Standardized hepatitis C virus RNA panels for nucleic acid testing assays. J Clin Virol 2001;20(1–2):35–40. [DOI] [PubMed] [Google Scholar]
- 43. Hou Y, Zhang H, Miranda L, Lin S. Serious overestimation in quantitative PCR by circular (supercoiled) plasmid standard: Microalgal pcna as the model gene. PLoS ONE 2010;5(3):e9545. [DOI] [PMC free article] [PubMed] [Google Scholar]